

A taxia Telangiectasia Mutated (ATM) kinase co-ordinates a wide spectrum of cellular processes through the phosphorylation of numerous substrates. ATM, which is implicated in cellular response to DNA double strand breaks (DSBs), plays a crucial role in anti-cancer DNA damage response (DDR) pathway, but also represents a key sensor to oxidative stress1 (Figure 1). Germinal ATM mutations lead to recessive inherited Ataxia telangiectasia syndrome characterized by neurodegenerative and immunological symptoms and an increased risk of tumor development.2 Somatic ATM inactivation is frequently found in lymphoproliferative diseases, contributing to elevated genomic instability and resulting in the defective activation of the p53 pathway and poor patient outcome. Thus, ATM defects represent an important prognostic and potentially predictive marker in chronic lymphocytic leukemia (CLL). Finally, ATM dysfunction also offers the opportunity to develop novel therapeutic strategies.
Figure 1.
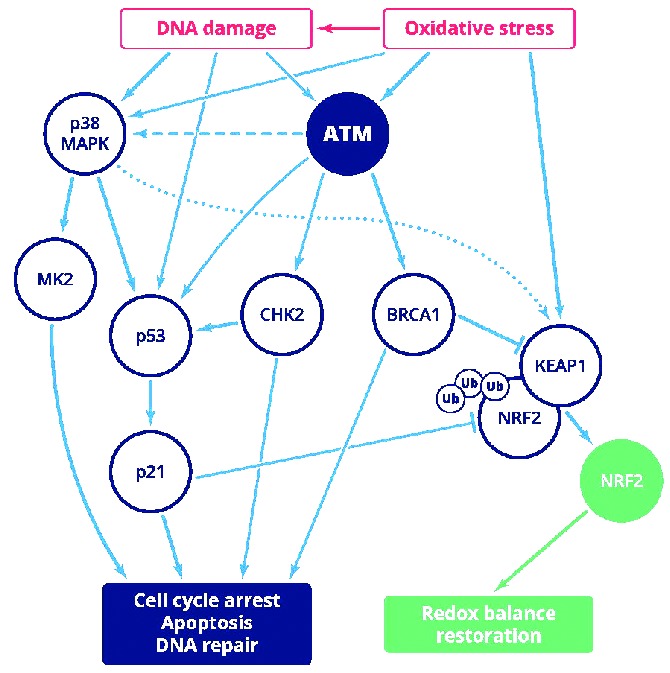
Schematic depiction of overlapping ATM role in DNA damage and oxidative stress response showing key molecules and cellular consequences. NRF2 activation could be ATM dependent or independent: ROS are able to directly activate NRF2 signaling via cysteine oxidation of NRF2 negative regulator KEAP1; indirect activation of NRF2 involves ATM-mediated block of NRF2 degradation. The role of p38 MAPK in NRF2 regulation remains to be fully elucidated (spotted line). p38 MAPK-mediated MK2 activation seems to be ATM-dependent (dashed line). ATR-mediated signaling is not activated in predominantly resting CLL cells and therefore is not shown in the figure.
ATM gene disruption and its functional and clinical consequences in CLL
A high occurrence of ATM alterations have been reported in lymphoid malignancies such as CLL, T-cell prolymphocytic leukemia, mantle cell lymphoma, and diffuse large B-cell lymphoma. However, ATM mutation types and their localization are different among these neoplasms.3 In CLL, ATM defects (11q deletion and/or mutation) occur in approximately 25% of patients at diagnosis and therefore represent the most frequent aberration.4 Biallelic inactivation is typically found in the majority of cases and results in impaired DDR in vitro, while sole 11q deletion (11q-) preserves ATM function.5 Besides DDR impairment, other important processes have also been shown to be affected in ATM-defective CLL cells. Britt-Compton et al.6 observed extremely short telomeres in ATM mutated and 11q-deleted patient cells, indicating the impact of ATM functionality on telomere length. The role of ATM deficiency in oxidative stress response nowadays seems to have great potential in CLL research, especially due to its possible therapeutic perspective.
The clinical manifestation of ATM alterations in CLL patients depends on the defect composition and thus on its functional impact. Gene mutations accompanied by 11q- have been proved to significantly reduce the time to first treatment and overall survival compared to sole 11q-,4,7 and patients with biallelic defects experienced shorter progression-free survival compared to monoallelic defects.8 Moreover, the impact of monoallelic ATM loss is not uniform. While sole 11q- probably influences patient outcome through the haploinsufficiency of other genes located in the 11q22-23 region, most single ATM mutations can still retain partial ATM activity leading to a milder effect.8 In addition to reduced patient survival, biallelic ATM alterations also result in resistance to cytotoxic chemotherapy.7,8 Thus, it is highly desirable to find an efficient therapy for ATM-defective patients who are refractory to the treatment currently available. Novel therapies such as chemoimmunotherapy and BCR signaling inhibition improved patient outcome including those with 11q-.9,10 A clonal evolution study by Shuch et al.11 revealed that a subclone with ATM biallelic loss gained a proliferative advantage following DNA damaging treatment but was abolished after chemoimmunotherapy. Nevertheless, the long-term treatment efficacy in ATM-defective patients is not completely clear. The great challenge now is to develop therapies that exploit the weakness of CLL cells with inactive ATM, i.e. impaired DDR or redox homeostasis.
Synthetic lethal approach in the context of ATM deficiency
ATM together with another member of the phosphatidyl inositol 3-kinase-related kinases family, Ataxia Telangiectasia and Rad3-related (ATR) kinase, constitutes canonical DDR signaling pathways, which operate through downstream effector kinases CHK2 and CHK1, respectively.12 Upon DNA damage, the ATM/ATR pathways mediate cell cycle checkpoint activation, DNA repair, senescence or apoptosis. Another stress-response kinase MAPK14 (p38 MAPK), with its downstream effector MAPKAP Kinase-2 (MK2), was shown to be involved in DDR, mainly by activating ATM/ATR-dependent G2/M checkpoint signaling13 (Figure 1).
Individual DDR pathway components have been reported to exhibit synthetic lethal interactions; i.e. cells defective in one key DDR molecule might be particularly sensitive to another DDR factor abrogation (or might exhibit specific drug cytotoxicity) and may not be able to overcome “suprathreshold” levels of genotoxic stress, resulting in cell death.13 Multiple experimental studies, as well as clinical experience, revealed synthetic lethality interaction between the ATM/CHK2 pathway and p53 disruption. In their seminal paper, Jiang et al.14 provided evidence that, while isolated ATM or p53 loss promotes tumor cell chemoresistance, ATM or CHK2 inhibition in a p53-deficient background sensitizes tumor cells to DNA-damaging chemotherapy. Another well-documented protein with synthetic lethal interaction is non-homologous end-joining kinase DNA-PK, whose activity was shown to be essential for DNA DSBs repair in the context of ATM dysfunction. DNA-PK inhibition leads to the inability of ATM-deficient cells to cope with DSB induced by doxorubicin and this resulted in the death of these cells.14 Regarding CLL, poly-(ADP-ribose) polymerase (PARP) inhibitor olaparib has been shown to efficiently target ATM-deficient CLL cells due to their defect in DSB repair15 (Figure 2).
Figure 2.
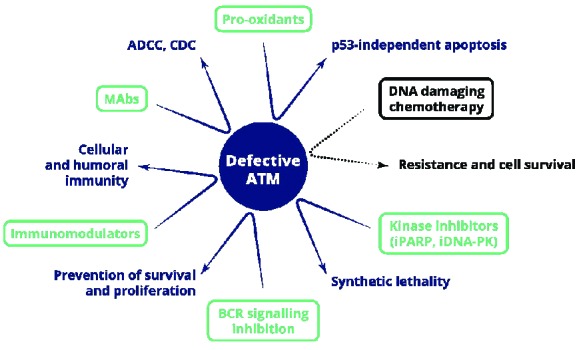
Potential therapeutic options for ATM-defective CLL patients with indicated mechanisms of action leading to p53-independent cell death. Ineffective therapy with DNA damaging chemotherapy is shown in black. MAbs: monoclonal antibodies; ADCC: antibody dependent cellular cytotoxicity; CDC: complement-dependent cytotoxicity; iPARP: inhibitor of poly (ADP-ribose) polymerase; iDNA-PK: inhibitor of DNA-dependent protein kinase.
The role of ATM in oxidative stress response: utilization in selective killing of ATM-defective CLL cells
Undoubtedly, reactive oxygen species (ROS) cause lipid, protein and DNA oxidation, contribute to genomic instability and additional mutation acquisition in cancer cells, and in that sense oxidants fuel tumor progression.16 On the other hand, increasing oxidative stress above a certain threshold may not be compatible with tumor cell survival and may result in cell death.17 In order to eliminate the detrimental effect of severe oxidative stress, cancer cell survival depends on the functional antioxidant defense involving ATM. Clear evidence for the role of ATM in the oxidative stress response comes from Ataxia telangiectasia patients with complete ATM inactivation who experience increased oxidative stress due to the elevated ROS level.1,18 Regarding the necessity to constantly regulate redox balance, an increase in oxidative stress of cancer cells renders them vulnerable to therapeutic interventions resulting in the further augmentation of ROS and oxidative damage.16 Collectively, these recent findings suggest that there is a possible therapeutic window for ATM-deficient CLL cells, in which a synthetic lethality-based approach can be exploited to selectively target these cells by escalating the oxidative stress to the level that induces cell death (Figure 2).
In this issue of Haematologica, Agathanggelou et al.19 propose a novel original p53 independent therapeutic strategy utilizing a defective antioxidant response in CLL patients with ATM inactivation. The authors demonstrated that pro-oxidant treatment (parthenolid or NAPQI) enhances oxidative stress resulting in cell death that bypasses the DDR pathway and acts in a p53 and caspase independent manner. They show that ATM-null cells are defective in NRF2 dependent antioxidant response, which results in their increased sensitivity to treatment with pro-oxidants both in vitro and in vivo. Indeed, these cells manifested a higher ROS level and simultaneously a lower antioxidant level. This strategy employs two main ATM roles that cannot be accomplished in ATM-null CLL cells, since ROS induce ATM-mediated DDR in addition to oxidative ATM response, which acts independently of DNA lesions (Figure 1). Another study indicates that bendamustine, used in CLL and other B-cell neoplasms, could serve as an alternative pro-oxidant drug because, in addition to the alkylating mechanism, it induces ROS production and p53-independent apoptosis.20 Nevertheless, further studies to confirm pro-oxidant effect on ATM-null cells in larger cohorts are warranted.
Future directions in targeting ATM-deficient CLL cells through elevated oxidative stress
Despite the indisputable importance and novelty of the investigation by Agathanggelou et al., 19 several issues need to be further clarified. One of the concerns is the pro-oxidant dosage to be used for selective ATM-null CLL cells targeting. In this study, pro-oxidants were deliberately used in levels below the limit known to cause oxidative damage in ATM-wild-type cells. However, this may be difficult to achieve consistently in clinical settings; therefore, a high dosage could lead to undesirable DNA damage and DDR activation in healthy cells. In this regard, the sensitivity to pro-oxidant therapeutics, as well as any possible side-effects, could also be influenced by additional ROS production triggered through ongoing inflammation in the CLL cell microenvironment, an aspect not addressed in the article by Agathanggelou et al. Another important question is how different CLL subclones existing in individual patients would respond to pro-oxidant therapy. Considering the fact that ROS and oxidative stress contribute to tumor development and progression, it is not clear how other co-existing clones harboring diverse genomic defects would react to the therapeutic augmentation of ROS. In some CLL subclones, increased oxidative stress resulting from pro-oxidant therapy could cause accelerated mutational rate and reinforced progression towards a more aggressive CLL. From a clinical point of view, it would be convenient to test pro-oxidants in combination with other p53-independent agents (e.g. monoclonal antibodies) to identify the most efficient treatment for ATM-defective CLL patients (potential therapeutic options are summarized in Figure 2). Collectively, the development of novel therapeutic strategies specifically targeting the ATM-null phenotype further emphasizes the importance of precise genetic characterization of CLL patients to allow the selection of individualized therapy.
Acknowledgements
The authors have been supported by research projects MSMT CR CZ.1.05/1.1.00/02.0068 (CEITEC) and CZ.1.07/2.3.00/30.0009 (SP, VN), and by grants IGA NT13576 from the Ministry of Health, Czech Republic (LRK, VD) and IGA MZ CR NT13493–4/2012 (SP).
Footnotes
Financial and other disclosures provided by the author using the ICMJE (www.icmje.org) Uniform Format for Disclosure of Competing Interests are available with the full text of this paper at www.haematologica.org.
References
- 1.Ambrose M, Gatti RA. Pathogenesis of ataxia-telangiectasia: the next generation of ATM functions. Blood. 2013;121(20):4036–4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meyn MS. Ataxia-telangiectasia, cancer and the pathobiology of the ATM gene. Clin Genet. 1999;55(5):289–304. [DOI] [PubMed] [Google Scholar]
- 3.Stankovic T, Stewart GS, Byrd P, Fegan C, Moss PA, Taylor AM. ATM mutations in sporadic lymphoid tumours. Leuk Lymphoma. 2002;43(8):1563–1571. [DOI] [PubMed] [Google Scholar]
- 4.Guarini A, Marinelli M, Tavolaro S, et al. ATM gene alterations in chronic lymphocytic leukemia patients induce a distinct gene expression profile and predict disease progression. Haematologica. 2012;97(1):47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Navrkalova V, Sebejova L, Zemanova J, et al. ATM mutations uniformly lead to ATM dysfunction in chronic lymphocytic leukemia: application of functional test using doxorubicin. Haematologica. 2013;98(7):1124–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Britt-Compton B, Lin TT, Ahmed G, et al. Extreme telomere erosion in ATM-mutated and 11q-deleted CLL patients is independent of disease stage. Leukemia. 2012;26(4):826–830. [DOI] [PubMed] [Google Scholar]
- 7.Austen B, Skowronska A, Baker C, et al. Mutation status of the residual ATM allele is an important determinant of the cellular response to chemotherapy and survival in patients with chronic lymphocytic leukemia containing an 11q deletion. J Clin Oncol. 2007;25(34):5448–5457. [DOI] [PubMed] [Google Scholar]
- 8.Skowronska A, Parker A, Ahmed G, et al. Biallelic ATM inactivation significantly reduces survival in patients treated on the United Kingdom Leukemia Research Fund Chronic Lymphocytic Leukemia 4 trial. J Clin Oncol. 2012;30(36):4524–4532. [DOI] [PubMed] [Google Scholar]
- 9.Byrd JC, Furman RR, Coutre SE, et al. Three-year follow-up of treatment-naïve and previously treated patients with CLL and SLL receiving single-agent ibrutinib. Blood. 2015;125(16):2497–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsimberidou AM, Tam C, Abruzzo LV, et al. Chemoimmunotherapy may overcome the adverse prognostic significance of 11q deletion in previously untreated patients with chronic lymphocytic leukemia. Cancer. 2009;115(2):373–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schuh A, Becq J, Humphray S, et al. Monitoring chronic lymphocytic leukemia progression by whole genome sequencing reveals heterogeneous clonal evolution patterns. Blood. 2012;120(20):4191–4196. [DOI] [PubMed] [Google Scholar]
- 12.Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell. 2007;28(5):739–745. [DOI] [PubMed] [Google Scholar]
- 13.Reinhardt HC, Jiang H, Hemann MT, Yaffe MB. Exploiting synthetic lethal interactions for targeted cancer therapy. Cell Cycle. 2009;8(19):3112–3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang H, Reinhardt HC, Bartkova J, et al. The combined status of ATM and p53 link tumor development with therapeutic response. Genes Dev. 2009;23(16):1895–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weston VJ, Oldreive CE, Skowronska A, et al. The PARP inhibitor olaparib induces significant killing of ATM-deficient lymphoid tumor cells in vitro and in vivo. Blood. 2010;116(22):4578–4587. [DOI] [PubMed] [Google Scholar]
- 16.Sabharwal SS, Schumacker PT. Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles’ heel¿ Nat Rev Cancer. 2014;14(11):709–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013;12(12):931–947. [DOI] [PubMed] [Google Scholar]
- 18.Guo Z, Kozlov S, Lavin MF, Person MD, Paull TT. ATM activation by oxidative stress. Science. 2010;330(6003):517–521. [DOI] [PubMed] [Google Scholar]
- 19.Agathanggelou A, Weston VJ, Perry T, et al. Targeting the Ataxia Telangiectasia Mutated-null phenotype in chronic lymphocytic leukemia with pro-oxidants. Haematologica. 2015;100(8):1076–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roué G, López-Guerra M, Milpied P, et al. Bendamustine is effective in p53-deficient B-cell neoplasms and requires oxidative stress and caspase-independent signaling. Clin Cancer Res. 2008;14(21):6907–6915. [DOI] [PubMed] [Google Scholar]