

Abstract
Next generation sequencing technologies have provided insights into the molecular heterogeneity of various myeloid neoplasms, revealing previously unknown somatic genetic events. In our cohort of 1444 cases analyzed by next generation sequencing, somatic mutations in the gene BRCA1-BRCA2-containing complex 3 (BRCC3) were identified in 28 cases (1.9%). BRCC3 is a member of the JAMM/MPN+ family of zinc metalloproteases capable of cleaving Lys-63 linked polyubiquitin chains, and is implicated in DNA repair. The mutations were located throughout its coding region. The average variant allelic frequency of BRCC3 mutations was 30.1%, and by a serial sample analysis at two different time points a BRCC3 mutation was already identified in the initial stage of a myelodysplastic syndrome. BRCC3 mutations commonly occurred in nonsense (n=12), frameshift (n=4), and splice site (n=5) configurations. Due to the marginal male dominance (odds ratio; 2.00, 0.84–4.73) of BRCC3 mutations, the majority of mutations (n=23; 82%) were hemizygous. Phenotypically, BRCC3 mutations were frequently observed in myelodysplastic syndromes and myelodysplastic/myeloproliferative neoplasms and associated with -Y abnormality (odds ratio; 3.70, 1.25–11.0). Clinically, BRCC3 mutations were also related to higher age (P=0.01), although prognosis was not affected. Knockdown of Brcc3 gene expression in murine bone marrow lineage negative, Sca1 positive, c-kit positive cells resulted in 2-fold more colony formation and modest differentiation defect. Thus, BRCC3 likely plays a role as tumor-associated gene in myelodysplastic syndromes and myelodysplastic/myeloproliferative neoplasms.
Introduction
Myelodysplastic syndromes (MDS) are characterized by clonal hematopoiesis, bone marrow failure, a propensity for progression to acute myeloid leukemia (AML) and a variety of molecular abnormalities, including chromosomal aberrations and somatic mutations.1 In recent years, a large number of somatic mutations affecting new classes of genes have been identified in MDS and related disorders, providing clues to the molecular pathogenesis of these diseases.2–7 These mutational events can be divided into those that are secondary and acquired during disease progression, and those that are founder in nature.8 Improved identification of genomic defects has substantiated the view that clinical disease heterogeneity is related to patho-molecular diversity. Clinically, evaluation of somatic defects in MDS may improve diagnosis, accuracy of prognoses, and treatments, i.e. may have implications.
Several novel classes of genes frequently affected by somatic mutations have been found in MDS, including genes involved in cohesin complexes9 and spliceosomes,4,10 genes related to methylation11 and genes of novel receptor tyrosine kinases.12,13 Because MDS and associated secondary AML (sAML) are diseases of the elderly, accumulation of mutations and alterations arising from DNA damage has been implicated in disease pathogenesis. Consequently, DNA repair defects may play important roles in maintenance of chromosomal integrity and predisposition to secondary molecular defects. Somatic mutations in DNA repair genes such as TP53 have thus been sought in cancers.14,15
Using unbiased sequencing approaches to identify molecular abnormalities in MDS, we probed the mutational status of a number of DNA repair genes. One of these, BRCA1-BRCA2-containing complex 3 (BRCC3), is a member of the JAMM/MPN+ family of zinc metalloproteases capable of cleaving Lys-63 linked polyubiquitin chains.16 BRCC3 codes for a component of two complexes, BRCA1-A and BRISC. Located in the nucleus, the BRCA1-A complex consists of UIMC1, FAM175A, BABAM1, BRE, BARD1, BRCC3 and BRCA1 and participates in DNA double-strand break (DSB) repair.17 DSBs are associated with genomic instability, cell death18 and carcinogenesis.19 To repair DSBs, cells use both homologous recombination (HR), which uses sister-chromatid alleles as templates in late S and G2, and non-homologous end joining (NHEJ) which can operate in all phases of the cell cycle but often leaves small deletions, possibly gene inactivating at the site of repair.20 The BRISC complex comprises the following proteins: FAM175B/ABRO1, BRCC3/BRCC36, BRE/BRCC45 and MERIT40/NBA1.
Here, we report that somatic defects of BRCC3 are acquired in various myeloid neoplasms and have consequences at the cellular, and perhaps the clinical level.
Methods
Patients
Bone marrow aspirates or blood were collected from the patients with various myeloid neoplasms seen at the Cleveland Clinic, University of Tokyo and Münchner Leukämielabor GmbH (MLL) (Online Supplementary Table S1). Informed consent for sample collection was obtained according to protocols approved by the institutional review boards in accordance with the Declaration of Helsinki. Diagnoses were confirmed according to 2008 World Health Organization classification criteria.21 A total of 1778 patients were enrolled in this study (Online Supplementary Table S1).
Next generation sequencing
Whole exome sequencing (WES) was performed as previously reported.4,6 Briefly, tumor DNAs were extracted from patients’ bone marrow cells. For germ-line controls, DNA was obtained from paired CD3+ T cells. For targeted detection of sequence alterations, confirmation of WES and assessment of variant allelic frequency (VAF), we applied deep sequencing to targeted exons, as previously described.6,22 Briefly, each targeted exon was amplified with each pair of primers, generating 200bp fragments on average. These amplicons were subjected to massive parallel sequencing (Illumina, San Diego, CA, USA) using TruSeq custom primers (Illumina) and SureSelect (Agilent, Santa Clara, CA, USA) with paired-end reads, according to the manufacture’s instruction. Some of these procedures were followed by confirmation using Sanger sequencing, as previously described.13 Annotations by NCBI reference numbers of the genes affected by somatic mutations are summarized in Online Supplementary Table S2.
Cytogenetics and single nucleotide polymorphism-array analyses
Technical details about sample processing and data analyses for single nucleotide polymorphism-array (SNP-A) have been previously described.23 Affymetrix 250K and 6.0 Kit (Affymetrix) were used. Germ-line copy number variants in our internal database or in a publicly available database (Database of Genomic Variants) (http://dgv.tcag.ca/dgv/app/home) were considered non-somatic variants and excluded. Results were analyzed with CNAG (v.3.0)24 or Genotyping Console (Affymetrix). All other lesions were confirmed as somatic or germ-line by analysis of CD3-sorted cells.
ShRNA knockdown and colony formation assay of LSK cells
Lineage-negative, Sca1-positive, c-kit-positive (LSK) cells were purified from C57BL/6 mice, as previously described.25 LSK cells were infected with pLKO.1 lentivirus containing shRNAs targeting Brcc3 (Targeting sequences: shRNA, 5′-CCCACCCT-CATATAACTGTTT-3′) or negative control shRNA (Sigma, SHC002). Lentiviral infections were performed twice by spinoculation. Colony formation assays were performed after another 24 h using 2×104 cells on IMDM methylcellulose medium supplemented with 15% horse serum, mouse SCF (100 ng/mL), IL-6 (6 ng/mL), IL-3 (3 ng/mL) and puromycin (2 mg/mL). Colony numbers were counted after seven days.
Statistical analysis
Overall survival was measured from the day of initial sampling to death from any cause (patients lost to follow up were censored) or last follow up and was summarized using Kaplan-Meier plots and analyzed using the Cox proportional hazard model. Results are for data collected as of May 2013. Pair-wise comparisons were performed by Wilcoxon test for continuous variables and by two-sided Fisher exact for categorical variables. Significance was determined at a two-sided alpha level of 0.05, except for P values in multiple comparisons, for which Bonferroni correction was applied. Analyses were performed using JMP10 (SAS Inc.).
Results
Identification of BRCC3 mutations in myeloid neoplasms
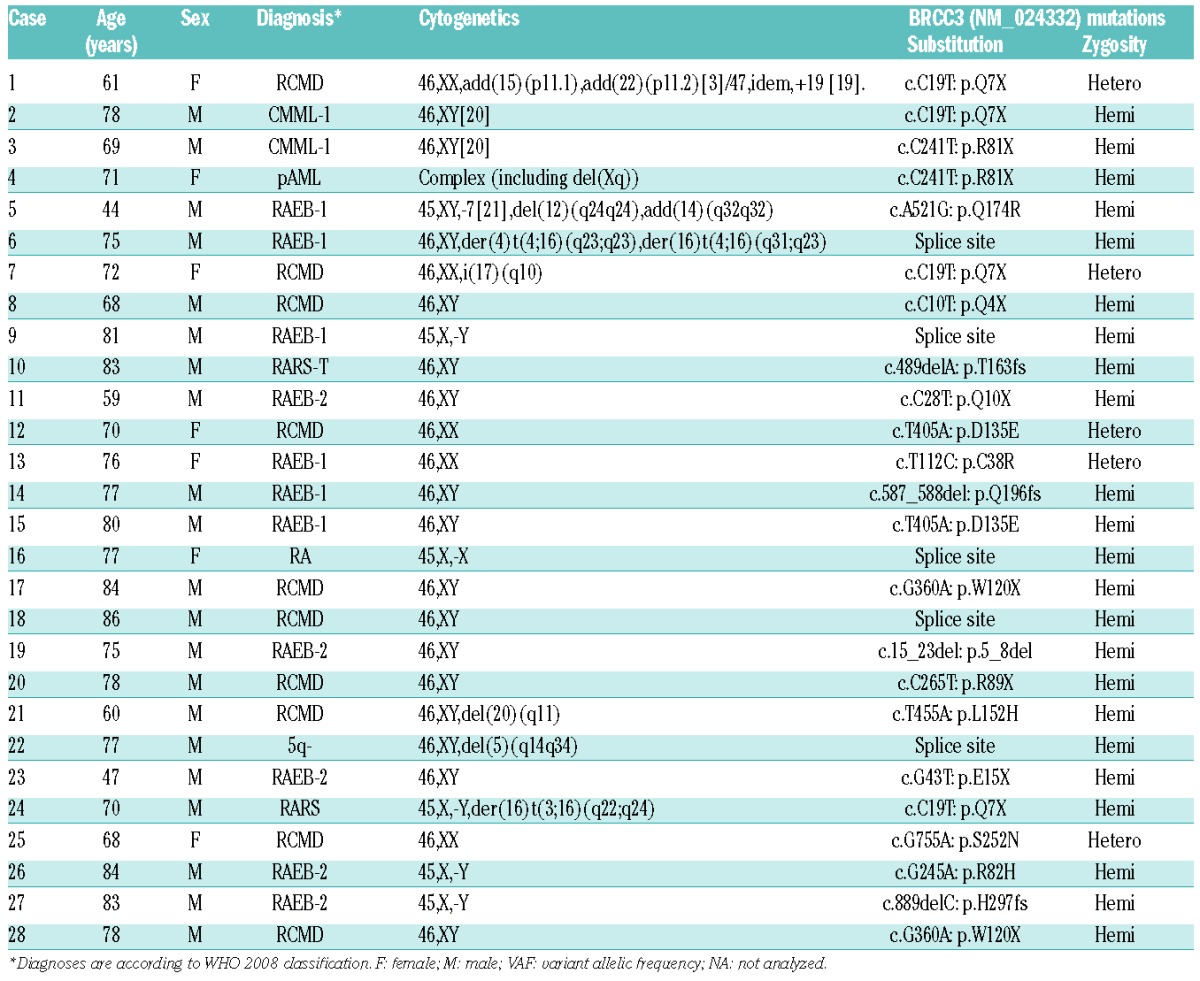
In our cohort of 149 cases analyzed by WES, 2 patients (1.3%) with refractory cytopenia with multilineage dysplasia (RCMD) and chronic myelomonocytic leukemia-1 (CMML-1) revealed 2 somatic recurrent BRCC3 mutations (c.C19T, p.Q7X). The somatic nature of these mutations was confirmed by Sanger and targeted deep DNA sequencing (Figure 1). When we expanded our study to the larger cohort for targeted deep sequencing (n=1295), 26 BRCC3 mutations were identified (2.0%), 11 with refractory anemia with excess blasts (RAEB), 9 with RCMD, 1 with refractory anemia, 1 with refractory anemia with ring sideroblasts, 1 with isolated 5q syndrome, 1 with CMML, 1 with RARS associated with thrombocytosis (RARS-T), and 1 with primary AML (Table 1). Four canonical mutations occurred in exon 1 (p.Q7X), 3 in exon 4 (p.R81X) and 2 in exon 5 (p.W120X). Thus, 28 (1.9%) of our 1444 patients with various myeloid neoplasms exhibited BRCC3 mutations.
Figure 1.
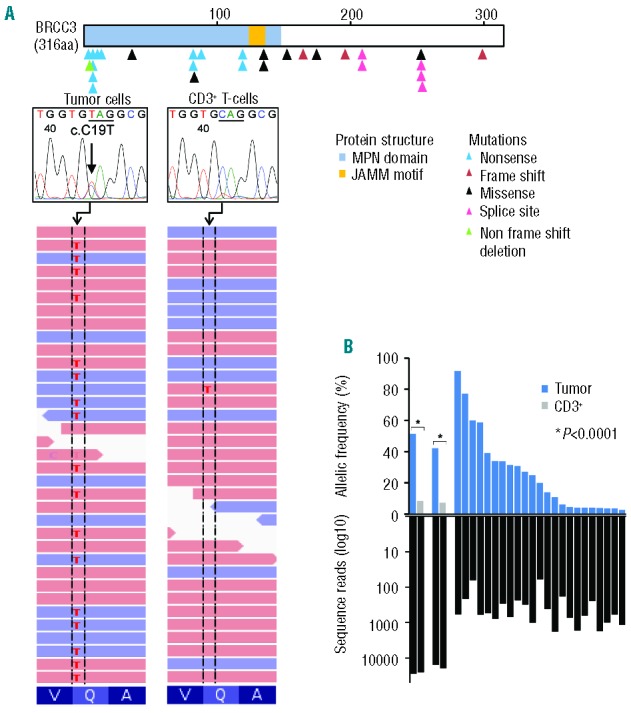
Somatic BRCC3 mutations as detected by next generation sequencing and Sanger sequencing. (A) Distribution of BRCC3 mutations identified in 28 out of 1444 myeloid neoplasms. Blue, red, black, pink, and green triangles indicate nonsense, frame shift, missense, splice site, and non-frame shift deletion mutations. On the right, a representative mutation (c.C19T, indicated by an arrow) confirmed by whole exome sequencing and Sanger sequencing. (B) (Top) Allelic frequencies in paired bone marrow and CD3+ T-cell samples (n=2) and not paired (n=23) samples as measured by deep sequencing. (Bottom) Depth of coverage of independent reads.
Table 1.
Characteristics of patients with BRCC3 mutations.
Genetic significance of BRCC3 mutations
The 28 BRCC3 mutations occurred in nonsense (n=12; 42.9%), missense (n=6; 21.4%), splice site (n=5; 17.9%), frameshift (n=4; 14.3%), and non-frameshift deletion (n=1; 3.6%) forms. Twenty-three (82.1%) were hemizygous due to either male sex or a deletion of chromosome X (-X) (BRCC3 is located on chromosome X). Evaluation of deep sequencing demonstrated that the mean value of variant allelic frequency of BRCC3 mutations was 30.1% (range 91.7–3.5). In one of these index cases, deep sequencing at two time points (Case #1 in Table 1) indicated that the BRCC3 mutation was present at RCMD diagnosis as well as throughout RAEB evolution (20 months later) (Figure 2A). This suggests that the BRCC3 mutation may have been an early event. By additional analyses on concomitant mutations of other functionally important genes in the cases with BRCC3 mutations, TET2 and SF3B1 mutations were observed most frequently among mutations of 23 genes detected (Figure 2B and C). SNP-A karyotyping, deletions involving the BRCC3 locus (Xq28) were detected in 7 out of 677 (1.0%) patients. For BRCC3 located on chromosome X, expression was assessed by comparison to sex-matched controls. Due to the small sample size, the impact of somatic deletion of chromosome X on BRCC3 expression could not be resolved (Online Supplementary Figure S1). In fact, in our cohort, there was no significant difference in BRCC3 expression between males and females (Online Supplementary Figure S2). In various hematopoietic cells, analysis of mRNA showed BRCC3 globally expressed in myeloid, erythroid and lymphoid cells (Online Supplementary Figure S3).
Figure 2.
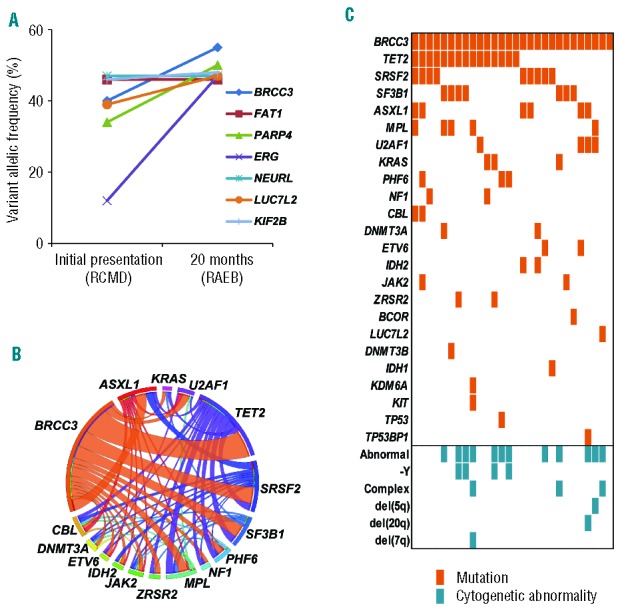
BRCC3 mutations and commonly associated other mutations. (A) Serial analysis of variant allelic frequencies (VAF) measured by targeted deep sequencing in a representative patient with a BRCC3 mutation at the diagnosis of RCMD and upon progression to RAEB. Co-existing gene mutation VAF shifts are indicated by corresponding lines. (B) A Circos plot illustrating co-existing mutations in 16 selected genes. (C) Co-existing mutations in the BRCC3-mutated cohort; 27 out of 28 cases were positive for other somatic mutations.
Clinical phenotype of the cases with BRCC3 mutations
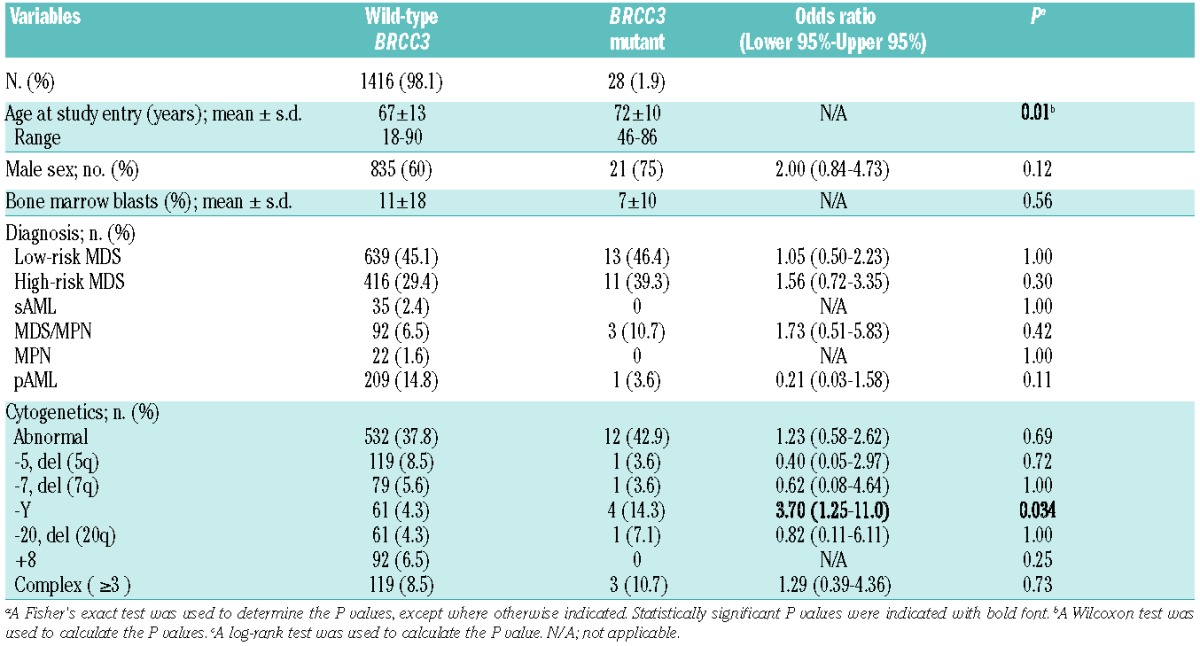
BRCC3 mutations were associated with higher age in the whole cohort [mean±standard deviation, 67±13 in wild-type (WT) and 72±10 in mutants; P=0.01]. There was a marginally significant male dominance of mutation frequency [odds ratio (OR) = 2.00; 95% confidence interval (CI) 0.84–4.73] (Table 2). With regard to cytogenetics, mutations of this gene were associated with -Y abnormality (OR; 3.70, 95%CI; 1.25–11.0; P=0.034), while all other karyotype abnormalities showed similar distributions between WT and mutant cases (Table 2). Phenotypically, BRCC3 mutations were identified in the low- (13 of 652; 2.0%) and high- (11 of 427; 2.6%) risk MDS groups and in myelodysplastic/myeloproliferative neoplasms (MDS/MPN) (3 of 94; 3.2%), but not in any sAML and myeloproliferative neoplasms (MPN) cases (Table 2). BRCC3 mutations had no impact on overall survival (Online Supplementary Figure S4).
Table 2.
Clinical characteristics of myeloid malignancies either with or without BRCC3 mutations.
Cellular consequences of BRCC3 lesions
Frequently, BRCC3 mutations detected in our cohort occurred in nonsense and frameshift configurations associated with myelodysplastic phenotypes. This suggests that BRCC3 is a candidate tumor suppressor gene in MDS. To clarify the functional consequences of BRCC3 defects, lentiviral shRNA mediated knockdown of Brcc3 expression was performed in mouse lineage-Sca-1+c-kit+ (LSK) cells, which are enriched for hematopoietic stem and early progenitor cells. Selection with puromycin for 24 h resulted in a pure population (>95%) of transduced cells. Brcc3 knockdown was confirmed by Western blotting analysis at 96 h after infection with a Brcc3-specific lentiviral shRNA. Colony formations significantly increased in LSK cells with Brcc3 knockdown compared to cells infected with control lentiviral shRNA (P<0.001) (Figure 3). Brcc3 knockdown also resulted in moderate differentiation block (Figure 3), which was confirmed by flow cytometry analyses showing increased expression of precursor marker c-Kit and reduced expression of differentiation marker Gr-1 with Brcc3 knockdown (Online Supplementary Figure S5).
Figure 3.
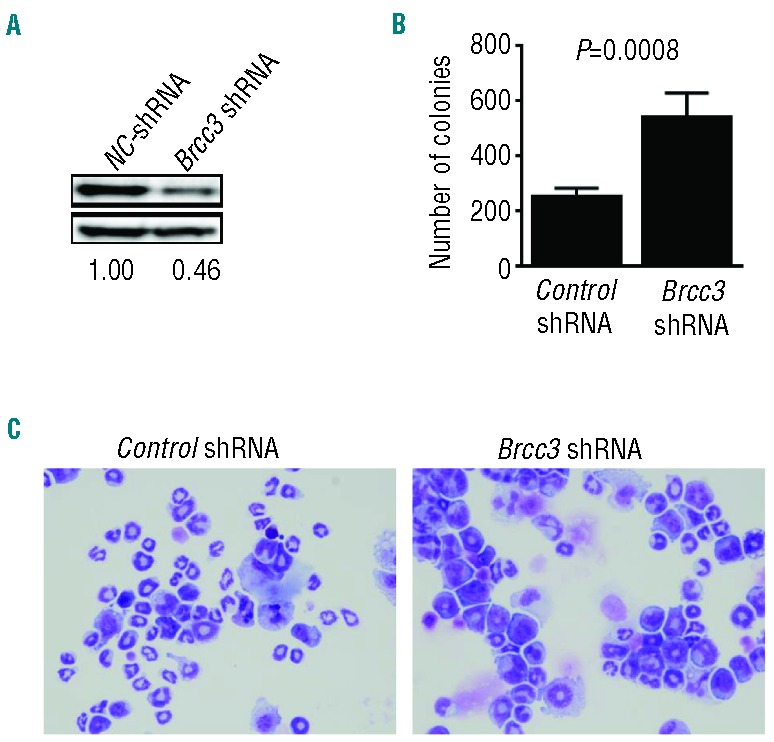
Cellular consequences of BRCC3 knockdown. (A) Immunoblot analysis of Brcc3 levels in LSK cells at 96 h after infection with lentivirus carrying Brcc3 -specific or control (NC) shRNA. (B) Brcc3 knockdown significantly increased colony formation capability of murine lineage− Sca-1+c-kit+ (LSK) cells. Columns correspond to mean ± SD of colony numbers formed by 5000 puromycin-resistant murine LSK cells in the presence of SCF, IL-3, and IL-6 at 48 h after infection with lentivirus carrying Brcc3-specific or control shRNA and subsequent selection with puromycin. (C) Wright-Giemsa staining of LSK cells in the presence of SCF, IL-3, and IL-6 at 7 days after infection with lentivirus carrying Brcc3-specific or control shRNA (X200).
Genetic defects of other component of BRCA1-A and BRISC complex
BRCC3 is a component of the BRCA1-A and BRISC complexes. We, therefore, analyzed other genes in these complexes in 149 cases and observed mutations in 3 patients (2.7%) (Online Supplementary Table S3). The mutations were in the genes UIMC1, FAM175A and BABAM1. By SNP-array karyotyping, deletions in BRCA1-A and BRISC component genes were found in 21 of 677 (3.1%) patients. UIMC1 deletions (5q35.2) were identified in 11 patients, FAM175A deletions (4q21.23) were present in 8 (Online Supplementary Figure S6) and those involving FAM175B and BABAM1 loci were each present in one patient (data not shown). Out of these genes affected in myeloid neoplasms, frequently deleted UIMC1 showed haploinsufficient expression (P<0.05) (Online Supplementary Figure S7).
Discussion
Here, we report the discovery of somatic mutations in the BRCC3 gene participating in the BRCA1-A and BRISC complexes in myeloid neoplasms. After the initial discovery of nonsense mutations in this gene by WES, our cohort for target deep sequencing was expanded and 28 mutations were discovered in various myeloid neoplasms. These mutations were frequently hemizygous and present in nonsense and frameshift forms. BRCC3 mutations were more common in MDS and MDS/MPN compared to primary AML and MPN, and were cytogenetically associated with -Y abnormality. Deep sequencing analysis of clonal architecture in serial samples indicated that BRCC3 mutations may constitute founder events, and Brcc3 knockdown in murine LSK cells resulted in approximately doubling clonogenic cells and modest differentiation block of hematopoietic progenitors transfected with Brcc3 shRNA. The pro-leukemogenic effects of BRCC3 mutations might be related to the impact of these lesions on tumor suppressive potential.
In addition to BRCC3, other genes encoding components of BRCA1-A and BRISC complexes were also affected by either somatic mutations or deletions. These components assemble to play a role in DSBs repair.17 For this process, canonical non-homologous end joining (C-NHEJ) is initiated by binding of the Ku70/80 heterodimer to DSB free ends; DNA-bound Ku protects the DSB free ends from nuclease digestion26 while it recruits DNA-PK catalytic subunit (DNA-PKcs) to the DSB. The resultant DNA-PK complex activates DNA-PK’s kinase activity27,28 which phosphorylates H2AX to form γH2AX. It also facilitates the recruitment of a ligation complex, which encompasses DNA LigIV and XRCC4 among other proteins. Compromised BRCC3 function in DSB repair, which is presumably to remove DSB repair proteins by degradation during DSB resolution, may cause an imbalance repair proteins globally, and as a result, genomic instability. In our cohort with BRCC3 mutations, multiple somatic driver mutations and cytogenetic abnormalities were identified, and one gave a cancer cell survival advantage that enabled genomic instability caused by BRCC3.
In analogy to somatic BRCC3 mutations, acquired mutations of other genes involved in DNA repair machinery have been reported in MDS and related conditions, for example, in TP53 and ATM. TP53-deficient cell lines show delayed γH2AX resolution post-irradiation.18,29 Similarly, mutant TP53 prevents the MRE11–RAD50–NBS1 complex from phosphorylating ATM, leading to impaired HR.30 ATM is activated by DSB and signals the cell-cycle checkpoint to slow the passage of cells through the cycle to allow time for DNA repair.31 Germ-line mutations in DNA repair machinery genes, including BRCA1 and BRCA2, TP53 and ATM convey a strong inherited predisposition to neoplasia.32–35 However, disease-prone germ-line variants of BRCC3 have not been reported so far.
Defects in DNA repair acquired in the malignant clone may create a leukemia cell-specific vulnerability, if it leads to a greater number of residual DSBs 24 h after their induction.18 Based on this theory, cells with partial loss of BRCC3 function could be sensitive to BRCC3 inhibition36 as they will have greater dependence on the remaining WT allele and thus be differentially more stressed by its inactivation. This suggests, however, a broader target, namely, any member of either the BRCA1-A or BRISC complexes might be capable of incapacitating the entire complex.
Knockdown of Brcc3 in normal hematopoietic cells resulted in the modest increase of colony formation and slightly less differentiation compared to WT experiments, which might support the evidence of the tumorigenic effects of human BRCC3 mutations. However, shRNA transductions were applied for Brcc3 knockdown with a single targeting sequence. Off target effects should always be considered when we perform knockdown experiments using a single shRNA. For validation of the effect, we showed the successful reduction of Brcc3 protein amount by immunoblot to confirm the suppressive effects of shRNA. Further rescue experiments and animal models are essential to authenticate these findings on BRCC3-associated leukemogenesis.
In summary, we have found new somatic mutations of BRCC3 and deletions of other BRCA1-A complex component in MDS and MDS/MPN. These results suggest that BRCC3 might be a tumor-associated gene in myeloid neoplasms.
Footnotes
The online version of this article has a Supplementary Appendix.
Funding
This work was supported by US National Institutes of Health (NIH) grants RO1 CA-143193 (Y.D.), by a Scott Hamilton CARES grant (H.M.) by a research fund from AA&MDS International Foundation (H.M.), by Grants-in-Aid from the Ministry of Health, Labor and Welfare of Japan and KAKENHI (23249052, 22134006 and 21790907; S.O.), the project for the development of innovative research on cancer therapies (p-direct; S.O.), the Japan Society for the Promotion of Science (JSPS) through the Funding Program for World-Leading Innovative R&D on Science and Technology, initiated by the Council for Science and Technology Policy (CSTP; S.O.) and Uniformed Services University of the Health Sciences Pediatrics grant KM86GI (Y.D.). The results presented here are partly based on data generated by The Cancer Genome Atlas (TCGA) pilot project established by the National Cancer Institute and the National Human Genome Research Institute. Information about TCGA and the investigators and institutions that constitute the TCGA research network can be found at http://cancergenome.nih.gov/.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Delhommeau F, Dupont S, Della Valle V, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009;360(22):2289–2301. [DOI] [PubMed] [Google Scholar]
- 3.Papaemmanuil E, Cazzola M, Boultwood J, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med. 2011;365(15):1384–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yoshida K, Sanada M, Shiraishi Y, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478(7367):64–69. [DOI] [PubMed] [Google Scholar]
- 5.Khan SN, Jankowska AM, Mahfouz R, et al. Multiple mechanisms deregulate EZH2 and histone H3 lysine 27 epigenetic changes in myeloid malignancies. Leukemia. 2013;27(6):1301–1309. [DOI] [PubMed] [Google Scholar]
- 6.Makishima H, Yoshida K, Nguyen N, et al. Somatic SETBP1 mutations in myeloid malignancies. Nat Genet. 2013;45(8):942–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Papaemmanuil E, Gerstung M, Malcovati L, et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. 2013;122(22):3616–3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walter MJ, Shen D, Ding L, et al. Clonal architecture of secondary acute myeloid leukemia. N Engl J Med. 2012; 366(12):1090–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kon A, Shih LY, Minamino M, et al. Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nat Genet. 2013;45(10):1232–1237. [DOI] [PubMed] [Google Scholar]
- 10.Makishima H, Visconte V, Sakaguchi H, et al. Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood. 2012;119(14):3203–3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ley TJ, Ding L, Walter MJ, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363(25):2424–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maxson JE, Gotlib J, Pollyea DA, et al. Oncogenic CSF3R mutations in chronic neutrophilic leukemia and atypical CML. N Engl J Med. 2013;368(19):1781–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Makishima H, Sugimoto Y, Szpurka H, et al. CBL mutation-related patterns of phosphorylation and sensitivity to tyrosine kinase inhibitors. Leukemia. 2012; 26(7):1547–1554. [DOI] [PubMed] [Google Scholar]
- 14.Levine AJ, Momand J, Finlay CA. The p53 tumour suppressor gene. Nature. 1991;351(6326):453–456. [DOI] [PubMed] [Google Scholar]
- 15.Bejar R, Stevenson K, Abdel-Wahab O, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med. 2011;364(26):2496–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feng L, Wang J, Chen J. The Lys63-specific deubiquitinating enzyme BRCC36 is regulated by two scaffold proteins localizing in different subcellular compartments. J Biol Chem. 2010;285(40):30982–30988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shao G, Lilli DR, Patterson-Fortin J, Coleman KA, Morrissey DE, Greenberg RA. The Rap80-BRCC36 de-ubiquitinating enzyme complex antagonizes RNF8-Ubc13-dependent ubiquitination events at DNA double strand breaks. Proc Natl Acad Sci USA. 2009;106(9):3166–3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Banath JP, Macphail SH, Olive PL. Radiation sensitivity, H2AX phosphorylation, and kinetics of repair of DNA strand breaks in irradiated cervical cancer cell lines. Cancer Res. 2004;64(19):7144–7149. [DOI] [PubMed] [Google Scholar]
- 19.Wu J, Liu C, Chen J, Yu X. RAP80 protein is important for genomic stability and is required for stabilizing BRCA1-A complex at DNA damage sites in vivo. J Biol Chem. 2012;287(27):22919–22926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coleman KA, Greenberg RA. The BRCA1-RAP80 complex regulates DNA repair mechanism utilization by restricting end resection. J Biol Chem. 2011;286(15):13669–13680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Swerdlow SH, Campo E, Harris NL, et al. WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues, Fourth Edition. Lyon: IARC Press, 2008. [Google Scholar]
- 22.Haferlach T, Nagata Y, Grossmann V, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014;28(2):241–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gondek LP, Tiu R, O’Keefe CL, Sekeres MA, Theil KS, Maciejewski JP. Chromosomal lesions and uniparental disomy detected by SNP arrays in MDS, MDS/MPD, and MDS-derived AML. Blood. 2008;111(3):1534–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nannya Y, Sanada M, Nakazaki K, et al. A robust algorithm for copy number detection using high-density oligonucleotide single nucleotide polymorphism genotyping arrays. Cancer Res. 2005;65(14):6071–6079. [DOI] [PubMed] [Google Scholar]
- 25.Oakley K, Han Y, Vishwakarma BA, et al. Setbp1 promotes the self-renewal of murine myeloid progenitors via activation of Hoxa9 and Hoxa10. Blood. 2012;119(25):6099–6108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jasin M. Genetic manipulation of genomes with rare-cutting endonucleases. Trends Genet. 1996;12(6):224–228. [DOI] [PubMed] [Google Scholar]
- 27.Meek K, Gupta S, Ramsden DA, Lees-Miller SP. The DNA-dependent protein kinase: the director at the end. Immunol Rev. 2004; 200:132–141. [DOI] [PubMed] [Google Scholar]
- 28.Kakarougkas A, Jeggo PA. DNA DSB repair pathway choice: an orchestrated handover mechanism. Br J Radiol. 2014;87(1035): 20130685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zheng H, Chen L, Pledger WJ, Fang J, Chen J. p53 promotes repair of heterochromatin DNA by regulating JMJD2b and SUV39H1 expression. Oncogene. 2013;33(6):734–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Song H, Hollstein M, Xu Y. p53 gain-of-function cancer mutants induce genetic instability by inactivating ATM. Nat Cell Biol. 2007;9(5):573–580. [DOI] [PubMed] [Google Scholar]
- 31.Somyajit K, Basavaraju S, Scully R, Nagaraju G. ATM- and ATR-mediated phosphorylation of XRCC3 regulates DNA double-strand break-induced checkpoint activation and repair. Mol Cell Biol. 2013; 33(9):1830–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Biesecker BB, Boehnke M, Calzone K, et al. Genetic counseling for families with inherited susceptibility to breast and ovarian cancer. Jama. 1993;269(15):1970–1974. [PubMed] [Google Scholar]
- 33.Narod S, Lynch H, Conway T, Watson P, Feunteun J, Lenoir G. Increasing incidence of breast cancer in family with BRCA1 mutation. Lancet. 1993;341(8852):1101–1102. [DOI] [PubMed] [Google Scholar]
- 34.Li FP, Fraumeni JF., Jr Rhabdomyosarcoma in children: epidemiologic study and identification of a familial cancer syndrome. J Natl Cancer Inst. 1969;43(6):1365–1373. [PubMed] [Google Scholar]
- 35.Vorechovsky I, Luo L, Lindblom A, et al. ATM mutations in cancer families. Cancer Res. 1996;56(18):4130–4133. [PubMed] [Google Scholar]
- 36.Seiberlich V, Goldbaum O, Zhukareva V, Richter-Landsberg C. The small molecule inhibitor PR-619 of deubiquitinating enzymes affects the microtubule network and causes protein aggregate formation in neural cells: implications for neurodegenerative diseases. Biochim Biophys Acta. 2012; 1823(11):2057–2068. [DOI] [PubMed] [Google Scholar]