

Survival in multiple myeloma (MM) has improved remarkably since the introduction of the proteasome inhibitors, (PI), bortezomib (BOR), carfilzomib (CAR), and the immune modulating cereblon-binding molecules (CBMs) thalidomide (THAL), lenalidomide (LEN), and pomalidomide (POM). BOR reversibly, and CAR irreversibly, inhibit proteasome function, thus inducing apoptosis in MM cells.1 CBMs bind to a specific pocket in the protein cereblon (CRBN)2–4 an interaction which enhances the E3-ubiquitin-ligase activity of the CRBN/Cul4A/Cul4B/DDB1/Roc1 complex. As a result, the downstream substrates of CRBN, Ikaros (IKZF1) and Aiolos (IKZF3), important regulators in T- and B-cell development, are more efficiently ubiquitinated and degradated.5,6 As a consequence specific downstream targets of IKZF1/3 including IRF4 and subsequently MYC are down-regulated and MM cell death is induced, while simultaneously transcription of Interleukin-2 is enhanced and the immune compartment activated.5
To better visualize IKZF1 degradation we created an adenoviral vector expressing IKZF1 fused to luciferase and used this vector to transfect 14 MM cell lines to investigate LEN induced IKZF1 degradation. We also utilized a lentiviral system to create two stable cell-lines (H929/IKZF1Luc and 8226/IKZF1Luc), both expressing an IKZF1-luciferase fusion gene and used them to assess the IKZF1 degradation capacity of other CBMs, as well as the synergistic and antagonistic effects of LEN-based combination therapies.
Specifically, we first created a plasmid (pCDHIKZF1Luc) containing a CMV promoter controlled luciferase-IKZF1 fusion gene, a control vector expressing luciferase and constructed an adenoviral vector, that expressed an IKZF1-luciferase fusion gene (AdIKLuc) as well as a luciferase only control vector (AdLuc). Next, we infected 14 MM cell lines with the adenoviral vector AdIKLuc or control vector and documented successful protein production. The cell lines were then treated with LEN for six days and their viability measured by MTT assay to document sensitivity or resistance to drug. Subsequently, we tested the ability of LEN to degrade IKZF1, by measuring luciferase activity 5 and 24 h after drug exposure and correlated this with drug sensitivity. Finally, we assessed the same response in the stable cell lines, 8226/IKZF1Luc and H929/IKZF1Luc, to other CBMs and the effects of combination treatment (LEN/DEX, LEN/BOR, LEN/CAR, and LEN/SAHA) on the degradation capacity of IKZF1 in 8226/IKZF1Luc cells.
Luciferase expression was examined after exposure to all three CBMs in 8226/IKZF1Luc (Figure 1A) and H929/IKZF1Luc (Figure 1B). With each of them IKZF-luciferase levels dramatically decreased and depletion was dose dependent. Using this surrogate marker cell-line assay luciferase is most efficiently degraded by POM which is twice as potent as LEN, but a thousand times more potent than THAL (ED50 for H929IZKF1Luc, POM: 4.9nM, LEN: 10.2 nM, and THAL: 4795 nM).
Figure 1.
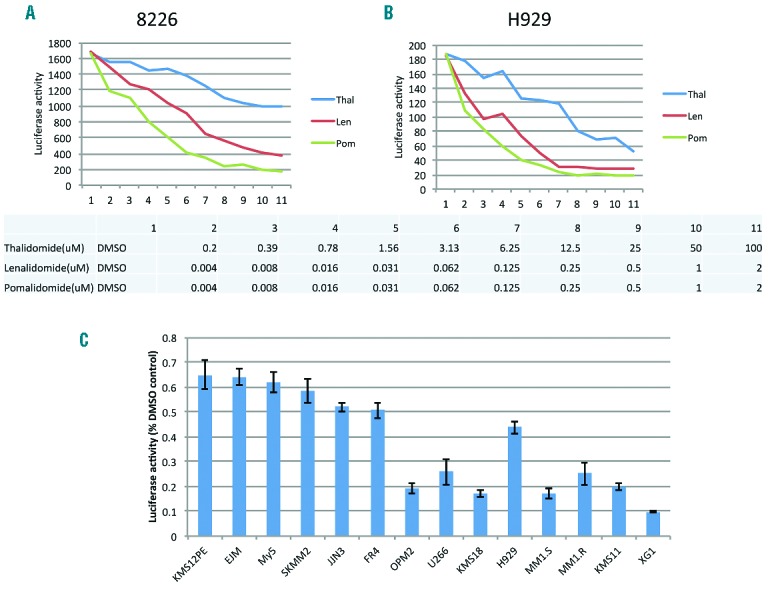
The capacity to degrade IKZF1 correlates with the anti-myeloma potency of THAL, LEN and POM. (A and B) 8226 and H929 cells, which stably express IKZF-luciferase fusion proteins were treated with the indicated dose of LEN, POM and THAL (see table below graph) for 24 h and luciferase activity in drug-exposed cell-lines were measured. The relative ability of drug to degrade IKZF1 is dose dependent and potency demonstrates THAL<LEN<POM. (C) 14 myeloma cell lines with variable sensitivity to LEN were infected with adenovirus expressing IKZF1-luciferase fusion proteins. Cells were treated with vehicle (DMSO) or LEN for 5 h and luciferase activities were measured. Cell lines are ranked from most LEN resistant on the left to most LEN sensitive on the right and luciferase percent degradation in response to LEN is shown in the bars. Data generated from each cell line were normalized to vehicle-treated control. Overall ability to degrade IKZF1 strongly correlates with drug responsiveness.
We next tested cytotoxicity in 14 MM cell lines after LEN treatment. Based on MTT (Online Supplementary Figure S1A), these cell lines can be divided into three groups including XG1, KMS11, MM1.S, MM1.R, and H929 (sensitive), OPM2, U266, KMS18 (intermediate response), and OCI-My5, SKMM2, KMS12PE, FR4, EJM and JJN3 (resistant). We infected these cell lines with an adenovirus expressing the IKZF1-luciferase fusion protein or a control expressing luciferase only, followed by treatment with LEN. After drug exposure a reduction of IKZF1-luciferase was observed in the positive control cell lines and minimal reduction was seen in the AdLuc negative controls (data not shown). As shown in Figure 1C, the degree of luciferase reduction observed 5 h after LEN treatment paralleled the cytotoxicity of MM cell lines to LEN. For example, luciferase activity dropped more than 80% in sensitive cell lines, while it declined less than 50% in all resistant cell lines. Similar effects were also observed at 24 h post treatment (Online Supplementary Figure S2). Of interest, all LEN-resistant cell lines were also unresponsive to THAL and POM treatment as measured by MTT and by ability to degrade IKZF1-luciferase (Online Supplementary Figure S3). Drug doses employed were those which are reliably cytotoxic in sensitive cells.
We next compared gene expression levels measured by RNAseq of the resistant versus the non-resistant cell lines and despite the small sample size and wide confidence intervals observed statistically significantly higher levels of IKZF3 (P=0.05) and a trend for higher KPNA2 (P=0.07), IKZF1 (P=0.1) and CRBN (P=0.44) levels in the sensitive cell lines (Online Supplementary Figure S4).
Since successful degradation of Ikaros and Aiolis require an intact proteasome, we then investigated whether proteasome inhibitors can alter CBM anti-MM activity. We treated 8226IKZF1Luc with LEN in combination with increasing doses of BOR or CAR for 24 h. Strikingly, both proteasome inhibitors almost totally blocked LEN-induced IKZF1-fusion protein degradation, at a dose of 6 nM for BOR treatment and 40 nM for CARF (Figure 2A and B). Similar results were achieved using H929IKZFLuc with LEN and increasing doses of BOR (Online Supplementary Figure S5). Of note, the blockage of IKZF1 degradation by BOR was time- and dose-dependent, for example, in 8226IKZF1Luc, the inhibition mediated by 100nM BOR only required 1 h of pre-treatment, but it took 3 h if a reduced dose of 40 nM of BOR was applied (Figure 2C). Interestingly, 3–5 nM concentrations of BOR did not fully block IKZF1-fusion protein degradation (Figure 2A), but were sufficient to mediate cytotoxicity (Figure 2D), suggesting the existence of a dosage window in which combined PI and CBM might still independently exert anti-MM effects.
Figure 2.
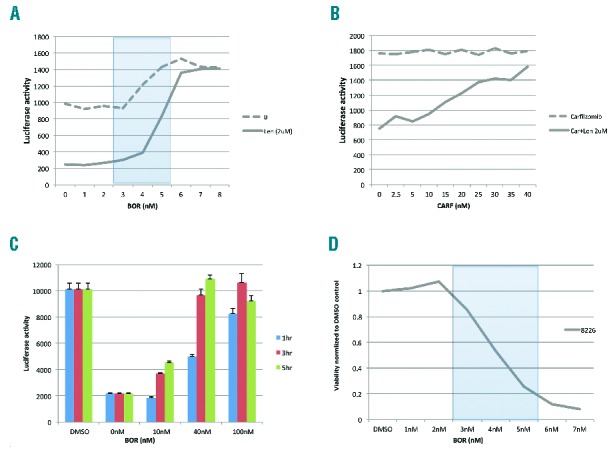
Proteasome inhibitors abrogate lenalidomide induced IKZF1 degradation in a dose- and time-dependent fashion. (A and B) Bortezomib and carfilzomib abolished lenalidomide–induced degradation of IKZF1. 8226/IKZF1Luc cells were treated with lenalidomide for 24 h alone or with the indicated doses of bortezomib (A) or carfilzomib (B), and luciferase activities as a measure of IKZF1 degradation were measured. (C) Blockage of lenalidomide induced IKZF1 activity by BOR is dose- and time-dependent. 8226/IKZF1Luc cells were pre-treated with the indicated doses of bortezomib for different times, then BOR was washed off. LEN (2 μM) was added and incubated for 24 h. (D) 8226 cell viability measured by MTT (normalized to DMSO), cells were treated with BOR for 72 h.
Based on this dose window observation, we explored lower doses of BOR below that which abrogate CBM activity, and we were able to confirm synergistic toxic effects by MTT of combined LEN and BOR treatment in both H929 and MM1.S (Online Supplementary Figure S6). Interestingly, in both we observed the highest synergy when LEN was administered prior BOR treatment (Online Supplementary Table S1) suggesting that the rapid degradation of IKZF1 may occur quickly enough to still allow for combination efficacy if dosing of BOR is delayed compared to LEN. To validate the specificity of our approach, we finally assessed the effect of combined LEN either in combination with DEX or the pan-HDAC inhibitor, SAHA. Neither significantly influenced LEN-induced IKZF1 degradation (Online Supplementary Figure S7). However, SAHA was shown to enhance both luciferase control and IKZF1-luciferase accumulation.
Anti-MM CBM action is mediated by proteasomal degradation of IKZF1 and IKZF3.5,6 BOR reversibly and CAR irreversibly block the proteasome, suggesting that combining both drugs might be counterproductive. However, clinical experience suggests that CBM and PI combination therapies in MM treatment lead to improved response rates and deep and durable remissions.7–12 To explore this paradox, we have described a model, in which the degradation capacity of IKZF1 can be measured by luciferase activity. We report that the IKZF1 degradation capacity of POM and LEN clearly outperformed that of THAL. Furthermore, POM showed an impressive 1000-fold higher IKZF1 degradation capacity than THAL, giving evidence that the clinical potency of CBMs is strongly related to their capacity to degrade IKZF1. The ability of drug-bound cereblon to promote degradation of IKZF1 is thus a hallmark of MM cellular response to CBMs.
We explored whether PI therapy can alter CBM induced IKZF1 degradation, and, strikingly, we were able to completely abrogate the CBM-induced IKZF1 degradation by concomitant PI treatment. We propose four possibilities that might explain the apparent biologic discrepancy with clinical results. First, and probably the most simple explanation, is that commonly used regimens administer CBMs continuously over a 21-day period whereas only 4 doses of BOR or 6 doses of CAR are given in a 21-day or 28-day cycle. These differences in the administration schedule might, therefore, allow dual action, if the proteasome inhibition is not complete or does not persist over the whole treatment cycle. Similarly, if a CBM is given prior to a PI then the rapidity of IKZF degradation may be such that proteasome inhibition comes too late to be of significant clinical consequence. Delivery of BOR or CAR prior to CBM would, however, be counterproductive. An alternative but speculative theory might be that IMiD and PI treatment are additive independent of proteasome function which would be possible if inactivation of IKZF proteins resulted from its polyubiquitination alone and the nonfunctional heavily ubiquitinated IKZF protein was sufficient to exert IMiD-based anti-MM properties. A final possibility is that we have noticed cellular cytotoxicity due to PI at levels of PI drug below those required to inhibit IKZF1 degradation (Figure 2C and Online Supplementary Figure S3). This finding suggests the potential existence of a therapeutic window in which both drugs can be used concurrently; however, such thresholds in humans are unknown and need to be determined.
Overall our data suggest that CBM administration on days of PI, and especially concurrent with or shortly after PI administration, might be ineffective. Consequently, clinical trials are needed to investigate whether CBM administration can be safely omitted on those days, reducing cost and potentially lowering toxicity, while maintaining treatment efficacy. Alternatively, such trials could potentially enhance activity by introducing a 6–12 h window after oral delivery of a CBM and the use of a PI. Formal pharmacokinetic and pharmacodynamics studies would be very helpful in widening our understanding.
In summary, we have demonstrated that CBM-induced IKZF1 degradation can be rapidly and completely abrogated by proteasome inhibitors. Our data suggest that the timing and dosing schedules of clinical use of CBMs in combination with PI treatment is critical, providing evidence that established treatment regimens need to be carefully reevaluated to maximize anti-tumor effects at the lowest costs and toxicity for the patient.
Acknowledgments
The authors would like to thank Dr. Trono, École Polytechnique Fédérale de Lausanne, Switzerland, for providing plasmids psPAX2 and pMD2.G and the National Cancer Institute for grant support (CA 183968).
Footnotes
Funding: KMK is supported by a research grant of the Deutsche Forschungsgemeinschaft (DFG, KO 4604/1-1).
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Kortuem KM, Stewart AK. Carfilzomib. Blood. 2013;121(6):893–897. [DOI] [PubMed] [Google Scholar]
- 2.Zhu YX, Braggio E, Shi CX, et al. Cereblon expression is required for the antimyeloma activity of lenalidomide and pomalidomide. Blood. 2011;118(18):4771–4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chamberlain PP, Lopez-Girona A, Miller K, et al. Structure of the human Cereblon-DDB1-lenalidomide complex reveals basis for responsiveness to thalidomide analogs. Nat Struct Mol Biol. 2014;21(9):803–809. [DOI] [PubMed] [Google Scholar]
- 4.Zhu YX, Braggio E, Shi CX, et al. Identification of cereblon-binding proteins and relationship with response and survival after IMiDs in multiple myeloma. Blood. 2014;124(4):536–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kronke J, Udeshi ND, Narla A, et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science. 2014;343(6168):301–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu G, Middleton RE, Sun H, et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science. 2014;343(6168):305–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Richardson PG, Xie W, Jagannath S, et al. A phase 2 trial of lenalidomide, bortezomib, and dexamethasone in patients with relapsed and relapsed/refractory myeloma. Blood. 2014;123(10):1461–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Richardson PG, Weller E, Lonial S, et al. Lenalidomide, bortezomib, and dexamethasone combination therapy in patients with newly diagnosed multiple myeloma. Blood. 2010;116(5):679–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cavo M, Tacchetti P, Patriarca F, et al. Bortezomib with thalidomide plus dexamethasone compared with thalidomide plus dexamethasone as induction therapy before, and consolidation therapy after, double autologous stem-cell transplantation in newly diagnosed multiple myeloma: a randomised phase 3 study. Lancet. 2010;376(9758): 2075–2085. [DOI] [PubMed] [Google Scholar]
- 10.Wang M, Delasalle K, Giralt S, Alexanian R. Rapid control of previously untreated multiple myeloma with bortezomib-lenalidomide-dexamethasone (BLD). Hematology. 2010;15(2):70–73. [DOI] [PubMed] [Google Scholar]
- 11.Niesvizky R, Martin TG, 3rd, Bensinger WI, et al. Phase Ib dose-escalation study (PX-171-006) of carfilzomib, lenalidomide, and low-dose dexamethasone in relapsed or progressive multiple myeloma. Clin Cancer Res. 2013;19(8):2248–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stewart AK, Rajkumar SV, Dimopoulos MA, et al. Carfilzomib, Lenalidomide, and Dexamethasone for Relapsed Multiple Myeloma. N Engl J Med. 2014;372(2):142–152. [DOI] [PubMed] [Google Scholar]