

Recurrent cytogenetic abnormalities in chronic lymphocytic leukemia (CLL), namely deletions of chromosomes 11q, 13q, 17p and trisomy 12 (+12), define subgroups of patients with different clinical behavior and response to treatment.1 We and others previously reported a minor proportion of CLL cases with co-existing trisomies of chromosomes 12 and 19 who share specific clinico-biological characteristics.2–4 However, since the cohort was small, no definitive conclusions could be drawn. Here, we analyzed a large, multi-institutional series. We confirm and significantly extend previous observations through the identification of subgroups of +12 CLL cases harboring particular concurrent trisomies demonstrating distinctive clinico-biological profiles. We analyzed an unselected cohort of 4486 CLL patients with available classic cytogenetic (n=4285) or high-density 250K single nucleotide polymorphism (SNP)-array (n=201) data. We identified 712 cases (16% of the cohort) carrying +12.5 Median time from diagnosis to cytogenetic/SNP analysis was 1.5 months (range 0–194); the majority of cases included in survival analysis were untreated prior to testing (94%). The study was approved by the local Ethics Review Committees. Details of the study cohort and the methodologies used are provided in the Online Supplementary Appendix.
Of the 712 +12 CLL cases, 86 [12% (or 2% of the entire cohort)] harbored multiple trisomies; 68 of these 86 cases [78% (or 1.5% of the entire cohort)] had co-existing +19 (+12+19 CLL), while the remaining 18 of 86 cases [22% (or 0.5% of the entire cohort)] were negative for +19 and instead carried other co-existing extra chromosome(s) (+12,+other-non19 CLL) (Online Supplementary Table S1 and Figure S1).
Amongst +12+19 cases, 49 of 68 (72%) harbored additional numerical and/or structural aberrations. Trisomy 18 was the predominant co-existing abnormality and was detected in 42 of 68 cases (62%) (Figure 1A). Comparison of +12+19 cases with/without +18 revealed no significant differences (regarding age and stage at diagnosis, sex, IGHV mutational status, CD38 expression and clinical outcome (data not shown), suggesting that +18 might represent a secondary event, probably related to clonal evolution. This claim is also supported by our unpublished FISH data using chromosome 12 and 18 centromeric probes in cases with +12+18+19, revealing cells with +12 alone, cells with +12+18, but not cells with +18 alone. Additional structural abnormalities, primarily concerning chromosome 13q, were observed in 12 of 63 (18%) cases, of whom 10 also carried +18. Only 1 of 23 (4%) and 3 of 59 (5%) cases with available data carried mutations in the NOTCH1 and TP53 genes, respectively;6,7 none of these cases carried +18.
Figure 1.

Genomic profile of: (A) +12,+19 and (B) +12,+other-non+19 CLL. The rows correspond to the specific lesion indicated while the columns represent individual patients. Positive (black), negative (dark gray), no data (light gray). del(13q): deletion of chromosome 13q detected by FISH, del(11q): deletion of chromosome 11q detected by FISH; TP53 abnormality: deletion of chromosome 17p and/or TP53 mutation.
The +12+19 CLL subgroup concerned relatively young patients (median age at diagnosis 59 years). In keeping with our previous report, all +12+19 CLL cases with available data (n=23) expressed surface IgG. Considering the low frequency of CLL cases with switched immunoglobulin (IG) in their B-cell receptors (BcR), this finding is highly suggestive of a particular immunopathogenetic process in this patient subgroup.8 This claim was further supported by the remarkable bias to lambda light chain expression in this particular cytogenetic subgroup [22 of 32 cases (69%) with available data], raising the intriguing possibility that the respective clonogenic progenitors may have been subject to light chain receptor editing.9,10
All but one +12+19 CLL case with available information (47 of 48, 98%) carried immunoglobulin heavy variable (IGHV) genes impacted to some degree by somatic hypermutation (SHM), leading to IGHV genes with less than 100% germline identity. Following the 98% germline identity cut-off value, only 2 of 48 (4%) cases were classified as IG-unmutated CLL (U-CLL), whereas the remaining 46 carried numerous SHMs and were classified as IG-mutated CLL (M-CLL). Despite this, and in keeping with our previous study, the majority of +12+19 cases (41 of 60, 68%) expressed CD38.2 Unexpectedly, 23 of 37 (62%) +12+19 cases were found to express monoclonal paraprotein detectable by immunofixation electrophoresis with identical heavy (gamma) and light chain to that present on the surface of the circulating CLL cells. This is significantly higher compared to generic CLL cohorts and merits further investigation,11 especially since, so far, paraproteinemia in CLL has not been associated with a particular cytogenetic aberration. Interestingly, no differences were identified regarding other clinico-biological features, including CD38 positivity, in +12+19 cases with/without paraproteinemia.
Turning to the +12+other-non+19 CLL subgroup, the co-existing cytogenetic aberrations were exclusively numerical: trisomy 3 predominated (7 of 18 cases, 39%), followed by trisomies 18 and 22 in 6 (33%) and 4 (22%) cases, respectively (Figure 1B). Similar to that reported above, most cases (7 of 9, 78%) belonged to the M-CLL category and expressed CD38 (11 of 17 cases, 65%). Recurrent gene mutations were very rare, indeed only 1 of 13 (8%) patients carried a TP53 mutation, whereas no NOTCH1 mutations were identified.6
Perhaps the most notable features of the +12+other-non+19 subgroup concerned the high incidence of clinical/laboratory autoimmune manifestations12 (4 of 9 cases with available information, 45%) and other malignancies13 (5 of 10 cases with additional information, 50%). High incidence of clinical/laboratory autoimmune manifestations included: autoimmune hemolytic anemia (n=2), a positive direct antiglobulin (n=1), and rheumatoid factor test (n=1); 3 of 4 of these cases never required treatment for their CLL. Other co-existing malignancies concerned: prostate cancer (n=2), bladder cancer (n=1), adenocarcinoma of the uterus (n=1), and basal cell carcinoma of the skin (n=1); one of these 5 cases never received any treatment for CLL, while 3 of the remaining 4 were diagnosed with the other malignancy prior to receiving any CLL-specific treatment. Altogether, the link with malignancy identified in our study is intriguing, however, it does require further investigation in larger patient cohorts.
Comparative assessment of the two subgroups reported above, namely +12+19 CLL and +12+other-non+19 CLL, revealed significant differences regarding heavy IgG-isotype expression (P<0.0001) as well as the incidence of del(13q) (P=0.001), autoimmune manifestations (P=0.013), and second malignancies (P=0.018) (Table 1). Comparisons were also made to 65 cases of the present series carrying isolated +12 as detected by classic cytogenetic analysis, illustrating that the two subgroups reported here display distinct subgroup-biased profiles (Table 1). In particular, both subgroups were enriched for M-CLL compared to isolated +12 CLL (P<0.001 and P=0.032 for +12+19 and +12+other-non+19, respectively). The +12+19 CLL subgroup also differed significantly from isolated +12 cases regarding age at diagnosis (younger in the +12+19 CLL subgroup), higher incidence of IgG-switched heavy and lambda light chains, CD38 positivity as well as higher incidence of co-existing del(13q) (P=0.001). As regards the +12+other-non+19 CLL subgroup, comparison to isolated +12 cases disclosed a significantly higher incidence of autoimmune manifestations and other malignancies. Finally, both subgroups experienced a more indolent clinical course compared to cases with isolated +12, reflected in a significantly longer overall survival (OS). In fact, whereas a median OS of 7.9 years was observed for cases with isolated +12, the median OS for +12+other-non+19 subgroup was 16 years, while that of the +12+19 subgroup had not yet been reached at the time of writing (P=0.033 and P=0.013, respectively) (Figure 2). These findings corroborate previous reports that cytogenetic complexity defined by solely numerical aberrations within CLL should not automatically be considered to be an unfavorable prognostic marker. That said, it should be acknowledged that the favorable clinical outcome within +12,+19 and +12,+other-non19 subgroups might be attributed to their enrichment for M-CLL (Online Supplementary Figure S2).
Table 1.
Comparison of +12+19 versus +12+other-non+19 versus isolated +12 cases.
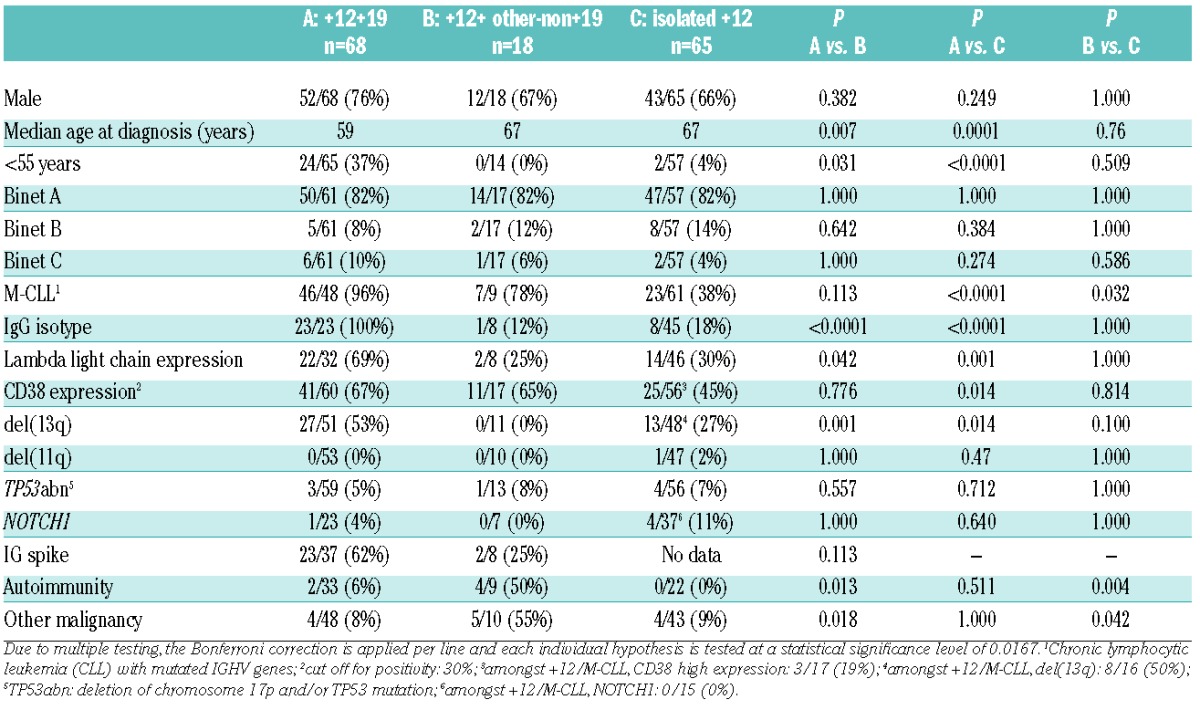
Figure 2.
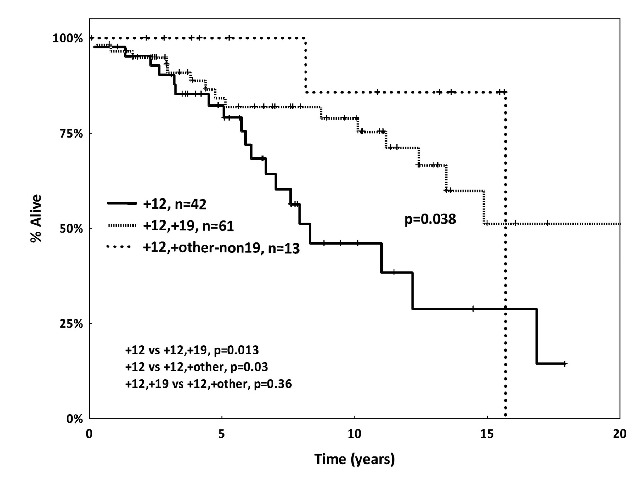
Kaplan-Meier curves for overall survival (OS). The +12,+19 and +12+other-non19 chronic lymphocytic leukemia (CLL) cases demonstrated significantly prolonged OS compared to CLL cases with isolated +12 on classic cytogenetic analysis.
Amongst various host- and tumor-related parameters assessed for their prognostic/predictive relevance, cytogenetic aberrations and recurrent gene mutations have attracted the greatest interest.7,14,15 Trisomy 12 is the second most frequent recurrent chromosomal aberration in CLL and is associated with clinical and biological heterogeneity, potentially linked to the presence of additional genomic aberrations. This concept is reinforced by our present findings regarding the biological background and clinical presentation/outcome of subgroups of +12 CLL patients defined by the presence of extra trisomies. These subgroups seem to differ from patients with isolated +12, while also exhibiting a constellation of biological and clinical features whose co-occurrence is not likely to be incidental.
This conclusion is also supported by our query for CLL cases with trisomy 19 in the Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer (available from: http://cgap.nci.nih.gov/Chromosomes/Mitelman) which retrieved 66 cases, of which 56 (85%) displayed a co-existing +12. Taking all the above into consideration, +19 in CLL appears to be heavily biased to patients carrying +12, suggesting a unique pathway of clonal evolution. As for the +12+other-non+19 subgroup, although caution is warranted due to the low number of cases, the lack of any structural chromosomal aberration and paucity of recurrent gene mutations is noteworthy.
In conclusion, we report the existence of subgroups within +12 CLL defined by the presence of extra trisomies, associated with subgroup-biased profiles of potential clinical relevance. The biological mechanisms underlying both the acquisition of additional chromosomes and, in particular, the specific phenotypes of these subgroups, still have to be clarified.
Acknowledgments
We would like to thank the members of the Spanish Cooperative Group for Hematological Cytogenetics for providing clinical and biological data, and Maria Gaitatzi, Zografia Lazarou, Olga Asteriou, Kristina Durechova and Eva Diviskova for performing cytogenetic and FISH analysis.
Footnotes
Funding: this project was supported in part by the ENosAI project (code 09SYN-13-880) co-funded by the EU and the General Secretariat for Research and Technology of Greece; the KRIPIS action, funded by the General Secretariat for Research and Technology of Greece; the EU Seventh Framework Programme under the “Capacities” specific programme; H2020 “AEGLE, An analytics framework for integrated and personalized healthcare services in Europe”, by the EU; H2020 “MEDGENET”, Medical Genomics and Epigenomics Network“, by the EU; the Swedish Cancer Society, the Swedish Research Council, the Lion’s Cancer Research Foundation, and Selander’s Foundation, Uppsala; research projects CEITEC CZ.1.05/1.1.00/02.68 and MZ CR projects NT13493-4/2012, AZV 15-31834A and AZV 15-30015A; Bloodwise (11052, 12036, 12050, 14027); the Kay Kendall Leukaemia Fund (873); Wessex Medical Research and the Bournemouth Leukaemia Fund, with infrastructure support from a Cancer Research-UK centre grant (C34999/A18087). TM is recipient of a Marie Sklodowska-Curie individual fellowship (grant agreement n. 702714), funded by the EU H2020 research and innovation program.
The online version of this letter has a Supplementary Appendix.
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Dohner H, Stilgenbauer S, Benner A, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000; 343(26):1910–1916. [DOI] [PubMed] [Google Scholar]
- 2.Ibbotson R, Athanasiadou A, Sutton LA, et al. Coexistence of trisomies of chromosomes 12 and 19 in chronic lymphocytic leukemia occurs exclusively in the rare IgG-positive variant. Leukemia. 2012; 26(1):170–172. [DOI] [PubMed] [Google Scholar]
- 3.Schwaenen C, Nessling M, Wessendorf S, et al. Automated array-based genomic profiling in chronic lymphocytic leukemia: development of a clinical tool and discovery of recurrent genomic alterations. Proc Natl Acad Sci USA. 2004;101(4):1039–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sellmann L, Gesk S, Walter C, et al. Trisomy 19 is associated with trisomy 12 and mutated IGHV genes in B-chronic lymphocytic leukaemia. Br J Haematol. 2007;138(2):217–220. [DOI] [PubMed] [Google Scholar]
- 5.Gunnarsson R, Mansouri L, Isaksson A, et al. Array-based genomic screening at diagnosis and during follow-up in chronic lymphocytic leukemia. Haematologica. 2011;96(8):1161–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Del Giudice I, Rossi D, Chiaretti S, et al. NOTCH1 mutations in +12 chronic lymphocytic leukemia (CLL) confer an unfavorable prognosis, induce a distinctive transcriptional profiling and refine the intermediate prognosis of +12 CLL. Haematologica. 2012;97(3):437–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baliakas P, Hadzidimitriou A, Sutton LA, et al. Recurrent mutations refine prognosis in chronic lymphocytic leukemia. Leukemia. 2015; 29(2):329–336. [DOI] [PubMed] [Google Scholar]
- 8.Vardi A, Agathangelidis A, Sutton LA, et al. IgG-switched CLL has a distinct immunogenetic signature from the common MD variant: ontogenetic implications. Clin Cancer Res. 2014;20(2):323–330. [DOI] [PubMed] [Google Scholar]
- 9.Belessi C, Stamatopoulos K, Hadzidimitriou A, et al. Analysis of expressed and non-expressed IGK locus rearrangements in chronic lymphocytic leukemia. Mol Med. 2005;11(1–12):52–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hadzidimitriou A, Darzentas N, Murray F, et al. Evidence for the significant role of immunoglobulin light chains in antigen recognition and selection in chronic lymphocytic leukemia. Blood. 2009; 113(2):403–411. [DOI] [PubMed] [Google Scholar]
- 11.Xu W, Wang YH, Fan L, et al. Prognostic significance of serum immunoglobulin paraprotein in patients with chronic lymphocytic leukemia. Leuk Res. 2011;35(8):1060–1065. [DOI] [PubMed] [Google Scholar]
- 12.Dearden C. Disease-specific complications of chronic lymphocytic leukemia. Hematology Am Soc Hematol Educ Program. 2008;450–456. [DOI] [PubMed] [Google Scholar]
- 13.Molica S. Second neoplasms in chronic lymphocytic leukemia: incidence and pathogenesis with emphasis on the role of different therapies. Leuk Lymphoma. 2005;46(1):49–54. [DOI] [PubMed] [Google Scholar]
- 14.Baliakas P, Iskas M, Gardiner A, et al. Chromosomal translocations and karyotype complexity in chronic lymphocytic leukemia: a systematic reappraisal of classic cytogenetic data. Am J Hematol. 2014; 89(3):249–255. [DOI] [PubMed] [Google Scholar]
- 15.Villamor N, Conde L, Martinez-Trillos A, et al. NOTCH1 mutations identify a genetic subgroup of chronic lymphocytic leukemia patients with high risk of transformation and poor outcome. Leukemia. 2013;27(5):1100–1106. [DOI] [PubMed] [Google Scholar]