

ABSTRACT
The Fanconi anemia (FA) pathway regulates DNA inter-strand crosslink (ICL) repair. Despite our greater understanding of the role of FA in ICL repair, its function in the preventing spontaneous genome instability is not well understood. Here, we show that depletion of replication protein A (RPA) activates the FA pathway. RPA1 deficiency increases chromatin recruitment of FA core complex, leading to FANCD2 monoubiquitination (FANCD2-Ub) and foci formation in the absence of DNA damaging agents. Importantly, ATR depletion, but not ATM, abolished RPA1 depletion-induced FANCD2-Ub, suggesting that ATR activation mediated FANCD2-Ub. Interestingly, we found that depletion of hSSB1/2-INTS3, a single-stranded DNA-binding protein complex, induces FANCD2-Ub, like RPA1 depletion. More interestingly, depletion of either RPA1 or INTS3 caused increased accumulation of DNA damage in FA pathway deficient cell lines. Taken together, these results indicate that RPA deficiency induces activation of the FA pathway in an ATR-dependent manner, which may play a role in the genome maintenance.
KEYWORDS: ATR, fanconi anemia, FANCD2 monoubiquitination, genome instability, replication protein A (RPA)
Introduction
DNA interstrand crosslinks (ICL) are DNA lesions where the bases of opposing DNA strands are covalently linked, preventing DNA replication fork progression. ICL are corrected by DNA repair proteins including components of the Fanconi anemia (FA) pathway.1,2 FA is a rare genetic disorder characterized by bone marrow failure, congenital malformations and cancer susceptibility,3 and currently 18 FA genes have been identified (FANC-A, -B, -C, -D1/BRCA2, -D2, -E, -F, -G, -I, -J/BRIP1, -L, -M, -N/PALB2, -O/RAD51C, -P/SLX4, -Q/XPF, -S/BRCA1 and –T/UBE2T).1,4-9 The FA pathway consists of 3 distinct functional groups: the FA core complex (FANCA, B, C, E, F, G, L, M and N), the ID complex (FANCI/FANCD2) and the downstream effector proteins (FANCD1, J, N, O, P, Q and S). In response to DNA damaging agents, and during S-phase of the cell cycle, ID complex is monoubiquitinated by FA core complex, a key step in the FA pathway.10-12 Monoubiquitinated ID complex is subsequently targeted into DNA repair foci containing downstream FA proteins as well as other DNA repair proteins.2
RPA is a single-stranded DNA (ssDNA)-binding protein complex composed of 3 subunits of p70 (RPA1), p34 (RPA2) and p14 (RPA3), and plays an essential role in DNA replication, repair and recombination.13,14 In response to replication stress, ATM and Rad3-related kinase (ATR) is recruited to RPA-bound ssDNA, generated at sites of stalled DNA replication forks and subsequently activated by additional ATR regulators including Rad17, Rad9 and TopBP1.15-17 Therefore, it is widely accepted that RPA is essential for ATR-mediated DNA damage response.
In response to various genotoxic stresses, many of the FA proteins are phosphorylated by ATM and ATR leading to proper cellular response to DNA damage.12,18-22 More importantly, it has been demonstrated that ATR or RPA is required for efficient FANCD2-Ub and nuclear foci assembly following exposure to DNA damaging agents,23 suggesting the FA pathway intersects with ATM and ATR-mediated DNA damage signaling.
Unexpectedly, here we found that RPA1 depletion induced FANCD2-Ub in the absence of DNA damaging agents. Following acute depletion of RPA1, increased chromatin association of FA core complex and FANCD2 nuclear foci were observed, indicating activation of the FA pathway. Moreover, RPA1 depletion induced phosphorylation of ATM/ATR substrates, including Chk1, Chk2, Rad 17 and TopBP1 with a concomitant increase of γ-H2AX. Notably, induction of FANCD2-Ub by RPA1 depletion was efficiently suppressed by ATR depletion, but not ATM. Lastly, we observed that single-stranded DNA-binding protein complex hSSB1/2-INTS3 deficiency also induced FANCD2-Ub. Furthermore, depletion of RPA1 or INTS3 induced higher level of γ-H2AX in FA pathway deficient cells. Taken together, our results suggest that RPA1 deficiency induces activation of the FA pathway in an ATR-dependent manner, which may contribute to protect genome stability under replication stress.
Results
Identification of siRNA targets inducing spontaneous genome instability and FANCD2 –Ub
The prevention of genome instability and cancer depends on the functions of a complex network of DNA damage response. It is well known that FA pathway plays an essential role in ICL repair,1,24 but more remains to be elucidated about its role in the preventing spontaneous genome instability. To further understand functional significance of the FA pathway in response to spontaneous DNA damage, we aimed to determine the level of FANCD2-Ub following depletion of gene products that cause spontaneous genome instability. U2OS cells transfected with siRNAs directed against cell cycle and DNA repair genes were analyzed by immunoblotting using FANCD2-monoubiquitination (FANCD2-Ub) and H2AX phosphorylation (γ-H2AX) as read-outs (Fig. 1 and Fig. S1). FANCD2-Ub serves as an indicator of the FA pathway activation and γ-H2AX serves as an early marker of genomic instability. The genes targeted in this study were previously demonstrated to mediate genome stability in human cells.25 The double bands of the FANCD2 blot correspond to FANCD2-L (monoubiquitinated FANCD2; upper band) and FANCD2-S (non-monoubiquitinated FANCD2; lower band), respectively. Notably, RPA1 depletion significantly increased the level of FANCD2-Ub compared to that in control siRNA-transfected cells (Fig. 1A, B, lane 2). The level of FANCD2-Ub after RPA1 depletion was correlated with the levels of Chk1 phosphorylation (Chk1 S345), Chk2 phosphorylation (Chk2 T68) and γ-H2AX, suggesting that DNA damage signaling was activated after RPA1 depletion. While RPA1 depletion induces FANCD2-Ub, we failed to observe any significant change in FANCD2-Ub after RPA2 depletion (another component of the RPA complex), which may be due to insufficient depletion of RPA2 (Fig. 1A, lane 3). Depletion of BACH1 (FANCJ), PALB2 (FANCN), and Chk1 significantly increased basal level of FANCD2-Ub (Fig. 1A, lanes 8, 11, 13), although the increases of FANCD2-Ub were much lower than that in RPA1 depletion. TopBP1 and Cdc2 have important roles in both DNA replication and checkpoint regulation. We did not observe any significant changes in FANCD2-Ub levels following depletion of either TopBP1 or Cdc2 (Fig. 1B, lanes 3, 5). In addition, we newly found that depletion of Timeless (Tim1), a replisome component that is required for efficient DNA replication,26,27 significantly increased the basal level of FANCD2-Ub (Fig. 1B, lane 10).
Figure 1.
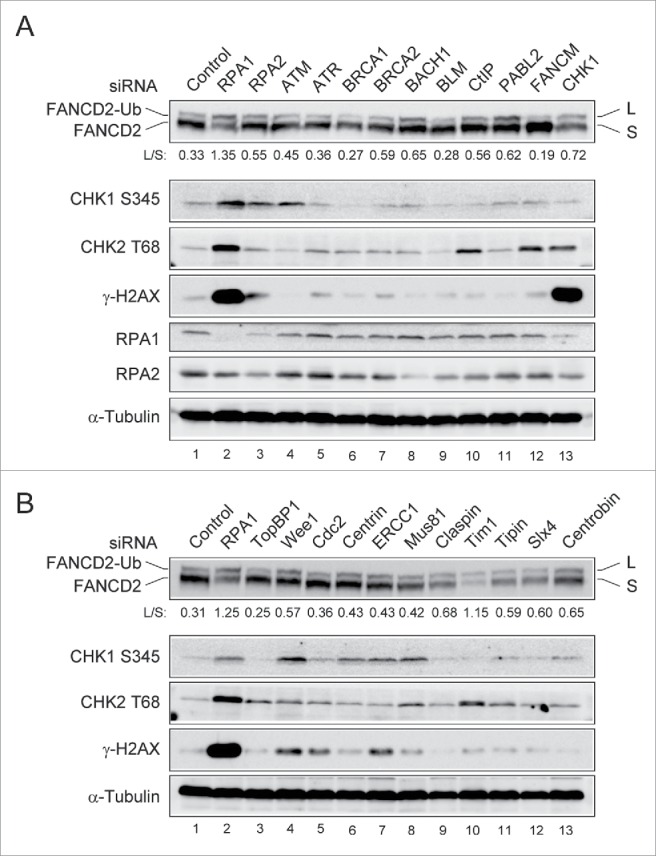
Identification of siRNA target inducing spontaneous genome instability and FANCD2-Ub. (A-B) U2OS cells were transfected with siRNAs targeting cell cycle and DNA repair genes. 72 hr after transfection, whole-cell lysates were prepared and immunoblotted using indicated antibodies. The levels of L (monoubiquitinated) and S (non-ubiquitinated) isoforms of FANCD2 were quantified using ImageJ software and are expressed as the L: S ratio below each sample.
RPA1 depletion induces FANCD2-Ub before the appearance of γ-H2AX
Since it has been demonstrated RPA is required for FANCD2-Ub in response to DNA damaging agents,23 the finding that RPA1 deficiency induced FANCD2-Ub was unexpected. To rule out off-target effects, different siRNA duplexes against RPA1 (RPA1 #1 and RPA1 #2) were tested for induction of FANCD2-Ub (Fig. 2A). At 24 h after transfection, the cell cycle profiles of RPA1-depleted cells were similar to that in control siRNA-transfected cells (Fig. S2A, B). At later time points, RPA1-depleted cells displayed late S and G2/M arrest. Immunoblotting showed that the expression of RPA1 was reduced by more than 90% within 48 h after transfection and RPA1 depletion apparently increased FANCD2-Ub (Fig. 2A, lanes 4-6). Interestingly, while the level of FANCD2-Ub peaked at around 48 h after transfection, overall level of γ-H2AX was still low at the time (Fig. 2A and Fig. S2C), indicating relatively low levels of DNA damage at 48h after RPA1 depletion. These results suggest that FANCD2-Ub may occur during early stages of DNA damage signaling in response to RPA1 depletion.
Figure 2.
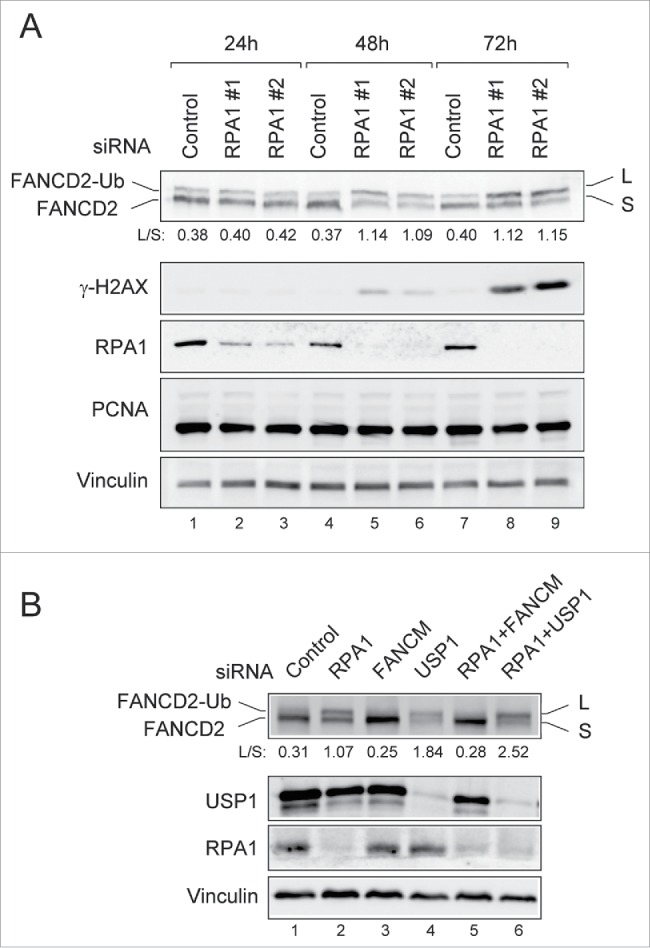
RPA1 depletion induces FANCD2-Ub in the absence of DNA damaging agents. (A) U2OS cells transfected with either control siRNA or RPA1-specific siRNAs were collected at 24, 48 h, and 72h after transfection. Whole cell lysates were analyzed by immunoblotting with indicated antibodies. The FANCD2-Ub: FANCD2 ratios (L: S) are indicated below each sample. (B) U2OS cells were transfected with indicated siRNAs. Whole-cell lysates were prepared 72 hr after transfection and analyzed with indicated antibodies. The FANCD2-Ub: FANCD2 ratios (L: S) are indicated below each sample.
RPA1 depletion-induced FANCD2-Ub is dependent on FA core complex
USP1 is a deubiquitinating enzyme of FANCD2 and PCNA, and USP1 depletion results in elevated levels of FANCD2-Ub and PCNA-Ub.28-31 We therefore tested if USP1 is affected in RPA1-depleted cells (Fig. 2B). However, we did not observe any significant differences in USP1 levels between control siRNA and RPA1 siRNA-treated cells. Moreover, no PCNA-Ub was detected after RPA1 depletion (Fig. 2A), suggesting that increased FANCD2-Ub in RPA1 depleted cells was not due to the deficiency of USP1. Contrary to this, co-depletion of RPA1 with FANCM, a component of FA core complex, reduced the level of FACND2-Ub in RPA1 depleted cells (Fig. 2B, lane 5), indicating that induction of FANCD2-Ub following RPA1 depletion requires FA core complex.
RPA1 depletion increases chromatin association of FA core complex
As we previously reported, chromatin localization of FA core complex is required for FANCD2-Ub.32,33 We next examined chromatin localization of FANCA, a component of FA core complex, by using biochemical cell fractionation (Fig. 3A). The “S (Sup)” fraction contains cytoplasmic and nucleoplasmic proteins, and the “P (Ppt)” fraction contains proteins bound to nuclear structure (eg. chromatin and nuclear matrix proteins). In RPA1- depleted cells, slightly increased level of FANCA was detected in the chromatin fraction compared to control siRNA-treated cells (Fig. 3B, compare lanes 1, 2 with lanes 3, 4), indicating increased chromatin association of the FA core complex in response to RPA1 depletion. As a positive control, treatment with mitomycin C (MMC), an ICL-inducing agent, displayed increased amount of FANCA and FANCD2-Ub in the chromatin fraction (Fig. 3B, lanes 5, 6). To further examine the chromatin localization of FA core complex after RPA1 depletion, Flag-tagged FANCL, a component of FA core complex, was ectopically expressed in RPA1 depleted cells (Fig. 3C). Cells were fractionated into “S1” (cytoplasmic and nucleoplasmic proteins) and “S2” (300 mM NaCl-extractable chromatin-bound proteins) fractions (Fig. 3A), then co-immunoprecipitated proteins with anti-FLAG antibody were analyzed (Fig. 3D). FANCM, in complex with FAAP24, is a platform for recruiting the rest of the FA core complex to chromatin.32 In control siRNA-transfected cells (Fig. 3D, lanes 1-4), an interaction of FANCA-FANCL proteins was detected in “S1” fraction (lane 3), and the interaction of FANCM-FANCA-FANCL proteins was preferentially detected in “S2” fraction (lane 4). After RPA1 depletion (lanes 5-8), the level of FANCM-FANCA-FANCL interaction in “S2” was significantly increased compared to control siRNA-treated cells (compare lane 4 with lane 8), whereas the level of FANCA-FANCL interaction in “S1” was conversely reduced (compare lane 3 with lane 7), indicating increased chromatin association of FANCA and FANCL in response to RPA1 depletion. A similar increase in the level of chromatin-bound FANCM-FANCA-FANCL interaction was detected in MMC-treated cells (lanes 9-12). Taken together, these results indicate that RPA1 depletion induces chromatin association and activation of the FA core complex that leads to FANCD2-Ub.
Figure 3.
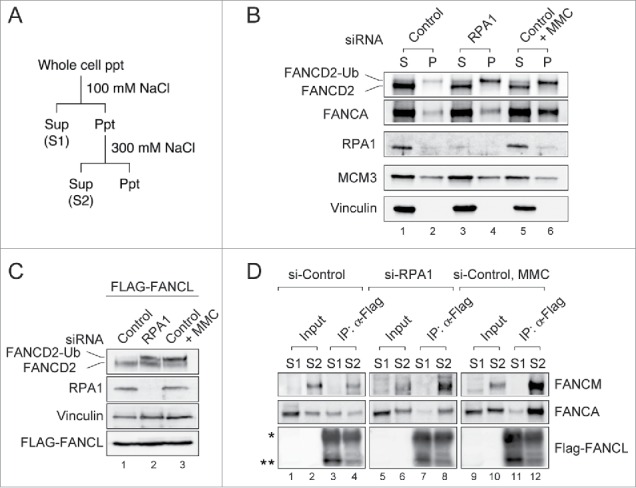
RPA1 depletion increases the chromatin association of FA core complex. (A) Experimental protocol used to prepare cell extracts described in panel B-D. (B) U2OS cells were transfected with either control or RPA1-specific siRNA. 72 hr after transfection, cells were fractionated into S (Sup) & P (Ppt) and immunoblotted for the indicated proteins. MCM3 and Vinculin were used as loading controls. (C-D) HeLa cells expressing FLAG-tagged FANCL were transfected with either control siRNA or RPA1-specific siRNA and cultured for 72hr. (C) Whole-cell lysates were prepared and analyzed with indicated antibodies. (D) Cells were fractionated into S1 and S2. Each fraction was immunoprecipitated with FLAG-M2 agarose beads, and the precipitates were immunoblotted with the indicated antibodies. The asterisk indicates the IgG heavy chain and the double asterisk shows the position of FLAG-tagged FANCL. Entire images come from the identical gel.
RPA1 deficiency forms FANCD2 nuclear foci that colocalize with CtIP
Monoubiquitinated FANCD2 and FANCI are targeted to chromatin and form DNA repair foci in response to DNA damage.10-12 Given that RPA1 depletion resulted in increased FANCD2-Ub, we next sought to examine whether RPA1 depletion-induced FANCD2-Ub is functionally intact. To this end, FANCD2 nuclear foci formation was examined after RPA1 depletion (Fig. 4A, B). Control siRNA-treated cells displayed only weak FANCD2 immunoreactivity with less than 3% of cells showing more than 10 FANCD2 foci. In contrast, RPA1depletion significantly increased the frequency of cells showing bright FANCD2 foci (Fig. 4A, upper panels), although the frequency of FANCD2 foci-positive cells and the number of foci per cell were still lower than those in MMC-treated cells (Fig. 4A, lower panels). Interestingly, this was in sharp contrast to USP1 depletion, which shows deficient FANCD2 foci formation despite increased level of FANCD2-Ub.28,30 Recent study demonstrated that CtIP mediates replication fork recovery in a FANCD2-regulated manner.34 We therefore tested whether FANCD2 and CtIP colocalize in nuclear foci after RPA1 depletion (Fig. 4C). In RPA1 depleted cells, FANCD2 and CtIP exhibited significant foci colocalization compared to control cells. Taken together, these results indicate that FANCD2-Ub induced by RPA1 depletion accumulates on chromatin and forms nuclear foci with other DNA repair proteins.
Figure 4.
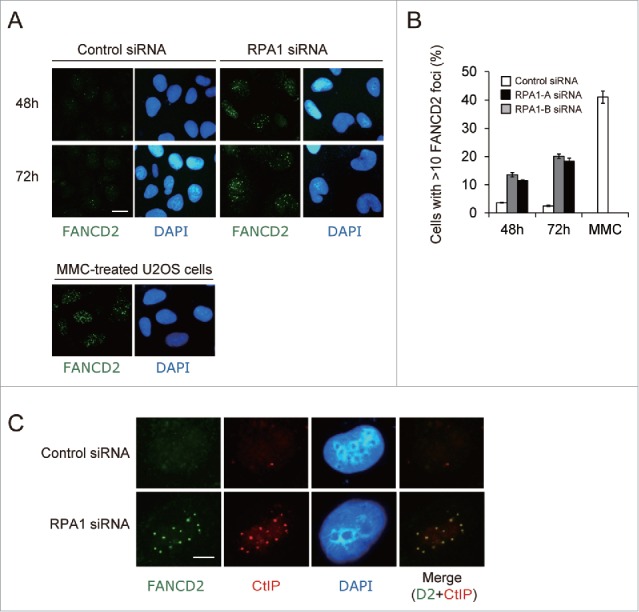
RPA1 depletion-induced FANCD2-Ub forms nuclear foci. (A) U2OS cells were transfected with either control siRNA or RPA1-specific siRNAs and then stained with anti-FAND2 antibody at 48 h and 72 hr after transfection. As a positive control for FANCD2 foci formation, U2OS cells were treated overnight with MMC (50 ng/ml). Scale bars, 10 μm. Representative fields are shown. (B) Quantification of cells with FANCD2 foci (>10 bright foci per cell). Values represent the mean ± SEM, examined at least 200 nuclei each in 3 independent experiments.(C) U2OS cells were transfected with either control siRNA or RPA1-specific siRNAs and then double stained with anti-FANCD2 antibody (green) and anti-CtIP antibody (red) at 72 hr after transfection. Scale bars, 5 μm. Representative fields are shown.
ATR activation in response to RPA1 depletion mediates FANCD2-Ub
It has been shown that RPA deficiency causes spontaneous DSBs, ATM-dependent G2/M arrest, and apoptotic cell death.35 Consistent with previous report, we observed increased phosphorylation of Chk1, Chk2, Rad17 and TopBP1 after RPA1 depletion, indicating ATM or ATR activation (Fig. 5A). Interestingly, we observed a significantly increased mobility shift of FANCM after RPA1 depletion, which may due to phosphorylation of FANCM. As it was recently reported that FANCM is phosphorylated in response to genotoxic stress and this effect is ATR-dependent,20 it is possible that ATR is activated in RPA1 depleted cells. We therefore attempted to identify the kinase(s) involved in FANCD2-Ub induced by RPA1 depletion. To this end, depletion of ATM or ATR was examined for its ability to inhibit RPA1 depletion-induced FANCD2-Ub (Fig. 5B, C). Co-depletion of either ATM or ATR with RPA1 efficiently inhibited RPA1 depletion-induced Chk1 S345 and Chk2 T68 (Fig. 5B, lanes 5, 6). In contrast, RPA1 depletion-induced FANCD2-Ub was selectively reduced by ATR depletion (more than 90% reduction) but not ATM depletion (Fig. 5B, C). These results suggest that ATR is the major kinase responsible for FANCD2-Ub induced by RPA1 depletion.
Figure 5.
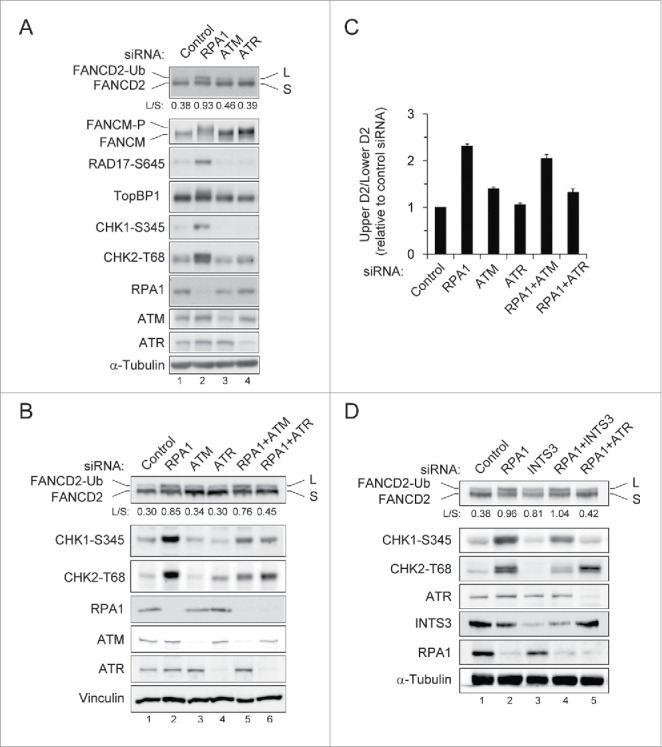
RPA1 depletion-induced FANCD2-Ub is dependent of ATR. (A) HeLa cells were transfected with indicated siRNAs. Whole-cell lysates were prepared 72 hr after transfection and analyzed with indicated antibodies. The FANCD2-Ub: FANCD2 ratios (L: S) are indicated below each sample. (B-C) U2OS cells were transfected with either control siRNA or siRNAs specific for ATM, ATR and RPA1, either individually or in combination. (B) Whole-cell lysates were prepared 72 hr after transfection and analyzed by immunoblotting with indicated antibodies. The FANCD2-Ub: FANCD2 ratios (L: S) are indicated below each sample. (C) Quantification of the data shown in B. The levels of FANCD2-Ub relative to those of control siRNA-transfected cells, which were set as 1.0, are shown in the bar graph. The values represent the mean ± SEM of 3 independent experiments. (D) U2OS cells were transfected with either control siRNA or siRNAs specific for INTS3 and RPA1, either individually or in combination. Whole-cell lysates were prepared at 72hr after siRNA transfection and then immunoblotted with indicated antibodies. The FANCD2-Ub: FANCD2 ratios (L: S) are indicated below each sample.
hSSB1/2-INTS3 is not involved in RPA1 depletion-induced FANCD2-Ub
It has been widely accepted that RPA is essential for ATR activity. How is ATR activated in RPA1-depleted cells? Interestingly, a recent study demonstrated that human ssDNA binding protein complex hSSB1/2-INTS3 can initiate ATR signaling in the absence of RPA.36 hSSB1 is known to function in the ATM-dependent DNA damage signaling and genomic stability.37 INTS3 forms a complex with hSSB1 and controls the hSSB1-mediated DNA damage response.38 We therefore tested if RPA1-induced FANCD2-Ub is affected in cells lacking hSSB1 or INTS3 (Fig 5D and Fig. S3). Depletion of hSSB1 showed a slight accumulation of S phase cells (Fig. S3A) and significantly increased FANCD2-Ub level in comparison to control cells (Fig. S3B, lane 3). Co-depletion of hSSB1 and ATR reduced FANCD2-Ub level (compare lanes 3 with 5), indicating hSSB1 depletion-induced FANCD2-Ub also requires ATR activity. However, depletion of hSSB1 with RPA1 did not change the levels of FANCD2-Ub, Chk1 S345, and Chk2 T68 induced by RPA1 depletion (compare lanes 2 with 4). To further investigate the involvement of hSSB1/2-INTS3 in RPA depletion-induced FANCD2-Ub, INTS3 were depleted along with RPA1 (Fig. 5D). INTS3 co-depletion reduced RPA1 depletion-induced Chk1 S345 and Chk2 T68 (compare lanes 2 with 4), as reported previously.36 However, any significant change in FANCD2-Ub level was not detected, indicating that hSSB1/2-INTS3 is not involved in RPA1 depletion-induced FANCD2-Ub. Therefore, it is unlikely that hSSB1/2-INTS3 compensates for RPA1 deficiency in induction of FANCD2-Ub. It seems more likely that residual low-level of RPA1 after siRNA depletion was sufficient to trigger basal ATR activity for activating the FA pathway.
RPA1 depletion causes an increased DNA damage response in FANCA-deficient patient cells
Given that RPA1 depletion induces the activation of the FA pathway, we speculated that FA pathway deficient cell lines might be hypersensitive to RPA1 inhibition. We therefore examined the effect of RPA1 depletion in FANCA deficient cell line (GM6914) and its isogenic FANCA corrected cell line (GM 6914+FANCA) (Fig. 6). Interestingly, FANCA-deficient cells showed higher levels of Chk1 S345, Chk2 T68 and γ-H2AX than FANCA corrected cells after RPA1 depletion (compare lanes 2 with 6). Notably, INTS3 depletion showed an enhanced level of γ-H2AX in the FANCA deficient cell line (compare lanes 4 with 8), suggesting FA pathway confer tolerance to genotoxic stress resulting from RPA1 or INTS3 depletion. These results therefore suggest that inhibition of RPA or INTS3 may be useful in treating cancers that have defects in FA DNA repair pathway, and further experiments should address this possibility. Taken together, our results collectively suggest that endogenous genome instability due to RPA depletion (insufficiency) leads to activation of the FA pathway through ATR-dependent pathway, which may play a role in genome maintenance.
Figure 6.
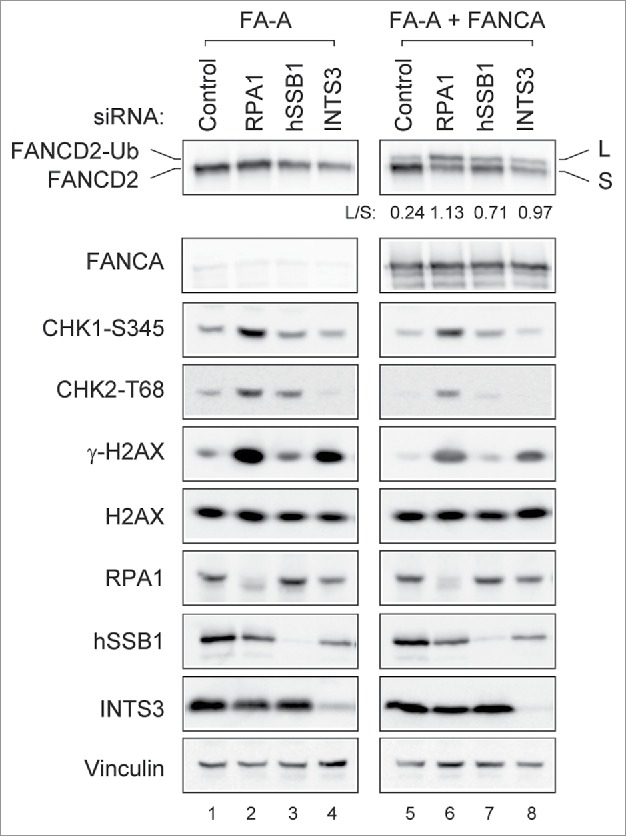
RPA1 depletion induces higher level of γ-H2AX in FA pathway deficient cells. FANCA deficient (GM6914) and isogenic corrected (GM6814+FANCA) cells were transfected with indicated siRNAs. Whole-cell lysates were prepared 72 hr after transfection and analyzed with indicated antibodies. The FANCD2-Ub: FANCD2 ratios (L: S) are indicated below each sample. Each pair of images was collected using the same exposure time for the comparison from identical gel.
Discussion
The FA pathway is required for efficient ICL repair and majority of our understanding has focused on its role in ICL-induced DNA damage response. In addition to its role in ICL repair, it has been recently demonstrated that the FA pathway also contributes to the maintenance of genome integrity by protecting against replication stress 33,39-41
RPA is an essential protein complex, and mediates both DNA replication and DNA repair pathways.13,14 It has been shown that RPA1 depletion causes spontaneous DNA damage and ATM-dependent G2/M checkpoint activation.35,42 In this study we have demonstrated that RPA1 depletion activates the FA pathway in ATR-dependent manner. RPA1 depletion induces chromatin association of FA core complex, FANCD2-Ub, and foci formation. Importantly, induction of FANCD2-Ub following RPA1 depletion was dependent of ATR activity, but not ATM. In addition, we found that second human single-stranded DNA-binding protein complex hSSB1/2-INTS3 deficiency induced FANCD2-Ub. More interestingly, depletion of either RPA1 or INTS3 caused increased accumulation of DNA damage in FA pathway deficient cell lines. Taken together, our results indicate that RPA depletion-induced genome instability (spontaneous replication stress) activates the FA pathway in an ATR-dependent manner, which may play a role in the genome maintenance.
The finding was surprising, since it has been shown that RPA is required for FANCD2-Ub following DNA damage.23 We observed that significantly increased FANCM phosphorylation after RPA1 depletion, perhaps mediated by ATR (Fig. 5A). Moreover, depletion of ATR efficiently suppressed RPA1 depletion-induced FANCD2-Ub (Fig. 5B, C), suggesting that ATR is activated following RPA1 depletion and mediates RPA1 depletion-induced FANCD2-Ub.
It has been widely accepted that RPA is essential for ATR activity. Then, how does RPA1 depletion activate ATR that triggers the activation of the FA pathway? Interestingly, a recent report presented evidence that second human ssDNA binding protein complex hSSB1/2-INTS3 can initiate ATR signaling in the absence of RPA,36 which may be an alternate mechanism that activates ATR signaling. As reported previously,36 we observed that INTS3 depletion reduced RPA1-induced Chk1 phosphorylation after combined depletion of INTS3 and RPA1 (Fig. 5D). However, we failed to detect a significant change of FANCD2-Ub, suggesting that hSSB1/2-INTS3 is not involved in RPA1 depletion-induced FANCD2-Ub. Alternatively, because of inherent problems associated with siRNA-based experiments, it seems more likely that a low threshold level of RPA1 following siRNA depletion was sufficient to activate ATR in our experiments.
ATR-dependent phosphorylation is essential for activation of the FA pathway.39,43 Both FANCD2 and FANCI are substrates of ATR; in particular, the phosphorylation of FANCI by ATR is important for the localization of ID complex to sites of ICLs.18 Besides its role in ICL repair, FANCD2 was recently shown to bind to MCM proteins in response to HU in an ATR-regulated manner, restraining DNA synthesis during the replication stress response.44 It also has been reported that ATR-mediated FANCI phosphorylation regulates dormant origin firing in response to replication stress.45 In addition to FANCD2 and FANCI, multiple components of the FA core complex including FANCM and FANCA are activated by ATR-mediated phosphorylation during replication stress, which is required for functional integrity of the FA pathway.20,22,46
A mouse cell line with a hypomorphic mutation in RPA1 (L230P) displays defective DNA repair and widespread chromosome instability.42 Haploinsufficiency of RPA1 has also been reported clinically: deletions within 17p13.3 chromosomal region, which harbors the RPA1 gene.47 We believe that in such physiological states, where increased genomic instability would be observed, FA pathway could be highly activated. Interestingly, we observed increased checkpoint activation and accumulation of DNA damage in FANCA-deficient patient cells following RPA1 depletion (Fig. 6). Recently, it has been shown that small molecule inhibitors targeting RPA display cytotoxic activity and synergic effects with chemotherapeutic DNA damaging agents such as cisplatin in lung cancer cells.48,49 Since FA pathway deficient cells are hypersensitive to cisplatin treatment, it is possible to think when RPA inhibition is combined with cisplatin treatment, enhanced cytotoxic effect could selectively eradicate the FA pathway-deficient tumor cells. Further experiments will be needed to verify this possibility in the near future.
In summation, we demonstrate that spontaneous replication stress caused by RPA1 depletion (insufficiency) activate the FA pathway in ATR-dependent manner, which may contribute to maintenance of genomic integrity.
Materials and methods
Cell culture and chemical
U2OS and HeLa cells were maintained with 10% FBS/DMEM. Isogenic FANCA deficient (GM6914) and proficient (GM6914+FANCA) cell lines were maintained with 10% FBS/DMEM and puromycin (1 μg/ml). Mitomycin C (MMC) was purchased from Sigma-Aldrich and dissolved in ethanol to make a stock solution of 10 mM.
Antibodies
Antibodies used in this study were as follows; anti-FANCD2, Vinculin, CtIP, Chk1, Wee1, ERCC1, Centrin antibodies (Santa Cruz), anti-α-tubulin and FLAG antibodies (Sigma Aldrich), anti-CHK2 T68, CHK1S345, and RAD17-S645 antibodies (Cell signaling), anti-ATM, TopBP1, MCM3, hSSB1, INTS3, Claspin, PALB2 and USP1 antibodies (Bethyl Laboratories), anti-ATR, RPA1, RPA2 and BRCA1 (Millipore), anti-γ-H2AX (Upstate), anti-Mus81 and Centrobin (Abcam), anti-BACH1 (Novus). Antibodies against FANCM and FANCA were kindly provided by the Fanconi Anemia Research Fund.
Immunofluorescence
Cells grown on the coverslips were pre-extracted with 0.3% Triton X-100 for 1 min, followed by fixation with 3.7% paraformaldehyde for 10 min and permeabilized with PBS + 0.5% Triton X-100 for 5 min. Cells were blocked with 5% BSA/PBS and were incubated with anti-FANCD2 (1:300), anti-CtIP (1:200) and anti-γ-H2AX (1:2000). Alexa 488 and Alexa 568-conjugated secondary antibodies (Invitrogen) were used at a dilution of 1:500. After counter-staining with DAPI, coverslips were mounted and analyzed by fluorescence microscopy.
siRNA transfection
Expression of targeted genes was knocked down by transient expression of siRNA against RPA1-#1 (5′-ccgtgtgacgatcccatgtta-3′), RPA1-#2 (5′ -aacactctatcctctttcatg-3′,17), USP1 (aaccagagacaaactagatca-3′,31), FANCM (aagctcataaagctctcggaa-3,32), ATR (5′-aacctccgtgatgttgcttga-3′,23), ATM (5′-aacatactactcaaagacatt,23), hSSB1 (5′-acccaacaaggcggtgcagaa-3′, INTS3 (5′-agggacaaagtactccagcta-3′ 36). Control siRNA is designed by Qiagen. SiRNA transfection was performed with RNAimax (Invitrogen). The target sequences for the other siRNA oligos used in the Figure 1 are listed in Supplementary Table S1.
Cell-cycle analysis
Samples were collected over indicated time points and fixed in 70% ethanol overnight. For cell cycle analysis, fixed cells were treated with RNase for 20 minutes before addition of 5μg/mL propidium iodide (Sigma-Aldrich). For BrdU incorporation analysis, BrdU was added to media to a final concentration of 10 μM for 1 h before harvesting the cells. Staining with FITC-conjugated α-BrdU antibody (Becton Dickinson) was done according to manufacturer′s protocol. Flow cytometric analysis was then performed using a FACSCalibur flow cytometer (Becton Dickinson).
Cell fractionation and immunoprecipitation
For whole-cell extracts, cell pellets were directly lysed with SDS sample buffer. For biochemical cell fractionation,32 cells were first lysed with 0.1% Triton X-100 CSK buffers for 5 minutes on ice. After centrifugation (1500g for 5 minutes), the supernatant was labeled “S (=S1).” The pellet was washed once more with the same buffer and labeled “P.” For immunoprecipitation, “P” was further extracted with 300 mM NaCl. After centrifugation (15 294g for 20 minutes), the supernatant was labeled “S2.”
S1 and S2 diluted to 150 mM NaCl before immunoprecipitation were incubated with 15 μL anti-FLAG M2 agarose overnight at 4°C on a rocker, and washed 3 times with 0.1% TX-150mCSK buffer.
Supplementary Material
Abbreviations
- FA
fanconi Anemia
- DSB
DNA double-strand break
- ICL
DNA inter-strand crosslink repair
- RPA
replication Protein A
- FANCD2
fanconi anemia complementation group D2
- Ub
ubiquitin
- PCNA
proliferating cell nuclear antigen
- USP1
ubiquitin specific protease 1
- MMC
mitomycin C
- ATM
ataxia telangiectasia mutated
- ATR
ATM and Rad3-related
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are grateful to A. D′Andrea for valuable tools and reagents for this study.
Funding
This work was supported by the National Research Foundation of Korea (Medical Research Council for Gene Regulation) funded by the Ministry of Science, ICT and Future Planning (2011-0030132). This work was supported in part by Basic science Research Program through the National Research Foundation of Korea funded by the Ministry of Education (2014R1A1A2054983).
Notes on contributors
S.W.J and J.K.J conducted all experiments and wrote the manuscript; J.M.K. designed the experiments and wrote the manuscript.
References
- [1].Wang W. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat Rev Genet 2007; 8:735-48; PMID:17768402; http://dx.doi.org/ 10.1038/nrg2159 [DOI] [PubMed] [Google Scholar]
- [2].Kim H, D'Andrea AD. Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev 2012; 26:1393-408; PMID:22751496; http://dx.doi.org/ 10.1101/gad.195248.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Joenje H, Patel KJ. The emerging genetic and molecular basis of Fanconi anaemia. Nat Rev Genet 2001; 2:446-57; PMID:11389461; http://dx.doi.org/ 10.1038/35076590 [DOI] [PubMed] [Google Scholar]
- [4].Bogliolo M, Schuster B, Stoepker C, Derkunt B, Su Y, Raams A, Trujillo JP, Minguillon J, Ramirez MJ, Pujol R, et al.. Mutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia. Am J Hum Genet 2013; 92:800-6; PMID:23623386; http://dx.doi.org/ 10.1016/j.ajhg.2013.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Hira A, Yoshida K, Sato K, Okuno Y, Shiraishi Y, Chiba K, Tanaka H, Miyano S, Shimamoto A, Tahara H, et al.. Mutations in the gene encoding the E2 conjugating enzyme UBE2T cause Fanconi anemia. Am J Hum Genet 2015; 96:1001-7; PMID:26046368; http://dx.doi.org/ 10.1016/j.ajhg.2015.04.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Rickman KA, Lach FP, Abhyankar A, Donovan FX, Sanborn EM, Kennedy JA, Sougnez C, Gabriel SB, Elemento O, Chandrasekharappa SC, et al.. Deficiency of UBE2T, the E2 Ubiquitin ligase necessary for FANCD2 and FANCI Ubiquitination, causes FA-T subtype of fanconi anemia. Cell Rep 2015; 12:35-41; PMID:26119737; http://dx.doi.org/ 10.1016/j.celrep.2015.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Vaz F, Hanenberg H, Schuster B, Barker K, Wiek C, Erven V, Neveling K, Endt D, Kesterton I, Autore F, et al.. Mutation of the RAD51C gene in a Fanconi anemia-like disorder. Nat Genet 2010; 42:406-9; PMID:20400963; http://dx.doi.org/ 10.1038/ng.570 [DOI] [PubMed] [Google Scholar]
- [8].Sawyer SL, Tian L, Kahkonen M, Schwartzentruber J, Kircher M, University of Washington Centre for Mendelian Genomics, FORGE Canada Consortium, Majewski J, Dyment DA, Innes AM, Boycott KM, Moreau LA, et al.. Biallelic mutations in BRCA1 cause a new Fanconi anemia subtype. Cancer Discov 2015; 5:135-42; PMID:25472942; http://dx.doi.org/ 10.1158/2159-8290.CD-14-1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Virts EL, Jankowska A, Mackay C, Glaas MF, Wiek C, Kelich SL, Lottmann N, Kennedy FM, Marchal C, Lehnert E, et al.. AluY-mediated germline deletion, duplication and somatic stem cell reversion in UBE2T defines a new subtype of Fanconi anemia. Hum Mol Genetics 2015; 24:5093-108; http://dx.doi.org/ 10.1093/hmg/ddv227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, D'Andrea AD. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell 2001; 7:249-62; PMID:11239454; http://dx.doi.org/ 10.1016/S1097-2765(01)00173-3 [DOI] [PubMed] [Google Scholar]
- [11].Sims AE, Spiteri E, Sims RJ, 3rd, Arita AG, Lach FP, Landers T, Wurm M, Freund M, Neveling K, Hanenberg H, et al.. FANCI is a second monoubiquitinated member of the Fanconi anemia pathway. Nat Struct Mol Biol 2007; 14:564-7; PMID:17460694; http://dx.doi.org/ 10.1038/nsmb1252 [DOI] [PubMed] [Google Scholar]
- [12].Smogorzewska A, Matsuoka S, Vinciguerra P, McDonald ER 3rd, Hurov KE, Luo J, Ballif BA, Gygi SP, Hofmann K, D'Andrea AD, et al.. Identification of the FANCI Protein, a Monoubiquitinated FANCD2 Paralog Required for DNA Repair. Cell 2007; 129:289-301; PMID:17412408; http://dx.doi.org/ 10.1016/j.cell.2007.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Iftode C, Daniely Y, Borowiec JA. Replication protein A (RPA): the eukaryotic SSB. Crit Rev Biochem Mol Biol 1999; 34:141-80; PMID:10473346; http://dx.doi.org/ 10.1080/10409239991209255 [DOI] [PubMed] [Google Scholar]
- [14].Wold MS. Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu Rev Biochem 1997; 66:61-92; PMID:9242902; http://dx.doi.org/ 10.1146/annurev.biochem.66.1.61 [DOI] [PubMed] [Google Scholar]
- [15].Flynn RL, Zou L. ATR: a master conductor of cellular responses to DNA replication stress. Trends Biochem Sci 2011; 36:133-40; PMID:20947357; http://dx.doi.org/ 10.1016/j.tibs.2010.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol 2008; 9:616-27; PMID:18594563; http://dx.doi.org/ 10.1038/nrm2450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003; 300:1542-8; PMID:12791985; http://dx.doi.org/ 10.1126/science.1083430 [DOI] [PubMed] [Google Scholar]
- [18].Ishiai M, Kitao H, Smogorzewska A, Tomida J, Kinomura A, Uchida E, Saberi A, Kinoshita E, Kinoshita-Kikuta E, Koike T, et al.. FANCI phosphorylation functions as a molecular switch to turn on the Fanconi anemia pathway. Nat Struct Mol Biol 2008; 15:1138-46; PMID:18931676; http://dx.doi.org/ 10.1038/nsmb.1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wang X, Kennedy RD, Ray K, Stuckert P, Ellenberger T, D'Andrea AD. Chk1-mediated phosphorylation of FANCE is required for the Fanconi anemia/BRCA pathway. Mol Cell Biol 2007; 27:3098-108; PMID:17296736; http://dx.doi.org/ 10.1128/MCB.02357-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Singh TR, Ali AM, Paramasivam M, Pradhan A, Wahengbam K, Seidman MM, Meetei AR. ATR-dependent phosphorylation of FANCM at serine 1045 is essential for FANCM functions. Cancer Res 2013; 73:4300-10; PMID:23698467; http://dx.doi.org/ 10.1158/0008-5472.CAN-12-3976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Taniguchi T, Garcia-Higuera I, Xu B, Andreassen PR, Gregory RC, Kim ST, Lane WS, Kastan MB, D'Andrea AD. Convergence of the fanconi anemia and ataxia telangiectasia signaling pathways. Cell 2002; 109:459-72; PMID:12086603; http://dx.doi.org/ 10.1016/S0092-8674(02)00747-X [DOI] [PubMed] [Google Scholar]
- [22].Collins NB, Wilson JB, Bush T, Thomashevski A, Roberts KJ, Jones NJ, Kupfer GM. ATR-dependent phosphorylation of FANCA on serine 1449 after DNA damage is important for FA pathway function. Blood 2009; 113:2181-90; PMID:19109555; http://dx.doi.org/ 10.1182/blood-2008-05-154294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Andreassen PR, D'Andrea AD, Taniguchi T. ATR couples FANCD2 monoubiquitination to the DNA-damage response. Genes Dev 2004; 18:1958-63; PMID:15314022; http://dx.doi.org/ 10.1101/gad.1196104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].D'Andrea AD, Grompe M. The Fanconi anaemia/BRCA pathway. Nat Rev Cancer 2003; 3:23-34; PMID:12509764; http://dx.doi.org/ 10.1038/nrc970 [DOI] [PubMed] [Google Scholar]
- [25].Paulsen RD, Soni DV, Wollman R, Hahn AT, Yee MC, Guan A, Hesley JA, Miller SC, Cromwell EF, Solow-Cordero DE, et al.. A genome-wide siRNA screen reveals diverse cellular processes and pathways that mediate genome stability. Mol Cell 2009; 35:228-39; PMID:19647519; http://dx.doi.org/ 10.1016/j.molcel.2009.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Chou DM, Elledge SJ. Tipin and Timeless form a mutually protective complex required for genotoxic stress resistance and checkpoint function. Proc Natl Acad Sci U S A 2006; 103:18143-7; PMID:17116885; http://dx.doi.org/ 10.1073/pnas.0609251103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Yoshizawa-Sugata N, Masai H. Human Tim/Timeless-interacting protein, Tipin, is required for efficient progression of S phase and DNA replication checkpoint. J Biol Chem 2007; 282:2729-40; PMID:17102137; http://dx.doi.org/ 10.1074/jbc.M605596200 [DOI] [PubMed] [Google Scholar]
- [28].Kim JM, Parmar K, Huang M, Weinstock DM, Ruit CA, Kutok JL, D'Andrea AD. Inactivation of murine Usp1 results in genomic instability and a Fanconi anemia phenotype. Dev Cell 2009; 16:314-20; PMID:19217432; http://dx.doi.org/ 10.1016/j.devcel.2009.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Nijman SM, Huang TT, Dirac AM, Brummelkamp TR, Kerkhoven RM, D'Andrea AD, Bernards R. The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway. Mol Cell 2005; 17:331-9; PMID:15694335; http://dx.doi.org/ 10.1016/j.molcel.2005.01.008 [DOI] [PubMed] [Google Scholar]
- [30].Oestergaard VH, Langevin F, Kuiken HJ, Pace P, Niedzwiedz W, Simpson LJ, Ohzeki M, Takata M, Sale JE, Patel KJ. Deubiquitination of FANCD2 is required for DNA crosslink repair. Mol Cell 2007; 28:798-809; PMID:18082605; http://dx.doi.org/ 10.1016/j.molcel.2007.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Huang TT, Nijman SM, Mirchandani K, Galardy PJ, Cohn MA, Haas W, Gygi SP, Ploegh HL, Bernards R, D'Andrea AD. Regulation of monoubiquitinated PCNA by DUB autocleavage. Nat Cell Biol 2006; 8:339-47; PMID:16531995 [DOI] [PubMed] [Google Scholar]
- [32].Kim JM, Kee Y, Gurtan A, D'Andrea AD. Cell cycle-dependent chromatin loading of the Fanconi anemia core complex by FANCM/FAAP24. Blood 2008; 111:5215-22; PMID:18174376; http://dx.doi.org/ 10.1182/blood-2007-09-113092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Longerich S, Li J, Xiong Y, Sung P, Kupfer GM. Stress and DNA repair biology of the Fanconi anemia pathway. Blood 2014; 124:2812-9; PMID:25237197; http://dx.doi.org/ 10.1182/blood-2014-04-526293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Yeo JE, Lee EH, Hendrickson EA, Sobeck A. CtIP mediates replication fork recovery in a FANCD2-regulated manner. Hum Mol Genetics 2014; 23:3695-705; http://dx.doi.org/ 10.1093/hmg/ddu078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Dodson GE, Shi Y, Tibbetts RS. DNA replication defects, spontaneous DNA damage, and ATM-dependent checkpoint activation in replication protein A-deficient cells. J Biol Chem 2004; 279:34010-4; PMID:15197179; http://dx.doi.org/ 10.1074/jbc.C400242200 [DOI] [PubMed] [Google Scholar]
- [36].Kar A, Kaur M, Ghosh T, Khan MM, Sharma A, Shekhar R, Varshney A, Saxena S. RPA70 depletion induces hSSB1/2-INTS3 complex to initiate ATR signaling. Nucleic Acids Res 2015; 43:4962-74; PMID:25916848; http://dx.doi.org/ 10.1093/nar/gkv369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Richard DJ, Bolderson E, Cubeddu L, Wadsworth RI, Savage K, Sharma GG, Nicolette ML, Tsvetanov S, McIlwraith MJ, Pandita RK, et al.. Single-stranded DNA-binding protein hSSB1 is critical for genomic stability. Nature 2008; 453:677-81; PMID:18449195; http://dx.doi.org/ 10.1038/nature06883 [DOI] [PubMed] [Google Scholar]
- [38].Skaar JR, Richard DJ, Saraf A, Toschi A, Bolderson E, Florens L, Washburn MP, Khanna KK, Pagano M. INTS3 controls the hSSB1-mediated DNA damage response. J Cell Biol 2009; 187:25-32; PMID:19786574; http://dx.doi.org/ 10.1083/jcb.200907026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Jones MJ, Huang TT. The Fanconi anemia pathway in replication stress and DNA crosslink repair. Cell Mol Life Sci 2012; 69:3963-74; PMID:22744751; http://dx.doi.org/ 10.1007/s00018-012-1051-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Raghunandan M, Chaudhury I, Kelich SL, Hanenberg H, Sobeck A. FANCD2, FANCJ and BRCA2 cooperate to promote replication fork recovery independently of the Fanconi Anemia core complex. Cell Cycle 2015; 14:342-53; PMID:25659033; http://dx.doi.org/ 10.4161/15384101.2014.987614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wang LC, Stone S, Hoatlin ME, Gautier J. Fanconi anemia proteins stabilize replication forks. DNA Repair (Amst) 2008; 7:1973-81; PMID:18786657; http://dx.doi.org/ 10.1016/j.dnarep.2008.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Wang Y, Putnam CD, Kane MF, Zhang W, Edelmann L, Russell R, Carrion DV, Chin L, Kucherlapati R, Kolodner RD, et al.. Mutation in Rpa1 results in defective DNA double-strand break repair, chromosomal instability and cancer in mice. Nat Genet 2005; 37:750-5; PMID:15965476; http://dx.doi.org/ 10.1038/ng1587 [DOI] [PubMed] [Google Scholar]
- [43].Moldovan GL, D'Andrea AD. To the rescue: the Fanconi anemia genome stability pathway salvages replication forks. Cancer Cell 2012; 22:5-6; PMID:22789533; http://dx.doi.org/ 10.1016/j.ccr.2012.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Lossaint G, Larroque M, Ribeyre C, Bec N, Larroque C, Decaillet C, Gari K, Constantinou A. FANCD2 binds MCM proteins and controls replisome function upon activation of s phase checkpoint signaling. Mol Cell 2013; 51:678-90; PMID:23993743; http://dx.doi.org/ 10.1016/j.molcel.2013.07.023 [DOI] [PubMed] [Google Scholar]
- [45].Chen YH, Jones MJ, Yin Y, Crist SB, Colnaghi L, Sims RJ 3rd, Rothenberg E, Jallepalli PV, Huang TT. ATR-mediated phosphorylation of FANCI regulates dormant origin firing in response to replication stress. Mol Cell 2015; 58:323-38; PMID:25843623; http://dx.doi.org/ 10.1016/j.molcel.2015.02.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Sobeck A, Stone S, Landais I, de Graaf B, Hoatlin ME. The Fanconi anemia protein FANCM is controlled by FANCD2 and the ATR/ATM pathways. J Biol Chem 2009; 284:25560-8; PMID:19633289; http://dx.doi.org/ 10.1074/jbc.M109.007690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Nagamani SC, Zhang F, Shchelochkov OA, Bi W, Ou Z, Scaglia F, Probst FJ, Shinawi M, Eng C, Hunter JV, et al.. Microdeletions including YWHAE in the Miller-Dieker syndrome region on chromosome 17p13.3 result in facial dysmorphisms, growth restriction, and cognitive impairment. J Medical Genetics 2009; 46:825-33; http://dx.doi.org/ 10.1136/jmg.2009.067637 [DOI] [PubMed] [Google Scholar]
- [48].Neher TM, Bodenmiller D, Fitch RW, Jalal SI, Turchi JJ. Novel irreversible small molecule inhibitors of replication protein A display single-agent activity and synergize with cisplatin. Mol Cancer Therapeutics 2011; 10:1796-806; http://dx.doi.org/ 10.1158/1535-7163.MCT-11-0303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Shuck SC, Turchi JJ. Targeted inhibition of replication protein a reveals cytotoxic activity, synergy with chemotherapeutic DNA-damaging agents, and insight into cellular function. Cancer Res 2010; 70:3189-98; PMID:20395205; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-3422 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.