

ABSTRACT
EZH2, the main catalytic component of the Polycomb Repressive Complex 2 (PRC2) is apparently upregulated in most solid tumors. Furthermore its expression generally associates with poor prognosis. It was proposed that this correlation reflects a causal event, EZH2 mediating the silencing of key tumor suppressor loci. In contrast, we recently showed that EZH2 is dispensable for solid tumor development and that its elevated expression reflects the abnormally high proliferation rate of cancer cells. Here, we investigate the functional association between EZH2 expression and silencing of key tumor suppressor loci and further illustrate the confounding effect of proliferation on EZH2′s association to outcome.
KEYWORDS: breast, EZH2, prostate, Polycomb, prognosis, PRC2, solid tumors, tumor suppressor
Introduction
Elucidating the molecular mechanisms controlling progressive malignant transformation of cancer cells enables the development of new therapeutic strategies to fight cancer. A standard approach to address this question has been to compare normal and cancer cells in search for cancer-specific alterations. However, a recurrent difficulty when interpreting such data is to dissect out driver (i.e., causal) from passenger (i.e., consequential) events. This is especially challenging when analyzing gene expression, whereby passenger events can represent either secondary effectors that are induced by driver events (e.g., transcriptional targets of the Myc oncogene) and/or genes whose apparent deregulation simply reflect a process that is exacerbated in the cancerous state (e.g., cell proliferation). Although most secondary deregulations may not be linked to cancer development, some might nonetheless be of interest as therapeutic targets. When prioritizing which alterations should be further studied, particular attention is given to those that can potentially be targeted such as cell-surface receptors that can be blocked by antagonistic molecules or enzymes whose activity can be modulated by small-molecule inhibitors.
In this respect, chromatin-modifying enzymes constitute attractive targets. Chromatin modifiers have been reported to be subject to various alterations (reviewed by ref. 1), including point mutations leading to either gain or loss of function, larger chromosomal rearrangements resulting in gene amplification or deletion, and translocations. In addition, altered expression of chromatin-modifying enzymes, leading to under- or over-expression is also frequent in cancer. However it is intriguing that in many cases, such variations are not paralleled by copy-number changes of the underlying DNA, suggesting that they may represent a secondary effect of transformation rather than a causal factor in the emergence of the cancerous state.2
Mechanistically, it has been hypothesized that deregulation of chromatin modifiers would lead to specific transcriptional defects such as overexpression of proto-oncogenes or silencing of tumor suppressor genes. Thus, it is hoped that targeting such enzymes could revert the cancerous state by restoring a normal transcriptome.
We focused on the role of EZH2 in solid tumor development. EZH2 is the main catalytic subunit of Polycomb Repressive Complex 2 (PRC2), a chromatin modifier responsible for the di- and tri-methylation of Lys27 of histone H3 (H3K27me2/3). H3K27me2 is broadly distributed; it is present on more than half of the histones and its function remains unclear. In contrast, H3K27me3 is found enriched at specific loci and is associated with a transcriptionally silent state. Other complexes are part of the Polycomb machinery and act together to ensure proper maintenance of transcriptional silencing.3 EZH2 has attracted a lot of attention due to its apparent overexpression across a wide range of solid tumor types. In breast and prostate cancer, EZH2 expression further increases in the most advanced stages and therefore constitutes a marker of poor prognosis.4,5 This, together with some in vitro functional assays, has lead to a broad acceptation that EZH2 plays an oncogenic role in solid tumors. While oncogenic pathways have been shown to activate EZH2 expression,6,7 several studies suggested that genomic gains/amplifications might further increase its expression.7 Yet the question of how overexpression of EZH2 promotes tumorigenesis remains enigmatic. Several reports indicate that the enzyme might become aberrantly recruited and repress tumor suppressors8–13 or sustain silencing of tumor suppressor loci such as INK4A/ARF and P21.14–17 Alternatively, recent reports have suggested that the action of EZH2 might be independent of its function within PRC2.18,19 Surprisingly, other studies have reported that, despite EZH2 overexpression, H3K27me3 levels are decreased in several solid tumor types and that low level of the histone mark is associated to poor prognosis18,20–22 thus prompting for a more thorough analysis of EZH2 involvement in tumor progression. We have previously shown that tumor development can occur in the absence of the enzyme.23 We demonstrated that the association of high EZH2 to poor prognosis is a consequence of a correlation between EZH2 expression and the rate of cell proliferation. Here, we investigate the association between EZH2, silencing of key tumor suppressors and disease outcome. We show that upon immortalization, Ezh2 is often dispensable for cell proliferation. This is linked to an Ezh2-independent regulation of the Ink4a/Arf locus. Similarly to what we found in breast cancer, we show that upon subtracting the effect of cell-proliferation, high EZH2 levels are no longer correlated to metastatic prostate cancer. We discuss the value of single marker versus metagene to account for cell proliferation and propose a general tumor-suppressive function for PRC2, possibly through the control of gene silencing stability.
Results
EZH2 is generally dispensable in immortalized cells
We investigated the role of Ezh2 in genetically defined mouse models of prostate and mammary tumorigenesis.23 Although Ezh2 expression is elevated in these models as compared to the corresponding normal tissues, we found that loss of the enzyme does not seem to impact prostate cancer development and results in a slight increase in the penetrance of mammary tumor development. We then analyzed the consequence of Ezh2 loss on the expression of p16, p19 (from the Ink4a/Arf locus) and p21 expression. We found that p16 and p19, already expressed in mouse prostate cancer cells, are unaffected by the loss of Ezh2. We hypothesize that immortalization might in some situations discharge Ezh2 from silencing the Ink4a/Arf locus. To test this hypothesis we have compared the impact of different immortalization strategies in mouse embryonic fibroblasts (MEFs), which normally rely on Ezh2 for silencing of Ink4a/Arf.14 As expected, Ndy1-immortalized MEFs rely on the presence of Ezh2 for proliferation and silencing of p16 and p19 genes (Fig. 1 and refs. 24, 25). However, upon c-Myc-mediated immortalization or inactivation of the P53 pathway, the enzyme becomes dispensable for both cell proliferation and silencing of the 2 genes. As in prostate cancer cells, Ezh2 still maintains silencing of the p21 locus. Consistently, in c-Myc iMEFs, H3K27me3 is essentially absent from the Ink4a/Arf locus while still being present along the p21 locus (Fig. 1C). Thus upon immortalization, silencing of the ink4a/Arf locus often becomes PRC2-independant.
Figure 1.

Context-dependent role of Ezh2 in the regulation of the Ink4a/Arf locus. (A) Western blot against Ezh2, H3K27me3 and Lamin B1 (loading control) of iMEF cells immortalized by c-Myc, P53-DN or Ndy1 in the presence or absence of OHT as indicated on top. (B) Top panels, proliferation curves of the 3 iMEF cell lines in the presence or absence of Ezh2 (mean ± SD, n = 3). Bottom panels, RT-qPCR analysis of p16, p19 and p21 genes in the corresponding cell lines. Values indicate relative expression of mutant cells compared to WT cells after normalization to the house keeping gene TATA-binding protein (TBP). ND, not detected. Mean ± SD, n = 3. (C) Snapshot of H3K27me3 ChIP-seq distribution in c-Myc iMEFs (C2 clone) Ezh2 WT or Ezh2 Δ/Δ at the level of the p16/p19 locus (left) and of the p21 locus (right).
Our results thus suggest that high levels of Ezh2 might be a secondary event resulting from tumorigenesis. We analyzed datasets of prostate and breast cancer transcriptome and found that EZH2 transcript clusters with genes associated to cell proliferation (e.g., cell cycle genes), suggesting that its expression in cancer is mostly under the control of proliferation cues.23 We showed that coupling of EZH2 expression to cell proliferation is required to counteract cell division mediated dilution of H3K27me3 and thus participates in homeostasis of the mark. Altogether, these results indicate that high EZH2 expression in cancers reflects the abnormally high proliferation rate of cancer cells rather than abnormal overexpression. Taking advantage of patient-derived breast xenografts treated with various drugs, we further investigated the link between cell proliferation, EZH2 expression and H3K27me3 abundance. As expected, EZH2 tightly correlates with the proliferation marker Ki67. In contrast, H3K27me3 is anti-correlated with both Ki67 and EZH2, suggesting that homeostatic maintenance of the mark is perturbed in breast cancer. Uncoupling Ezh2 expression from proliferation can experimentally create a similar situation; in such a case, increased proliferation leads to decreased H3K27me3 abundance. We propose that insufficient coupling of PRC2 activity to proliferation might be a key explanation to the antagonistic variations of EZH2 and H3K27me3 levels reported in various solid tumor types including breast cancer.
Proliferation is a confounding factor in the association between high EZH2 expression and poor outcome
In view of the above-mentioned observations, we expected that the prognostic value of EZH2 rely mostly on its correlation to proliferation. Surprisingly, we found that in breast cancer, subtracting the impact of cell proliferation in fact reverts the association between EZH2 expression and outcome: high EZH2 expression relative to cell proliferation now becomes associated to a good prognosis. Thus, this analysis establishes that the prognostic value of EZH2 is the sum of several potentially opposite components; the positive association of EZH2 to poor outcome stems from proliferation being the major factor driving its expression. Similarly in prostate cancer, EZH2 transcript levels are significantly higher in metastatic cancer than in primary tumors; however this association is cancelled-out when subtracting the impact of cell proliferation (Fig. 2). Accordingly, a few studies found that EZH2 is not an independent prognostic marker when proliferation is taken into account (e.g., refs. 26, 27). We note that a number of reports concluding that EZH2 is an independent prognostic factor did not investigate the impact of proliferation. This is surprising given the well-established association of proliferation to poor outcome, previously shown to constitute a confounding factor for many gene expression signatures (e.g., ref. 28). In some studies however, EZH2 remained an independent prognostic marker even when proliferation was taken into account (see ref. 29 for an example). This might come from the method used to evaluate the impact of proliferation that often relies on single measurements such as mitotic count or a proliferation marker. Instead we used a metagene consisting of the median value of 52 proliferation-associated genes.30 This latter method allows to cancel-out the error associated with each individual measurement. The difference between metagene vs. single marker in the normalization for cell proliferation is illustrated in Figure 3 with the example of ORC6 transcript. While the prognostic power of ORC6 is completely lost after adjusting its expression to the proliferation metagene, it still retains a significant association to outcome if only Ki67 expression is used to account for cell proliferation.
Figure 2.

The association of EZH2 transcript levels to metastatic prostate cancer is driven by proliferation. Plots of EZH2 transcript levels in primary and metastatic prostate cancer (data extracted from ref. 49) before (left) and after (right) adjustment to the proliferation metagene (as described by ref. 30). p-value of unpaired t-tests are displayed.
Figure 3.

Comparison of multiple markers vs. single proliferation marker to adjust for the impact of cell proliferation. Kaplan-Meier plots of breast cancer specific survival for patients of the METABRIC cohort having primary tumors with high (above median) or low (under median) ORC6 transcript levels before (left) and after adjustment of its expression with Ki67 (middle) or the proliferation metagene (right). Hazard ratio (HR) between highest and lowest survival groups and p-value of the Log-rank (Mantel-Cox) test are displayed on the plots.
Discussion
PRC2, a constraint for tumor development?
In addition to establishing that proliferation underlies the association between high EZH2 levels and poor prognosis, our analyses have revealed that low expression of EZH2 relative to proliferation, which is underpinned by genetic loss of the gene, is associated to poor outcome.23 Along these lines, analyzing of a small cohort of breast cancer metastases, we uncovered several mutations in PRC2 genes including a truncating mutation of EZH2 in a hemizygous context, predicting acute loss of the protein. Furthermore, analysis of the TCGA breast cancer cohort revealed that although rare in primary tumors (3% of all tumors), mutations in PRC2 genes are associated with poor prognosis. Using CRISPR/CAS9-mediated genome engineering, we genetically inactivated EZH2 in a commonly used human cancer cell line (MDA-MB-231). Strikingly, this led to increased tumor growth in vivo and enhanced 3D migration in vitro. This latter result was confirmed using a specific PRC2 inhibitor. Therefore, if anything, EZH2 seems to constrain breast tumorigenesis.
A recent study reached a similar conclusion;22 using a mouse model of mammary tumorigenesis caused by Brca1 loss, the authors found that concomitant loss of Ezh2 shortens the latency of tumor development. Beyond breast cancer, a restrictive role for PRC2 in tumor development is emerging as a common theme in distinct tumor types. Recurrent loss-of-function mutations in EZH2 have been reported in myelodysplastic syndromes31 and T-cell acute lymphoblastic leukemia.32,33 Less frequent hypomorphic mutations in SUZ12 and EED, encoding 2 PRC2 core components, also occur in myeloid malignancies.34 In contrast, complete PRC2 inactivation occurs in malignant peripheral nerve sheath tumors through mutation of SUZ12 or EED.35–37 It is likely that inactivation of PRC2 also occurs in other sarcomas in view of frequent H3K27me3 loss.38 In pediatric high-grade gliomas (pHGG), point mutation of the K27 residue of H3.3 or H3.1 histone variants into methionine (H3K27M) lead to a global reduction of H3K27me3.39–41 This decrease is accompanied by focal gains of H3K27me3, but the functional significance of such gains remains unclear. Analysis of mouse models of pancreas cancer,42 glioblastoma43 and lung cancer44 similarly point to a tumor suppressive role for PRC2. Hence, with the exception of some subtypes of lymphomas, where EZH2 gain of function has been described, partial to complete loss of PRC2 function seems to be favored in cancers. It is however noticeable that the different mutations leading to impaired PRC2 activity are unlikely to be functionally equivalent considering their tumor-type specificity and the extent to which they impair PRC2 activity. Using a model cell line to compare the consequences of partial versus complete PRC2 inhibition, we have shown that lower PRC2 activity selectively impairs silencing of genes that have a low density of H3K27me3 in their promoter region.23 This suggests that H3K27me3 enrichment directly relates to the robustness of gene silencing. Hence, genes for which the mark is present at a low density are expected to be more sensitive to subtle variations of PRC2 activity. This result might help understand the differences in PRC2 mutation spectrum observed across different tumor types.
Impact of PRC2 impairment on the stability of gene silencing
In the last part of our study, we investigated how impaired PRC2 activity affects the stability of gene silencing. This question is not only relevant in the context of PRC2 alterations but also more generally, regarding the impact of mutations in chromatin modifiers and therefore their contribution to tumor development. Upon inactivation of a chromatin modifier, only a fraction of the genes to which it was bound become transcriptionally deregulated, presumably due to the redundancy within gene-regulatory mechanisms. Hence, PRC2-independent recruitment of PRC1 could potentially compensate for loss of PRC2. In addition, it has been proposed that relieving Polycomb-mediated transcriptional silencing is ineffectual in the absence of pertinent cell-type specific transcriptional activators.45 The function of the genes that become activated upon loss of PRC2 is therefore tightly linked to the identity of the cells where the mutation occurs. In the case of transformed cells, pro-oncogenic pathways could also influence this transcriptional response as previously suggested in tumors caused by oncogenic NOTCH1 signaling33 or by hyperactive RAS pathway.36
In addition to this deterministic transcriptional response, it is possible that mutations of chromatin modifiers, by disturbing a layer of gene regulation, lead to a more global transcriptional instability. Our single cell analyses suggest that deletion of Ezh2 indeed results in stochastic reactivation of many Polycomb targets. These transcriptional events are too infrequent to be detectable at the population level. Whether stochastic gene activation events, once initiated, are stably maintained still remains to be tested. Since many Polycomb target genes encode key cell fate regulators (e.g, transcription factors), it is possible that even transient activation bursts could translate into long-lasting reprograming of downstream gene regulatory networks, leading to an epigenetic drift between distinct sub-clones of the same tumor. Hence, we speculate that transcriptional instability of Polycomb-target genes could participate in the diversification of intra-tumor phenotypes, as previously hypothesized.46
Altogether, we propose that mutations in PRC2 genes lead to a dual impact on gene expression: first, the robust de-regulation of a fraction of genes and second defects in the transcriptional stability of a larger number of genes. We hypothesize that these 2 responses might respectively promote reprogramming of cancer cells' transcriptome and diversification of gene expression programs within tumors (Fig. 4). Such variations could lead to increased intra-tumor heterogeneity, followed by the selection of the “epigenotypes” that confer a selective advantage. This hypothesis is supported by independent observations. A first study reported that prolonged knock-down of EZH2 in glioblastoma results in the emergence of “escaper” tumors due to cell-fate switching into a more aggressive phenotype.43 In a second study, inhibition of PRC2 was found to promote bromodomain and extra terminal protein (BET) inhibitor resistance by facilitating rewiring of oncogenic gene regulatory networks.47
Figure 4.
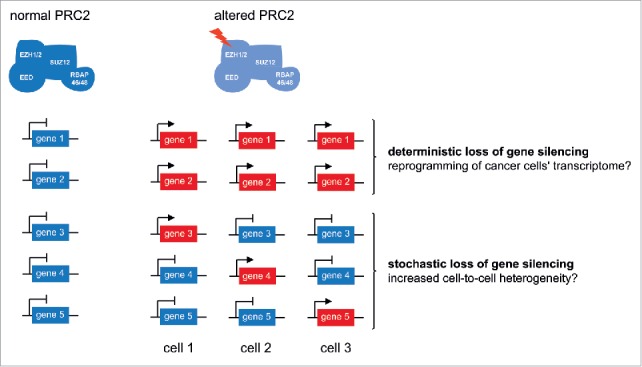
Proposed model of how deterministic and stochastic transcriptional responses in PRC2-altered cells could lead to increased tumorigenicity.
Conclusion
With the advent of EZH2 inhibitors in clinical trials, it is urgent to precisely define the tumor types that might benefit from such targeted therapies. Although there is a strong case to use EZH2 inhibitors against tumors harboring EZH2 gain-of-function mutations (DLBCL and FL), several studies including ours question the usefulness and safety of such molecules in solid tumors in which EZH2 is unaltered at the genetic level. Recent studies have suggested inhibiting EZH2 as a synthetic lethal approach in solid tumors harboring inactivating mutations in SWI/SNF nucleosome remodelers and BAP1 histone deubiquitinase. However, even in such cases, although an immediate benefit might be expected due to favorable transcriptional reprogramming of cancer cells, long-term consequences should be carefully evaluated.
Materials and methods
Cell lines
iMEF cells were grown in DMEM medium supplemented with 10% FCS, 100 mM non essential amino acids, 1 mM L-Glutamine. Ezh2 flox/flox;ROSA26-CreERT2 MEF cells were isolated from 13.5 d old embryos and subsequently infected with the following retroviral constructs: pMXs-hc-MYC (addgene 17220) to generate c-Myc iMEFs, pBABE-hygro p53 DD (addgene 9058) to generate p53-DN iMEFs, Ndy1-MigR1 (kindly provided by Philip N. Tsichlis) to generate Ndy1 iMEFs.
Cell growth assay
20000 cells were plated in 6 well dishes in triplicates and counted every 24 hours over 4 d using a Vi-cells counter (Beckman-coulter).
Antibodies
Antibodies against Ezh2, Lamin B1 and H3K27me3 were previously described.23
RT-qPCR
Total RNA was isolated using TRIzol reagent (Invitrogen). cDNA was synthetized using High Capacity cDNA RT kit (4368814-Applied Biosystems) and quantitative PCR was performed with technical triplicate using SYBR green reagent (Roche) on a ViiA7 equipment (Applied Biosystems). At least 3 biological independent experiments were performed for each assay. Primers sequences were previously described.23
Gene expression, copy-number and survival analysis of primary breast tumors
This study makes use of data generated by the Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) and first described in ref. 48. Funding for the project was provided by Cancer Research UK and the British Columbia Cancer Agency Branch. Upon access request, SNP 6.0 copy-number and Illumina HT-12 expression data for nearly 2000 primary breast tumors were available through European Genome-Phenome Archive (http://www.ebi.ac.uk/ega/), under accession number EGAS00000000083. Survival analyses were performed as previously described.23
Adjusting gene expression for proliferation
To adjust gene expression values to a single marker or a metagene (median expression of 52 proliferation associated genes as described in ref. 30), residuals, i.e. distance of gene expression values to the best-fit curve of the linear regression, were calculated and used as such for subsequent analyses.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This study was supported by the European Research Council (ERC-Stg, REPODDID). M.W. was a recipient of a post-doctoral fellowship from the Association pour la Recherche contre le Cancer (ARC).
References
- [1].Plass C, Pfister SM, Lindroth AM, Bogatyrova O, Claus R, Lichter P. Mutations in regulators of the epigenome and their connections to global chromatin patterns in cancer. Nat Rev Genet 2013; 14:765-80; PMID:24105274; http://dx.doi.org/25484917 10.1038/nrg3554 [DOI] [PubMed] [Google Scholar]
- [2].Shah MA, Denton EL, Arrowsmith CH, Lupien M, Schapira M. A global assessment of cancer genomic alterations in epigenetic mechanisms. Epigenetics Chromatin 2014; 7:29; PMID:25484917; http://dx.doi.org/ 10.1186/1756-8935-7-29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Simon JA, Kingston RE. Mechanisms of Polycomb gene silencing: knowns and unknowns. Nat Rev Mol Cell Biol 2009; 10:1-12; http://dx.doi.org/ 10.1038/nrn2589 [DOI] [PubMed] [Google Scholar]
- [4].Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, Ghosh D, Sewalt RGAB, Otte AP, Hayes DF, et al.. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci U S A 2003; 100:11606-11; PMID:14500907; http://dx.doi.org/ 10.1073/pnas.1933744100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, Ghosh D, Pienta KJ, Sewalt RGAB, Otte AP, et al.. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002; 419:624-9;; PMID:12374981; http://dx.doi.org/ 10.1038/nature01075 [DOI] [PubMed] [Google Scholar]
- [6].Koh CM, Iwata T, Zheng Q, Bethel C, Yegnasubramanian S, De Marzo AM. Abstract 263: MYC enforces overexpression of EZH2 in early prostatic neoplasia via transcriptional and posttranscriptional mechanisms. Cancer Res 2011; 71:263-263;; http://dx.doi.org/ 10.1158/1538-7445.AM2011-263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bracken AP, Pasini D, Capra M, Prosperini E, Colli E, Helin K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J 2003; 22:5323-35; PMID:14532106; http://dx.doi.org/ 10.1093/emboj/cdg542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ren G, Baritaki S, Marathe H, Feng J, Park S, Beach S, Bazeley PS, Beshir AB, Fenteany G, Mehra R, et al.. Polycomb protein EZH2 regulates tumor invasion via the transcriptional repression of the metastasis suppressor RKIP in breast and prostate cancer. Cancer Res 2012; 72:3091-104;; PMID:22505648; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-3546 [DOI] [PubMed] [Google Scholar]
- [9].Beke L, Nuytten M, Van Eynde A, Beullens M, Bollen M. The gene encoding the prostatic tumor suppressor PSP94 is a target for repression by the Polycomb group protein EZH2. Oncogene 2007; 26:4590-5; PMID:17237810; http://dx.doi.org/ 10.1038/sj.onc.1210248 [DOI] [PubMed] [Google Scholar]
- [10].Taniguchi H, Jacinto F V, Villanueva A, Fernandez AF, Yamamoto H, Carmona FJ, Puertas S, Marquez VE, Shinomura Y, Imai K, et al.. Silencing of Kruppel-like factor 2 by the histone methyltransferase EZH2 in human cancer. Oncogene 2012; 31:1988-94; PMID:21892211; http://dx.doi.org/ 10.1038/onc.2011.387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Fujii S, Ito K, Ito Y, Ochiai A. Enhancer of zeste homologue 2 (EZH2) down-regulates RUNX3 by increasing histone H3 methylation. J Biol Chem 2008; 283:17324-32;; PMID:18430739; http://dx.doi.org/ 10.1074/jbc.M800224200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Yu J, Cao Q, Wu L, Dallol A, Li J, Chen G, Grasso C, Cao X, Lonigro RJ, Varambally S, et al.. The neuronal repellent SLIT2 is a target for repression by EZH2 in prostate cancer. Oncogene 2010; 29:5370-80; PMID:20622896; http://dx.doi.org/ 10.1038/onc.2010.269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Chen H, Tu SW, Hsieh JT. Down-regulation of human DAB2IP gene expression mediated by polycomb Ezh2 complex and histone deacetylase in prostate cancer. J Biol Chem 2005; 280:22437-44; PMID:15817459; http://dx.doi.org/ 10.1074/jbc.M501379200 [DOI] [PubMed] [Google Scholar]
- [14].Bracken AP, Kleine-Kohlbrecher D, Dietrich N, Pasini D, Gargiulo G, Beekman C, Theilgaard-Mönch K, Minucci S, Porse BT, Marine J-C, et al.. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev 2007; 21:525-30; PMID:17344414; http://dx.doi.org/ 10.1101/gad.415507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Seward S, Semaan A, Qazi AM, Gruzdyn OV, Chamala S, Bryant CC, Kumar S, Cameron D, Sethi S, Ali-Fehmi R, et al.. EZH2 blockade by RNA interference inhibits growth of ovarian cancer by facilitating re-expression of p21waf1/cip1 and by inhibiting mutant p53. Cancer Lett 2013; 336:53-60; PMID:23603558; http://dx.doi.org/ 10.1016/j.canlet.2013.04.012 [DOI] [PubMed] [Google Scholar]
- [16].Batchu RB, Qazi AM, Gruzdyn OV, Semaan A, Seward SM, Chamala S, Dhulipala VB, Bouwman DL, Weaver DW, Gruber SA. EZH2-shRNA-mediated upregulation of p21waf1/cip1 and its transcriptional enhancers with concomitant downmodulation of mutant p53 in pancreatic ductal adenocarcinoma. Surgery 2013; 154:739-46; discussion 746–7; PMID:24074410; http://dx.doi.org/ 10.1016/j.surg.2013.06.041 [DOI] [PubMed] [Google Scholar]
- [17].Bai J, Chen J, Ma M, Cai M, Xu F, Wang G, Tao K, Shuai X. Inhibiting enhancer of zeste homolog 2 promotes cellular senescence in gastric cancer cells SGC-7901 by activation of p21 and p16. DNA Cell Biol 2014; 33:337-44; PMID:24588771; http://dx.doi.org/ 10.1089/dna.2014.2340 [DOI] [PubMed] [Google Scholar]
- [18].Xu K, Wu ZJ, Groner AC, He HH, Cai C, Lis RT, Wu X, Stack EC, Loda M, Liu T, et al.. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science 2012; 338:1465-9; PMID:23239736; http://dx.doi.org/ 10.1126/science.1227604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lee ST, Li Z, Wu Z, Aau M, Guan P, Karuturi RKM, Liou YC, Yu Q. Context-Specific Regulation of NF-κB Target Gene Expression by EZH2 in Breast Cancers. Mol Cell 2011; 43:798-810; PMID:21884980; http://dx.doi.org/22766277 10.1016/j.molcel.2011.08.011 [DOI] [PubMed] [Google Scholar]
- [20].Holm K, Grabau D, Lövgren K, Aradottir S, Gruvberger-Saal S, Howlin J, Saal LH, Ethier SP, Bendahl P-O, Stål O, et al.. Global H3K27 trimethylation and EZH2 abundance in breast tumor subtypes. Mol Oncol 2012; 6:494-506; PMID:22766277; http://dx.doi.org/ 10.1016/j.molonc.2012.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wei Y, Xia W, Zhang Z, Liu J, Wang H, Adsay N V, Albarracin C, Yu D, Abbruzzese JL, Mills GB, et al.. Loss of trimethylation at lysine 27 of histone H3 is a predictor of poor outcome in breast, ovarian, and pancreatic cancers. Mol Carcinog 2008; 47:701-6; PMID:18176935; http://dx.doi.org/ 10.1002/mc.20413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Bae WK, Yoo KH, Lee JS, Kim Y, Chung I-J, Park MH, Yoon JH, Furth PA, Hennighausen L. The methyltransferase EZH2 is not required for mammary cancer development, although high EZH2 and low H3K27me3 correlate with poor prognosis of ER-positive breast cancers. Mol Carcinog 2015; 54:1172-80; PMID:25043748; http://dx.doi.org/ 10.1002/mc.22188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wassef M, Rodilla V, Teissandier A, Zeitouni B, Gruel N, Sadacca B, Irondelle M, Charruel M, Ducos B, Michaud A, et al.. Impaired PRC2 activity promotes transcriptional instability and favors breast tumorigenesis. Genes Dev 2015; 29:1-16; PMID:25561492; http://dx.doi.org/ 10.1101/gad.253682.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Pfau R, Tzatsos A, Kampranis SC, Serebrennikova OB, Bear SE, Tsichlis PN. Members of a family of JmjC domain-containing oncoproteins immortalize embryonic fibroblasts via a JmjC domain-dependent process. Proc Natl Acad Sci U S A 2008; 105:1907-12; PMID:18250326; http://dx.doi.org/ 10.1073/pnas.0711865105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Tzatsos A, Pfau R, Kampranis SC, Tsichlis PN. Ndy1/KDM2B immortalizes mouse embryonic fibroblasts by repressing the Ink4a/Arf locus. Proc Natl Acad Sci U S A 2009; 106:2641-6; PMID:19202064; http://dx.doi.org/ 10.1073/pnas.0813139106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Laitinen S, Martikainen PM, Tolonen T, Isola J, Tammela TLJ, Visakorpi T. EZH2, Ki-67 and MCM7 are prognostic markers in prostatectomy treated patients. Int J Cancer 2008; 122:595-602; PMID:17943722; http://dx.doi.org/21592298 10.1002/ijc.23145 [DOI] [PubMed] [Google Scholar]
- [27].Tolonen TT, Tammela TLJ, Kujala PM, Tuominen VJ, Isola JJ, Visakorpi T. Histopathological variables and biomarkers enhancer of zeste homologue 2, Ki-67 and minichromosome maintenance protein 7 as prognosticators in primarily endocrine-treated prostate cancer. BJU Int 2011; 108:1430-8; PMID:21592298; http://dx.doi.org/ 10.1111/j.1464-410X.2011.10253.x [DOI] [PubMed] [Google Scholar]
- [28].Venet D, Dumont JE, Detours V. Most Random Gene Expression Signatures Are Significantly Associated with Breast Cancer Outcome. PLoS Comput Biol 2011; 7:e1002240; PMID:22028643; http://dx.doi.org/ 10.1371/journal.pcbi.1002240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Bachmann IM. EZH2 Expression Is Associated With High Proliferation Rate and Aggressive Tumor Subgroups in Cutaneous Melanoma and Cancers of the Endometrium, Prostate, and Breast. J Clin Oncol 2005; 24:268-73; PMID:16330673; http://dx.doi.org/ 10.1200/JCO.2005.01.5180 [DOI] [PubMed] [Google Scholar]
- [30].Nagalla S, Chou JW, Willingham MC, Ruiz J, Vaughn JP, Dubey P, Lash TL, Hamilton-Dutoit SJ, Bergh J, Sotiriou C, et al.. Interactions between immunity, proliferation and molecular subtype in breast cancer prognosis. Genome Biol 2013; 14:R34; PMID:23618380; http://dx.doi.org/ 10.1186/gb-2013-14-4-r34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ernst T, Chase AJ, Score J, Hidalgo-Curtis CE, Bryant C, Jones A V, Waghorn K, Zoi K, Ross FM, Reiter A, et al.. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet 2010; 42:722-6; PMID:20601953; http://dx.doi.org/ 10.1038/ng.621 [DOI] [PubMed] [Google Scholar]
- [32].Simon C, Chagraoui J, Krosl J, Gendron P, Wilhelm B, Lemieux S, Boucher G, Chagnon P, Drouin S, Lambert R, et al.. A key role for EZH2 and associated genes in mouse and human adult T-cell acute leukemia. Genes Dev 2012; 26:651-6; PMID:22431509; http://dx.doi.org/ 10.1101/gad.186411.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ntziachristos P, Tsirigos A, Van Vlierberghe P, Nedjic J, Trimarchi T, Flaherty MS, Ferres-Marco D, da Ros V, Tang Z, Siegle J, et al.. Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nat Med 2012; 18:298-303; PMID:22237151; http://dx.doi.org/ 10.1038/nm.2651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Score J, Hidalgo-Curtis C, Jones AV, Winkelmann N, Skinner A, Ward D, Zoi K, Ernst T, Stegelmann F, Döhner K, et al.. Inactivation of polycomb repressive complex 2 components in myeloproliferative and myelodysplastic/myeloproliferative neoplasms. Blood 2012; 119:1208-13; PMID:22053108; http://dx.doi.org/ 10.1182/blood-2011-07-367243 [DOI] [PubMed] [Google Scholar]
- [35].Lee W, Teckie S, Wiesner T, Ran L, Prieto Granada CN, Lin M, Zhu S, Cao Z, Liang Y, Sboner A, et al.. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat Genet 2014; 46:1227-32; PMID:25240281; http://dx.doi.org/ 10.1038/ng.3095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].De Raedt T, Beert E, Pasmant E, Luscan A, Brems H, Ortonne N, Helin K, Hornick JL, Mautner V, Kehrer-Sawatzki H, et al.. PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature 2014; 514:247-51; Available from: http://www.nature.com/doifinder/ 10.1038/nature13561; PMID:25119042 [DOI] [PubMed] [Google Scholar]
- [37].Zhang M, Wang Y, Jones S, Sausen M, McMahon K, Sharma R, Wang Q, Belzberg AJ, Chaichana K, Gallia GL, et al.. Somatic mutations of SUZ12 in malignant peripheral nerve sheath tumors. Nat Genet 2014; 46:1170-2; PMID:25305755; http://dx.doi.org/ 10.1038/ng.3116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Cleven AH, Sannaa GA, Briaire-de Bruijn I, Ingram DR, van de Rijn M, Rubin BP, de Vries MW, Watson KL, Torres KE, Wang W, et al.. Loss of H3K27 tri-methylation is a diagnostic marker for malignant peripheral nerve sheath tumors and an indicator for an inferior survival. Mod Pathol 2016; 29:582-90; http://dx.doi.org/ 10.1038/modpathol.2016.45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, Qu C, Ding L, Huether R, Parker M, et al.. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet 2012; 44:251-3; PMID:22286216; http://dx.doi.org/ 10.1038/ng.1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Lewis PW, Müller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, Garcia BA, Muir TW, Becher OJ, Allis CD. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 2013; 340:857-61; PMID:23539183; http://dx.doi.org/ 10.1126/science.1232245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Chan K-M, Fang D, Gan H, Hashizume R, Yu C, Schroeder M, Gupta N, Mueller S, James CD, Jenkins R, et al.. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev 2013; 27:985-90; PMID:23603901; http://dx.doi.org/ 10.1101/gad.217778.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Mallen-St. Clair J, Soydaner-Azeloglu R, Lee KE, Taylor L, Livanos A, Pylayeva-Gupta Y, Miller G, Margueron R, Reinberg D, Bar-Sagi D. EZH2 couples pancreatic regeneration to neoplastic progression. Genes Dev 2012; 26:439-44; PMID:22391448; http://dx.doi.org/ 10.1101/gad.181800.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].de Vries NA, Hulsman D, Akhtar W, de Jong J, Miles DC, Blom M, van Tellingen O, Jonkers J, van Lohuizen M. Prolonged Ezh2 Depletion in Glioblastoma Causes a Robust Switch in Cell Fate Resulting in Tumor Progression. Cell Rep 2015; 10:383-97; http://dx.doi.org/ 10.1016/j.celrep.2014.12.028 [DOI] [PubMed] [Google Scholar]
- [44].Serresi M, Gargiulo G, Proost N, Siteur B, Cesaroni M, Koppens M, Xie H, Sutherland KD, Hulsman D, Citterio E, et al.. Polycomb Repressive Complex 2 Is a Barrier to KRAS-Driven Inflammation and Epithelial-Mesenchymal Transition in Non-Small-Cell Lung Cancer. Cancer Cell 2016; 29:17-31; PMID:26766588; http://dx.doi.org/ 10.1016/j.ccell.2015.12.006 [DOI] [PubMed] [Google Scholar]
- [45].Ezhkova E, Pasolli HA, Parker JS, Stokes N, Su I, Hannon G, Tarakhovsky A, Fuchs E. Ezh2 Orchestrates Gene Expression for the Stepwise Differentiation of Tissue-Specific Stem Cells. Cell 2009; 136:1122-35; PMID:19303854; http://dx.doi.org/ 10.1016/j.cell.2008.12.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Timp W, Feinberg AP. Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nat Rev Cancer 2013; 13:497-510; PMID:23760024; http://dx.doi.org/26367798 10.1038/nrc3486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Rathert P, Roth M, Neumann T, Muerdter F, Roe J-S, Muhar M, Deswal S, Cerny-Reiterer S, Peter B, Jude J, et al.. Transcriptional plasticity promotes primary and acquired resistance to BET inhibition. Nature 2015. [cited 2016January28]; 525:543-7; PMID:26367798; http://dx.doi.org/ 10.1038/nature14898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Curtis C, Shah SP, Chin S-F, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, et al.. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012; 486:346-52; Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artidD3440846&toolDpmcentrez&rendertypeDabstract; PMID:22522925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, et al.. Integrative Genomic Profiling of Human Prostate Cancer. Cancer Cell 2010; PMID:20579941; 18:11-22; http://dx.doi.org/ 10.1016/j.ccr.2010.05.026 [DOI] [PMC free article] [PubMed] [Google Scholar]