

ABSTRACT
NUP98 is a recurrent partner gene in translocations causing acute myeloid leukemias and myelodisplastic syndrome. The expression of NUP98 fusion oncoproteins has been shown to induce mitotic spindle defects and chromosome missegregation, which correlate with the capability of NUP98 fusions to cause mitotic checkpoint attenuation. We show that NUP98 oncoproteins physically interact with the APC/CCdc20 in the absence of the NUP98 partner protein RAE1, and prevent the binding of the mitotic checkpoint complex to the APC/CCdc20. NUP98 oncoproteins require the GLEBS-like domain present in their NUP98 moiety to bind the APC/CCdc20. We found that NUP98 wild-type is a substrate of APC/CCdc20 prior to mitotic entry, and that its binding to APC/CCdc20 is controlled via phosphorylation of a PEST sequence located within its C-terminal portion. We identify S606, within the PEST sequence, as a key target site, whose phosphorylation modulates the capability of NUP98 to interact with APC/CCdc20. We finally provide evidence for an involvement of the peptidyl-prolyl isomerase PIN1 in modulating the possible conformational changes within NUP98 that lead to its dissociation from the APC/CCdc20 during mitosis. Our results provide novel insight into the mechanisms underlying the aberrant capability of NUP98 oncoproteins to interact with APC/CCdc20 and to interfere with its function.
KEYWORDS: acute myeloid leukemia, anaphase promoting complex, cell cycle, mitotic checkpoint, myelodisplastic syndrome, NUP98, nucleoporins, oncogenes
Introduction
The NUP98 gene is recurrently involved in chromosomal translocations that cause acute myeloid leukemias (AML) and myelodisplastic syndrome (MDS)1,2 and lead to the formation of fusion oncoproteins with over 20 different partner proteins.3,4 NUP98 codes for a nucleoporin, whose primary function, as a component of the nuclear pore complex, is the transport of proteins and RNA across the nuclear membrane.5-8 NUP98, however, has been implicated, like other nucleoporins, in several additional processes, such as transcriptional regulation and mitotic progression.2,9 The NUP98 protein contains 2 main functional domains: a GLFG repeat region, which serves as a nuclear transport receptor docking surface7,10; and a GLEBS-like domain, which mediates the interaction with the RAE1 mRNA nuclear export factor.11 Both domains are located within the N-terminal half of the NUP98 protein, spanning from amino acids 1 to ∼470, which is present in essentially all NUP98 fusion oncoproteins.1,4 A GLEBS domain is also found in the mitotic checkpoint factor BubR1 where it mediates the interaction with Bub3, also a regulator of mitosis.12 Bub3, in turn, shares extensive homology with the Rae1 protein, which led to hypothesize a role of NUP98 and Rae1 as regulators of mitosis.13 Indeed, in addition to their function in nucleocytoplasmic transport, NUP98, together with Rae1, have been shown to modulate the function of the anaphase promoting complex/cyclosome (APC/C).14
The APC/C regulates mitosis and cell cycle progression by targeting a series of key substrates for degradation. One of these is securin,15 an anaphase inhibitor protein that blocks the action of the cohesin-degrading protease separase.16 In mitosis, APC/C activity is regulated by the spindle assembly checkpoint (SAC), which senses the correct attachment of chromosome kinetochores (spindle attachment sites) to spindles and blocks APC/C function, preventing sister chromatid separation, until all kinetochores are properly attached (reviewed in17). The effector of the SAC is the mitotic checkpoint complex (MCC). It is composed of the SAC proteins Mad2, BubR1(Mad3), Bub3 and Cdc20, which interact together to assemble the MCC. The MCC can diffuse freely within cells to interact with the APC/C and block its function until the SAC is satisfied (reviewed in17).
We reported that the exogenous expression of NUP98 fusion oncoprotein causes SAC attenuation, and as a consequence chromosome missegregation. We demonstrated that NUP98 oncoproteins, unlike wild type NUP98, physically interact with the Cdc20 APC/C regulator, thus suggesting their direct interference with APC/C function, the mechanism and the APC/C components involved, however, remain still unclear.18
In this work we dissected the molecular mechanisms underlying the interference of NUP98 oncoproteins with APC/CCdc20. We found that NUP98 oncoproteins physically interact with the APC/CCdc20 in the absence of the NUP98 partner protein RAE1, and prevent binding of the mitotic checkpoint complex (MCC) in the presence of an unsatisfied SAC, thus justifying their SAC-attenuating action. We show that NUP98 oncoproteins require the NUP98 GLEBS-like domain for interaction with the APC/CCdc20. NUP98 wt, while being unable to interact with APC/CCdc20 during mitosis, was found to bind, and to be a substrate of APC/CCdc20 prior to mitotic entry. Binding of NUP98 to APC/CCdc20 was found to be controlled by the phosphorylation state of a PEST sequence located within the C-terminal portion of NUP98. We identify S606, located within the PEST sequence, as a key target site, whose phosphorylation affects the capability of NUP98 to interact with APC/CCdc20. Finally, we provide evidence for an involvement of the peptidyl-prolyl isomerase PIN1 in modulating the possible conformational changes, ensuing NUP98 phosphorylation, that lead to its dissociation from the APC/CCdc20. A model is discussed, which provides a justification of the aberrant association of NUP98 fusion oncoproteins with APC/CCdc20 based on the potential of NUP98 wt to conditionally interact with APC/CCdc20 as a target.
Results
NUP98 fusion oncoproteins physically interact with the APC/C during mitosis preventing MCC binding
We previously-reported the aberrant physical interaction, in mitosis-arrested cells, between NUP98 fusion oncoproteins and the Cdc20 APC/C regulator.18 In mitosis-arrested cells Cdc20 can be both part of an active mitotic checkpoint complex (MCC) and part of a MCC-containing, inactive APC/C (APC/CMCC, see17), we therefore started by determining whether NUP98 fusion oncoproteins could be co-immunoprecipitated with three different APC/C core components: APC3, APC4, and APC10. The fusion proteins tested originate all from AML-associated chromosomal translocations involving the NUP98 gene, located on chromosome 11. They all contain the N-terminal portion of the NUP98 protein including the GLFG and GLEBS motifs.18 NUP98-HOXD13 represents a fusion with the C-terminal homeodomain DNA-binding moiety of the HOXD13 protein.19 The NUP98-LOC348801 fusion (hereafter indicated as NUP98-LOC) contains the C-terminal portion of a polypeptide of unknown function, encoded by the LOC348801 gene.20 Finally, NUP98-HHEX is a fusion protein that incorporates the homeodomain of the hematopoietically expressed homeobox gene (HHEX).21
HEK293 cells exogenously expressing NUP98, NUP98-HOXD13, NUP98-LOC, or NUP98-HHEX were arrested in mitosis by treatment with the microtubule-depolymerizing, SAC-activating drug nocodazole, and total extracts were prepared for immunoprecipitation with specific antibodies against APC3, APC4, and APC10. The NUP98-HOXD13, NUP98-LOC348801, and NUP98-HHEX oncoprotein fusions were all coimmunoprecipitated with the anti-APC3, the anti-APC4, and the anti-APC10 antibodies (Fig. 1A). Moreover, as expected, since NUP98 was previously shown to be part of a Cdh1-containing APC/C complex in mitosis,14 NUP98 wild type was also coimmunoprecipitated with APC3, APC4, and APC10 (Fig. 1A).
Figure 1.
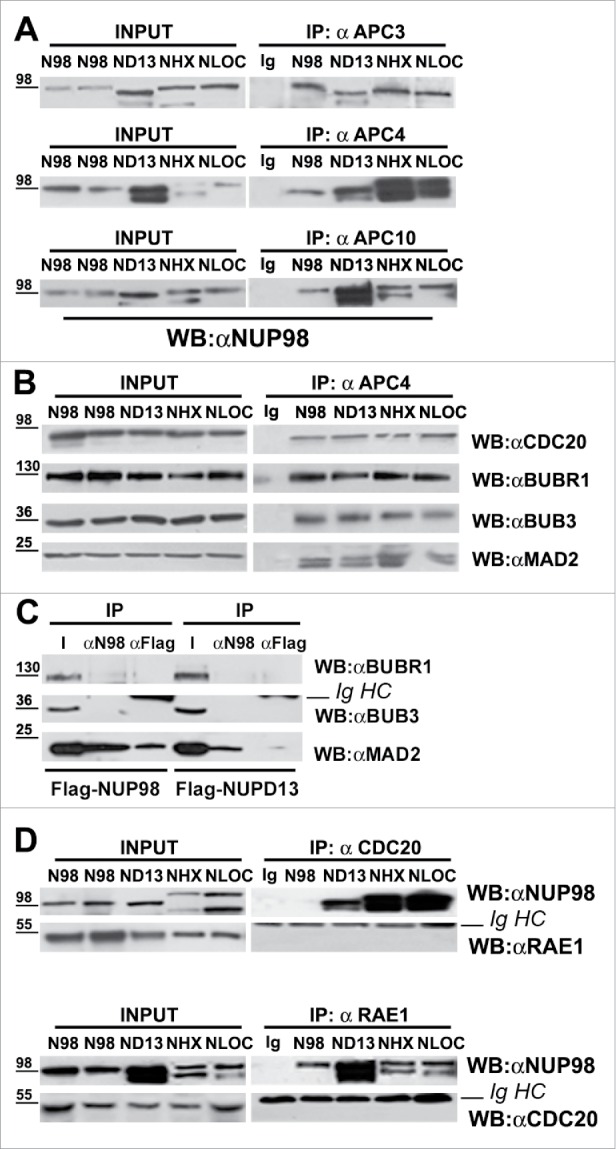
NUP98 oncoproteins bind the APC/C and prevent MCC binding. A), HEK293 cells were transfected with expression constructs for NUP98, NUP98-HOXD13, NUP98-LOC, NUP98-HHEX (ND13, NLOC, NHHX). Mitotic extracts were obtained from nocodazole-treated cells and subjected to immunoprecipitation (IP) with the indicated antibodies. Anti-IgG (Ig) was used as a negative control. Immunoprecipitated proteins were revealed by immunoblot analysis (WB) with an anti-NUP98 antibody (αNUP98). B), HEK293 were transfected and mitotic extracts were obtained as in A). Samples were immunoprecipitated (IP) with the anti-APC4 antibody (αAPC4). Anti-IgG (Ig) was used as a negative control. Immunoprecipitated proteins were revealed by immunoblotting using the indicated antibodies. C), HEK293 were transfected with expression constructs for Flag-NUP98 or Flag-NUP98-HOXD13, and mitotic extracts were obtained as in A). Immunoprecipitated proteins were revealed by immunoblotting using the indicated antibodies. D), HEK293 were transfected and mitotic extracts were obtained as in A). Immunoprecipitated proteins were revealed by immunoblotting using the indicated antibodies. IgHC, immunoglobulin heavy chains.
We next tested whether the APC/C, in mitosis-arrested cells that express NUP98 oncoproteins, would also contain components of the mitotic checkpoint complex (MCC). Total extracts from HEK293 cells expressing NUP98-HOXD13, NUP98-LOC, or NUP98-HHEX, arrested in mitosis, were immunoprecipitated with anti-APC4 antibodies, and the presence of the MCC components CDC20, BUBR1, BUB3 and MAD2 was verified by immunoblotting (Fig. 1B). All MCC components tested, efficiently coimmunoprecipitated with APC4 (Fig. 1B), suggesting that at least part of the cellular APC/C pool is bound, as expected, by MCC components, or that NUP98 fusions interact with APC/CMCC, without significantly displacing any of the MCC components. To discriminate between these two possibilities, mitotic extracts from cells expressing Flag-tagged NUP98 or NUP98-HOXD13 were immunoprecipitated with anti-Flag or anti-NUP98 antibodies to analyze bound MCC components. None of the three MCC components tested coimmunoprecipitated with NUP98-HOXD13 (Fig. 1C), while NUP98 wt interacted only with the MAD2 MCC component, but not with BUBR1 and BUB3 (Fig. 1C). This result indicates that NUP98 oncoproteins interact with a fraction of the cellular APC/C that is not bound to the MCC.
NUP98 was shown to interact with the APC/CCdh1, together with its heterodimerization partner RAE1, and to regulate its activity during mitosis.14 We reported that NUP98 fusion oncoproteins can still potentially interact with RAE1,18 and therefore wanted to verify whether RAE1 is also part of the NUP98 oncoprotein-bound APC/C. Extracts from mitotic-arrested HEK293 cells expressing NUP98, NUP98-HOXD13, NUP98-LOC, or NUP98-HHEX were thus immunoprecipitated with anti-CDC20 antibodies or, reciprocally, with anti-RAE1 antibodies and analyzed by immunoblotting. RAE1 did not coimmunoprecipitate with CDC20 and, reciprocally CDC20 did not coimmunoprecipitate with RAE1 in all of the tested extracts (Fig. 1D). Together these results indicate that NUP98 oncoproteins physically interact, in the absence of RAE1, with the APC/CCdc20 during mitosis, and preclude its interaction with the MCC in the presence of an unsatisfied SAC.
The NUP98 GLEBS-like domain is required for the binding of fusion oncoproteins to the APC/CCdc20
We next wanted to determine which portion of the NUP98 oncoproteins was necessary for their interaction with the APC/CCdc20. To this end we generated deletion mutants involving the NUP98 moiety of the NUP98-HOXD13 fusion or the NUP98 protein (Fig. S1), and tested them for their capability to be coimmunoprecipitated by the CDC20 component of APC/C from mitotic extracts of HEK293 cells. We first tested a NUP98 mutant derivative, NUP98(1–469), which represents a deletion of the C-terminal portion of NUP98, and corresponds to the portion of NUP98 that is present in essentially all NUP98 oncoproteins.1,4 Surprisingly, as NUP98 full length does not interact with CDC20 (see e. g. Fig. 1D), the NUP98(1–469) mutant was efficiently coimmunoprecipitated by the anti-CDC20 antibody (Fig. 2A). In accordance, NUP98(1–469) coimmunoprecipitated also with the APC3 APC/C component (Fig. S2). The reciprocal deletion mutant, representing the C-terminal portion of NUP98 wt, NUP98(469–920), did not interact with CDC20 (Fig. 2A) and did also not coimmunoprecipitate APC3 (Fig. S2). This result confirmed that the region required for binding the APC/CCdc20 is located within the N-terminal portion of NUP98 that is present in all NUP98 oncoproteins.1,4 It moreover suggested that the presence of the NUP98 C-terminal portion prevents NUP98 from interacting with APC/CCdc20.
Figure 2.
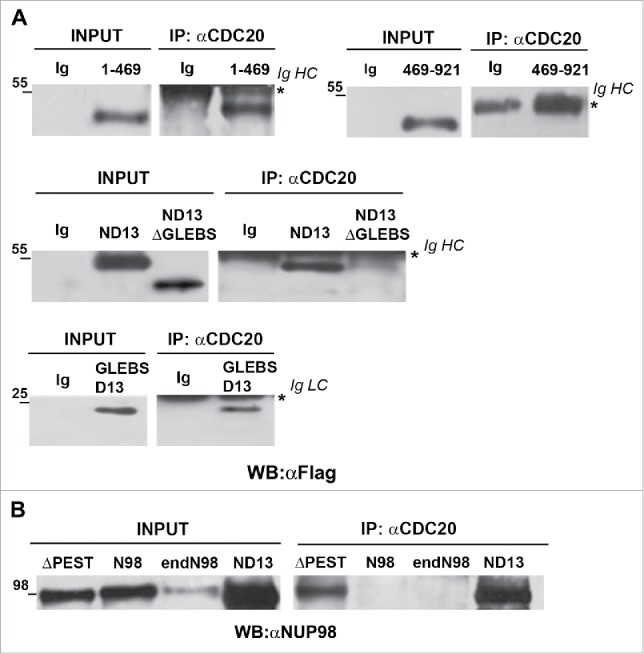
The NUP98 GLEBS-like domain is required for interaction with the APC/CCdc20. HEK293 were transfected with expression constructs for NUP98, NUP98-HOXD13, NUP98(1–469), NUP98(469–920), NUP98-HOXD13ΔGLEBS (ND13ΔGLEBS), NUP98ΔPEST (ΔPEST), and GLEBSD13. Mitotic extracts were obtained from nocodazole-treated cells and were subjected to immunoprecipitation (IP) with the indicated antibodies. Anti-IgG (Ig) was used as a negative control. Immunoprecipitated proteins were revealed by immunoblot analysis (WB) with an anti-Flag antibody (αFlag) in A), or with an anti-NUP98 antibody (αNUP98) in B). IgHC and IgLC, immunoglobulin heavy and light chains, respectively.
We thus tested a NUP98 mutant, NUP98ΔPEST, in which a previously identified functional domain within the C-terminal portion of the protein is altered. It represents the internal deletion of a PEST sequence, which was found to be located between amino acids 599–616.18 NUP98ΔPEST, was coimmunoprecipitated with CDC20, while exogenously-expressed NUP98 and endogenous NUP98 (endoNUP98) were not (Fig. 2B).
Finally, to identify which part of the NUP98 N-terminal region was required for NUP98 oncoprotein binding to APC/CCdc20, we tested an internal deletion of the NUP98 GLEBS-like domain in the context of the NUP98-HOXD13 oncoprotein (NUP98D13ΔGLEBS). The NUP98 GLEBS-like domain appeared to us as a good candidate region, as it was shown to mediate the interaction with the RAE1 protein.11 The NUP98D13ΔGLEBS (ND13ΔGLEBS) mutant indeed failed to coimmunoprecipitate with CDC20 (Fig. 2A, middle panel), whereas the NUP98-HOXD13 (ND13) oncoprotein readily interacted with CDC20 (Fig. 2A), indicating that the GLEBS-like domain of NUP98 is necessary for the interaction between NUP98 fusion oncoproteins and the APC/CCdc20.
To test for the sufficiency of the NUP98 GLEBS-like domain in the interaction with APC/CCdc20, we generated a fusion protein containing solely the NUP98 GLEBS-like domain fused to the HOXD13 homeodomain (GLEBSD13), and analyzed its capability to interact with CDC20. The GLEBSD13 fusion coimmunoprecipitated with CDC20 (Fig. 2A bottom panel), confirming that the NUP98 GLEBS-like domain is potentially sufficient for the interaction between NUP98 oncoproteins and APC/CCdc20.
Taken together, these data reveal that the NUP98 GLEBS-like domain is necessary for the interaction of NUP98 oncoproteins with the APC/CCdc20, but its presence is apparently not sufficient to allow the binding of NUP98 wt to the APC/CCdc20. The removal, such as in NUP98(1–469), or the alteration, such as in NUP98ΔPEST, of the C-terminal portion of NUP98, however, causes the resulting mutant proteins to mimic the aberrant capability of NUP98 oncoproteins to interact with the APC/CCdc20. This led us to speculate that possible structural changes within the NUP98 N-terminus, ensuing the generation of the oncogenic fusions, promote the availability of the GLEBS-like domain for an interaction with the APC/CCdc20.
CDC20 and MAD2 overexpression rescues spindle assembly checkpoint attenuation by NUP98 oncoproteins
As exogenous expression of NUP98 oncoproteins was found to reduce the fraction of BubR1 bound to APC/CCdc20,18 and BUBR1, BUB3 and MAD2 proved to be not associated with NUP98 oncoproteins (Fig. 1C), we sought to determine whether the overexpression of MCC components would rescue SAC attenuation caused by NUP98 oncoprotein expression. HEK293 cells expressing NUP98-HOXD13 were transfected with expression vectors for BUBR1, CDC20, and MAD2, and arrested in mitosis using nocodazole. These were subsequently harvested at different time points and analyzed for SAC attenuation by assessing securin protein levels in immunoblottings of total cell extracts. The overexpression of CDC20 or MAD2 abolished NUP98-HOXD13-induced securin degradation, while BUBR1 overexpression did not (Fig. 3A see also Fig. S3). These results suggest that NUP98 oncoproteins and MCC components compete for binding to the APC/C, likely via similar or overlapping docking surfaces, and that, most importantly, the effect of NUP98 oncoproteins on the APC/CCdc20 is potentially reversible.
Figure 3.
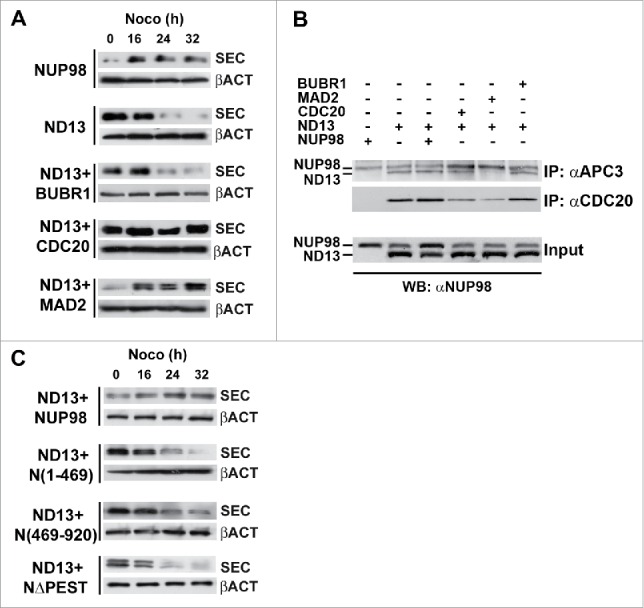
CDC20 and MAD2 rescue SAC attenuation by NUP98 oncoproteins. A), Immunoblots showing securin (SEC) degradation in extracts prepared from Nocodazole-arrested HEK293 cells exogenously expressing the indicated proteins (left). Samples were collected at the indicated times after incubation. Loading control: Anti-ßactin (ßACT). B), HEK293 were transfected with expression constructs for the indicated proteins, mitotic extracts were obtained from nocodazole-treated cells and subjected to immunoprecipitation (IP) with the indicated antibodies. Immunoprecipitated proteins were revealed by immunoblot analysis (WB) with an anti-NUP98 antibody (αNUP98). C), Immunoblots showing securin (SEC) degradation. Mitotic extracts were prepared from nocodazole-arrested HEK293 cells, expressing the indicated proteins (left), as in A).
We thus next wanted to determine whether the overexpression of BUBR1, MAD2, and CDC20 antagonized NUP98-HOXD13 oncoprotein activity by causing its dissociation from the APC/C. We therefore overexpressed in HEK293 cells BUBR1, MAD2, and CDC20 together with NUP98-HOXD13 and verified the coimmunoprecipitation of NUP98-HOXD13 with anti-APC3 or anti-CDC20 antibodies. The overexpression of BUBR1 or of NUP98 wt did not affect the amount of NUP98-HOXD13 coimmunoprecipitated with APC3, and the overexpression of CDC20 only slightly reduced the amount of NUP98-HOXD13 coimmunoprecipitated with APC3 (Fig. 3B). Conversely, the overexpression of MAD2 nearly abolished the interaction of NUP98-HOXD13 with the APC/C (Fig. 3B). Immunoprecipitations with anti-CDC20 confirmed that overexpression of CDC20 only weakly reduces the interaction between NUP98-HOXD13 and APC/CCdc20, whereas the overexpression of MAD2 significantly reduces the fraction of NUP98-HOXD13 bound to APC/CCdc20 (Fig. 3B). Together these results suggest that the rescue of NUP98 oncoprotein-induced SAC attenuation by MAD2 overexpression rests on the dissociation of NUP98 oncoproteins from APC/CCdc20, while rescue by CDC20 overexpression relies only partially on NUP98 oncoproteins displacement from the APC/CCdc20.
Interestingly, the overexpression of NUP98 wt, while not leading to a displacement of NUP98-HOXD13 from the APC/CCdc20 (Fig. 3B), could also efficiently rescue NUP98-HOXD13-induced securin degradation (Fig. 3C). In the same conditions, the overexpression of NUP98 mutant derivatives, such as NUP98(1–469), NUP98(469–920), and NUP98ΔPEST did not cause a reversal of NUP98-HOXD13-induced securin degradation (Fig. 3C).
To further explore this surprising finding, we sought to determine whether NUP98 wt was able to physically interact with the APC/C in phases of the cell cycle other than mitosis, and could thus provide an explanation for the observed antagonism with NUP98-HOXD13.
NUP98 interacts with CDC20 prior to but not during mitosis
To verify whether NUP98 interacts with the APC/CCdc20 during interphase, extracts from asynchronous or mitotically-arrested HEK293 cells were immunoprecipitated with anti-CDC20 antibodies, and co-immunoprecipitation of endogenous NUP98 was assayed by immunoblotting (Fig. 4A). In asynchronous HEK293 cells, a lower, faster-migrating form of NUP98 was indeed found to physically interact with the CDC20 APC/C component (Fig. 4A). Similarly, in HeLa cells, both asynchronous and G2-arrested, a faster-migrating form of NUP98 was efficiently co-immunoprecipitated by anti-CDC20 antibodies. Conversely, as expected, no interaction with CDC20 was detectable in mitotically arrested HeLa cells (Fig. 4A). These results indicate that NUP98 actually interacts with APC/CCdc20, albeit exclusively prior to mitosis, and that a slower-migrating, possibly modified form of NUP98, present in mitosis, is unable to bind APC/CCdc20.
Figure 4.
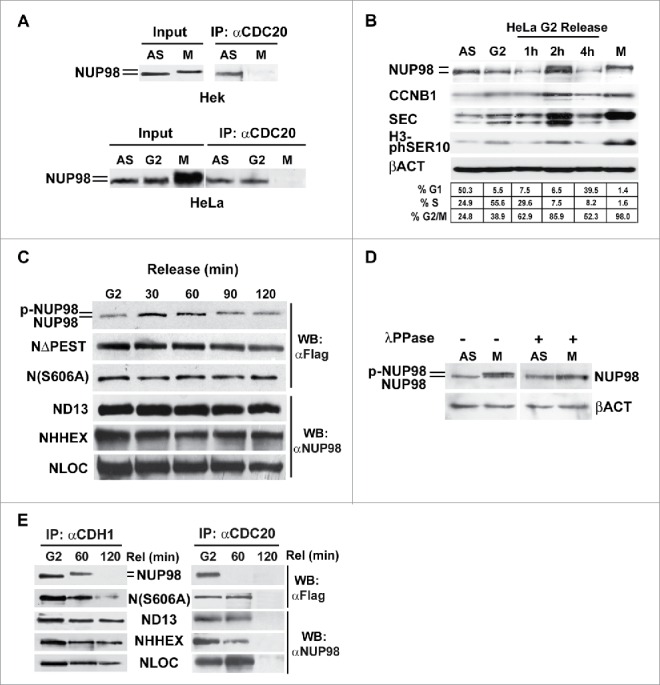
NUP98 binds to CDC20 during interphase. A), Extracts were obtained from asynchronous (AS) HEK293 or HeLa cells, from HEK293 cells mitotically arrested by nocodazole-treatment (M), from HeLa cells mitotically arrested by nocodazole-treatment (M), or from G2-phase enriched HeLa cells collected after shake-off of mitotic cells (G2). Samples were subjected to immunoprecipitation (IP) with the anti-CDC20 antibody (αCDC20). Immunoprecipitated proteins were revealed by immunoblot analysis (WB) with an anti-NUP98 antibody (αNUP98). B), Extracts were obtained from asynchronous (AS) HeLa cells, or from G2-phase enriched (G2) HeLa cells collected after shake-off of mitotic cells (M), and from cells released after 1, 2, and 4 hrs. Immunoblot analysis was performed with an anti-NUP98 antibody. C), HEK293 cells were transfected with expression constructs for Flag-tagged NUP98, NUP98ΔPEST (NΔPEST), and NUP98(S606A) (N(S606A)), or for NUP98-HOXD13 (ND13), NUP98-LOC (NLOC), NUP98-HHEX (NHHX). Extracts were obtained from nocodazole-treated cells after shake-off of mitotic cells and analyzed using a cell free system recapitulating mitotic progression (see text). Immunoblot analysis (WB) was performed with an anti-NUP98 (αNUP98) or with an anti-Flag (αFlag) antibody. D), Extracts obtained from asynchronous (AS) or from mitotically arrested (M) HEK293 cells were treated with lambda protein phosphatase (λPPase) Immunoblot analysis (WB) was performed with an anti-NUP98 antibody. Loading control: Anti-ßactin (ßACT). E), HEK293 cells were transfected with expression constructs for Flag-tagged NUP98, and NUP98(S606A) (N(S606A)), or for NUP98-HOXD13 (ND13), NUP98-LOC (NLOC), NUP98-HHEX (NHHX). Extracts from G2-enriched HEK293 cells, obtained after nocodazole treatment and shake-off of mitotic cells (G2), were released in vitro, and subjected at two different time points to immunoprecipitation (IP) with anti-CDH1 (αCDH1) or anti-CDC20 (αCDC20) antibodies. Immunoblot analysis (WB) was performed with an anti-NUP98 (αNUP98) or with an anti-Flag (αFlag) antibody.
We next wanted to further investigate if the appearance of the slower-migrating form of NUP98 correlated with mitotic entry. We therefore analyzed G2 phase-enriched HeLa cells, obtained after nocodazole treatment, removal of mitotic cells by shake-off, and subsequent release of the remaining adherent cells from nocodazole block by adding fresh medium. Cell extracts were prepared at 1, 2, and 4 hours after release and subjected to immunoblotting. After 2 hours from the release, NUP98 was detectable as a band migrating slower than the form detected in G2 or in asynchronously growing cells, and of the same size of that detected in mitotic cells (Fig. 4B), suggesting that NUP98 is modified early upon M phase entry.
We then verified whether the mitotic modification of NUP98 was detectable also in NUP98 fusion oncoproteins. HEK293 cells expressing the NUP98-HOXD13, NUP98-LOC, and NUP98-HHEX were treated with nocodazole and analyzed after shake-off using a cell-free system that recapitulates mitotic checkpoint events.18,22 While NUP98 wt displayed a mobility shift after 90 min following release, no differences in electrophoretic mobility were observed for NUP98-HOXD13, NUP98-LOC, and NUP98-HHEX even at 120 minutes after release (Fig. 4C). Similarly, no differences in mobility were observed for the NUP98ΔPEST mutant (Fig. 4C). Together these results suggest that the modification of NUP98 occurring at the onset of mitosis requires the C-terminal portion of the protein and, more specifically, the NUP98 PEST domain, which are missing in NUP98 oncoproteins.
A number of phosphorylation sites have been identified within NUP98, whose phosphorylation has been shown to be crucial to nuclear pore disassembly at the onset of mitosis.23 We generated a NUP98 mutant, NUP98(S606A), which affects one of the reported phosphorylation sites, S606, that lies within the PEST domain (see Fig. S4). No differences in electrophoretic mobility were observed for the NUP98(S606A) mutant, even after 120 minutes of release (Fig. 4C). These results indicate that the different forms of NUP98 observed in interphase and in mitosis likely reflect differential phosphorylation statuses of NUP98 during the cell cycle. Indeed, treatment of mitotic cell extracts with lambda protein phosphatase (λPPase) was sufficient to completely eliminate the difference in mobility (Fig. 4D).
As NUP98 has been shown to interact with the CDH1-bound form of APC/C already in early mitosis,14 we wanted to confirm whether the mitotic form of NUP98 was capable of differentially interacting with APC/CCdh1 versus APC/CCdc20. Extracts from G2-enriched HEK293 cells, obtained after nocodazole treatment and shake-off of mitotic cells, were released in vitro as described in,22 and subjected, at two different time points, to immunoprecipitation with anti-CDH1 or anti-CDC20 antibodies. In G2-arrested cells NUP98 co-immnuoprecipitated both with CDH1 as well as with CDC20 (Fig. 4E), whereas after 60 min of release the mitotic form of NUP98 co-immunoprecipitated only with CDH1. At a later time point (120 min), NUP98 was not detected in both anti-CDH1 and anti-CDC20 immunoprecipitates (Fig. 4E). Conversely, NUP98 oncoproteins interacted both with CDH1 and with CDC20 at 60 minutes after release (Fig. 4E). At 120 after the release all 3 NUP98 oncoproteins tested dissociated from CDC20, while interaction with CDH1 persisted (Fig. 4E). Notably, the NUP98(S606A) mutant displayed a behavior superimposable to that of NUP98 oncoproteins, being bound both to CDH1 and to CDC20 at 60 minutes after release and dissociating from CDC20 120 minutes after the release (Fig. 4E). NUP98(S606A), moreover, displayed an intracellular half-life superimposable to that of the NUP98-HOXD13 oncoprotein and of the NUP98ΔPEST mutant (Fig. S5), suggesting that the knocking-out of this phosphorylation target site is sufficient to confer NUP98 at least some of the aberrant properties of NUP98 fusion oncoproteins.
NUP98 is a substrate of APC/CCdc20 prior to the onset of mitosis
We next sought to determine the functional significance of the observed interaction between NUP98 and the APC/C prior to mitosis. One possible explanation envisaged NUP98 as a substrate of APC/C. We thus set out to assess possible variations of NUP98 protein levels during the cell cycle. HeLa cells, arrested in late G1 phase by double thymidine block, were subsequently released and collected at different time points to prepare total cell extracts (Fig. 5A). NUP98 levels remained relatively low with respect to asynchronously growing cells (AS), until 9 hrs after release, when they reached their maximum, which coincided with the majority of cells being in G2/M phase (Fig. 5A). At 12 hrs after release, NUP98 levels sharply declined as cells were exiting mitosis and re-entering G1, concomitantly with the persistence of high levels of CDC20 (Fig. 5A). Similarly, in human primary fibroblasts, synchronized by serum starvation and subsequently released, NUP98 levels were relatively low throughout the cell cycle, reaching their peak only when a large fraction of the cells approached mitosis (Fig. S6).
Figure 5.
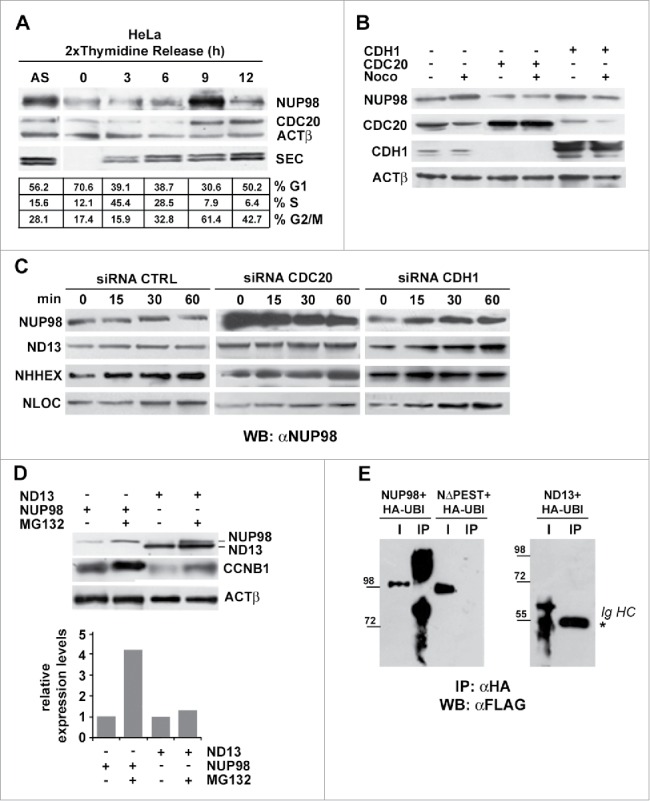
NUP98 is a substrate of APC/CCdc20 prior to the onset of mitosis. A), Extracts were obtained from asynchronous (AS) HeLa cells, or from HeLa cells arrested in late G1 phase by double thymidine block and subsequently released and collected at the indicated time points. Immunoblot analysis was performed using an anti-NUP98 (NUP98), an anti-CDC20 (CDC20), or an anti-Securin (SEC) antibody. Loading control: Anti-ßactin (ßACT). B), HEK293 were transfected with expression constructs for CDH1 or CDC20. Extracts were prepared from nocodazole-arrested (noco) or from control cells and subjected to immunoblot analysis with anti-NUP98, anti-CDH1, anti-CDC20 antibodies. Loading control: Anti-ßactin (ßACT). C), Knock down experiments, using a control scrambled siRNA (CTRL), siRNA directed against CDH1, or against CDC20, in cells expressing NUP98, NUP98-HOXD13 (ND13), NUP98-LOC (NLOC), or NUP98-HHEX (NHHX). Immunoblot analysis (WB) was performed with an anti-NUP98 (αNUP98) antibody. D), HEK293 cells were arrested with nocodazole, transfected with expression constructs for NUP98 or NUP98-HOXD13, and treated with the proteasome inhibitor MG132. Immunoblot analysis was performed with an anti-NUP98 (αNUP98) or with an anti-cyclinB1 (CCNB1) antibody. Loading control: Anti-ßactin (ßACT). E), Ubiquitination assays in HEK293 cells. HEK293 cells were transfected with expression constructs for NUP98, NUP98-HOXD13 (ND13), or NUP98ΔPEST (NΔPEST) and for HA-tagged Ubiquitin (HA-UBI). Cell extracts were subjected to immunoprecipitation (IP) with the anti-HA antibody (αHA). Immunoprecipitated proteins were revealed by immunoblot analysis (WB) with an anti-FLAG antibody (αFLAG). IgHC, immunoglobulin heavy chains.
To determine whether the APC/CCdc20, the APC/CCdh1, or both forms of APC/C were responsible for NUP98 degradation, we overexpressed in HEK cells the CDH1 or the CDC20 APC/C co-activators and determined NUP98 levels. While CDH1 overexpression did not affect NUP98 protein levels, both in nocodazole-treated and in untreated cells, CDC20 overexpression led to a visible reduction in NUP98 levels, both in the presence or absence of nocodazole (Fig. 5B). These results suggested that NUP98 is mainly targeted by APC/CCdc20 for degradation. We thus sought to confirm APC/CCdc20 involvement in NUP98 protein degradation by siRNA-mediated knockdown of CDC20 or of CDH1. CDC20 knockdown led to a substantial increase in NUP98 levels, while control siRNA left NUP98 amounts unaffected (Fig. 5C). Conversely, siRNA-mediated knockdown of CDH1 did not cause any changes in NUP98 levels (Fig. 5C). In the same experiments, NUP98 fusion oncoprotein levels did not show any variation, indicating that these are targeted neither by the APC/CCdc20 nor by the APC/CCdh1 (Fig. 5C).
To further substantiate the conclusion that NUP98 protein turnover occurs via its ubiquitynation and proteasome-mediated degradation, we analyzed NUP98 and NUP98-HOXD13 protein levels in the presence of the MG132 proteasome inhibitor. MG132 treatment led to a considerable increase in NUP98 levels (Fig. 5D), while left NUP98-HOXD13 levels essentially unchanged (Fig. 5D).
Finally, we verified NUP98 ubiquitination by overexpressing HA-tagged ubiquitin in HEK cells and by immunoprecipitating HA-ubiquitinated proteins from whole cell extracts. Immunoblotting using anti-FLAG antibodies showed the presence of ubiquitinated and degraded forms of NUP98 (Fig. 5E). Conversely, in the same conditions, the NUP98ΔPEST mutant was not ubiquitinated (Fig. 5E).
Together, these results show that the wt NUP98 protein is a target of the CDC20-activated form of APC/C during G2 phase, but not during mitosis, thus accounting for the observed physical interaction of NUP98 with CDC20 during interphase. These results furthermore suggest that phosphorylation of NUP98, at the onset of mitosis, at a critical residue(s), such as S606, could trigger conformational changes that would prevent its interaction with the APC/CCdc20, thus causing its accumulation.
PIN1 promotes the stability of NUP98 but not of NUP98 fusion oncoproteins
The peptidyl-prolyl isomerase PIN1 has recently emerged as a critical player in many cellular processes including cell cycle control.24 PIN1 is known to affect the stability and function of phosphoproteins, by catalyzing structural changes at phosphorylated Ser or Thr residues within pSer/Thr-Pro sequences. As S606 within the NUP98 PEST motif matches these requirements (see Fig S4), PIN1 appeared to us as a potential candidate for a modulator of NUP98 protein interaction with APC/CCdc20. We therefore tested whether the overexpression of PIN1 would affect endogenous NUP98 protein levels both in cycling and in mitotically-arrested cells. PIN1 enforced expression caused indeed a substantial increase in endogenous NUP98 protein amounts in both cycling and nocodazole-arrested cells (Fig. 6A), suggesting its direct effect on NUP98 protein stability. We further wanted to verify whether PIN1 overexpression would also affect the stability of the NUP98-HOXD13 oncoprotein, or of the NUP98ΔPEST and NUP98S606A mutants. PIN1 enforced expression did not alter NUP98-HOXD13 levels, nor did it affect the levels of NUP98ΔPEST and NUP98S606A (Fig. 6B).
Figure 6.
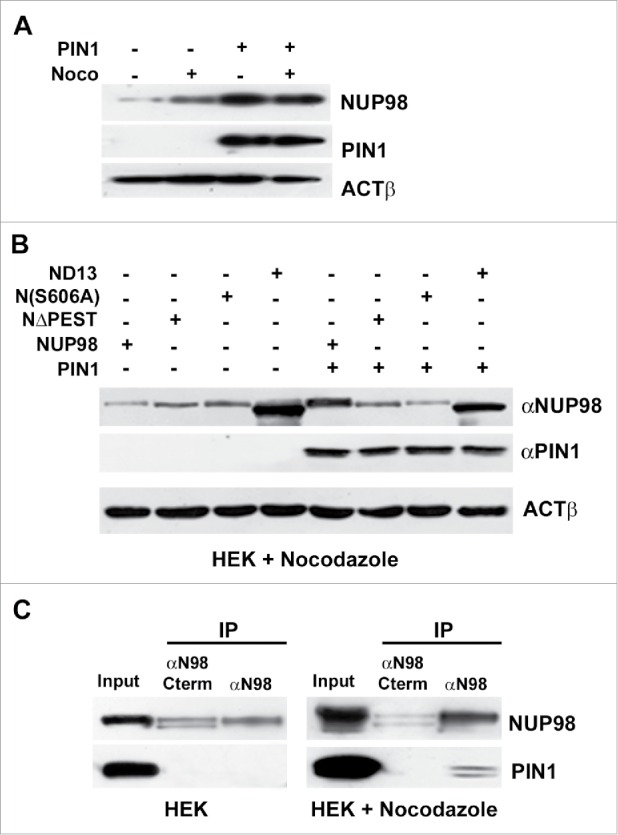
PIN1 interacts with and promotes the stability of NUP98. A), HEK293 cells were transfected with an expression construct for PIN1. Extracts were obtained from nocodazole-treated cells (noco) or from control cells. Immunoblot analysis was performed using an anti-NUP98 (NUP98) or an anti-PIN1 (PIN1) antibody. Loading control: Anti-ßactin (ßACT). B), HEK293 cells were transfected or co-transfected with expression constructs for NUP98, NUP98ΔPEST (NΔPEST), NUP98(S606A) (N(S606A)), NUP98-HOXD13 (ND13), or PIN1. Immunoblot analysis was performed using an anti-NUP98 (NUP98) or an anti-PIN1 (PIN1) antibody. Loading control: Anti-ßactin (ßACT). C), HEK293 cells were transfected with an expression construct for HA-tagged PIN1. Extracts were obtained from nocodazole-treated cells or from control cells and subjected to immunoprecipitation (IP) with an antibody directed against the C-terminal portion of NUP98 (αN98C-term), which recognizes only the non-phosphorylated form of NUP98, or with an antibody directed against the N-terminus of NUP98 (αN98), which recognizes both phosphorylated and non-phosphorylated NUP98. Immunoblot analysis was performed using an anti-NUP98 (NUP98) or an anti-PIN1 (PIN1) antibody.
Finally, we sought to determine whether PIN1 and NUP98 physically interact in mitotically arrested cells. To this end, we immunoprecipitated exogenously-expressed NUP98 from whole-cell extracts of asynchronous or nocodazole-arrested HEK cells with two different antibodies, one directed against the C-terminal portion of NUP98, which recognizes only the non-phosphorylated form of NUP98,23 the other directed against the N-terminus of NUP98, which recognizes both phosphorylated and non-phosphorylated NUP98. Exogenously-expressed PIN1 co-immunoprecipitated with NUP98 only in mitotically arrested cells and only using the antibody recognizing both phosphorylated and non-phosphorylated NUP98 (Fig. 6C). No interaction was detected in asynchronous cells (Fig. 6C).
Together, these results point to a role of PIN1 in modulating the stability of NUP98 during the cell cycle. More specifically, they suggest a direct action of PIN1 on the phosphorylated, mitotic form of NUP98, as the intracellular levels of the phosphorylation defective mutant NUP98S606A were not affected by PIN1 overexpression and the non-phosphorylated form of NUP98 did not physically interact with PIN1.
Discussion
The nucleoporin NUP98 is involved in the formation of fusion oncoproteins with over 20 different partner proteins, as a result of chromosomal translocations that cause AML and MDS.1,2 The functional characterization of the components of oncogenic fusion proteins is crucial to understand the transformation process and to allow the tailoring of specific therapeutic approaches. To explain the leukemogenic action of the wide array of NUP98 oncoproteins, several models have been proposed, mainly centered on their capability to perturb at the transcriptional level the normal differentiation program of haematopoietic progenitor cells.4 We recently reported an additional mechanism centered on the NUP98 moiety, thus potentially common to all NUP98 fusion oncoproteins,18 which stems from the increasing appreciation that NUP98, like other nucleoporins, is implicated is several processes, in addition to nucleocytoplasmic transport, including mitotic progression see.9 We demonstrated that NUP98 oncoproteins interfere with the function of the anaphase-promoting complex (APC/C), leading to an attenuation of the mitotic checkpoint (or spindle assembly checkpoint, SAC), ultimately resulting in whole chromosome instability.18 Chromosomal instability due to deregulation of the mitotic checkpoint has been shown to increase the predisposition to malignant transformation,25-27 and, more specifically, the misregulation of APC/C components has been reported to be associated with a number of human malignancies.28-31
In this work, we explored the molecular mechanisms underlying the perturbation of SAC function by NUP98 oncoproteins, as these remained still to be clarified. Our evidence points to a mechanism for the interference of NUP98 oncoproteins with APC/CCdc20 function, which rests on the aberrant occupancy by NUP98 oncoproteins of a docking surface(s) for MCC components within the APC/CCdc20, thus preventing inhibition by the MCC in the presence of an unsatisfied SAC. We show namely that NUP98 oncoproteins physically interact with the CDC20 protein in the context of the APC/C, as they could be also coimmunoprecipitated with three major APC/C components, APC3, APC4, and APC10. The presence of NUP98 oncoproteins within the APC/CCdc20 prevents its association with key components of the mitotic checkpoint complex (MCC), such MAD2, BUBR1, and BUB3, in the course of an unsatisfied SAC, thus “locking” the APC/CCdc20 in an active form and hence ultimately resulting in an attenuation of the mitotic checkpoint.
The interaction of NUP98 oncoproteins with the APC/CCdc20 differs from that of NUP98 wt, which, together with RAE1 inhibits APC/CCdh1 function at the onset of mitosis.14 NUP98 oncoproteins, while being potentially still capable of heterodimerizing, via their NUP98 portion, with RAE1,18 were not found to bind the APC/CCdc20 in association with RAE1.
The interference of NUP98 oncoproteins with APC/CCdc20 revealed to be reversible, as the overexpression of MAD2 and CDC20 could rescue SAC attenuation induced by NUP98-HOXD13 and caused partial (CDC20) or complete (MAD2) dissociation of NUP98-HOXD13 from APC/CCdc20. Interestingly, BubR1 overexpression was ineffective, indicating that among MCC components CDC20 and MAD2 are those that become limiting in a NUP98 oncoprotein-attenuated SAC. MAD2 is considered as a crucial factor in the assembly and function of the MCC. MAD2 directly competes with the coactivator function of CDC20 by binding to one of the two CDC20 molecules associated to APC/CCdc20. And dissociation of MAD2 with the concomitant ubiquitin-dependent degradation of CDC20 is held to be a major checkpoint silencing mechanism, leading to APC/C re-activation upon checkpoint satisfaction (reviewed in17). Further work will be required to determine the structure of the NUP98 oncoprotein-bound APC/CCdc20, to reveal its exact composition, and to determine the position occupied by NUP98 oncoproteins within the complex.
By generating and testing mutant derivatives of the NUP98-HOXD13 oncoprotein and of NUP98 wt, we were able to establish that for NUP98 oncoproteins, the domain required for interaction with the APC/CCdc20 resides within the GLEBS-like domain present in the NUP98 moiety. Surprisingly, we also found that the interaction of NUP98 oncoproteins with the APC/CCdc20 could be mimicked by NUP98 mutants affecting the C-terminal portion of the protein, such as NUP98(1–469), which corresponds to the portion of NUP98 present in fusion oncoproteins, and NUP98ΔPEST, which represents a deletion of a PEST sequence,18 located within the C-terminal portion of NUP98. This observation prompted us to speculate that wild-type NUP98, under certain conditions, may interact with APC/CCdc20, possibly as a substrate, and that the NUP98 C-terminal portion, and in particular the PEST sequence, could exert a control over the capability of NUP98 to interact with, and be a substrate of the APC/CCdc20. We found that NUP98 indeed interacts with APC/CCdc20, albeit only prior to, but not during mitosis, and that NUP98 association with APC/CCdc20 results in the ubiquitination of NUP98, contributing to its turnover during interphase. The lack of interaction between NUP98 and APC/CCdc20 in mitosis correlated with the presence, in mitotically-arrested cells, of a slower-migrating, phosphorylated form of NUP98. Several potential phosphorylation sites have been reported to be targeted by Ser/Thr kinases upon entry in mitosis, concomitantly with nuclear pore disassembly.23 We focused on S606, as this evolutionarily conserved phosphorylation target site, is located within the NUP98 PEST sequence, and was shown to be phosphorylatable by CDK1 in vitro.23 A phosphodeficient mutant, NUP98(S606A), could indeed mimic the aberrant interaction with APC/CCdc20 displayed by NUP98 oncoproteins and by the NUP98ΔPEST mutant, confirming that phosphorylation at S606 is relevant for the lack of association of NUP98 with APC/CCdc20 and for its accumulation at the onset of mitosis. Interestingly, the NUP98(S606A) mutant, despite its capability to interact also in mitosis with the APC/CCdc20, proved to be as stable as the NUP98-HOXD13 oncoprotein and the NUP98ΔPEST mutant, suggesting that the substitution of S606 results in addition in a lack of ubiquitination by APC/CCdc20. NUP98 wt can thus be regarded as a conditional target of APC/CCdc20 depending on the phosphorylation state of its PEST sequence, and in particular of S606, whose phosphorylation state appears to affect the conformation of NUP98.
The NUP98 S606 residue is part of a peptide sequence, Ser-Pro, which represents, in its phosphorylated form (pSer-Pro), a potential target for the activity of peptidyl-prolyl isomerases. These are enzymes known to affect the stability and function of phosphoproteins by catalyzing the isomerization of phosphor-Ser-Pro or phosphor-Thr-Pro peptide bonds, thus inducing conformational changes (reviewed in24,32). Because the PIN1 peptidyl-prolyl isomerase has recently emerged as a relevant player in cell cycle control,32 it appeared to us as a good candidate for a modulator of NUP98 protein conformation. Our results show that PIN1 overexpression indeed leads to an increase in NUP98 protein levels, possibly via its stabilization, while no effect is observed on NUP98 oncoproteins or on the NUP98ΔPEST and NUP98(S606A) mutants. Further work will be required to characterize the role of PIN1 on NUP98 function.
The finding that NUP98 can be a target of APC/CCdc20 provides important clues about the cause of the aberrant interaction of NUP98 oncoproteins with APC/CCdc20. Based on our results, we propose that the ability of NUP98 oncoproteins to bind APC/CCdc20 via their NUP98 moiety reflects the potential of NUP98 to bind APC/CCdc20 as a substrate during interphase. However, while the interaction of wild-type NUP98 with APC/CCdc20 is controlled by its PEST sequence, via phosphorylation and the possible subsequent PIN1-mediated isomerization at pSer606-Pro, NUP98 fusion oncoproteins, which lack the NUP98 C-terminal portion altogether, and thus the PEST sequence, are constitutively bound to APC/CCdc20. NUP98 oncoproteins, though, are not effective substrates of APC/CCdc20, probably because they lack the PEST sequence, and thus remain bound to APC/CCdc20, preventing its regulation by the MCC in the presence of an unsatisfied mitotic checkpoint. NUP98 oncoproteins could thus be considered as de facto pseudosubstrates, which by lingering in the APC/CCdc20 would “lock” it in an active form, ultimately leading to an attenuation of the spindle assembly checkpoint.
Materials and methods
Plasmid constructs
The expression vectors for pSG5-FlagNUP98(N98) pSG5-FlagNUPHOXD13 (Flag-ND13), EGFP-NUPD13 (ND13), EGFP-NUPLOC348801(isoform1) (NLOC) and EGFP-NUP98HHEX (NHHEX) have already been described in Refs.18 and 21. NUP98 deletion mutants were obtained by cloning the N-terminal (amino acids 1–469, NUP98(1–469)) or the C-terminal (amino acids 469–920, NUP(469–920)) portion of NUP98 into the pSG5 expression vector. Expression constructs for the NUP98ΔPEST and NUP98(S606A) mutants were obtained by PCR-mediated mutagenesis using pSG5-NUP98 as a template by deleting the aa from 599 to 616 of NUP98 cDNA or by substituting the serine 606 residue (S) with an alanine residue (A). Mutants of pSG5-Flag ND13 were obtained by deleting the N-terminal portion of the protein from aa 34 to 262, (ND13DGLEBS) or by fusion of the NUP98 GLEBS domain (aa 154 to 221) in frame with ND13 homeodomain portion (ND13GLEBS-HD).
Cell culture, retroviral transduction and transfection
HEK-293 (ATCC# CRL-1573) human embryonic kidney cells, HeLa (ATCC# CCL-2) human epithelial cervix cells and human primary fibroblasts (hPF) were cultured in Dulbecco's modified Eagle's medium (DMEM) (Celbio ECB7501L-50). Transfection experiments in HEK 293 or Hela cells were carried out by CaPO4 precipitation and cells were collected 48 hours after transfection. The cells were transfected with 20 µg of the expression vectors for Flag-NUP98-D13, Flag-NUP98, Flag-NUP98ΔPEST, Flag-NUP98(S606A), Flag-NUP98(1–469), Flag-NUP98(469–920), Flag-NUP98-D13ΔGLEBS, Flag-NUP98-D13GLEBS-HD, pSGNUP98-D13, pSGNUP98LOC, pSGNUP98HHEX per 10 cm dish.
Cell synchronisation and cytofluorimetric analysis
HEK293 cells and human primary fibroblasts were synchronized in G2/M-phase by treatment with 0.2 mg/ml Nocodazole (Sigma) and collected at the indicated times. HeLa cells were synchronized in G2/M phase by mitotic shake off following Nocodazole treatment. Primary fibroblasts were starved by growing highly confluent cultures in DMEM without fetal bovine serum (FBS) for 24 hours. Cells were then reseeded in DMEM plus 20% FBS and collected at the indicated time points. HeLa cells were synchronized in G1/S-phase by double thymidine block: cells were incubated with 2 mM thymidine for 18 hours, released in fresh DMEM for 9 hours and then treated again with thymidine for 15 hours. Cells were collected for cell cycle profile analysis and western blot at different time points after thymidine release. Protein synthesis inhibition in HEK cells was obtained by adding cycloheximide (CHX) at a final concentration of 100 µg/ml. After 3, 6, 12, 24 and 36 h of treatment cells were collected. In HEK 293 cells transiently transfected before Nocodazole or CHX treatments, the synchronization procedure started 24 hours after transfection. HEK293 treatment with the proteasome inhibitor MG132 was conducted for 16 hours at a final of concentration of 1µM. For cell cycle analysis, measure of DNA content in cells was performed by permeabilization of cells with Tryton-X100 0,1% and staining with 50 mg/ml of propidium iodide in 1X PBS.
Proteins depletion with siRNA
siRNA duplexes targeting the coding sequence of human CDH1, CDC20, and siRNA control duplexes were synthesized by Invitrogen (StealthR siRNA duplexes, 25 MER and Stealth RNAi Negative Control Duplexes, (12935–400). Two different siRNA duplexes were designed targeting the CDC20 (CDC20HSS101650, CDC20HSS101651), or CDH1 (FZR1HSS122071, FZR1HSS122072) mRNAs. siRNA transient transfection in Hek 293 cells was performed using 40–pM of RNAi duplexes per 6–cm wells, using LipofectamineTM 2000 (Invitrogen, 11668–027) following the manufacturer's protocol. Under these conditions, proteins depletion was detected by western blot analyses 48 hours after siRNA transfection.
Co-immunoprecipitation, immunoblotting and antibodies
For immunoblotting analysis of HEK 293 and human primary fibroblasts, total cell extracts were prepared by resuspending cells in lysis buffer (50 mM TrisHCl ph7.8, 400 mM NaCl, 1% NP40). Thirty µg of extracts were loaded in 10% poly-acrilamide gel and immunoblotting was performed using the indicated antibody(ies).
For co-immunoprecipitation experiments, cells were collected after nocodazole treatment, lysed in extraction buffer (20 mM Hepes ph7.9, 5 mMKCl, 1 nM DTT) and sonicated for 10.” For each immunoprecipitation, 250 µg were incubated overnight with 5 µg of antibody and precipitated with 10 µl of protein G agarose for 2 hours, then washed three times with extraction buffer. Half of the immunoprecipitated matherial was resolved by SDS-PAGE and detected by immunoblotting using the indicated antibody(ies). For co-immunoprecipitation of NUP98 and PIN1, cells were cross-linked with dithio-bis-(succinimidylproprionate) (DSP; Sigma) for 30 min at room temperature. The cross-linking was stopped by the addition of glycine at a 0.2 m final concentration for 15 min. 500 µg of cell extracts were then sonicated for 10” and subjected to immunoprecipitation as indicated.
Antibodies against Securin (Ab-3305), CDH1 (DSC-226), BUBR1 (Ab-70544) and RAE1 Ab-124783) were from Abcam. Antibodies against NUP98 (2288 S), Cyclin B (4135) BUB3 (Ab-8195), and HA epitope (2367) were from Cell Signaling Technology. The antibody against Flag-epitope was from sigma (F3165). Antibodies against NUP98 C-terminus (sc-74553), APC10 (sc-166790), CDC20 (sc-13162), and β–actin (sc-1616) were from Santa Cruz. Antibodies against APC3 (A301–183–A), APC4 (A301–176 A), and MAD2 (A300–301 A)) were from Bethyl.
Mitotic check point release in vitro
Nocodazole-arrested cell extracts preparation from HEK293 cells were obtained as described in22 with the exception that cells were sonicated 10” after lysis. A reaction mix (160 mg of each extract) was prepared and incubated with an ATP-regenerating system (5 mM MgCl2, 10 mM Phosphocreatine, 0,1–mg/ml Creatine phosphokinase, 1 mM ATP) at 30°C to induce mitotic release. At the indicated time points an aliquot from the mix corresponding to 30 µg of protein extract was collected for the immunoblot experiments.
Ubiquitination assays
HEK293 cells were transfected with pBluescript HA6xUbiquitine,33 Flag-NUP98-D13, Flag-NUP98 or Flag-NUP98ΔPEST. HEK293 were treated with 1 mM MG132 for 16 hours, harvested 48 hours after transfection and lysed in RIPA buffer supplemented with 1 mM PMSF, PIC, 2.5 mM sodium fluoride (NaF), 1 mM sodium orthovanadate (Na3VO4), 0.5 mM EDTA, 10 mM Iodoacetamide, 2 mM MG132 and 1 mM G5, Ubiquitin Isopeptidase Inhibitor I. 500 µg of cell extracts were used for co-immunoprecipitation using 10 mg of anti HA antibody.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
Thanks are due to Dr. Susanna Molinari for the kind gift of the expression construct for the PIN1 protein, to Prof. Carlo Pincelli for the kind gift of human primary fibroblasts, and to J. van Deursen for the kind gift of the pUH10SNUP98, K. Humphries for MIGR1-NUPD13, and C. Mecucci for the NUP98-LOC and NUP98-HHEX expression constructs.
Funding
This work was supported by grants from the Italian Association for Cancer Research (AIRC).
References
- [1].Moore MA, Chung KY, Plasilova M, Schuringa JJ, Shieh JH, Zhou P, Morrone G. NUP98 dysregulation in myeloid leukemogenesis. Ann N Y Acad Sci 2007; 1106:114-42; PMID:17442773; http://dx.doi.org/ 10.1196/annals.1392.019 [DOI] [PubMed] [Google Scholar]
- [2].Takeda A, Yaseen NR. Nucleoporins and nucleocytoplasmic transport in hematologic malignancies. Semin Cancer Biol 2014; 27:3-10; PMID:24657637; http://dx.doi.org/ 10.1016/j.semcancer.2014.02.009 [DOI] [PubMed] [Google Scholar]
- [3].Slape C, Aplan PD. The role of NUP98 gene fusions in hematologic malignancy. Leuk Lymphoma 2004; 45:1341-50; PMID:15359631; http://dx.doi.org/ 10.1080/10428190310001659325 [DOI] [PubMed] [Google Scholar]
- [4].Gough SM, Slape CI, Aplan PD. NUP98 gene fusions and hematopoietic malignancies: common themes and new biologic insights. Blood 2011; 118:6247-57; PMID:21948299; http://dx.doi.org/ 10.1182/blood-2011-07-328880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Fontoura BM, Blobel G, Matunis MJ. A conserved biogenesis pathway for nucleoporins: proteolytic processing of a 186-kilodalton precursor generates Nup98 and the novel nucleoporin, Nup96. J Cell Biol 1999; 144:1097-112; PMID:10087256; http://dx.doi.org/ 10.1083/jcb.144.6.1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Powers MA, Forbes DJ, Dahlberg JE, Lund E. The vertebrate GLFG nucleoporin, Nup98, is an essential component of multiple RNA export pathways. J Cell Biol 1997; 136:241-50; PMID:9015297; http://dx.doi.org/ 10.1083/jcb.136.2.241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Radu A, Moore MS, Blobel G. The peptide repeat domain of nucleoporin Nup98 functions as a docking site in transport across the nuclear pore complex. Cell 1995; 81:215-22; PMID:7736573; http://dx.doi.org/ 10.1016/0092-8674(95)90331-3 [DOI] [PubMed] [Google Scholar]
- [8].Iwamoto M, Asakawa H, Hiraoka Y, Haraguchi T. Nucleoporin Nup98: a gatekeeper in the eukaryotic kingdoms. Genes Cells 2010; 15:661-9; PMID:20545767; http://dx.doi.org/ 10.1111/j.1365-2443.2010.01415.x [DOI] [PubMed] [Google Scholar]
- [9].Chatel G, Fahrenkrog B. Dynamics and diverse functions of nuclear pore complex proteins. Nucleus 2012; 3:162-71; PMID:22555605; http://dx.doi.org/ 10.4161/nucl.19674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Bayliss R, Littlewood T, Stewart M. Structural basis for the interaction between FxFG nucleoporin repeats and importin-beta in nuclear trafficking. Cell 2000; 102:99-108; PMID:10929717; http://dx.doi.org/ 10.1016/S0092-8674(00)00014-3 [DOI] [PubMed] [Google Scholar]
- [11].Pritchard CE, Fornerod M, Kasper LH, van Deursen JM. RAE1 is a shuttling mRNA export factor that binds to a GLEBS-like NUP98 motif at the nuclear pore complex through multiple domains. J Cell Biol 1999; 145:237-54; PMID:10209021; http://dx.doi.org/ 10.1083/jcb.145.2.237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wang X, Babu JR, Harden JM, Jablonski SA, Gazi MH, Lingle WL, de Groen PC, Yen TJ, van Deursen JM. The mitotic checkpoint protein hBUB3 and the mRNA export factor hRAE1 interact with GLE2p-binding sequence (GLEBS)-containing proteins. J Biol Chem 2001; 276:26559-67; PMID:11352911; http://dx.doi.org/ 10.1074/jbc.M101083200 [DOI] [PubMed] [Google Scholar]
- [13].Matsuoka Y, Takagi M, Ban T, Miyazaki M, Yamamoto T, Kondo Y, Yoneda Y. Identification and characterization of nuclear pore subcomplexes in mitotic extract of human somatic cells. Biochem Biophys Res Commun 1999; 254:417-23; PMID:9918853; http://dx.doi.org/ 10.1006/bbrc.1998.9953 [DOI] [PubMed] [Google Scholar]
- [14].Jeganathan KB, Malureanu L, van Deursen JM. The Rae1-Nup98 complex prevents aneuploidy by inhibiting securin degradation. Nature 2005; 438:1036-9; PMID:16355229; http://dx.doi.org/ 10.1038/nature04221 [DOI] [PubMed] [Google Scholar]
- [15].Zou H, McGarry TJ, Bernal T, Kirschner MW. Identification of a vertebrate sister-chromatid separation inhibitor involved in transformation and tumorigenesis. Science 1999; 285:418-22; PMID:10411507; http://dx.doi.org/ 10.1126/science.285.5426.418 [DOI] [PubMed] [Google Scholar]
- [16].Uhlmann F, Lottspeich F, Nasmyth K. Sister-chromatid separation at anaphase onset is promoted by cleavage of the cohesin subunit Scc1. Nature 1999; 400:37-42; PMID:10403247; http://dx.doi.org/ 10.1038/21831 [DOI] [PubMed] [Google Scholar]
- [17].Musacchio A. The Molecular Biology of Spindle Assembly Checkpoint Signaling Dynamics. Curr Biol 2015; 25:R1002-18; PMID:26485365; http://dx.doi.org/ 10.1016/j.cub.2015.08.051 [DOI] [PubMed] [Google Scholar]
- [18].Salsi V, Ferrari S, Gorello P, Fantini S, Chiavolelli F, Mecucci C, Zappavigna V. NUP98 Fusion Oncoproteins Promote Aneuploidy by Attenuating the Mitotic Spindle Checkpoint. Cancer Res 2014; 74(4):1079-90; PMID:24371226; http://dx.doi.org/ 10.1158/0008-5472.CAN-13-0912 [DOI] [PubMed] [Google Scholar]
- [19].Raza-Egilmez SZ, Jani-Sait SN, Grossi M, Higgins MJ, Shows TB, Aplan PD. NUP98-HOXD13 gene fusion in therapy-related acute myelogenous leukemia. Cancer Res 1998; 58:4269-73; PMID:9766650 [PubMed] [Google Scholar]
- [20].Gorello P, Brandimarte L, La Starza R, Pierini V, Bury L, Rosati R, Martelli MF, Vandenberghe P, Wlodarska I, Mecucci C. t(3;11)(q12;p15)/NUP98-LOC348801 fusion transcript in acute myeloid leukemia. Haematologica 2008; 93:1398-401; PMID:18603550; http://dx.doi.org/ 10.3324/haematol.12945 [DOI] [PubMed] [Google Scholar]
- [21].Jankovic D, Gorello P, Liu T, Ehret S, La Starza R, Desjobert C, Baty F, Brutsche M, Jayaraman PS, Santoro A, et al.. Leukemogenic mechanisms and targets of a NUP98/HHEX fusion in acute myeloid leukemia. Blood 2008; 111:5672-82; PMID:18388181; http://dx.doi.org/ 10.1182/blood-2007-09-108175 [DOI] [PubMed] [Google Scholar]
- [22].Braunstein I, Miniowitz S, Moshe Y, Hershko A. Inhibitory factors associated with anaphase-promoting complex/cylosome in mitotic checkpoint. Proc Natl Acad Sci U S A 2007; 104:4870-5; PMID:17360335; http://dx.doi.org/ 10.1073/pnas.0700523104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Laurell E, Beck K, Krupina K, Theerthagiri G, Bodenmiller B, Horvath P, Aebersold R, Antonin W, Kutay U. Phosphorylation of Nup98 by Multiple Kinases Is Crucial for NPC Disassembly during Mitotic Entry. Cell 2011; 144:539-50; PMID:21335236; http://dx.doi.org/ 10.1016/j.cell.2011.01.012 [DOI] [PubMed] [Google Scholar]
- [24].Lu Z, Hunter T. Prolyl isomerase Pin1 in cancer. Cell Res 2014; 24:1033-49; PMID:25124924; http://dx.doi.org/ 10.1038/cr.2014.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Baker DJ, Chen J, van Deursen JM. The mitotic checkpoint in cancer and aging: what have mice taught us? Curr Opin Cell Biol 2005; 17:583-9; PMID:16226453; http://dx.doi.org/ 10.1016/j.ceb.2005.09.011 [DOI] [PubMed] [Google Scholar]
- [26].Dai W, Wang Q, Liu T, Swamy M, Fang Y, Xie S, Mahmood R, Yang YM, Xu M, Rao CV. Slippage of mitotic arrest and enhanced tumor development in mice with BubR1 haploinsufficiency. Cancer Res 2004; 64:440-5; PMID:14744753; http://dx.doi.org/ 10.1158/0008-5472.CAN-03-3119 [DOI] [PubMed] [Google Scholar]
- [27].Ricke RM, van Ree JH, van Deursen JM. Whole chromosome instability and cancer: a complex relationship. Trends Genet 2008; 24:457-66; PMID:18675487; http://dx.doi.org/ 10.1016/j.tig.2008.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kim JM, Sohn HY, Yoon SY, Oh JH, Yang JO, Kim JH, Song KS, Rho SM, Yoo HS, Kim YS, et al.. Identification of gastric cancer-related genes using a cDNA microarray containing novel expressed sequence tags expressed in gastric cancer cells. Clin Cancer Res 2005; 11:473-82; PMID:15701830 [PubMed] [Google Scholar]
- [29].Li D, Zhu J, Firozi PF, Abbruzzese JL, Evans DB, Cleary K, Friess H, Sen S. Overexpression of oncogenic STK15/BTAK/Aurora A kinase in human pancreatic cancer. Clin Cancer Res 2003; 9:991-7; PMID:12631597 [PubMed] [Google Scholar]
- [30].Singhal S, Amin KM, Kruklitis R, DeLong P, Friscia ME, Litzky LA, Putt ME, Kaiser LR, Albelda SM. Alterations in cell cycle genes in early stage lung adenocarcinoma identified by expression profiling. Cancer Biol Ther 2003; 2:291-8; PMID:12878869; http://dx.doi.org/ 10.4161/cbt.2.3.399 [DOI] [PubMed] [Google Scholar]
- [31].Wang CX, Fisk BC, Wadehra M, Su H, Braun J. Overexpression of murine fizzy-related (fzr) increases natural killer cell-mediated cell death and suppresses tumor growth. Blood 2000; 96:259-63; PMID:10891459 [PubMed] [Google Scholar]
- [32].Lin CH, Li HY, Lee YC, Calkins MJ, Lee KH, Yang CN, Lu PJ. Landscape of Pin1 in the cell cycle. Exp Biol Med (Maywood) 2015; 240:403-8; PMID:25662955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Badodi S, Baruffaldi F, Ganassi M, Battini R, Molinari S. Phosphorylation-dependent degradation of MEF2C contributes to regulate G2/M transition. Cell Cycle 2015; 14:1517-28; PMID:25789873; http://dx.doi.org/ 10.1080/15384101.2015.1026519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Corpet F. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res 1988; 16:10881-90; PMID:2849754; http://dx.doi.org/ 10.1093/nar/16.22.10881 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.