

ABSTRACT
p53 is a central factor in tumor suppression as exemplified by its frequent loss in human cancer. p53 exerts its tumor suppressive effects in multiple ways, but the ability to invoke the eradication of damaged cells by programmed cell death is considered a key factor. The ways in which p53 promotes cell death can involve direct activation or engagement of the cell death machinery, or can be via indirect mechanisms, for example though regulation of ER stress and autophagy. We present here another level of control in p53-mediated tumor suppression by showing that p53 activates the glycosidase, FUCA1, a modulator of N-linked glycosylation. We show that p53 transcriptionally activates FUCA1 and that p53 modulates fucosidase activity via FUCA1 up-regulation. Importantly, we also report that chemotherapeutic drugs induce FUCA1 and fucosidase activity in a p53-dependent manner. In this context, while we found that over-expression of FUCA1 does not induce cell death, RNAi-mediated knockdown of endogenous FUCA1 significantly attenuates p53-dependent, chemotherapy-induced apoptotic death. In summary, these findings add an additional component to p53s tumor suppressive response and highlight another mechanism by which the tumor suppressor controls programmed cell death that could potentially be exploited for cancer therapy.
KEYWORDS: cell death, chemotherapy, FUCA1, glycosylation, p53
Introduction
The removal of damaged cells by programmed cell death is one of the major mechanisms of tumor suppression.1 Cell death in this context can be achieved by multiple mechanisms, but a key regulator of this response is the tumor suppressor p53.2 The importance of this protein in the prevention of tumor development is highlighted by the fact that it is lost in approximately 50% of humans cancers and that mice deficient in p53 develop tumors by 6 months of age3,4. While p53 modulates multiple cellular pathways that are important for tumor suppression, studies have shown that its regulation of programmed cell death is of high importance.5 The protein primarily functions as a transcription factor and has been shown to activate a spectrum of genes involved in cell death regulation.6 These genes include a number that encode pro-apoptotic members of the Bcl-2 family such as Puma and Noxa which directly engage the intrinsic cell death pathway at mitochondria.7-9 In addition, p53 has also been reported to activate genes that stimulate the extrinsic apoptotic pathway.10 Moreover, in additional to these transactivation-dependent mechanisms, p53 has also been shown to regulate cell death more directly by binding to and repressing anti-apoptotic members of the Bcl-2 family resulting in mitochondrial outer membrane permeabilisation (MOMP) and release of apoptogenic factors into the cytosol.11-13
In addition to its role in tumor suppression, p53-induced programmed cell death is also an important factor in the response to chemotherapy.14 Many chemotherapeutic drugs induce DNA-damage which is a potent activation signal for p53.15-17 In unstimulated cells, p53 is restrained by its target gene Mdm2 which promotes p53 degradation. However, upon exposure to genotoxic agents, this negative feedback loop is alleviated and the levels of p53 increase and the protein becomes activated.16,17 The importance of this response in the effectiveness of chemotherapeutic agents is reflected by the decreased cell death that is observed upon drug exposure of p53-deficient cells.14 Moreover, the fact that cell death pathways can be perturbed during tumor development - including those caused by loss of p53 - has important implications for resistance to many chemotherapeutic drugs.
The perturbation of cell death pathways is only one facet of tumor progression.1 For the development of cancer, cells must attain a number of attributes which include for example, deregulated proliferation, evasion of immune surveillance and immortality.1 One characteristic on many cancers, however, that is relatively under-investigated involves changes in glycosylation. There are 2 principle forms of protein glycosylation and both have been reported to be perturbed in human cancer.18 O-linked glycosylation involves attachment of sugars to proteins via serine or threonine, whereas N-linked glycosylation involves attachment via asparagine.18 Interestingly, while increases and changes in glycosylation are considered common events in human cancer,19 very few connections have been reported between the control of glycosylation and the signaling pathways that either promote or repress tumor development. In this regard, we report here that p53 can transcriptionally activate the gene encoding the glycosidase FUCA1 which cleaves fucose linked moieties in N-linked glycans. Moreover, we show that chemotherapeutic drugs activate FUCA1 via p53 and that FUCA1 is a contributing factor to chemotherapy-induced cell death.
Materials and methods
Cell lines, cell culture and treatments
Saos2, RKO and U2OS cell lines were purchased from the American Type Culture Collection and maintained in DMEM (Gibco BRL, Paisley, UK) containing 10% FBS, 4.5 g l−1 glucose, 1 mM L-glutamine, 0.11 g l−1 pyruvate and maintained at 37°C in 5% CO2 atmosphere. p53wt and mutant 273H inducible cell lines have been previously described.20 Expression of p53 was induced using 1 μg/ml of Doxycycline (Dox). RKO-LMP-Scr, RKO-LMP-p53, U2OS-LMP-SCR, U2OS-LMP-p53 were generated by infection of RKO and U2OS cells with LMP-Scr and LMP-p53 respectively.21 RKO-pRS-Scr, RKO-pRS-p53, U2OS-pRS-SCR, U2OS-pRS-p53 were previously described (Crighton 2006 and Crighton 2007). HCT116 p53−/− cells were kind gift from Bert Vogelstein. E1a expressing U2OS were previously described.22 TetOn-TAp73α Saos-2, TetOn-TAp73β Saos-2, TetOn-TAp73γ Saos-2 were kind gifts from Gerry Melino and previously described.1 Expression of p73 was induced with 1 μg/ml Dox. Where indicated, cells were treated with Actinomycin D (ActD) (Sigma Aldrich St Louis, MI, USA); Adriamycin, Cisplatin or etoposide (Calbiochem).
siRNA, plasmids and transfections
FUCA1 knockdown was performed using pre-designed on target plus siRNAs purchased from Dharmacon (Lafayette, CO, USA). siRNAs directed against FUCA1 were transfected using Oligofectamine (Invitrogen, Life Technologies Paisley, UK) according to the manufacturer's instructions for 48 h before treatment.
pcDNA3-FUCAwt-Myc/His was generated by PCR and cloned in to sites of pcDNA3.1MycHisA (Invitrogen, Life Technologies Paisley, UK). pcDNA3-FUCA1M1-Myc/His was generated by site-directed mutagenesis. pCMV-CD20 has been previously described.23
Transient transfections into Saos2 cells were undertaken by calcium phosphate precipitation using 5 µg of indicated plasmid together with 1µg pCMV-CD20.
qRT-PCR
qRT-PCR analysis was undertaken as previously described.22 FUCA1 and FUCA2 primers are QuantiTect primers from QIAGEN. All samples were normalized to 18S rRNA and expressed as relative mRNA expression.
Fuca1 enzymatic activity
The enzymatic activity of alpha-L-fucosidase was assessed as previously described (Rapoport and Pendu 1999). Briefly, cells were lysed in 0.2 M acetate buffer pH5, containing 1% triton-X 100 (TTX), 0.1% SDS and protease inhibitor. Twenty-five µg of protein in 100 µl of 0.2 M acetate buffer pH5 were incubated in a 96 well plate together with 100 µl of 0.2mM 4-methylumbelliferyl alpha-l-fucopyranoside (Sigma Aldrich St Louis, MI, USA) for 90 minutes at 37°C.
Western blotting
Cells were lysed in buffer containing 1% TTX, 0.1 % SDS, 50 mM HEPES pH 7.5, 150 mM NaCl, 100 mM NaF, 10 mM EDTA, 10 mM Na4P2O7 and protease inhibitors (Roche) as previously described.24 Protein concentrations were determined by BCA assay (Sigma Aldrich St Louis, MI, USA). Cell lysates were separated by SDS-PAGE and transfer into Immobilon®-P membranes (Millipore). Membranes were probed with anti-p53, anti-p21 (sc-397, Santa Cruz, CA, USA), anti-HDM2, anti-FUCA1, cleaved caspase-3, anti-PARP (Cell Signaling Technology Beverly, MA, USA), Myc –tag (4A6) (Upstate Biotechnology), β-actin (ab8227, Abcam, Cambridge, UK) or Hsp90 (D-19) (Santa Cruz, CA, USA) antibodies.
Cell death analysis and caspase 3 activity
Cell death was evaluated by flow cytometry (FACScalibur, Becton Dickinson San Jose, CA, USA) as previously described.25 The percentage of cells with sub-G1 DNA content was taken as a measure of apoptotic rate.26
Cells which had been transfected with the pCMV-CD20 were stained with a FITC-conjugated CD20 antibody, sorted for fluorescence isothiocynate fluorescence, and analyzed for DNA content.25
Clonogenic survival assays were performed on Saos2 cells transfected with the indicated plasmids. 48 hours after transfection, cells were selected with 600 μg/ml G418 (Invitrogen, Life Technologies Paisley, UK) for 2 to 3 weeks and then stained with Giemsa (Sigma).
Caspase 3 activation was assessed by flow cytometry. Cells were fixed and permeabilized with Cytofix/Cytoperm, then incubated for 30 min with anti-active caspase-3-FITC antibody.
Chromatin immunoprecipitation
Chromatin was prepared from Saos2 cells treated with or without Dox. ChIP assays were performed using the ChIP-Assay kit (Merck Millipore) according to manufacturer's instructions. Chromatin was immunoprecipitated with 10 µg of anti-human p53 (clone DO-7, PharMingen) or anti-adenovirus E1A (PharMingen) as a negative control. PCR amplifications of FUCA1 region containing the consensus p53-binding sites, were performed using the specific primers,
FUCA1 (i) GAGAACAAGGGTGCAAAAGG
TCGGTTTGCATAGTGGTCTTT
FUCA1 (ii) TGGGACTTCCTCAAATCTGC
TCATGTGGTTTTGCTGTCCACAG
Control primers have been previously described27. The amount of coprecipitating DNA was normalized to inputs.
Luciferase reporter assay
Saos-2 cells were co-transfected with luciferase reporter constructs containing potential p53 binding sites and either control vector or a p53-WT-expressing construct. Twenty-four hours post-transfection, cells were lysed in 200 μl of 1 × Luciferase Cell Culture Lysis Reagent (Promega, Southampton, UK) and assayed for luciferase activity using the Veritas Microplate Luminometer.
Luciferase constructs containing potential p53 binding site i: AGGCTAGTCTCCAACTTGTGG within the promoter region (chromosome 1: 23,868, 672) was generated by PCR using the specific primer:
Forward(i): CTGGGATTACAGGCGCACCCCATG
Reverse(i): CCGGAGCCCGCATCGCTACCCTCAGCG
Luciferase constructs containing the potential p53 binding site ii: GGGCAAGTTCATGCAAGTTC within intron 1 (chromosome 1: 23.865,667) was generated by PCR using the specified primers
Forward(ii): TCGTTCCTACCAGAAGTGTTGAAG
Reverse(i): CCTGTGGACAGCAAAACCACATGAGC
Results
The glycosidase FUCA1 is a p53 target gene
As an approach to understand the mechanisms by which p53 directs programmed cell death, our group has previously performed microarrays using p53-inducible cells to identify novel p53 target genes. These microarray screens lead to the identification of DRAM1 and ADORA2B as genes activated by p53 in response to cellular stress.22,28 With the aim of identifying other factors regulated by p53 which contribute to its cell death response, we once again scrutinized these microarray data. Since little is known about the role of glycosylation in cancer, we were drawn to the fact that the mRNA for the glycosidase FUCA1 had increased levels when p53 was switched on in this inducible system (array data not shown).
To examine the relationship between p53 and FUCA1 in more detail, we performed qPCR on RNA from cells containing a tetracycline-inducible (TetOn) transgene for either wild-type p53 or a tumor-derived mutant of p53 in which amino acid 273 is changed from arginine to histidine. In confirmation of our microarray data we found that induction of wild-type p53 by treatment with doxycycline (Dox) could induce FUCA1 as well as 2 previously described target genes, HDM2 and p21 (Fig. 1A and B).16,17,29 By contrast, the 273H mutant of p53 had no impact on FUCA1 expression, nor did it affect p21 and HDM2 (Fig. 1A and B). Interestingly, p53 (either wild-type or mutant) had no significant impact on the expression of the FUCA1-related gene, FUCA2 (Fig. S1A). Moreover, we also found that FUCA1 was not induced by isoforms of the p53 family member p73 (Fig. S1B), indicating that unlike many other genes induced by p53, the induction of FUCA1 is to some level p53-specific.
Figure 1.
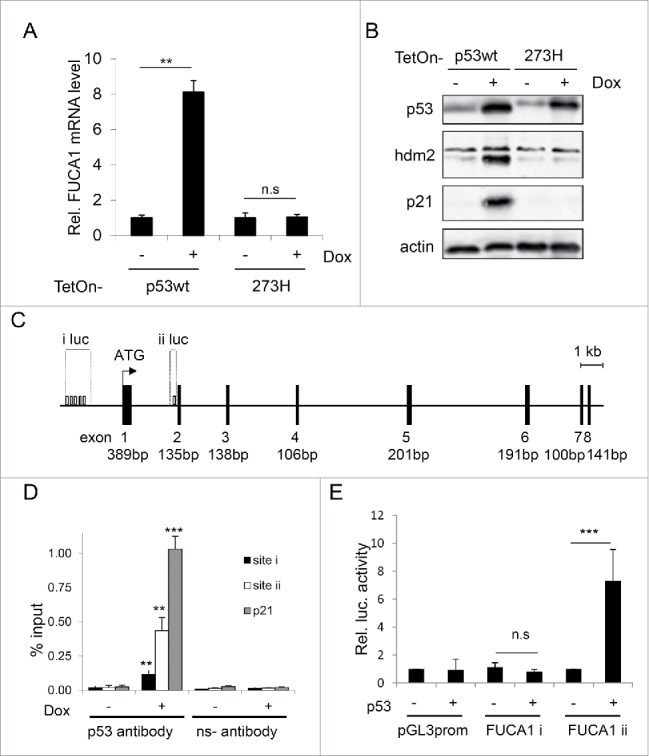
FUCA1 is a direct p53 target gene. (A-B) TetOn-p53wt and TetOn-p53273H Saos2 cells were treated with doxycycline (Dox) for 24 hours. FUCA1 mRNA levels were quantified by qRT-PCR and expressed as Relative Fuca1 mRNA level and represented as mean ± SD (n = 2 independent experiments, 6 replicates, one way Anova *** p < 0.0001; n.s = non significant)(A). p53, Hdm2 and p21expression were assessed by western blotting. Immunoblot against actin was used as a loading control (B). (C) Identification of 2 potential p53 binding site in the sequence of FUCA1 gene: one in the promoter region (i) the other one in the first intron (ii). (D) Chromatin immunoprecipitation (ChIP) was performed on TetOn-p53 cells treated with Dox for 24 hr. Immunoprecipitations were carried out with anti-sera against p53 or a nonspecific antibody (Ns). The % input of coprecipitating DNAs were calculated by quantitative PCR and presented as mean ± SD (n = 3 independent experiements, one way Anova: Site i **p = 0.0053, Site ii**p = 0.0006, p21 ***p < 0.0001). (E) Saos-2 cells were co-transfected with either an empty pcDNA3 vector or a wild-type-p53-expressing construct along with luciferase reporter constructs containing potential p53 binding sites i–ii (pGL3-FUCA1 i-ii) or vector control (pGL3 prom). Luciferase activity was analyzed 24 h after transfection and expressed as the relative fold increase relative to vector control (n = 4 independent experiements, one way Anova: Site i n.s p = 0.6751, Site ii ***p < 0.0001).
We considered that FUCA1 may be a direct target gene of p53 and so we examined the FUCA1 gene for p53 binding sites using the previously described MH algorithm.30 This revealed a number of potential p53 binding in the FUCA1 promoter and one potential site in FUCA1 Intron 1 (Fig. 1C). Examination of these potential binding sites using chromatin immuno-precipitations with a p53-specific antibody revealed that p53 binds to the site within FUCA1 Intron 1 (shown as site ii in Fig. 1C), whereas p53 binding could only be detected at a low level at the cluster of potential binding sites identified in the FUCA1 promoter (Fig. 1D). To test if the binding site in FUCA1 Intron 1 is functionally significant, we cloned this site into a luciferase reporter construct. This construct was then transiently transfected into p53-null Saos-2 cells either with or without an expression construct for p53. As can be seen in Figure 1E, expression of p53 caused an approximate 6-fold induction of this reporter construct, whereas p53 had no effect on a reporter construct which contained the cluster of potential p53 binding sites from the FUCA1 promoter (Fig. 1E).
As our data indicated that FUCA1 was a direct p53 target gene, we next examined if chemotherapeutic drugs which are known to induce p53-dependent responses also lead to induction of FUCA1. To test this we treated HCT116, U2OS and RKO cells, each of which contain endogenous wild-type p53, with actinomycin D, Adriamycin (doxorubicin) or etoposide. RNA was then isolated from these cells and the levels of FUCA1 mRNA determined by qPCR. This revealed that each of the chemotherapeutic agents increased the levels of FUCA1 mRNA with varying degrees in the cell types (Fig. 2A, C, E). Analysis of a p53-deficient variant of HCT116 cells,31 revealed that a large component of FUCA1 induction in response to Adriamycin and actinomycin D was p53-dependent (Fig. 2A and B). In contrast, knockdown of p53 by RNA interference in RKO cells revealed that induction of FUCA1 in these cells was also driven by p53, but only in response to Adriamycin and etoposide (Fig. 2C and D). In U2OS cells, similar to RKO cells, FUCA1 mRNA levels were induced in a p53-dependent manner in response to Adriamycin and etoposide (Fig. 2E and F). In summary, we conclude that FUCA1 is a direct p53 target gene that is induced by several classes of chemotherapeutic drugs. In many cases this effect involves a marked aspect of p53-dependency, but clearly other factors are also involved the regulation of FUCA1 in response to these drugs.
Figure 2.
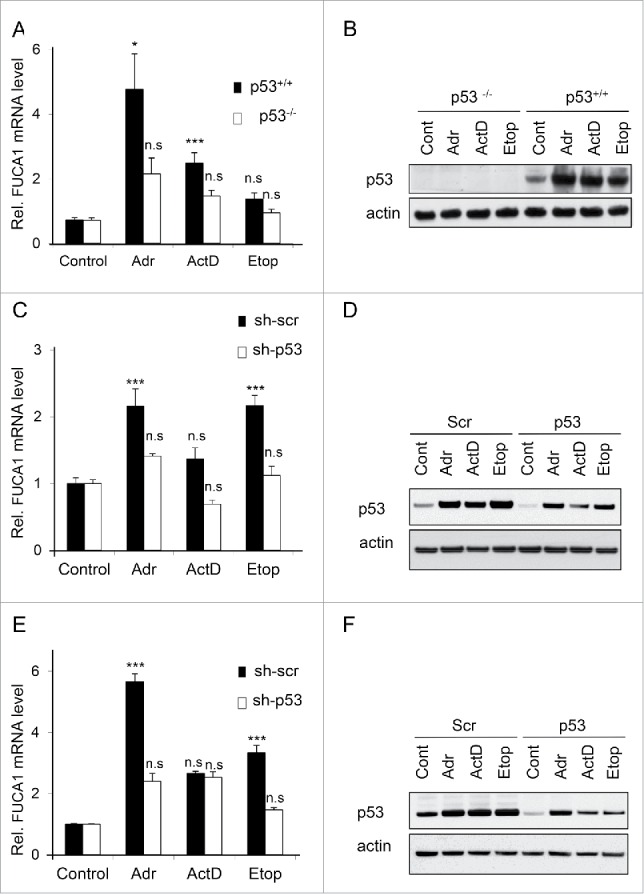
FUCA1 is induced by gentoxic stress in a p53 dependent manner. (A-F) p53 null HCT116 (−/−p53) and WT HCT116 (+/+p53) (A-B), RKO expressing scrambled (LMP-Scr) or p53-specific (LMP-p53) shRNAs (C-D) and U2OS expressing scrambled (LMP-Scr) or p53-specific (LMP-p53) shRNAs (E-F) were treated with either Adriamycin (Adr)(0.5 μg/ml), ActinomycinD (ActD)(2nM ) or Etoposide (Etop) (20 μm), for 24 hr prior to qRT-PCR quantification of FUCA1 (A,C,E). Data are expressed as relative FUCA1 mRNA level and represented as mean ± SD (n = 2 or 3 independent experiements, 5 to 9 replicates, one way Anova: *p < 0.005 **p < 0.001, p21 ***p < 0.0001). (B,D,F) p53 expression were assessed by western blotting. Immunoblot against actin was used as a loading control.
p53 and chemotherapeutic drugs induce fucosidase activity
FUCA1 encodes for a glycosidase which cleaves fucose moieties from N-linked glycan.32,33 As fucosidase activity is believed to be central to FUCA1 function,34,35 we next sought to determine if p53 could modulate fucosidase activity and whether this was dependent on FUCA1. To this end, we adapted an in vitro enzymatic assay using the fluorogenic substrate 4-Methylumbelliferyl-α-L-fucopyranoside (4-MU).36 4-MU has the property to fluoresce when it is excited at 365nm and this is directly proportional to FUCA1activity. Examination of lysates from TetOn-p53 cells that had been incubated in either the absence or presence of Dox (to induce p53) revealed that expression of p53 induces a gradual increase in fucosidase activity over time that is coincident with an increase in FUCA1 protein levels (Fig. 3A–B). To test if the increase in fucosidase activity induced by p53 is FUCA1-dependent, we induced p53 in Saos-2 cells that had been transfected with 3 different FUCA1-targeting siRNAs. In line with a role for endogenous FUCA1 in this response, each of these siRNAs markedly impaired the ability of p53 to induce both FUCA1 levels and fucosidase activity (Fig. 3C–D).
Figure 3.

FUCA1 protein level and activity increase following p53 expression. (A-B) TetOn-p53wt Saos2 cells were treated with doxycycline (Dox) for 24 and 48 hours. Fuca1 activity (A) was measured following Dox treatment and expressed as arbitrary units (A.U.) per 25 µg of protein (n = 3 independent experiement, 9 replicates, one way Anova ***p < 0.0001). (B) FUCA1 and p53 expression was assessed by western blotting and immunoblot against actin was used as a loading control. (C-D) TetOn-p53wt Saos2 cells were transfected with 3 different siRNA directed against FUCA1, prior to doxycycline treatment. Fuca1 enzymatic activities were assessed 48 hours after p53 induction, expressed as arbitrary unit (A.U) per 25 µg of protein and represented as mean ± SD. The graph shown represnts data from one representative experiment done with 3 technical replicates. p53 and Fuca1 expression were assessed by Western blotting and immunoblot against Hsp90 was used as a loading control. RKO expressing scrambled (pRS-Scr) or p53-specific (pRS-p53) shRNAs were either Adriamycin (Adr)(0.5 μg/ml) (E-F), Cisplatin (Cis) (20 µM ) (G-H) or Etoposide (Etop) (20 μm) (I-J) for 48 hr. FUCA1 activity was assessed and expressed as arbitrary units (A.U.) per 25 µg of protein (n = 3 independent experiement, 9 replicates, one way Anova **p = 0.0008, ***p < 0.0001). (E,G,I). FUCA1 and p53 expression was assessed by western blotting and immunoblot against actin was used as a loading control (F,H,J).
As we discovered that p53 can induce fucosidase activity, we were interested to know if chemotherapeutic drugs can also induce the activity of this enzyme. We therefore treated RKO cells with either Adriamycin, cisplatin or etoposide for 48h prior to examination of cell lysates for fucosidase activity using our enzymatic assay. This indeed revealed that these 3 chemotherapeutic drugs induce fucosidase activity when administered to cells (Fig. 3E–J). Moreover, using a matched cell line containing a p53-targeting shRNA, we found that this increase in fucosidase activity was virtually entirely p53 dependent (Fig. 3E–J). We conclude therefore that fucosidase activity may be important not only for the activity of p53 during tumor suppression, but also for the therapeutic response of certain tumors containing wild-type p53.
FUCA1 expression does not induce cell death, but FUCA1 is involved in p53's apoptotic response
As the purpose of our microarray screens was to identify factors involved in p53-mediated programmed cell death, we examined if FUCA1 has a role in the regulation of cell viability. Firstly, we over-expressed FUCA1 in Saos-2 cells which are known to undergo a pronounced apoptotic response following expression of p53. In line with previous reports, expression of p53 in these cells indeed caused a large proportion of cells to undergo apoptosis as measured by analysis of cells with sub-G1 DNA content by flow cytometry (Fig. 4A).26 By contrast, expression of FUCA1 did not seem to affect apoptosis as measured by this assay (Fig. 4A–B). Cell death was also not induced by a mutant of FUCA1 that is associated with the lysosomal storage disorder, fucosidosis.37,38
Figure 4.
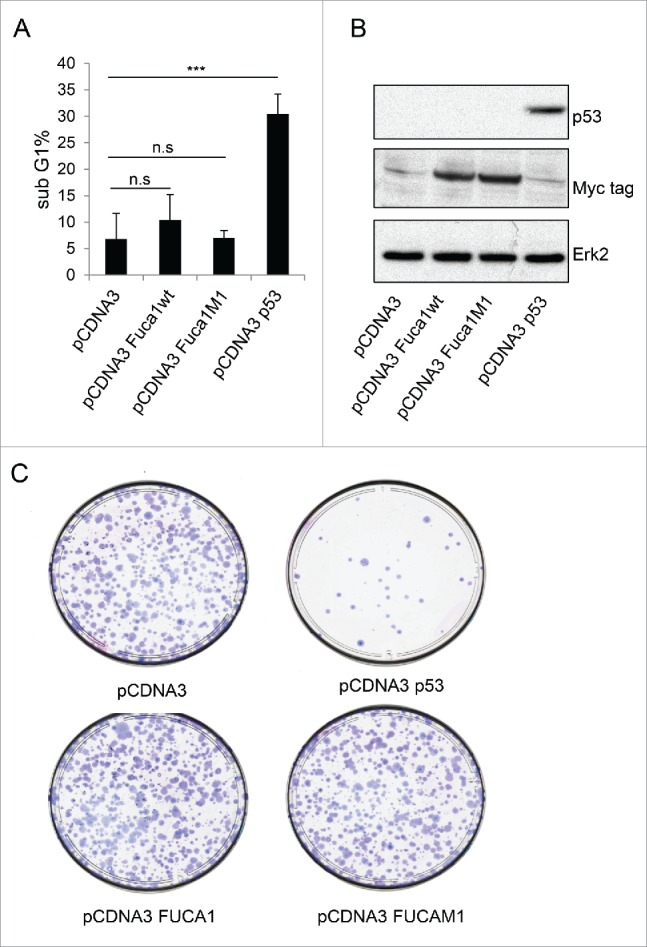
FUCA1 overexpression is not sufficient to induce cell death. Saos 2 cells were transiantly co-transfected with either an empty pcDNA3 vector, a wild-type-Fuca1wt (pCDNA3 FUCA1wt), an enzymatically inactive mutant of FUCA1 (pCDNA3 FUCA1M1), or a wild-type-p53-expressing construct together with pCMV-CD20. After 72 h, the transfected cells were identified by staining for CD20 and analyzed for sub-G1 DNA content by flow cytometry (n = 4 independent experiement, one way Anova ***p < 0.0001) (A). (B) 24 hours post transfection p53 and Fuca1 expression were assessed by western blotting using respectively an anti-p53 antibody and an anti-Myc-tag antibody. Immunoblot against actin was used as a loading control. (C) Saos-2 cells were transfected with either an empty pcDNA3 vector, a wild-type-FUCA1wt (pCDNA3 FUCA1wt), an enzymatically inactive mutant of FUCA1 (pCDNA3 FUCA1M1), or a wild-type-p53-expressing construct. Following selection, cells were assessed for clonogenic survival using Giemsa staining (C).
Since FUCA1 overexpression does not appear to induce apoptosis, we considered that FUCA1 may affect cell viability by other mechanisms. To test this, we performed clonogenic assays which act as readout for all events that can affect cell viability or growth. As would be expected, expression of p53 significantly affected the clonogenic potential of the cells (Fig. 4C). Expression of FUCA1 however, appeared not to have any impact in this assay with FUCA1-expressing cells having a clonogenic potential equivalent to cells transfected with empty plasmid (pcDNA3) as control (Fig. 4C).
Although over-expression of FUCA1 did not affect cell viability, we considered that it may still have an effect on cell death as a part of a p53 response in which other cell death factors are induced or regulated. We therefore utilized FUCA1-targeting siRNAs to knockdown FUCA1 in TetOn-p53 cells and examined the effect this had on cell death following p53 induction. As can be seen in Figure 5A, each of the FUCA1-targeting siRNA reduced FUCA1 expression and also reduced the percentage of cells with sub-G1 DNA content following induction of p53 expression by treatment with Dox (Fig. 5A and B). To confirm if the effect on sub-G1 DNA content was via modulation of p53-mediated apoptosis, we also examined the effect of FUCA1 knockdown on the ability of p53 to affected caspase activation and PARP cleavage – a substrate of caspase activation.39 In both cases, it was clearly evident that knockdown of FUCA1 impaired p53's ability to increase the activity of the effector caspase, caspase 3 and reduced the extent of p53-induced PARP cleavage (Fig. 5C and D). These collective results therefore show that while FUCA1 does not affect cell viability when expressed alone, it has a clear role in p53's apoptotic response.
Figure 5.
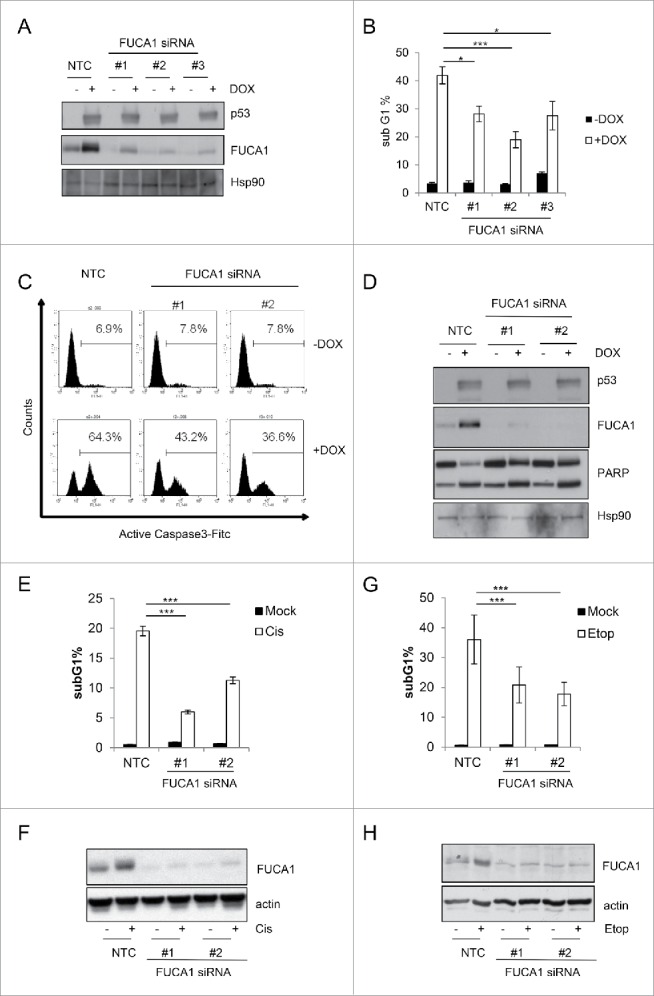
FUCA1 expression contribute to Chemotherapeutic-induced cell death. (A-D). TetOn-p53wt Saos2 cells were were transfected with independent siRNA directed against FUCA1 prior to doxycycline treatment (DOX) for 48 hours. (A) P53 and Fuca1 expression were assessed by Western blotting and immunoblot against Hsp90 was used as a loading control. Cell death was analyzed by flow cytometry, measuring the percentage of cells with sub-G1 DNA content (n = 3 independent experiement, one way Anova *p < 0.01,***p < 0.0001) (B), Caspase 3 activation using an anti-active caspase 3 antibody (C), and western blotting assessing PARP cleavage (D). (E-H) U2OS E1a were trasfected with 2 different siRNA directed against FUCA1 prior treatment with 20 µM Cisplatin (Cis) (E-F) or 20 µM Etoposide (Etop) (G-H) for 48 hours. Cells were assessed for cell death by flow cytometry measuring sub-G1 DNA content content (n = 2 independent experiement, 6 replicates one way Anova,***p < 0.0001) (E-G) and expression of Fuca1 by western blotting. Actin was used as a loading control (F-H).
Since we had discovered that FUCA1 and fucosidase activity is induced by chemotherapeutic drugs in a p53-dependent manner (Figs. 2A, B, and 3E–J), we were naturally intrigued to know if FUCA1 plays a role in the apoptotic response to these agents. To this end, we once again employed FUCA1-targeting siRNAs to knockdown FUCA1 in RKO and U2OS cells. These cells were then treated with either cisplatin (U2OS) or etoposide (U2OS and RKO) and the levels of FUCA1 and apoptosis were assessed by western blotting and flow cytometry respectively (Fig. 5E–H and Fig. S2). Following treatment with either drug, the extent of FUCA1 induction was greatly reduced in cells transfected with FUCA1-targeting siRNAs when compared to controls (Fig. 5F, H and Fig. S2B). In addition and in line with a role for FUCA1 in p53-mediated cell death, the extent of apoptosis induced by etoposide or cisplatin was also markedly reduced by knockdown of FUCA1 (Fig. 5E, 5G and Fig. S2A).
Discussion
In this study we report that the gene encoding the fucosidase FUCA1 is induced by the tumor suppressor p53. We show that p53 mediates this effect by direct transcriptional activation through binding of p53 to FUCA1 at a site within intron 1 of the gene. Moreover, we also report that FUCA1 is induced by different classes of chemotherapeutic drugs in a p53-dependent manner indicating that the induction of FUCA1 is a function of endogenous p53. Interestingly, for a gene that is so widely induced by p53, we were surprised to find that FUCA1 is not induced by the p53 family member, p73. In contrast, many key components of p53-mediated tumor suppression including p21 and PUMA have also been reported to be induced by p73.40-42 So, what does this tell us about the selective activation of FUCA1 by p53? It could be considered that FUCA1 is a relatively ‘weak’ target gene that is only moderately induced by p53 and as a result not induced by other p53-related proteins. Comparison, however, of the levels of FUCA1 and other p53 targets studied in this TetOn system or in response to chemotherapeutic drugs, such as DRAM1 and ADORA2B, which are also induced by p73,43,44 shows that the induction of FUCA1 by p53 is at least equivalent. So, does this tell us that the function of p53 is indeed something specific to p53? Since p53 has a greater impact on tumor suppression compared to p73, it is tempting to speculate that the regulation of FUCA1 represents a component of this p53-specific effect and future studies in this area would certainly be worthwhile.
Although beyond the scope of the present study, another question arising from our findings relates to how FUCA1 contributes to p53-mediated apoptotic death. Intriguingly, we found that expression of FUCA1 alone does not induce cell death indicating that unlike the induction of pro-apoptotic members of the Bcl2 family e.g. PUMA, NOXA and Bax,7-9,45 FUCA1 does not contribute to cell death via direct engagement of the cell death machinery. It is therefore clear that p53 must induce other target genes that work in conjunction with FUCA1 in order to execute a cell death response. It seems likely though that this would not be one target gene, but a number of other p53 targets making their identification a considerable challenge. As a result, perhaps future studies should focus on what functions of FUCA1 contribute to this effect. In this regard, we also report in this study that p53 and chemotherapeutic drugs not only induce FUCA1, but they also induce fucosidase activity. When this is coupled with the fact that naturally occurring mutations in FUCA1 that lead to fucosidosis all involve impairment of fucosidase activity,37,38 it seems conceivable that FUCA1 also contributes to p53-induced cell death through this enzymatic activity. If this is the case, then FUCA1 would function by the removal of fucose moieties from selective target proteins. As a result, the levels or activities of these proteins would change in a way that together with the action of other p53 target genes would push cells toward elimination by apoptotic death. Currently, however, no substrates of fucosidase activity have been identified making this a large area for further investigation.
Since p53 and programmed cell death are both important components of tumor suppression, perhaps the biggest question arising from this work regards the role of fucosylation control in cancer. Interestingly, several previous studies have reported that glycosylation and more specifically fucosylation are perturbed during tumor development.18 Moreover, increased levels of a fucosylated protein are even used as an FDA-approved biomarker for the detection of hepatocellular carcinoma.46,47 However, whether these changes in glycosylation actually contribute to tumor development is still an open question. The evidence we provide in this study undoubtedly adds weight to this possibility, but the field is currently lacking appropriate animal models with which to address this issue. The development of mice where FUCA1 and as a result fucosidase activity could be deleted during the genesis of cancer as would be a great step forward in this area. In addition, since we show that FUCA1 and fucosidase activity are induced by chemotherapeutic drugs, the development of mice in which the relative levels of fucosylation could be modulated in established tumors may well lead to the identification of novel targets for treatment of tumors associated with changes in glycosylation.
Supplementary Material
Disclosure of potential conflicts of interest
The authors do not have any financial, personal or professional interests to declare that could be construed to influence this paper.
Acknowledgments
We would like to thank Scott Lowe for providing RNAi constructs to target p53 and Bert Vogelstein for providing p53−/− HCT116 cells.
Funding
This work was supported by Worldwide Cancer Research and Cancer Research UK.
References
- [1].Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646-74; PMID:21376230; http://dx.doi.org/ 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- [2].Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature 2000; 408:307-10; PMID:11099028; http://dx.doi.org/ 10.1038/35042675 [DOI] [PubMed] [Google Scholar]
- [3].Beroud C, Soussi T. The UMD-p53 database: new mutations and analysis tools. Human mutation 2003; 21:176-81; PMID:12619103; http://dx.doi.org/ 10.1002/humu.10187 [DOI] [PubMed] [Google Scholar]
- [4].Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA Jr., Butel JS, Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992; 356:215-21; PMID:1552940; http://dx.doi.org/ 10.1038/356215a0 [DOI] [PubMed] [Google Scholar]
- [5].Attardi LD. The role of p53-mediated apoptosis as a crucial anti-tumor response to genomic instability: lessons from mouse models. Mutat Res 2005; 569:145-57; PMID:15603759; http://dx.doi.org/ 10.1016/j.mrfmmm.2004.04.019 [DOI] [PubMed] [Google Scholar]
- [6].Bieging KT, Attardi LD. Deconstructing p53 transcriptional networks in tumor suppression. Trends Cell Biol 2012; 22:97-106; PMID:22154076; http://dx.doi.org/ 10.1016/j.tcb.2011.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell 2001; 7:683-94; PMID:11463392; http://dx.doi.org/ 10.1016/S1097-2765(01)00214-3 [DOI] [PubMed] [Google Scholar]
- [8].Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell 2001; 7:673-82; PMID:11463391; http://dx.doi.org/ 10.1016/S1097-2765(01)00213-1 [DOI] [PubMed] [Google Scholar]
- [9].Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi T, Tanaka N. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 2000; 288:1053-8; PMID:10807576; http://dx.doi.org/ 10.1126/science.288.5468.1053 [DOI] [PubMed] [Google Scholar]
- [10].Owen-Schaub LB, Zhang W, Cusack JC, Angelo LS, Santee SM, Fujiwara T, Roth JA, Deisseroth AB, Zhang WW, Kruzel E, et al.. Wild-type human p53 and a temperature-sensitive mutant induce Fas/APO-1 expression. Mol Cell Biol 1995; 15:3032-40; PMID:7539102; http://dx.doi.org/ 10.1128/MCB.15.6.3032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M, Green DR. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004; 303:1010-4; PMID:14963330; http://dx.doi.org/ 10.1126/science.1092734 [DOI] [PubMed] [Google Scholar]
- [12].Leu JI, Dumont P, Hafey M, Murphy ME, George DL. Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat Cell Biol 2004; 6:443-50; PMID:15077116; http://dx.doi.org/ 10.1038/ncb1123 [DOI] [PubMed] [Google Scholar]
- [13].Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, Moll UM. p53 has a direct apoptogenic role at the mitochondria. Mol Cell 2003; 11:577-90; PMID:12667443; http://dx.doi.org/ 10.1016/S1097-2765(03)00050-9 [DOI] [PubMed] [Google Scholar]
- [14].Lowe SW, Ruley HE, Jacks T, Housman DE. p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell 1993; 74:957-67; PMID:8402885; http://dx.doi.org/ 10.1016/0092-8674(93)90719-7 [DOI] [PubMed] [Google Scholar]
- [15].Crighton D, Ryan KM. Splicing DNA-damage responses to tumour cell death. Biochim Biophys Acta 2004; 1705:3-15; PMID:15585169 [DOI] [PubMed] [Google Scholar]
- [16].Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature 1997; 387:299-303; PMID:9153396; http://dx.doi.org/ 10.1038/387299a0 [DOI] [PubMed] [Google Scholar]
- [17].Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature 1997; 387:296-9; PMID:9153395; http://dx.doi.org/ 10.1038/387296a0 [DOI] [PubMed] [Google Scholar]
- [18].Pinho SS, Reis CA. Glycosylation in cancer: mechanisms and clinical implications. Nat Rev Cancer 2015; 15:540-55; PMID:26289314; http://dx.doi.org/ 10.1038/nrc3982 [DOI] [PubMed] [Google Scholar]
- [19].Varki A, Kannagi R, Toole BP. Glycosylation Changes in Cancer In: Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME, eds. Essentials of Glycobiology. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; The Consortium of Glycobiology Editors, La Jolla, California, 2009. [PubMed] [Google Scholar]
- [20].Ryan KM, O'Prey J, Vousden KH. Loss of nuclear factor-kappaB is tumor promoting but does not substitute for loss of p53. Cancer Res 2004; 64:4415-8. [DOI] [PubMed] [Google Scholar]
- [21].Dickins RA, Hemann MT, Zilfou JT, Simpson DR, Ibarra I, Hannon GJ, Lowe SW. Probing tumor phenotypes using stable and regulated synthetic microRNA precursors. Nat Genet 2005; 37:1289-95; PMID:16200064 [DOI] [PubMed] [Google Scholar]
- [22].Long JS, Crighton D, O'Prey J, Mackay G, Zheng L, Palmer TM, Gottlieb E, Ryan KM. Extracellular adenosine sensing-a metabolic cell death priming mechanism downstream of p53. Molecular Cell 2013; 50:394-406; PMID:23603120; http://dx.doi.org/ 10.1016/j.molcel.2013.03.016 [DOI] [PubMed] [Google Scholar]
- [23].Zhu L, van den Heuvel S, Helin K, Fattaey A, Ewen M, Livingston D, Dyson N, Harlow E. Inhibition of cell proliferation by p107, a relative of the retinoblastoma protein. Genes Dev 1993; 7:1111-25; PMID:8319904; http://dx.doi.org/ 10.1101/gad.7.7a.1111 [DOI] [PubMed] [Google Scholar]
- [24].Baudot AD, Haller M, Mrschtik M, Tait SW, Ryan KM. Using enhanced-mitophagy to measure autophagic flux. Methods 2015; 75:105-11; PMID:25498004; http://dx.doi.org/ 10.1016/j.ymeth.2014.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ryan KM, Ernst MK, Rice NR, Vousden KH. Role of NF-kappaB in p53-mediated programmed cell death. Nature 2000; 404:892-7; PMID:10786798; http://dx.doi.org/ 10.1038/35009130 [DOI] [PubMed] [Google Scholar]
- [26].Pellicciari C, Manfredi AA, Bottone MG, Schaack V, Barni S. A single-step staining procedure for the detection and sorting of unfixed apoptotic thymocytes. Eur J Histochem 1993; 37:381-90; PMID:7510545 [PubMed] [Google Scholar]
- [27].Tanikawa C, Matsuda K, Fukuda S, Nakamura Y, Arakawa H. p53RDL1 regulates p53-dependent apoptosis. Nat Cell Biol 2003; 5:216-23; PMID:12598906; http://dx.doi.org/ 10.1038/ncb943 [DOI] [PubMed] [Google Scholar]
- [28].Crighton D, Wilkinson S, O'Prey J, Syed N, Smith P, Harrison PR, Gasco M, Garrone O, Crook T, Ryan KM. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006; 126:121-34; PMID:16839881; http://dx.doi.org/ 10.1016/j.cell.2006.05.034 [DOI] [PubMed] [Google Scholar]
- [29].el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell 1993; 75:817-25; PMID:8242752; http://dx.doi.org/ 10.1016/0092-8674(93)90500-P [DOI] [PubMed] [Google Scholar]
- [30].Hoh J, Jin S, Parrado T, Edington J, Levine AJ, Ott J. The p53MH algorithm and its application in detecting p53-responsive genes. Proc Natl Acad Sci U S A 2002; 99:8467-72; PMID:12077306; http://dx.doi.org/ 10.1073/pnas.132268899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998; 282:1497-501; PMID:9822382; http://dx.doi.org/ 10.1126/science.282.5393.1497 [DOI] [PubMed] [Google Scholar]
- [32].Johnson SW, Alhadeff JA. Mammalian alpha-L-fucosidases. Comp Biochem Physiol B 1991; 99:479-88; PMID:1769200; http://dx.doi.org/ 10.1016/0305-0491(91)90327-A [DOI] [PubMed] [Google Scholar]
- [33].Tsay GC, Dawson G, Sung SS. Structure of the accumulating oligosaccharide in fucosidosis. J Biol Chem 1976; 251:5852-9; PMID:972144 [PubMed] [Google Scholar]
- [34].Winchester B. Lysosomal metabolism of glycoproteins. Glycobiology 2005; 15:1r-15r; PMID:15647514; http://dx.doi.org/ 10.1093/glycob/cwi041 [DOI] [PubMed] [Google Scholar]
- [35].Yamashita K, Tachibana Y, Takada S, Matsuda I, Arashima S, Kobata A. Urinary glycopeptides of fucosidosis. J Biol Chem 1979; 254:4820-7; PMID:438217 [PubMed] [Google Scholar]
- [36].Rapoport E, Pendu JL. Glycosylation alterations of cells in late phase apoptosis from colon carcinomas. Glycobiology 1999; 9:1337-45; PMID:10561459; http://dx.doi.org/ 10.1093/glycob/9.12.1337 [DOI] [PubMed] [Google Scholar]
- [37].Willems PJ, Gatti R, Darby JK, Romeo G, Durand P, Dumon JE, O'Brien JS. Fucosidosis revisited: a review of 77 patients. Am J Med Genet 1991; 38:111-31; PMID:2012122; http://dx.doi.org/ 10.1002/ajmg.1320380125 [DOI] [PubMed] [Google Scholar]
- [38].Willems PJ, Seo HC, Coucke P, Tonlorenzi R, O'Brien JS. Spectrum of mutations in fucosidosis. Eur J Hum Genet 1999; 7:60-7; PMID:10094192; http://dx.doi.org/ 10.1038/sj.ejhg.5200272 [DOI] [PubMed] [Google Scholar]
- [39].Lazebnik YA, Kaufmann SH, Desnoyers S, Poirier GG, Earnshaw WC. Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature 1994; 371:346-7; PMID:8090205; http://dx.doi.org/ 10.1038/371346a0 [DOI] [PubMed] [Google Scholar]
- [40].Melino G, Bernassola F, Ranalli M, Yee K, Zong WX, Corazzari M, Knight RA, Green DR, Thompson C, Vousden KH. p73 Induces apoptosis via PUMA transactivation and Bax mitochondrial translocation. J Biol Chem 2004; 279:8076-83; PMID:14634023; http://dx.doi.org/ 10.1074/jbc.M307469200 [DOI] [PubMed] [Google Scholar]
- [41].Kaghad M, Bonnet H, Yang A, Creancier L, Biscan JC, Valent A, Minty A, Chalon P, Lelias JM, Dumont X, et al.. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell 1997; 90:809-19; PMID:9288759; http://dx.doi.org/ 10.1016/S0092-8674(00)80540-1 [DOI] [PubMed] [Google Scholar]
- [42].Jost CA, Marin MC, Kaelin WG Jr. p73 is a simian [correction of human] p53-related protein that can induce apoptosis. Nature 1997; 389:191-4; PMID:9296498; http://dx.doi.org/ 10.1038/38298 [DOI] [PubMed] [Google Scholar]
- [43].Crighton D, O'Prey J, Bell HS, Ryan KM. p73 regulates DRAM-independent autophagy that does not contribute to programmed cell death. Cell Death Differ 2007; 14:1071-9; PMID:17304243; http://dx.doi.org/ 10.1038/sj.cdd.4402108 [DOI] [PubMed] [Google Scholar]
- [44].Long JS, Schoonen PM, Graczyk D, O'Prey J, Ryan KM. p73 engages A2B receptor signalling to prime cancer cells to chemotherapy-induced death. Oncogene 2015; 34:5152-62; PMID:25659586; http://dx.doi.org/ 10.1038/onc.2014.436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 1995; 80:293-9; PMID:7834749; http://dx.doi.org/ 10.1016/0092-8674(95)90513-8 [DOI] [PubMed] [Google Scholar]
- [46].Mehta A, Block TM. Fucosylated glycoproteins as markers of liver disease. Dis Markers 2008; 25:259-65; PMID:19126969; http://dx.doi.org/ 10.1155/2008/264594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Warnes TW, Smith A. Tumour markers in diagnosis and management. Bailliere's Clin Gastroenterol 1987; 1:63-89; PMID:2437983 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.