

Abstract
Endothelial cell (EC) dysfunction is a critical mediator of the acute respiratory distress syndrome (ARDS). Recent studies have demonstrated that stromal cell–derived factor 1α (SDF-1α) promotes EC barrier integrity. Our previous studies used a SDF-1α analogue CTCE-0214 (CTCE) in experimental sepsis and demonstrated that it attenuated vascular leak and modulated microRNA (miR) levels. We examined the hypothesis that CTCE improves EC function in lipopolysaccharide (LPS)-induced ARDS through increasing miR-126 expression. Human microvascular endothelial cells (HMVECs) were treated with thrombin to disrupt the EC integrity followed by incubation with CTCE or SDF-1α. Barrier function was determined by trans-endothelial electrical resistance assay. CTCE-induced alterations in miRNA expression and signaling pathways involved in barrier function were determined. Thrombin-induced vascular leak was abrogated by both CTCE and SDF-1α. CTCE also prevented thrombin-induced decreases of vascular endothelial (VE)-cadherin cell surface expression and expansion of the intercellular space. CTCE increased miR-126 levels and induced activation of AKT/Rac 1 signaling. Cotreatment with a miR-126 inhibitor blocked the protective effects of CTCE on AKT activation and endothelial permeability. In subsequent in vivo studies, ARDS was induced by intratracheal instillation of LPS. Intravenous injection of CTCE diminished the injury severity as evidenced by significant reductions in protein, immune cells, inflammatory cytokines and chemokines in the bronchoalveolar lavage fluid, increased miR-126 expression and decreased pulmonary vascular leak and alveolar edema. Taken together, our data show that CTCE improves endothelial barrier integrity through increased expression of miR-126 and activation of Rac 1 signaling and represents an important potential therapeutic strategy in ARDS.
INTRODUCTION
The acute respiratory distress syndrome (ARDS) characterized by lung inflammation, disruption of the alveolar-capillary barrier and resultant pulmonary edema is a frequent cause of respiratory failure in critically ill patients (1,2). Despite improvements in supportive care, mortality from ARDS remains high as no effective pharmacologic therapies are available to treat this syndrome (2–4).
During ARDS, endothelial cells (ECs) are activated by inflammatory stimuli including interleukin (IL)-1, IL-6, tumor necrosis factor-α (TNFα) and oxidative stress (5,6). Activated ECs amplify local inflammatory responses by releasing complement and proinflammatory cytokines that propagate focal and ongoing microvascular injury (7). This injury disrupts EC junctions comprised of tight junctions and adherens junctions, allowing protein, inflammatory cell and fluid extravasation into lungs with resulting alveolar edema and dysfunction (8). If uncorrected, this syndrome impairs oxygen transport culminating in cellular hypoxia, acute organ dysfunction and death (9). Thus, therapeutic approaches, which preserve the microvascular endothelial function and barrier integrity, may provide significant improvements in ARDS morbidity and mortality.
Recent studies have demonstrated that stromal cell–derived factor 1 (SDF-1α) also known as C-X-C motif chemokine 12 (CXCL12) can modulate EC barrier integrity through binding to its respective G-protein-coupled receptor, C-X-C chemokine receptor type 4 (CXCR4) (10). In a murine model of shiga-toxin exposure, the CXCL12/CXCR4 interaction appeared to increase endothelial permeability (11), whereas this same interaction has also been shown to attenuate thrombin-induced endothelial permeability through phosphoinositide 3-kinase (PI3K) and Rac1 activation (10). Our previous studies have demonstrated that the analogue SDF-1α cyclic peptide known as CTCE 0214 (CTCE) improved endothelial barrier integrity in the cecal ligation and puncture (CLP) model of murine sepsis (12,13). Mice treated with intravenous CTCE 24 h after CLP experienced less vascular leak, organ failure and death. In addition, CTCE treatment modulated plasma levels of microRNAs (miRNAs) known to promote endothelial homeostasis (12).
MicroRNAs are 19- to 25-nucleotide noncoding RNAs that regulate gene expression at the post-transcriptional level (14). MicroRNAs facilitate cell-to-cell communication via circulating exosomes (15) and play critical roles in the regulation of inflammation and barrier function (16,17). MicroRNA-126 is highly expressed in ECs (18,19) and regulates barrier integrity and inflammatory response (20). Genomic ablation of miR-126 has been shown to increase vascular permeability (16,17,19,21). MicroRNA-126 targets PIK3R2 and Spred-1 leading to stabilization of vascular endothelial (VE)-cadherin via AKT and ERK 1/2 signaling cascades (22,23). CTCE injections result in significantly increased plasma miR-126 levels in CLP-induced sepsis suggesting a potential mechanism by which it reduces endothelial permeability (12).
The current study tested the hypothesis that CTCE improves EC function in lipopolysaccharide (LPS)-induced ARDS through altering miR-126 expression. The data demonstrate that CTCE mitigated thrombin-induced endothelial permeability by modulating miR-126 expression and PI3K/AKT/Rac 1 signaling in human microvascular endothelial cells (HMVECs). Further, in vivo treatment with CTCE ameliorated LPS-induced ARDS in mice. The results raise the possibility that both CTCE and miR-126 may have novel therapeutic potential in ARDS.
MATERIALS AND METHODS
Culture and Treatment
The HMVECs were a Lonza product and were used between passages 3 and 6. Cells were cultured in endothelial cell basal medium-2 (EBM-2) supplemented with EGM-2 MV SingleQuot (Lonza) containing 5% fetal bovine serum and 2% penicillin/streptomycin. They were cultured in tissue culture flasks (Corning) at 37°C with 5% CO2 in humidified air. The culture medium was changed every other day until 90% confluence of the cells was reached (4–6 d). HMVECs were detached with a trypsin (0.05% wt/vol) solution and subcultured.
For miRNA expression, HMVECs were serum starved for 6 h and treated with SDF-1α (100 pg/mL) or CTCE (10 μg/mL) for 24 h. MicroRNAs were isolated with miRNeasy kits (Qiagen) and subsequently analyzed by real-time polymerase chain reaction (PCR) with miScript SYBR Green PCR kits and primers specific to miR-126-5p (Qiagen). Hs-RNU6-2 miRNA was used as internal control.
For signaling protein activation, HMVECs were serum starved for 6 h and treated with CTCE (10 μg/mL) for different time intervals. Cells were washed twice with ice-cold phosphate-buffered saline (PBS) and lysed with RIPA buffer (pH7.4) for 40 min at 4°C. Cell lysates were subjected to Western blot for signaling protein activation.
Transfection of HMVEC
HMVECs were transfected with a miR-126-5p inhibitor and their corresponding negative controls using HiPerFect Transfection Reagent (Qiagen) according to the manufacturer’s Supplementary Transfection Protocol.
Trans-endothelial Electrical Resistance Assay
The trans-endothelial electrical resistance (TEER) assay was performed using the electric cell-substrate impedance sensing (ECIS) system as described previously (24). Briefly, HMVECs were plated on fibronectin-coated ECIS standard 8-well arrays (8W10E, Applied Biophysics) at 1.5 × 105 cells per well. After attachment to the array, cells were transfected or left alone and allowed to equilibrate in serum-free medium (EBM-2, Lonza) for 6 h. The arrays were placed in the ECIS 1600R sensing system (Applied Biosystems). After a stable baseline was reached, thrombin (0.01 U), CTCE (10 μg/mL), SDF-1α (250 pg/mL) or combinations were added directly to the wells at the indicated times. Electrical impedance of the HMVECs was measured every 5 min for 4 h, and impedance values were normalized by dividing each value by the level of impedance measured just prior to the addition of reagents. Statistical analysis using repeated-measures analysis of variance (ANOVA) was performed on impedance values for each condition.
Western Blotting
The cell lysates were mixed with Laemmli sample buffer (Bio Rad) and boiled for 5 min. Equal amounts of protein sample were resolved on 12% sodium dodecylsulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene difluoride membranes. Blots were blocked in Tris-buffered saline–Tween 20 buffer containing 5% nonfat milk and probed with primary antibodies for phospho-AKT (1:1,000 dilution) and total AKT (Cat#9272, Cell Signaling 1:1,000 dilution) overnight at 4°C or at room temperature (RT) for 1 h. Blots were then incubated with electrochemiluminescence (ECL) peroxidase-labeled secondary antibodies at RT for 1 h. The protein bands were visualized with ECL Plus Western Blotting Detection System (GE Healthcare) and exposed to X-ray film. The relative intensity of each protein band was quantified with ImageJ software.
Rac 1 Activity Assay
The GTP-bound active Rac 1 assay was performed using a Rac 1 Activation Assay Kit (Cat#80501, NewEast Biosciences) according to the manufacturer’s instructions. Briefly, HMVECs were stimulated with CTCE (10 μg/mL) for indicated periods of time and lysed at 4°C in the lysis buffer for 20 min. After centrifugation at 12,000 × g for 10 min, anti-active Rac 1 mouse monoclonal antibody was incubated with cell lysates containing Rac 1-GTP at 4°C for 1 h. The active Rac 1 was pulled down by protein A/G agarose. The precipitated active Rac 1 was detected by immunoblotting with antiRac 1 rabbit polyclonal antibody.
Cell Staining
HMVECs were seeded on a fibronectin-coated chamber slide and grown to confluence. After stimulation with or without CTCE and thrombin for 30 min, cells were fixed with 4% paraformaldehyde (PFA) for 10 min. Cells were then permeabilized with 0.1% Triton X-100 for 15 min and blocked with 3% bovine serum albumin (BSA) for 30 min. Cells were treated with rabbit anti-VE-cadherin antibody (Cat#2500, Cell Signaling, 1:400 dilution) overnight at 4°C. Goat antirabbit secondary IgG antibody tagged with Alexa Fluor 488 (Cat#A11008, Thermo Fisher Scientific, 1:200 dilution) was then applied for 1 h at RT. Slides were mounted with ProLong Gold antifade reagent with DAPI (Thermo Fisher Scientific) then covered by a coverslip. All the images were captured using a Leica DMR Fluorescent Phase Contrast microscope (Leica).
Intratracheal Instillation of LPS
ARDS was induced by administering a single intratracheal dose of LPS using the tongue-pull maneuver in mice as described previously (25). Briefly, CD-1 mice (male, 7–8 wks of age) were obtained from Harlan laboratories. Investigations conformed to the Guidelines for the Care and Use of Laboratory Animals used by the National Institutes of Health and were approved by the Institutional Animal Care and Use Committee. After anesthesia by isoflurane inhalation, mice underwent intratracheal instillation of saline control or LPS (25 μg in 75 μL of saline) through a catheter. Mice received CTCE (10 mg/kg) or saline control via intravenous injection at 2 h after LPS instillation. At 24 h after LPS, mice were euthanized and lung tissue and bronchoalveolar lavage (BAL) were collected. Lung miRNAs were isolated with miRNeasy kits(Qiagen) and subsequently analyzed by real-time PCR with primers specific to miR-126-5p (Qiagen). Hs-RNU6-2 miRNA was used as internal control.
BAL and Cytokine/Chemokine Array
After euthanasia, the thorax was opened to expose the trachea. The trachea was cannulated with a 20 g angiocatheter and lavaged with cold PBS (0.75 mL) using a 1-mL syringe. This was performed four times to obtain an adequate BAL. The BAL fluid was centrifuged at 600 × g for 5 min to pellet cells. The supernatant was separated into aliquots and frozen at –80°C until use for cytokine and chemokine array analyses. Mouse cytokine array and chemokine array 31-plex were performed and analyzed by Eve Technologies. Protein levels in the supernatant were determined by Dc protein assay (Bio Rad). The cell pellet was resuspended in 500 μL 1×RBC lysis buffer and centrifuged at 600g for 5 min. Cell pellets were resuspended in 500 μL PBS, and immune cells were quantified using a hemacytometer.
Assessment of Pulmonary Microvascular Permeability Using Evans Blue Dye
Twenty-four hours after intratracheal instillation of LPS, mice were injected with Evans blue (30 mg/kg, 100 μL) via the tail vein. After 40 min, mice were euthanized and perfused through the heart with 10 mL PBS and lung tissue was collected. Lung tissue was weighed and homogenized in 1 mL formamide and then incubated at 60°C for 16 h. The homogenate was centrifuged at 10,000 × g for 10 min, and the supernatant was collected. Evans blue dye was quantified in the supernatant by measuring absorbance at 620 nm. Concentrations of Evans blue dye were determined from a standard curve and expressed as nanograms per milligram lung weight.
Determination of Lung Water Content
After mice were sacrificed, lungs were excised, weighed and dried for 48 h at 70°C. Lung water content was calculated as (wet wt – dry wt)/(wet wt) × 100.
Statistical Analysis
Data are expressed as the mean ± standard error of the mean. Statistical significance was determined by ANOVA with Fisher probable least-squares difference test, repeated-measures ANOVA or Student t test using GraphPad Prism software. A p value <0.05 value was considered statistically significant.
All supplementary materials are available online at www.molmed.org.
RESULTS
CTCE and SDF-1α Inhibit Thrombin-Induced Permeability in HMVECs
HMVECs were treated with thrombin (0.01 U), SDF-1α (250 ng/mL), CTCE (10 μg/mL) or their combinations. Thrombin-induced vascular leak was evidenced by a significant reduction in the electrical impedance of HMVEC monolayers with a maximal reduction of 55 ± 3%. Concomitant treatment with either SDF-1α or CTCE significantly (p < 0.05) attenuated thrombin-induced endothelial permeability with maximal reductions in electrical impedance of 26 ± 7% and 31 ± 8%, respectively (Figure 1).
Figure 1.

SDF-1α and CTCE inhibited thrombin-induced permeability in HMVECs. HMVECs were treated with thrombin (0.01 U), SDF-1α (250 ng/mL), CTCE (10 μg/mL) or a combination of thrombin with SDF-1α or CTCE. HMVEC permeability was determined by measuring electrical impedance using an ECIS system. Thrombin treated cells are significantly different (*p < 0.05) from control cells, and thrombin combined with SDF-1α or CTCE groups are significantly (#p < 0.05) different from thrombin group. Data were analyzed using repeated-measures ANOVA. N = 4–5 independent experiments.
CTCE Decreased Thrombin-Induced Intercellular Space in HMVECs
HMVECs were treated with thrombin (0.01 U), CTCE (10 μg/mL) or a combination of thrombin with CTCE for 30 min. The intercellular adhesion molecule VE-cadherin was located at cell–cell contact areas under resting conditions. The unstimulated and CTCE-treated HMVECs expressed VE-cadherin and did not exhibit any intercellular space. Cells stimulated with thrombin demonstrated decreased peripheral VE-cadherin expression and increased intercellular space. Cotreatment with CTCE significantly mitigated this reduction in monolayer integrity (Figure 2).
Figure 2.

CTCE decreased thrombin-induced intercellular space in HMVECs. HMVECs were treated with thrombin (0.01 U), CTCE (10 μg/mL) or a combination of thrombin with CTCE for 30 min. Typical images of immunostaining of VE-cadherin (green) staining. Scale bar is 20 μm. The results presented are representative of three experiments.
CTCE Increased miR-126 Levels and AKT-Rac1 Signaling in HMVECs
HMVECs were treated with SDF-1α (100 ng/mL) or CTCE (10 μg/mL) and miR-126-5p levels were determined by real-time PCR. SDF-1α and CTCE significantly increased miR-126-5p by 2.0 ± 0.1-fold and 2.6 ± 0.5-fold, respectively (p < 0.05) (Figure 3). MiR-126-5p levels were also increased by SDF-1α or CTCE with or without presence of thrombin (Supplementary Figure S1). HMVECs were also treated with CTCE (10 μg/mL) for different time intervals. AKT and Rac 1 activation were determined by Western blot and Rac 1 activation kits. Thirty minutes after CTCE treatment, there was a significant (p < 0.05) increase in AKT and Rac 1 activation. P-AKTS473 level increased 5.2 ± 1.6-fold at 30 min and 5.3 ± 1-fold at 45 min (Figure 4A), Rac-1 GTP levels increased 13 ± 5-fold at 30 min (Figure 4B). HMVECs were also transfected with miR-126 inhibitors or control inhibitors and treated with CTCE (10 μg/mL) for 30 min. AKT activation was determined by Western blot. CTCE-induced AKT activation was blocked by miR-126 inhibitors (Figure 4C).
Figure 3.
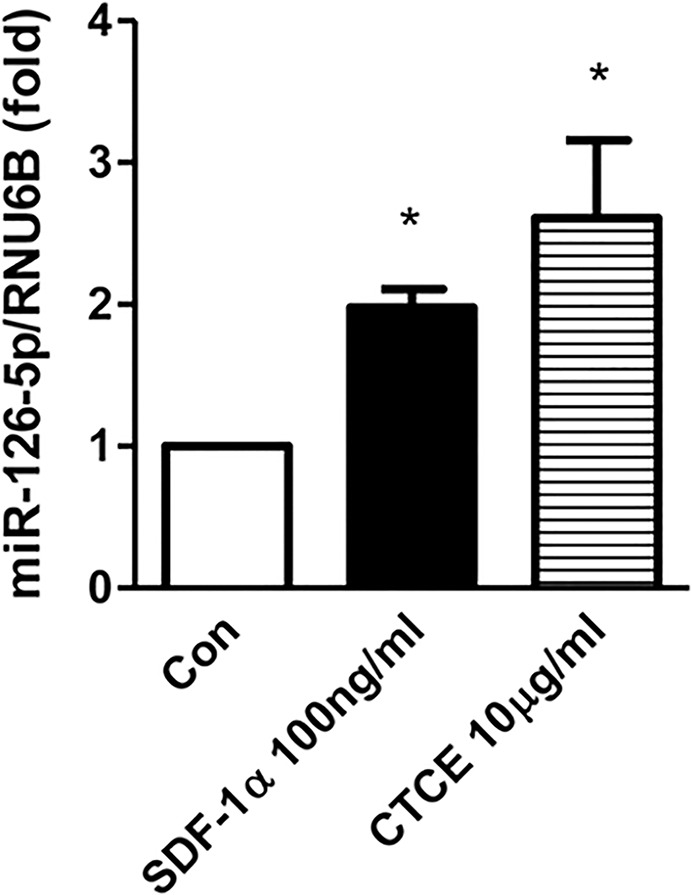
SDF-1α and CTCE increased miR-126 levels in HMVECs. HMVECs were treated with SDF-1α (100 ng/mL) or CTCE (1 or 10 μg/mL) for 24 h. MicroRNA-126-5p levels were determined with real-time PCR. *p < 0.05 compared with control group. N = 4–5 independent experiments.
Figure 4.

CTCE increased AKT/Rac 1 signaling in HMVECs. HMVECs were treated with CTCE (10 μg/mL) for different time intervals. AKT activation (A) and Rac 1 activation (B) were determined by Western blot and Rac 1 activation assay kit. HMVECs were also transfected with miR-126 inhibitors or control inhibitors and CTCE-induced AKT activation was determined (C). *p < 0.05 compared with control group. N = 3–5 independent experiments.
CTCE and SDF-1α Inhibited Thrombin-Induced Vascular Leak Partially Through miR-126 Signaling
HMVECs were transfected with control and miR-126 inhibitors and treated with thrombin (0.01 U), SDF-1α (250 ng/mL), CTCE (10 μg/mL) or a combination of thrombin with SDF-1α or CTCE. In isolation, transfection with the miR-126 inhibitor had no effect on thrombin-induced permeability in HMVECs. However, the miR-126 inhibitor partially, but significantly (p < 0.05, Figure 5) blocked the protective effect of SDF-1α and CTCE on thrombin-induced permeability.
Figure 5.


SDF-1α and CTCE inhibited thrombin-induced permeability through miR-126. HMVECs were transfected with miR-126 inhibitors or control inhibitors and treated with thrombin (0.01 U), SDF-1α (250 ng/mL), CTCE (10 μg/mL) or a combination of thrombin with SDF-1α (A) or CTCE (B). HMVEC permeability was determined by measuring electrical impedance using an ECIS system. Thrombin decreased HMVEC monolayer integrity (*p < 0.05) and this effect was significantly reduced by treatment with SDF-1α (A) and CTCE (B) (#p < 0.05). Transfection with a miR-126 inhibitor alone did not affect thrombin-induced permeability; however, concomitant treatment with SDF-1α (A) or CTCE (B) demonstrated that the miR-126 inhibitor partially but significantly blocked their protective effects ($p < 0.05). Repeated-measures ANOVA used for all comparisons. N = 5 independent experiments.
CTCE Decreased LPS-Induced Pulmonary Vascular Leak and Inflammation In Vivo in Mice
CD-1 mice underwent intratracheal instillation of LPS (25 μg in 75 μL of saline) and intravenous treatment with CTCE (10 mg/kg intravenously) at 2 h post–LPS instillation. BAL protein levels, cell counts, cytokine and chemokine levels were determined. LPS significantly increased BAL protein levels and cell counts, which were decreased by CTCE (–48 ± 13% and –52 ± 8%, respectively, p < 0.05, Figure 6). LPS also significantly (p < 0.05) increased the inflammatory cytokines IL-1α, IL-1α, IL-6, TNFα, G-CSF, IL-12 (p70) as well as the chemokines IP-10, KC, MIG, RANTES, MIP-1α, MIP-1α, MIP-2 and growth factor VEGF. CTCE treatment significantly (p < 0.05) abrogated these increases (Figure 7). In separate experiments, CTCE’s effect on lung vascular leak was determined by Evans blue dye and the wet-to-dry weight ratio of lung tissue. LPS significantly increased lung vascular leak (Figure 8A) and lung water content (Figure 8B) which were both significantly reduced by CTCE. The effects of LPS and CTCE treatment on miR-126 expression in lung tissue were also determined. LPS decreased miR-126 expression in lung tissue, which was reversed by CTCE treatment (Figure 8C).
Figure 6.

CTCE decreased BAL protein levels and total cells in LPS-induced ARDS. CD-1 mice were given an intratracheal instillation of LPS (25 μg in 75 μL of saline) and treated with CTCE at 2 h post–LPS instillation. BAL protein levels (A) and total cell counts (B) were determined. *p < 0.05 compared with control group, #p < 0.05 compared with LPS groups. N = 6–10 mice/group.
Figure 7.
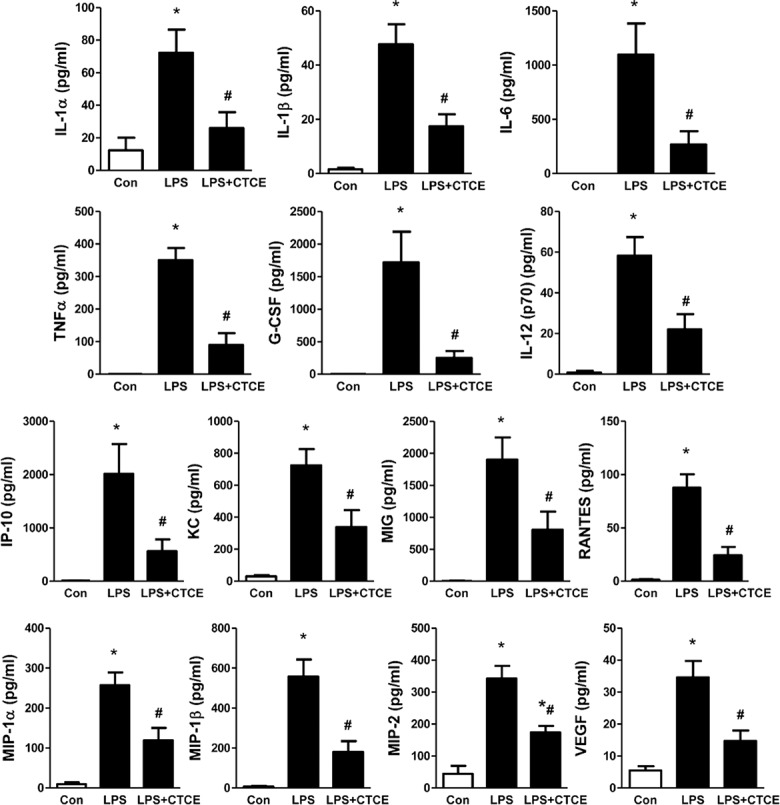
CTCE decreased BAL cytokine and chemokine levels in LPS-induced ARDS. See Figure 6 for method. BAL cytokine and chemokine and growth factor levels were determined by mouse cytokine and chemokine array. *p < 0.05 compared with control group, #p < 0.05 compared with LPS groups. N = 6–9 mice/group.
Figure 8.

CTCE decreased lung vascular leak and water content and increased miR-126 levels in LPS-induced ARDS. ARDS was induced by intratracheal instillation of LPS. Evans blue dye was injected to determine lung vascular leak (A). Lung tissue wet/dry ratio was determined as an index of lung water content (B). MicroRNA-126 expression was determined by real-time PCR of whole lung homogenate (C). *p < 0.05 compared with control group, #p < 0.05 compared with LPS groups. N = 5–6 mice/group.
DISCUSSION
Our findings demonstrated that the SDF-1α analogue CTCE improves EC barrier integrity through increased expression of miR-126 and activation of Rac 1 signaling. Our previous studies have demonstrated that CTCE improves endothelial progenitor cell and neutrophil function and improves survival in a murine model of sepsis (12). However, its effect on vascular permeability had not previously been well characterized. The present study determined the effect of CTCE on thrombin-induced permeability in HMVECs in vitro. We found that cotreatment with CTCE inhibited thrombin-induced reductions in TEER and paracellular gap formation in these cells. As impaired vascular barrier function is critical in the pathogenesis of ARDS (26,27), we investigated whether CTCE protects barrier integrity in LPS-induced ARDS. We found that CTCE decreased LPS-induced protein, inflammatory cell, cytokine and chemokine levels in BAL as well as lung permeability and alveolar edema. These data suggest that CTCE may represent an important potential therapeutic approach in ARDS.
In addition to analysis of electrical impedance, we monitored changes in the endothelial-specific adhesion molecule, VE-cadherin, using fluorescence microscopy. VE-cadherin is vital for EC contacts and the prevention of vascular leak (28,29). As expected, thrombin-induced leak in HMVEC cultures was associated with a significant decline in peripheral VE-cadherin expression and increased intercellular spaces. However, cotreatment with CTCE prevented these thrombin-mediated declines in peripheral VE-cadherin. These results provide a mechanistic link whereby SDF-1α downstream signaling events may prevent EC dysfunction.
MicroRNAs constitute a recently discovered class of noncoding RNAs that play key roles in the regulation of gene expression at a post-translational level (14,30). MicroRNA-126 is the most abundant miRNA expressed during EC differentiation. It has previously been shown to have pleiotropic effects during EC differentiation and its genetic absence is known to augment vascular permeability (22,31). A potential mechanism by which miR-126 may decrease vascular permeability includes its targeted inhibition of PIK3R2 (32). PIK3R2 suppresses AKT signaling (32,33), which has been shown to play a critical role in modulating cytoskeletal rearrangement and maintaining barrier integrity (34). Thus, increases in miR-126 levels may be expected to augment AKT signaling and enhance vascular barrier integrity while its absence would generate the opposite effect. Rac1 is a signaling G protein and pleiotropic regulator of many cellular processes, including the cell cycle, cell–cell adhesion, motility and differentiation (35). Rac1 causes cytoskeletal rearrangement and endothelial barrier enhancement mediated by PI3K/Akt signaling (36,37). As the effects of cytoskeletal rearrangement on EC permeability are well established (38,39), miR-126 may be a key modulator of endothelial permeability through its effects on AKT and Rac1 signaling. We determined miR-126 levels and Rac1 activation levels in HMVECs after treatment with CTCE. Our data showed that CTCE increased miR-126 levels and AKT-Rac1 signaling in HMVECs. Further, when we suppressed miR-126 levels with a miR-126 inhibitor, CTCE’s effect on AKT activation and thrombin-induced permeability was attenuated, indicating miR-126 is at least partially responsible for CTCE’s effect on endothelial barrier integrity. Thus, the role of miR-126 in vascular barrier integrity merits future investigation in addition to other recently described mediators of vascular permeability in ARDS (40,41).
Importantly, our data have also shown that CTCE improves vascular barrier integrity in vivo in a clinically relevant murine model of ARDS. This effect may be the result of CTCE’s direct impact on endothelial permeability as demonstrated in our in vitro studies and by our observation that LPS-induced decreases in lung miR-126 expression were reversed by CTCE treatment. However, as we have previously observed that CTCE reduces inflammatory cytokine production in LPS-activated macrophages (42), it is also possible that CTCE impacts vascular permeability through immune modulation. Accordingly, we measured inflammatory cytokines and chemokines in the alveolar space of the injured lung and verified that CTCE treatment does modulate their expression in vivo. The exact cells in which this effect takes place is not yet known but, regardless, the net result is a reduction in inflammatory activation expected to reduce microcirculatory disruption and trafficking of immune cells to the alveoli. Future studies utilizing various pathway inhibitors may help to clarify the relative contributions of the direct impact of CTCE on EC junctions versus its impact on immune activation.
CONCLUSION
The SDF-1α analogue CTCE reduces EC permeability through modulation of miR-126 expression and the resultant impact on its downstream target AKT. Further, CTCE protects against pulmonary edema in the setting of ARDS while simultaneously modulating inflammatory cytokine and chemokine production. These observations provide evidence that CTCE has therapeutic potential for inflammation-related vascular leak and warrants further investigation as a novel therapeutic in ARDS.
Supplementary Material
ACKNOWLEDGMENTS
The authors acknowledge the technical support of Ms Sarah Ashton. This work was supported in part by GM113995 (H Fan), GM27673 (J Cook, P Halushka), GM67202 (B Zingarelli), KL2 TR000060, UL1 TR 000062 (A Goodwin) and UL1TR001450 (P Halushka).
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
The authors declare they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
Cite this article as: Guo C, et al. (2016) A stromal cell–derived factor 1α analogue improves endothelial cell function in lipopolysaccharide-induced acute respiratory distress syndrome. Mol. Med. 22:115–23.
REFERENCES
- Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342:1334–49. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- Rubenfeld GD, et al. Incidence and outcomes of acute lung injury. N Engl J Med. 2005;353:1685–93. doi: 10.1056/NEJMoa050333. [DOI] [PubMed] [Google Scholar]
- National Heart, Lung, and Blood Institute ARDS Clinical Trials Network et al. Rosuvastatin for sepsis-associated acute respiratory distress syndrome. N Engl J Med. 2014;370:2191–200. doi: 10.1056/NEJMoa1401520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spragg RG, et al. Recombinant surfactant protein C-based surfactant for patients with severe direct lung injury. Am J Respir Crit Care Med. 2011;183:1055–61. doi: 10.1164/rccm.201009-1424OC. [DOI] [PubMed] [Google Scholar]
- Trzeciak S, et al. Resuscitating the microcirculation in sepsis: The central role of nitric oxide, emerging concepts for novel therapies, and challenges for clinical trials. Acad Emerg Med. 2008;15:399–413. doi: 10.1111/j.1553-2712.2008.00109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerrits H, et al. Early postnatal lethality and cardiovascular defects in CXCR7-deficient mice. Genesis. 2008;46:235–45. doi: 10.1002/dvg.20387. [DOI] [PubMed] [Google Scholar]
- van den Berg BM, Vink H, Spaan JA. The endothelial glycocalyx protects against myocardial edema. Circ Res. 2003;92:592–4. doi: 10.1161/01.RES.0000065917.53950.75. [DOI] [PubMed] [Google Scholar]
- Lu Q, et al. Sustained adenosine exposure causes lung endothelial barrier dysfunction via nucleoside transporter-mediated signaling. Am J Respir Cell Mol Biol. 2012;47:604–13. doi: 10.1165/rcmb.2012-0012OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aird WC. The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood. 2003;101:3765–77. doi: 10.1182/blood-2002-06-1887. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, et al. Stromal cell-derived factor-1α/C-X-C chemokine receptor type 4 axis promotes endothelial cell barrier integrity via phosphoinositide 3-kinase and Rac1 activation. Arterioscler Thromb Vasc Biol. 2014;34:1716–22. doi: 10.1161/ATVBAHA.114.303890. [DOI] [PubMed] [Google Scholar]
- Petruzziello-Pellegrini TN, et al. The CXCR4/CXCR7/SDF-1 pathway contributes to the pathogenesis of Shiga toxin-associated hemolytic uremic syndrome in humans and mice. J Clin Invest. 2012;122:759–76. doi: 10.1172/JCI57313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan S, et al. Combined treatment with a CXCL12 analogue and antibiotics improves survival and neutrophil recruitment and function in murine sepsis. Immunology. 2014;144:405–11. doi: 10.1111/imm.12382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan H, et al. Endothelial progenitor cells and a stromal cell-derived factor-1α analogue synergistically improve survival in sepsis. Am J Respir Crit Care Med. 2014;189:1509–19. doi: 10.1164/rccm.201312-2163OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–54. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- Valadi H, et al. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9:654–9. doi: 10.1038/ncb1596. [DOI] [PubMed] [Google Scholar]
- Staszel T, et al. Role of microRNAs in endothelial cell pathophysiology. Pol Arch Med Wewn. 2011;121:361–6. [PubMed] [Google Scholar]
- Wei Y, Nazari-Jahantigh M, Neth P, Weber C, Schober A. MicroRNA-126, -145, and -155: a therapeutic triad in atherosclerosis? Arterioscler Thromb Vasc Biol. 2013;33:449–54. doi: 10.1161/ATVBAHA.112.300279. [DOI] [PubMed] [Google Scholar]
- Lorenzen JM. Vascular and circulating microRNAs in renal ischaemia-reperfusion injury. J Physiol. 2015;593:1777–84. doi: 10.1113/JP270318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, et al. The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev Cell. 2008;15:261–71. doi: 10.1016/j.devcel.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques-Rocha JL, et al. Noncoding RNAs, cytokines, and inflammation-related diseases. FASEB J. 2015;29:3595–611. doi: 10.1096/fj.14-260323. [DOI] [PubMed] [Google Scholar]
- Meng S, et al. Downregulation of microRNA-126 in endothelial progenitor cells from diabetes patients, impairs their functional properties, via target gene Spred-1. J Mol Cell Cardiol. 2012;53:64–72. doi: 10.1016/j.yjmcc.2012.04.003. [DOI] [PubMed] [Google Scholar]
- Pepini T, Gorbunova EE, Gavrilovskaya IN, Mackow JE, Mackow ER. Andes virus regulation of cellular microRNAs contributes to hantavirus-induced endothelial cell permeability. J Virol. 2010;84:11929–36. doi: 10.1128/JVI.01658-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suurna MV, et al. Cofilin mediates ATP depletion-induced endothelial cell actin alterations. Am J Physiol Renal Physiol. 2006;290:F1398–407. doi: 10.1152/ajprenal.00194.2005. [DOI] [PubMed] [Google Scholar]
- Wilkerson BA, Grass GD, Wing SB, Argraves WS, Argraves KM. Sphingosine 1-phosphate (S1P) carrier-dependent regulation of endothelial barrier: High density lipoprotein (HDL)-S1P prolongs endothelial barrier enhancement as compared with albumin-S1P via effects on levels, trafficking, and signaling of S1P1. J Biol Chem. 2012;287:44645–53. doi: 10.1074/jbc.M112.423426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah D, Romero F, Stafstrom W, Duong M, Summer R. Extracellular ATP mediates the late phase of neutrophil recruitment to the lung in murine models of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2014;306:L152–61. doi: 10.1152/ajplung.00229.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, et al. Blockade of PDE4B limits lung vascular permeability and lung inflammation in LPS-induced acute lung injury. Biochem Biophys Res Commun. 2014;450:1560–7. doi: 10.1016/j.bbrc.2014.07.024. [DOI] [PubMed] [Google Scholar]
- Tauseef M, et al. TLR4 activation of TRPC6-dependent calcium signaling mediates endotoxin-induced lung vascular permeability and inflammation. J Exp Med. 2012;209:1953–68. doi: 10.1084/jem.20111355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian X, et al. Asef controls vascular endothelial permeability and barrier recovery in the lung. Mol Biol Cell. 2015;26:636–50. doi: 10.1091/mbc.E14-02-0725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JA, et al. Regulation of vascular leak and recovery from ischemic injury by general and VE-cadherin-restricted miRNA antagonists of miR-27. Blood. 2013;122:2911–9. doi: 10.1182/blood-2012-12-473017. [DOI] [PubMed] [Google Scholar]
- Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T. Identification of novel genes coding for small expressed RNAs. Science. 2001;294:853–8. doi: 10.1126/science.1064921. [DOI] [PubMed] [Google Scholar]
- Kane NM, et al. Derivation of endothelial cells from human embryonic stem cells by directed differentiation: Analysis of microRNA and angiogenesis in vitro and in vivo. Arterioscler Thromb Vasc Biol. 2010;30:1389–97. doi: 10.1161/ATVBAHA.110.204800. [DOI] [PubMed] [Google Scholar]
- Zhang J, et al. microRNA 126 inhibits the transition of endothelial progenitor cells to mesenchymal cells via the PIK3R2-PI3K/Akt signalling pathway. PLoS One. 2013;8:e83294. doi: 10.1371/journal.pone.0083294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie ZC, et al. MicroRNA-126 is down-regulated in human esophageal squamous cell carcinoma and inhibits the proliferation and migration in EC109 cell via PI3K/AKT signaling pathway. Int J Clin Exp Pathol. 2015;8:4745–54. [PMC free article] [PubMed] [Google Scholar]
- Umapathy SN, et al. Adenosine A1 receptors promote vasa vasorum endothelial cell barrier integrity via Gi and Akt-dependent actin cytoskeleton remodeling. PLoS One. 2013;8:e59733. doi: 10.1371/journal.pone.0059733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Ambrosi N, Rossi S, Gerbino V, Cozzolino M. Rac1 at the crossroad of actin dynamics and neuroinflammation in Amyotrophic Lateral Sclerosis. Front Cell Neurosci. 2014;8:279. doi: 10.3389/fncel.2014.00279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunduz D, et al. Insulin stabilizes microvascular endothelial barrier function via phosphatidylinositol 3-kinase/Akt-mediated Rac1 activation. Arterioscler Thromb Vasc Biol. 2010;30:1237–45. doi: 10.1161/ATVBAHA.110.203901. [DOI] [PubMed] [Google Scholar]
- Singleton PA, et al. Akt-mediated transactivation of the S1P1 receptor in caveolin-enriched microdomains regulates endothelial barrier enhancement by oxidized phospholipids. Circ Res. 2009;104:978–86. doi: 10.1161/CIRCRESAHA.108.193367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek SM, Garcia JG. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol (1985) 2001;91:1487–500. doi: 10.1152/jappl.2001.91.4.1487. [DOI] [PubMed] [Google Scholar]
- Kasa A, Csortos C, Verin AD. Cytoskeletal mechanisms regulating vascular endothelial barrier function in response to acute lung injury. Tissue Barriers. 2015;3:e974448. doi: 10.4161/21688370.2014.974448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong H, et al. HIF2α signaling inhibits adherens junctional disruption in acute lung injury. J Clin Invest. 2015;125:652–64. doi: 10.1172/JCI77701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barabutis N, et al. p53 protects against LPS-induced lung endothelial barrier dysfunction. Am J Physiol Lung Cell Mol Physiol. 2015;308:L776–87. doi: 10.1152/ajplung.00334.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan H, et al. Beneficial effect of a CXCR4 agonist in murine models of systemic inflammation. Inflammation. 2012;35:130–7. doi: 10.1007/s10753-011-9297-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.