

TO THE EDITOR
Acro-cardio-facial syndrome (ACFS) is a rare condition that has primarily been diagnosed based on presence of a constellation of clinical findings including ectrodactyly, heart defects, cleft lip and palate, ear anomalies, dysmorphic facial features, and intellectual disability. In the past, autosomal recessive inheritance has been suggested following observation of parental consanguinity and affected siblings in few families [Guion-Almeida et al., 2000; Mingarelli et al., 2005; Digilio and Dallapiccola, 2010]. More recently Toschi et al. [2012] proposed that ACFS may be a new microdeletion syndrome based on a 6q21–22.3 microarray deletion identified in an affected child. The authors reviewed reported cases of chromosome deletions in the 6q region, and found the clinical spectrum of anomalies consistent with ACFS [Toschi et al., 2012]. However, most of the reviewed cases shared either no or only little overlap with their reported 6q21–22.3 deletion. The only case with significant overlap involved a deletion that was more than twice as large.
Here we report on an infant with a clinical presentation consistent with ACFS found to have an interstitial deletion of 6q21q22.1 on chromosomal microarray encompassing a segment very similar to the previously reported 6q21–22.3 deletion. As delineated in our report ACFS shares many of the clinical and molecular characteristics of a contiguous gene deletion syndrome, confirming the possibility that 6q21–22.3 microdeletion is a mechanism causing this disorder.
The female patient is the second child born to healthy, non-consanguineous parents. A 2-year-old full sibling is also healthy. She was the product of an uncomplicated pregnancy with vaginal delivery at 41 weeks. The patient was small for gestational age with a birth weight of 2.665 kg (3rd centile) a birth length of 49.5 cm (25th centile), and a head circumference of 33 cm (10–25th centile). At delivery, she was noted to have bilateral ectrodactyly, an unusual facial morphology including mild brachycephaly, flat forehead, hypertelorism, downslanting palpebral fissures, mild micrognathia, and asymmetric ears. The left ear showed deficiency of the crus of the helix and an attached lobule. The right ear had a very tightly attached lobule and in addition to the deficiency of the crus of the helix, there was also prominence of the inferior crus horizontally across the helix. The limb anomalies consisted of bilateral hand ectrodactyly with a more central ray deficiency on the left. The 4th digit was absent bilaterally. Thumbs and 5th fingers were normal (Figs. 1 and 2). Postnatal echocardiogram revealed an atrial septum defect (ASD). The child had some early feeding difficulties, and at 2 months of age, showed hypotonia and specific weakness of the neck muscles. Ophthalmological evaluation showed strabismus and a high degree of hyperopia. EEG demonstrated abnormal brain wave patterns without evidence for seizure activity. Brain imaging and abdominal ultrasounds have not been completed to date.
FIG. 1.
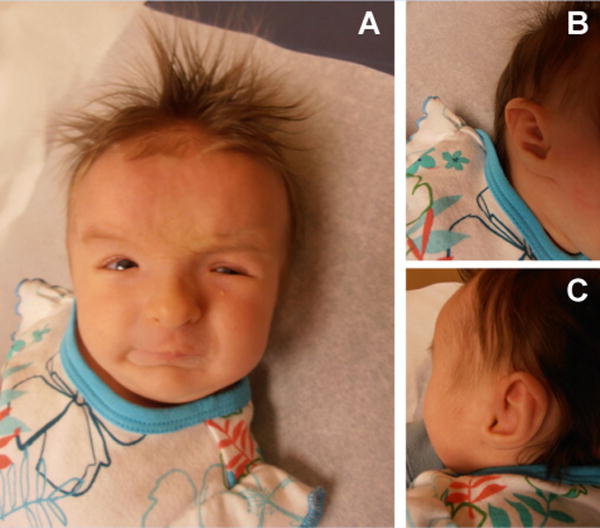
A: Face with high forehead, brachycephaly, flat forehead, hypertelorism, downward slanting palpebral fissures, mild micrognathia; B: Right ear with tightly attached lobule and deficiency of the crus of the helix; C: Left ear with deficiency of the crus of the helix and an attached lobule.
FIG. 2.
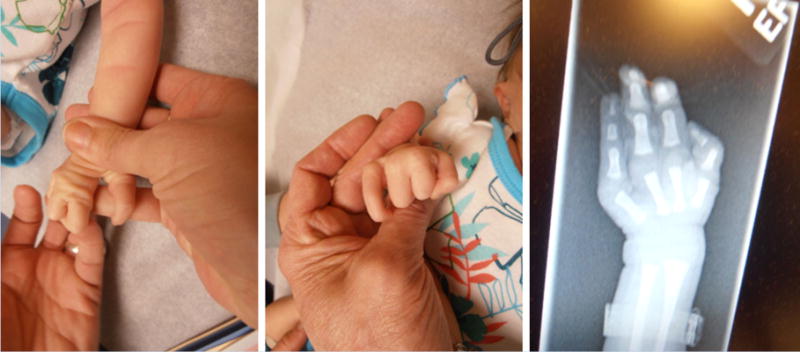
A: Ectrodactyly left hand; B: Ectrodactyly right hand; C: X-ray left hand.
Chromosome analysis (G-banding, 500 band level) in peripheral lymphocytes was normal 46, XX. Following DNA isolation from peripheral blood, an Affymetrix CytoScan SNP/CNV microarray was performed according to the manufacturer’s protocol, and was analyzed by ChAS 2.0 software. We routinely investigate for gains/losses at or below 25 kilobases (kb) near genes and for gains and losses in general of 40 kb or above. A deletion of chromosome 6q21–22 from position chr11:105,930,050–117,483,097 (hg19, GRCh37) was observed (Fig. 3). The deletion was confirmed by FISH (fluorescent in situ hybridization) with the probe RP1-142L7 targeting to 6q21 from (BlueGnome, Inc., Cambridge, UK) (Fig. 4). Parental FISH results with the same probe set were normal indicating that this is a de novo deletion.
FIG. 3.
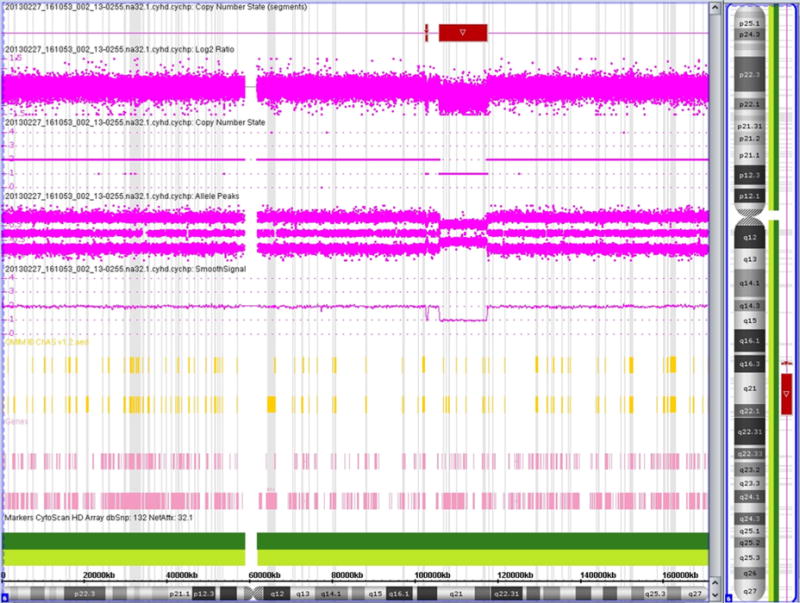
Deletion present in 6q21.
FIG. 4.
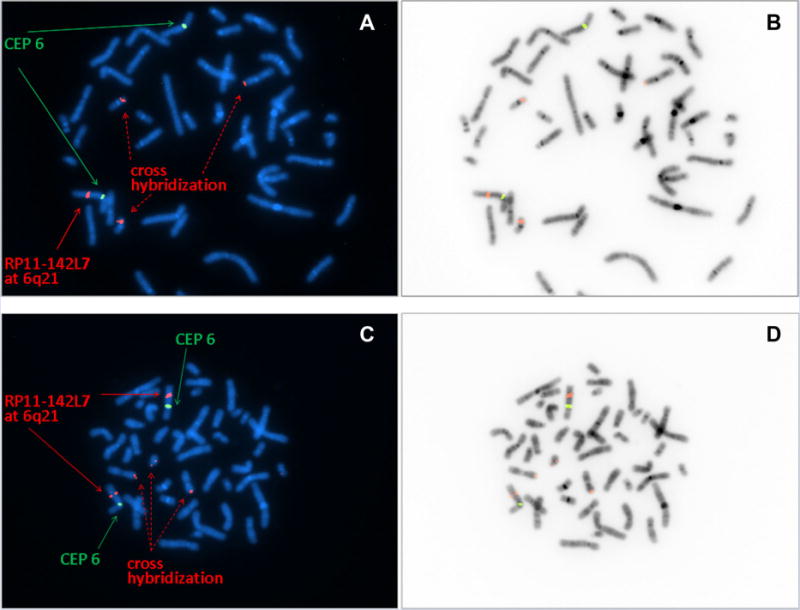
FISH confirmation of 6q21 deletion FISH analysis was performed for the patient specimen and a control specimen with RP1-142L7 (red) targeting to 6q21 and a CEP 6 probe (green) targeting the centromere of chromosome 6. Some cross hybridizations signals for RP1-142L7 were observed both in the patient and control specimen. Color images for patient specimen and control specimen are presented in panel A and C, respectively. B and D present inverted images for A and C, respectively. Loss of one RP1-142L7 signal at 6q21 was seen in all metaphases from this patient specimen (A, B), but not seen in metaphases from the control specimen (C, D).
This deletion encompasses approximately 11.15 megabases and deletes one allele for at least 73 genes of which 45 genes are listed in OMIM including more than 17 OMIM disorders (Table I). Notable among these disorders are coenzyme Q10 deficiency, subtypes of polycystic liver disease, osteopetrosis, amyotrophic lateral sclerosis, Charcot-Marie-tooth disease, a few Spondylometaphyseal dysplasias (perhaps with the gene ColA10 involved in the skeletal phenotype), sudden infant death syndrome with dysgenesis of the testes, and a ciliary dyskenesia subtype. Given the potential for treatment, perhaps these patients should be tested for CoQ10 deficiency.
TABLE I.
Listing of Deleted Genes and OMIM Conditions
| 73 Genes deleted | OMIM conditions |
|---|---|
| PRDM1, ATG5, AIM1, RTN4IP1, QRSL1, LOC100422737, C6orf203, BEND3, PDSS2, SOBP, SCML4, SEC63, OSTM1, NR2E1, SNX3, LACE1, FOXO3, NCRNA00222, ARMC2, SESN1, CEP57L1, CCDC162, CD164, PPIL6, SMPD2, MICAL1, ZBTB24, AKD1, FIG4, GPR6, WASF1, CDC40, C6orf186, DDO, SLC22A16, CDK19, AMD1, GTF3C6, RPF2, GSTM2P1, SLC16A10, KIAA1919, REV3L, LOC643749, TRAF3IP2, FYN, WISP3, TUBE1, C6orf225, LAMA4, RFPL4B, MARCKS, FLJ34503, HDAC2, HS3ST5, FRK, TPI1P3, NT5DC1, COL10A1, TSPYL4, TSPYL1, DSE, FAM26F, BET3L, FAM26E, FAM26D, RWDD1, RSPH4A, ZUFSP, KPNA5, FAM162B, GPRC6A, RFX6 | Oculocutaneous albinism type IV (AIM1), Skin hair eye pigmentation 5 dark fair skin (AIM1), Skin hair eye pigmentation 5 black nonblack hair (AIM1), Skin hair eye pigmentation 5 dark light eyes (AIM1), Coenzyme Q10 deficiency (PDSS2), Polycystic liver disease (SEC63), Osteopetrosis autosomal recessive 5 (OSTM1), Amyotrophic lateral sclerosis 11 (Fig. 4), Charcot-Marie-Tooth disease type 4J (Fig. 4), Spondyloepiphyseal dysplasia tarda with progressive arthropathy (WISP3), Arthropathy progressive pseudorheumatoid of childhood (WISP3), Spondylometaphyseal dysplasia (COL10A1), Metaphyseal chondrodysplasia (COL10A1), Sudden infant death with dysgenesis of the testes syndrome (TSPYL1), Ciliary dyskinesia primary 11 (RSPH4A) |
This is the second report of a patient with a clinical diagnosis of ACFS associated with a microarray deletion of 6q21q22.1, confirming the possibility that the spectrum of ACFS is caused by microdeletions in this region. Previous authors have suggested an autosomal recessive inheritance pattern for this rare syndrome [Guion-Almeida et al., 2000; Mingarelli et al., 2005; Digilio and Dallapiccola, 2010]. However, this assumption was based on one case of parental consanguinity and few observations of intrafamilial recurrence. Notably, no molecular studies were performed in these cases to confirm a unifying genetic etiology. Here, we present supportive evidence for a contiguous gene syndrome as an etiology of ACFS. A number of candidate genes for cardinal features of ACFS are located within the deleted 6q21q22 segment reported here and by Toschi et al. [2012]. Previous reports have identified 6q21as a candidate region for split-hand/split foot malformation (SHFM). [Tsukahara et al., 1997; Niedrist et al., 2009]. A candidate gene for SHFM proposed by Niedrist et al. [2009] is SNX3. Viljoen and Smart [1993] reported a patient with ectrodactyly, micropthalmia, prognathism, and intellectual disability with a de novo reciprocal t(6;13)(q21;q12) translocation. In a subsequent report, Vervoort et al. [2002] mapped the translocation breakpoints determining that SNX3 was disrupted; therefore concluding it to be a candidate gene for SHFM.
The 6q21–23 region was also found to harbor a number of candidate susceptibility genes for cleft lip and/or palate, another phenotypic feature of ACFS. Vieira et al. [2008] found statistical significance for many genes in the 6q21–23 region, including LAMA4 and SCML4, which are deleted in our patient. In addition, LAMA4 has been implicated in hypospadia, a manifestation of ACFS observed in previously reported patients [Giannotti et al., 1995; Kariminejad et al., 2008]: A recent study using genome-wide DNA methylation profiling of CpG islands found an association between epigenetic modification of CpG islands at several loci including LAMA4 locus and hypospadias [Choudhry et al., 2012]. Heterozygous mutations in LAMA4 cause a form of dilated cardiomyopathy, whereas a GJA1, a gene located downstream the distal breakpoint of the 6q21–22.1 deletion (Genomic coordinates, GRCh37: 6:121,756,744–121,770,872), is associated with several cardiac phenotypes including ASD and hypoplastic left heart syndrome 1 (HLHS1) [Knöll et al., 2007].
We reviewed the other OMIM genes located within the deleted segment. Most cause recessive conditions expect to manifestations a result of haploinsufficiency caused by the deletion (PDSS2, SOBP, OSTM1, ZBTB24, TSPYL1, DSE, RSPH4A, RFX6; Table I). Several genes were dominant including those causing polycystic liver disease and metaphyseal chondrodysplasia, but polycystic liver disease is adult onset and of reduced penetrance, and there is evidence that metaphyseal chondrodysplasia is not a result of haploinsufficiency [Wallis et al., 1996]. Mutations in FIG4 are associated with both dominant (amyotrophic lateral sclerosis 11) and recessive (Charcot-Marie-Tooth disease, type 4J; Yunis-Varon syndrome) conditions.
The deleted segments identified in our case (chr6:105,930,050–117,483,097; hg19, GRCh37) and reported by Toschi et al. [2012] (chr6:106,543,543–119,109,372; hg19, GRCh37) are very similar with respect to the OMIM gene content reflecting the closely resembling phenotypes characterized by cardinal features of ACFS and supportive of a contiguous gene deletion syndrome. Similarly, previously reported patients harboring 6q deletions of less overlap with 6q21–22.1 have shown only some of the features ACFS spectrum. Although ACFS may be genetically heterogeneous, the lack of microarray studies in earlier case reports suggests the possibility of a microdeletion present in at least some of these patients. The possibility of a deletion “unmasking” an autosomal recessive condition when combined with a mutation of a 6q gene in trans that would manifest in the observed phenotype cannot be excluded.
The differential diagnosis of ectrodactyly syndromes includes Oculo-cerebro-acral syndrome, Focal dermal hypoplasia, and Ectrodactyly, ectodermal dysplasia, and cleft lip/palate syndrome (EEC1 and EEC3). Oculo-cerebro-acral syndrome has some overlap, but our patient does not have microphthalmia or syndactyly seen in this condition. Focal dermal hypoplasia includes reports of ectrodactyly, but more often syndactyly as well as more involved dermal findings, which were not found in our patient, EEC1 and EEC3 are associated with ectrodactyly and clefts, but not heart defects. Our patient did not have any findings suggestive of ectodermal dysplasia.
In order to further elucidate the etiology of ACFS, as well as provide families with recurrence risk information, it would be important for patients with this phenotype to have microarray testing to determine the presence and extend of a 6q microdeletion.
Based on the similarity of phenotypes and microarray deletions in this case compared to Toschi et al. [2012] (Fig. 5), we suggest there is sufficient evidence to establish ACFS as a new microdeletion syndrome.
FIG. 5.
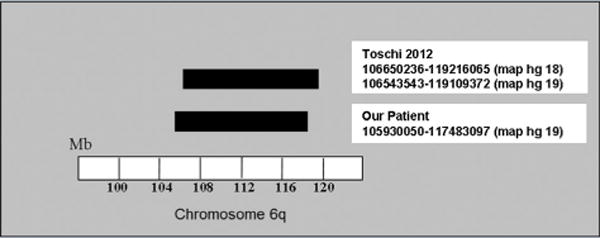
Microarray deletion comparison of ACFS patient described by Toschi et al. [2012] and our patient.
References
- Digilio MC, Dallapiccola B. Acro-cardio-facial syndrome. Orphanet J Rare Dis. 2010;5:25. doi: 10.1186/1750-1172-5-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhry S, Deshpande A, Qiao L, Beckman K, Sen S, Baskin LS. Genome-wide DNA methylation profiling of CpG islands in hypospadias. J Urol. 2012;188:1450–1455. doi: 10.1016/j.juro.2012.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannotti A, Digilio MC, Mingarelli R, Dallapiccola B. An autosomal recessive syndrome of cleft palate, cardiac defect, genital anomalies, and ectrodactyly (CCGE) J Med Genet. 1995;32:72–74. doi: 10.1136/jmg.32.1.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guion-Almeida ML, Zechi-Ceide RM, Richieri-Costa A. Cleft lip/palate, abnormal ears, ectrodactyly, congenital heart defect, and growth retardation: Definition of the acro-cardio-facial syndrome. Clin Dysmorphol. 2000;9:269–272. doi: 10.1097/00019605-200009040-00007. [DOI] [PubMed] [Google Scholar]
- Kariminejad A, Bozorgmehr B, Sedighi Gilani MA, Almadani N, Kariminejad MH. Clinical variability in acro-cardio-facial syndrome. Am J Med Genet. 2008;146A:1977–1979. doi: 10.1002/ajmg.a.32052. [DOI] [PubMed] [Google Scholar]
- Knöll R, Postel R, Wang J, Krätzner R, Hennecke G, Vacaru AM, Vakeel P, Schubert C, Murthy K, Rana BK, Kube D, Knöll G, Schäfer K, Hayashi T, Holm T, Kimura A, Schork N, Toliat MR, Nürnberg P, Schultheiss HP, Schaper W, Schaper J, Bos E, Den Hertog J, van Eeden FJ, Peters PJ, Hasenfuss G, Chien KR, Bakkers J. Laminin-alpha4 and integrin-linked kinase mutations cause human cardiomyopathy via simultaneous defects in cardiomyocytes and endothelial cells. Circulation. 2007;116:515–525. doi: 10.1161/CIRCULATIONAHA.107.689984. [DOI] [PubMed] [Google Scholar]
- Mingarelli R, Zuccarello D, Digilio MC, Dallapiccola B. A new observation of acro-cardio-facial syndrome substantiates interindividual clinical variability. Am J Med Genet A. 2005;136A:84–86. doi: 10.1002/ajmg.a.30770. [DOI] [PubMed] [Google Scholar]
- Niedrist D, Lurie IW, Schinzel A. 4q32-q35 and 6q16-q22 are valuable candidate regions for split hand/foot malformation. Eur J Hum Genet. 2009;17:1086–1091. doi: 10.1038/ejhg.2009.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toschi B, Valetto A, Bertini V, Congregati C, Cantinotti M, Assanta N, Simi P. Acro-cardio-facial syndrome: A microdeletion syndrome? Am J Med Genet A. 2012;158A:1994–1999. doi: 10.1002/ajmg.a.35444. [DOI] [PubMed] [Google Scholar]
- Tsukahara M, Yoneda J, Azuma R, Nakashima K, Kito N, Ouchi K, Kanehara Y. Interstitial deletion of 6q21-q23 associated with split hand. Am J Med Genet. 1997;69:268–270. doi: 10.1002/(sici)1096-8628(19970331)69:3<268::aid-ajmg10>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Vervoort VS, Viljoen D, Smart R, Suthers G, DuPont BR, Abbott A, Schwartz CE. Sorting nexin 3 (SNX3) is disrupted in a patient with a translocation t(6;13)(q21;q12) and microcephaly, microphthalmia, ectrodactyly, prognathism (MMEP) phenotype. J Med Genet. 2002;39:893–899. doi: 10.1136/jmg.39.12.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira AR, McHenry TG, Daack-Hirsch S, Murray JC, Marazita ML. Candidate gene/loci studies in cleft lip/palate and dental anomalies finds novel susceptibility genes for clefts. Genet Med. 2008;10:668–674. doi: 10.1097/GIM.0b013e3181833793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viljoen DL, Smart R. Split-foot anomaly, microphthalmia, cleft-lip and cleft-palate, and mental retardation associated with a chromosome 6;13 translocation. Clin Dysmorphol. 1993;2:274–277. [PubMed] [Google Scholar]
- Wallis GA, Rash B, Sykes B, Bonaventure J, Maroteaux P, Zabel B, Wynne-Davies R, Grant ME, Boot-Handford RP. Mutations within the gene encoding the alpha 1 (X) chain of type X collagen (COL10A1) cause metaphyseal chondrodysplasia type Schmid but not several other forms of metaphyseal chondrodysplasia. J Med Genet. 1996;33:450–457. doi: 10.1136/jmg.33.6.450. [DOI] [PMC free article] [PubMed] [Google Scholar]