

Abstract
The complex function and regulation of nuclear receptors cannot be fully understood without a thorough knowledge of the receptor-associated coregulators that either enhance (coactivators) or inhibit (corepressors) transcription. While nuclear receptors themselves have garnered much attention as therapeutic targets, the clinical and etiological relevance of the coregulators to human diseases is increasingly recognized. Aberrant expression or function of coactivators and corepressors has been associated with malignant and metabolic disease development. Many of them are key epigenetic regulators and utilize enzymatic activities to modify chromatin through histone acetylation/deacetylation, histone methylation/demethylation or chromatin remodeling. In this review, we showcase and evaluate coregulators with the most promising therapeutic potential based on their physiological roles and involvement in various diseases that are revealed thus far. We also describe the structural features of the coactivator and corepressor functional domains and highlight areas that can be further explored for molecular targeting.
Keywords: p160/SRC, PGC-1, ANCCA, SMRT, NCoR, histone methylation, histone acetylation, cancer, diabetes, obesity
1. Introduction
The significance of understanding transcriptional regulation by the nuclear hormone receptors (NRs) is underscored by the diverse diseases, such as cancer, where numerous aberrations in hormone signaling pathways are uncovered. Transcriptional regulation by NRs involves ordered and dynamic protein-protein interactions between the receptor, associated coregulators, and the RNA polymerase II transcriptional machinery at the chromatin of target genes. Many coregulators possess enzymatic activities or recruit multi-subunit enzymatic protein complexes to mediate specific chromatin modifications that result in either activate or repress transcription (see below).
Coregulators can be divided into two general classes, namely, coactivators and corepressors. Coactivators are generally characterized by their ability to enhance NR transactivation by interacting with the N-terminus and/or the C-terminal ligand binding domain (LBD) domain of agonist-bound NRs. The counterparts of coactivators, called corepressors, were identified as mediators for selectively repressing NR-dependent gene transcription through interaction with unliganded or antagonist-bound (or in some cases, agonist-bound) NRs on their target genes [1].
Because many coregulators influence the activity of multiple nuclear hormone receptors and thus the transcriptional output of many gene networks, it is not surprising that disruption of their normal function or expression can contribute to a vast spectrum of physiological abnormalities and diseases. Many studies using mouse models support this notion and have contributed to our understanding of coregulators for normal biological function. For example, gene knockout studies have demonstrated important roles for individual members of the p160 family of coactivators (see below) in hormonal responses and organ developments during reproduction, metabolism, and growth [2]. Furthermore, the phenotypic defects were not restricted to hormone-regulated tissues and processes, reflecting in some circumstances a more general role played by the coregulators in control of gene expression. While we acknowledge that many coregulators associate with and mediate the functions of NRs besides steroid receptors, their aberrant expression and function appear to be best understood so far in the steroid hormone responsive human malignancies such as breast cancer and prostate cancer (e.g. the p160/SRC family) or metabolism and energy homeostasis (e.g. PGC-1). Here, we outline the structure and function of coregulators that can thus be considered as highly attractive and promising, new therapeutic targets.
2. Coactivators – Structure, function and therapeutic implications
Enzymatic modification of chromatin structure is at the heart of gene regulation at the transcriptional level. Coactivators can promote gene-specific NR-mediated transcription via several activities. Changes in post-translational modifications on core histone tails – particularly acetylation and methylation – serve as a crucial step in the remodeling of chromatin structure during gene expression. Loss-of-function experiments revealed that the histone acetylase (HAT) activities of the general coactivator CBP and its associated factor p/CAF are important for enhancing NR transactivation [3]. However, transactivation in vivo requires their indirect association with NRs through the p160 coactivators. The intrinsic HAT activity of p160 coactivators in some cases may not be essential for NR-dependent transactivation and, as such, the p160 proteins may primarily serve as adaptors for recruiting HAT containing coactivator complexes in a promoter- and NR-specific context [3].
Other notable classes of histone modifying coactivators that enhance transactivation of NRs include methyltransferases (CARM1 and PRMT1) and demethylases (LSD1, JMJD2a, JMJD2c). Like CBP and p/CAF, CARM1 acts as a secondary coactivator through interaction with the 160 family. SWI/SNF/BRG complexes interact directly with NRs and utilize the energy of ATP hydrolysis to remodel local chromatin structure a key step for transcription [1]. The histone modifying, chromatin remodeling, and RNAPII activating mediator (DRIP/TRAP) coactivator complexes, with their different mechanisms of action, may work in concert to facilitate and enhance NR mediated gene transcription [4].
2.1. The p160/SRC family – key mediators of NR function
2.1.1. The structural determinants for p160/SRC interaction with the NRs
The p160/SRC genes were among the first gene families characterized as coactivators for NRs and include SRC-1, TIF2/GRIP-1, and ACTR/AIB1/RAC3/SRC-3/TRAM-1. The p160s serve as a platform for the assembly of coactivator complexes on the regulatory regions of genes that are targeted by ligand-bound NRs. They thus act as bridging factors between the receptor and other coregulators such as HAT proteins p300/CBP and p/CAF and the mediator complex. Proteins encoded by the p160s family possess conserved homologous domains – consisting of 50-55% sequence similarity - that confer these common functions.
The central receptor interaction domain (RID) of the p160s contains three LXXLL motifs that form amphipathic alpha-helices that are responsible for direct association with the LBD of NRs. The structural determinants of interaction and binding affinity can vary for each p160, depending on the receptor. On the receptor side, the interaction usually requires a ligand-dependent formation of a hydrophobic cleft in the LBD and sometimes the participation of AF-1 and AF-2 [5-6]. On the coactivator side, the interaction requires at least one LXXLL motif. Receptor-specific or preferential utilization of LXXLL motifs dependent, in part, on amino acid sequences adjacent to the motis [2, 7-8]. Although they do not bind DNA directly, the p160s contain two intrinsic transcriptional activation domains (AD1 and AD2) in the C-terminal region [2]. Interestingly, AD1 contains three LXXLL motifs and is responsible for interaction with CBP/p300 and p/CAF. AD2, on the other hand, interacts with CARM1 and PRMT1 and partly confers the HAT activity of SRC-1 and ACTR. Intriguingly, the HAT domain of ACTR shares several sequence motifs with that of CLOCK, a key circadian regulator and a HAT, including a glutamine-rich (Q-rich) feature [9].
The molecular determinants of p160 interaction with estrogen receptor-α (ERα) have been intensively investigated in the context of ligand specificity and therapeutic modulation. It should be noted that the overall structural folding of the LBD is highly conserved among NRs, and that ligand binding induces similar conformational changes [10-11]. The LBD comprises twelve alpha-helices and two beta-sheets that form a hydrophobic binding pocket for ligands. Helices 3, 4, 5, and 12 together form a hydrophobic groove that interacts with the LXXLL amphipathic alpha-helix of the RID in p160s when agonist such as estradiol or diethylstilbestrol is bound [5]. The positioning of helix 12 as part of the hydrophobic groove, in particular, has been revealed to be critical for formation of the coactivator binding surface. Structural studies show that ERα undergoes a different conformational change in the AF-2, which lies within the LBD and contains helix 12, depending on whether it binds agonists, antagonists or other selective ER modulators (SERMs). Binding by 4-hydroxy-tamoxifen or raloxifene renders helix 12 positioning in a way that prevents association with coactivators and instead favors association with corepressors.
Aside from ligand specificity, the context of receptor target gene regulatory region to which a NR binds, particularly the sequence of the hormone response elements, may also modulate receptor conformation and affinity for specific coregulators. This concept has been demonstrated in vitro using ERα and EREs from various estrogen target genes [12-13]. Thus, at least three factors – namely ligand, DNA sequence, and presence of coactivators and corepressors – likely influence the transcriptional activation status of NRs and their recruitment of coregulator complexes in a promoter and cell type-specific manner.
2.1.2. The p160/SRC family involvement in cancer
The aberrant expression of p160 family members in various cancers - as well as their roles and functions in tumorigenesis and disease progression – is an ongoing area of intense research. Gene amplification and/or overexpression of SRC-1, TIF2 and ACTR/AIB1 have been independently reported in clinical samples from breast, prostate and ovarian cancer patients (summarized in reference 14). These findings are not restricted to hormone-responsive malignancies, however, which again reflects the actions of p160s in a diverse range of tissues [14] . Significant clinicopathological correlations were observed for overexpression with high grade, advanced disease stage, and lower disease-free survival for subsets of the cancer patients. In some cohorts of breast and prostate cancers, high SRC-1, TIF2 and ACTR/AIB1 expression have been linked to greater disease recurrence after hormone deprivation therapies [15-23]. Notably, elevated co-expression of ACTR/AIB1 with members of the HER/ErbB family (HER1, HER2, or HER3) was associated with poor response to tamoxifen therapy in invasive breast tumors [19], while coexpression with ER was associated with improved response [24-25].
While clinical studies highlight the potential value of p160s as prognostic indicators, studies using mouse and tissue culture models have revealed their tumor-promoting functions and their roles in disease progression, including disease recurrence after hormone depletion in breast and prostate cancer ([14] and references therein). Overexpression of ACTR/AIB1 or RNAi-mediated depletion of p160 coactivators has demonstrated that their expression is critical for the proliferation of hormone-dependent and –independent breast and prostate cancer cells and tamoxifen resistance [22, 26-29]. In addition to the classical ligand-mediated pathway, ER and androgen receptor (AR) can be activated to increase expression of cell proliferation genes through growth factor/kinase mediated signaling pathways that elicit phosphorylation of the receptors and their coactivators. Whether this non-classical pathway is critically involved in hormone independence and resistance to hormone blockade therapies is still being investigated.
MAPK phosphorylation of SRC-1 and TIF2 at specific residues, for example, is required for optimal androgen-independent activation of AR by IL-6 and EGF [30-32]. Schiff et al reported that HER2/Neu phosphorylation and/or activation of downstream kinases is required for ACTR recruitment to the promoters of ER target genes and to enhance the ER agonist activity of tamoxifen in a resistant clone of MCF-7 breast cancer cells [33]. However, subsequent in vivo study suggests that non-genomic activation of EGFR/HER2 by ER is the predominant mechanism of acquired tamoxifen resistance and that ER target genes continue to be repressed despite agonistic effect on tumor growth [34]. More recent studies showed that RNAi-mediated disruption of ACTR inhibits agonist properties of tamoxifen [35] as well as EGF-induced proliferation in tamoxifen resistant ER positive breast cancer cell lines that express high levels of ACTR and HER1/HER2 [36], supporting a crucial role for this coactivator in acquired tamoxifen resistance [27].
2.1.3. Therapeutic modulation of ER signaling through altered coregulator interaction – a model for targeting NR function
The preclinical and clinical studies above attest to the important contribution of p160 coactivators in ER-positive breast cancer cell proliferation through receptor signaling, a concept that is further supported by anti-estrogen actions on ER-coregulator interactions in vitro (see section 2.1.1). However, SERMs such as tamoxifen and raloxifene display partial agonist or antagonist activity in vivo depending on tissue type likely involving selective recruitment of coactivator or corepressor complexes to target genes [37]. Such swing of effect is believed to be influenced by the relative contribution of coactivators and corepressors that are expressed in different tissues. For example, high levels of SRC-1 in endometrial cancer cells may mediate the agonist activity of tamoxifen in this specific cell type [38].
While AF-1 contributes to tissue-selective activation by SERMs, the positioning of helix 12 induced by SERMs to the LBD hydrophobic groove is believed to influence the affinity of the receptor for the coregulators and to depend on the specific SERM ligand. In the case of tamoxifen, helix 12 is positioned in a way that mimics the interaction of coactivator LXXLL-containing peptide with the LBD [39]. This and other work suggests that helix 12 actually competes with corepressor for binding in the presence of SERM agonist, while removal of helix 12 from the hydrophobic cleft in the presence of a full antagonist such as ICI182,780 promotes corepressor binding [37].
While all known SERMs and ER antagonists function by binding to the ER LBD, the identification of coactivator binding inhibitors has emerged as an alternative approach to effectively inhibit estrogen action. The bulk of this work has relied heavily on chemical and cell based screening of small molecules that contain substituents mimicking the structure of coactivator LXXLL alpha-helical consensus sequence, and that directly compete with the coactivator for high affinity (at low micromolar range) binding to the coactivator binding groove in the ER LBD [40-47]. Given that the inhibitors do not significantly affect receptor ligand binding, it is possible that these new peptide inhibitors or functionally equivalent small molecule compounds could be developed for use in conjunction with first-line antagonists such as tamoxifen to prevent or treat hormone refractory breast cancer in susceptible patients. In support of this idea, studies suggest that disruption of ER interactions with coactivator proteins can inhibit cell growth of endocrine-resistant breast cancer [48-49]. Importantly, it has been shown that analogous structural mimetic strategies can be applied to target other NRs including androgen receptor and thyroid hormone receptor [50-51].
2.2. PGC-1α – a tissue-specific coactivator for metabolic regulation and energy homeostasis
2.2.1. Physiological roles and involvement in metabolic diseases
PGC-1α represents an example in which a primary regulation of transcriptional programs by NRs is at the level of a transcriptional coactivator. PGC-1α is one of three members of the PGC-1 gene family that possess highly homologous functional domains including a transcriptional activation domain with NR interacting LXXLL motifs in the N-terminus and an RNA-binding motif in the C-terminus. Although they have not been demonstrated to possess any enzymatic activity, full transcriptional activation by PGC-1 likely depends on recruitment of several classes of coactivators, including Mediator, SWI/SNF complex and p160/SRC members, through distinct interaction domains at the PGC-1 [52-54]. While PGC-1α shares some common functions with the PGC-1β isoform, such as regulating mitochondrial biogenesis, energy metabolism and fatty acid oxidation, PGC-1α is unique in that its expression is upregulated by cold, fasting and exercise, and in that it stimulates adaptive thermogenesis, heme biosynthesis and liver gluconeogenesis [55].
The diverse, tissue-specific metabolic functions of PGC-1α are believed to be mediated through coactivation of particular transcription factors that regulate gluconeogenic and mitochondrial genes in response to environmental stimuli. For example, UCP-1, a mitochondrial gene expressed specifically in brown adipocytes that is critical for activating cold-induced thermogenesis, is induced through PGC-1α coactivation of PPAR-γ and TR [56]. By uncoupling oxidative phosphorylation through the mitochondrial respiratory chain, UCP-1 contributes to the generation of heat through brown adipose tissue. By coactivating PPAR-α and ERR-α, PGC-1α regulates several genes involved in mitochondrial biogenesis, respiration and fatty acid oxidation in cardiac and skeletal muscle [57-58], which likely explains its important role in maintaining normal oxidative capacity and energy metabolism in these tissues [59-61]. PGC-1α also enhances transcriptional activity of glucocorticoid receptor and hepatocyte nuclear factor-4α, two NRs that regulate expression of key gluconeogenic genes in liver [62]. It is believed that the two separate metabolic pathways of gluconeogenesis and fatty acid oxidation, integrated through PGC-1α, function to maintain energy and nutrient homeostasis during fasting [55].
In agreement with its role as a coordinator of diverse metabolic processes, altered PGC-1 expression levels in various tissues have been implicated in diseases that involve impaired mitochondrial function, including type 2 diabetes (T2D). In skeletal muscle, a key site for insulin action in vivo, PGC-1α induces expression of the insulin-sensitive glucose transporter GLUT4 and enhances glucose uptake through coactivation of the muscle-selective transcription factor MEF2C [63]. Reduced PGC-1α expression in skeletal muscle or adipose tissue was reportedly associated with T2D patients or with morbidly obese and insulin resistant subjects, respectively [64-66]. The finding that genes involved in oxidative phosphorylation are also downregulated in T2D patients [64, 67] supports the hypothesis that PGC-1α and mitochondrial dysfunction constitute a cellular mechanism for insulin resistance and T2D pathology. In several murine models of insulin-deficiency or insulin-resistance, hepatic PGC-1α mRNA was dramatically elevated [62]. Liver-specific expression of PGC-1α transgene increased blood glucose and insulin levels in normal non-fasting animals [62], while PGC-1 deficiency in the liver enhanced insulin sensitivity [68]. Thus, both increased and decreased PGC-1α expression in different metabolic tissues may contribute to the metabolic disturbances characteristic of T2D. Furthermore, animal models suggest that decreased PGC-1α in the heart and brain may promote cardiac hypertrophy and failure, and neurodegenerative diseases, two conditions that are accompanied by critical defects in metabolism [69].
2.2.2 PGC-1α regulators as potential therapeutic targets
As an integrator of diverse metabolic processes that influence health and disease, PGC-1α poses as an attractive target for the prevention and treatment of metabolic disorders. The aforementioned pre-clinical and clinical studies imply that modulation of PGC-1α activity could impact mitochondrial function in T2D subjects. Much of the evidence that supports this prospect has focused on SIRT-1, an NAD(+)-dependent histone deacetylase of the sirtuin family, that enhances PGC-1α activity through deacetylation of multiple lysine residues in response to cellular nutrient signals [70]. Elevated pyruvate and NAD+ increase SIRT-1 protein levels and activity in the fasting liver where it is required for PGC-1α upregulation of gluconeogenic genes to increase glucose output. Subsequently, modulation of SIRT-1 activity was found to impact mitochondrial function and whole-body metabolism. Enhancing SIRT-1 activity through supplementation of the polyphenol resveratrol increased mitochondrial biogenesis, muscle function, and insulin sensitivity through PGC-1α transcriptional programs in mouse models of diet-induced obesity [71-72]. Increased oxidative capacity in skeletal muscle and improved glucose and lipid metabolism in the liver were also observed in these studies. However, more recent work in vitro suggests that resveratrol’s effects are not mediated through direct activation of SIRT-1 [73].
Whether indirect activation of PGC-1α through resveratrol or other small molecules with similar activity presents a valid approach to treat T2D is still an active area of investigation. More recent studies have established that resveratrol reverses hyperglycemia in a diabetic rat model. This observation was accompanied by positive physiological changes including improved plasma insulin levels and liver glycogen content, with the latter effect associated with modulation of enzymatic activities that control gluconeogenesis [74-75]. These findings, together with the overall improvement in glucose homeostasis seen in mice, suggest that resveratrol may not promote PGC-1α enhancement of liver gluconeogenesis, a property that would be highly desirable as an effective treatment for T2D. Furthermore, the enhancement of insulin sensitivity in muscle through resveratrol supports the hypothesis that induction of PGC-1α activity can be translated to positive therapeutic outcomes for diabetic patients. Since mitochondrial dysfunction has been linked to other diseases such as cardiomyopathy and neurodegeneration, therapeutic targeting of PGC-1α may extend beyond diabetes.
2.3. Coactivators with histone and chromatin modifying activities
Mounting evidence suggests that dynamic multi-level changes in chromatin structure that are mediated by transcriptional coregulators strongly impact cellular processes including differentiation, cell death, inflammation, and oncogenesis [76-79]. Consequently, epigenetic aberrations in gene expression attributed to deregulated coregulators may play important roles in various human diseases. This notion is underscored by the observation, for example, that many histone demethylases are inactivated or overexpressed in various human malignancies [80]. Furthermore, inhibitors of DNA methyltransferases and histone deacetylases are currently touted as potential therapeutic compounds for their ability to derepress epigenetically silenced tumor suppressor genes (see section 3.3). Here, we will highlight recently identified coactivators, some of which have been linked to cancer, that directly interact with and/or mediate chromatin histone modifications to drive hormone-responsive or -independent gene expression.
2.3.1. Histone demethylases – important mediators in AR-dependent gene transcription
LSD1
As a member of the amine oxidase family of histone demethylases, lysine-specific histone demethylase 1 (LSD1) catalyzes the cleavage of substrate alpha-carbon bond through a three-step reaction involving reduction of cofactor FAD and generation of byproducts hydrogen peroxide and formaldehyde [81-82]. The C-terminal amine oxidase-like (AOL) domain comprises the catalytic core and binds both the peptide substrate and FAD cofactor. The central Tower domain binds the corepressor CoREST which promotes stability, efficient nucleosomal association and demethylase specificity for H3K4 [83]. As methylation of H3K4 typically marks transcriptionally active genomic regions, LSD1 was initially identified and characterized as a corepressor. However, more recent studies revealed that LSD1 also targets methylated H3K9, a marker that has been found in both the promoters of silenced genes and the coding regions of actively transcribed genes [84-86]. Thus, LSD1 can act as a coactivator or corepressor depending on the chromatin context.
Association with NRs, particularly the AR, has been shown to switch the substrate specificity of LSD1 to H3K9 in vitro and in vivo. While H3K4 methylation levels were unaffected by androgen in prostate cancer cells, repressive methylation of H3K9 in the enhancer region of PSA promoter was diminished by androgen treatment of cells. LSD1 knockdown or inhibition blocked AR-dependent gene expression and H3K9 demethylation [87]. Thus, in this context, LSD1 acts as a coactivator to drive AR-mediated transcription. Similar findings have been found during ligand-stimulated transcription of ERα target genes in breast cancer cells. However, this observation was accompanied by LSD1 dependent H3K4 demethylation in the enhancer and promoter regions after loss of receptor from chromatin or under hormone starvation [88]. Collectively, these studies suggest that LSD1 is a key player in regulating hormone-responsive genes through modulation of H3 lysine methyl marks, leading to dynamic transcriptional derepression and, in the case of ER, repression in a receptor-dependent manner.
Small molecule inhibitors of LSD1 have been characterized, prompting investigation into the potential for LSD1 as a therapeutic target. These include compounds, some of which are used to treat depression, that inhibit structurally homologous monoamine and polyamine oxidases. Identified LSD1 inhibitors have been shown to increase global H3K4 methylation in colon carcinoma and P19 embryonic carcinoma cells [89-90]. In colon cancer, this finding was accompanied by selective derepression of aberrantly silenced genes as well as decreased H3K9 methylation and increased H3K9 acetylation at the specific gene promoters [89]. Other factors may contribute to selective gene reexpression resulting from LSD1 inhibition, however, and the less well-characterized consequences of increased global H3K4 methylation suggest that further studies are required to evaluate the therapeutic efficacy and specificity of targeting LSD1 in different cell and tissue context. In addition, LSD1 knockdown derepresses hTERT expression - which is silenced in normal cells - and delays stabilization of p53 upon DNA damage [91-92]. LSD1 knockdown also represses p53 transcriptional activity by demethylating K370 on this tumor suppressor substrate [93].
An alternative strategy could employ inhibition of binding between LSD1 and specific transcription factors. Prostate cancer presents a particularly promising model to assess this targeted approach as the structural domains involved in LSD1 interaction with AR have been identified. Furthermore, LSD1 knockdown blocks androgen-dependent proliferation of prostate cancer cells [87]. High expression of LSD1 in primary prostate tumors has been correlated with high grade and disease recurrence after radical prostatectomy [94], suggesting additional involvement of LSD1 in androgen-independent receptor activation and tumor progression.
JumonjiC domain-containing proteins
Histone lysines can be mono-, di-or tri-methylated, and removal of all three forms of methylation on H3K9 is required for ligand-induced AR dependent transcription. However, the demethylation reaction catalyzed by LSD1 requires a lone pair of electrons on the substrate nitrogen, limiting its activity to mono-or dimethylated lysines and indicating the involvement of a H3K9 tri-demethylase which does not require protonated substrate nitrogen. Two separate groups identified several members of the JmjC domain-containing protein family that are capable of removing trimethyl group from H3K9 and enhancing AR-mediated transcriptional activation [95-96]. As bona fide coactivators of AR, JMJD2A, JMJD2C, and JMJD2D interact with the receptor in a ligand dependent manner. Like LSD1, JMJD2C binds PSA promoter chromatin in a ligand-independent manner where it forms a complex with ligand-activated AR and is required for H3K9 demethylation. Interestingly, JMJD2C is also associated with LSD1 on the PSA promoter, and the two coactivators can cooperatively stimulate AR-dependent transactivation [95].
The JmjC family of histone demethylases utilize a hydroxylation reaction involving Fe(II) and α-ketoglutarate as cofactors to remove substrate methyl groups, generating formaldehyde as a byproduct. As the name implies, catalytic activity is dependent on the conserved Jumonji C domain, although other functional domains not present in all members are also crucial for the demethylase activity of some JmjC proteins. The JMJD2A-C subfamily contains C-terminal PHD and Tudor domains that are involved in binding methylated histones, which are not present in the JMJD2D-F subfamily. Mutation of critical residues in the JmjC domain disrupted stimulation of AR transactivation, indicating that catalytic activity is required for the coactivation function [95-97]. Despite sequence similarities in the JmjC domain, individual characterized demethylases have very distinct specificity with regard to both methylation site and state. Other AR coactivators belonging to the JmjC family include JHDM2A, which specifically demethylates mono- and di-methylated H3K9, and JARID1B, which removes mono-, di-, and tri-methylated forms of H3K4 [97-98]. In contrast to JMJD2C, JHDM2A recruitment to AR target gene promoters was hormone-induced, although LSD1 remained critical for optimal demethylation of H3K9 and transactivation by AR [98].
In context of their involvement in transcriptional activation of AR target genes, the aforementioned JmjC coactivators can be considered potential therapeutic targets. Knockdown of JMJD2C in LNCaP cells blocked androgen-induced cell proliferation [95], and cancer gene expression profiling analysis indicated that JMJD2C expression is upregulated in prostate cancer compared to benign prostate hyperplasia [97]. These studies suggest that the involvement of JmjC proteins in prostate cancer development and progression warrants further investigation. Furthermore, preliminary data suggest that some JmjC proteins can coactivate other NRs besides AR [95].
2.3.2. AAA+ Nuclear Coregulator Cancer-Associated (ANCCA)
We and others recently identified ANCCA, a member of the AAA+ (ATPases associated with various cellular activities) family, as a novel transcriptional coactivator for ERalpha and AR as well as c-Myc [99-101]. Members of this functionally diverse ATPase family possess conserved AAA+ ATP-binding domains and assemble into characteristic ring-shaped hexamers constituting the active ATPase holoenzyme. ATP binding sites are formed at the interface between adjacent AAA+ protein subunits. The AAA+ domain contains several structural motifs, particularly within the key SRH (second region of homology) element, believed to coordinate ATP hydrolysis and the propagation of conformational changes throughout the enzyme assembly. The dynamic coupling of these two events then drives conformational changes in substrate proteins during cellular processes that involve protein unfolding and protein-complex remodeling, such as proteolysis, membrane fusion, DNA replication, and microtubule sliding [102-105]. Based on structure-function analysis of several AAA+ proteins, it has been proposed that residues extending from each subunit into the central pore formed by the hexamer are involved in substrate binding and translocation through the pore during processing [106-108].
The predicted structure of ANCCA includes a bromodomain located C-terminal to two centrally situated AAA+ domains that are most closely related in sequence homology to the so-called classic clade of AAA+ proteins such as p97/VCP [99-100]. Our work suggest that ANCCA is involved in the recruitment or assembly of transcriptionally active protein complexes including CBP on target genes and hence the histone modifications mediated by these complexes [99-100]. ANCCA is recruited to the promoters of specific subsets of ER or AR target genes with known functions in cell proliferation and survival, and its depletion impairs their hormone-induced expression. In the case of the ER target gene promoters, ANCCA is also necessary for estrogen stimulated CBP recruitment and chromatin histone H4 hyperacetylation. Interestingly, expression of ANCCA itself is robustly induced by estrogen and androgen in breast and prostate cancer cells respectively [99-100].
Although the mechanism by which ANCCA contributes to tumorigenesis is still largely unexplored, our studies and others support the notion that ANCCA plays important roles in the proliferation and survival of cancer cells, likely through its direct regulation of target genes. RNAi-mediated knock-down studies demonstrated that ANCCA promotes G1-S cell cycle progression of estrogen-stimulated breast cancer cells and survival of androgen-dependent or hormone-refractory prostate cancer cells [99-100]. Furthermore, high levels of ANCCA have been found in a significant percentage of breast and prostate cancer specimens, and its overexpression is associated with higher grade and disease recurrence [100-101, 109 and Kalashnikova E. et. al., Cancer Research, 2010, in press]. These findings, along with other studies identifying ANCCA as part of an aberrant gene expression profile in breast tumors, underscore the value of examining ANCCA as a potential therapeutic target [110-114].
Potential strategies for targeting ANCCA in cancer will likely depend on further structural and functional characterization, as well as elucidating the roles of relevant domains in NR coactivation. We demonstrated that ANCCA binds and hydrolyzes ATP [99] and our preliminary data suggests that ANCCA forms multimers (unpublished data). Interestingly, mutating key residues of the Walker A and Walker B motifs in the first AAA+ domain disrupt ANCCA function as an ER coactivator, indicating the critical involvement of ATPase activity [99]. Integrity of the bromodomain, which is often found in both HAT containing and ATP-dependent chromatin remodeling proteins [115], also affects the ability of ANCCA to function as a coactivator (Revenko A. et. al., Molecular and Cellular Biology, 2010, in press). Based on other AAA+ proteins, mutation of a key arginine residue in the conserved SRH element impairs oligomerization and ATPase activity [116]. This approach may be appropriate to determine whether ANCCA multimer assembly is required for its coactivator function. Alternatively, structural determinants responsible for ANCCA-NR interaction could be identified and targeted for specific disruption of ANCCA-mediated coactivation. For example, ANCCA directly associates with the DBD-hinge region of AR primarily through an N-terminal region that lies outside the first AAA+ domain [100].
3. Corepressors – Structure, function and therapeutic implications
3.1. NCoR and SMRT – Repressors of unliganded and antagonist-bound NRs
The first NR corepressors were identified based on their ability to mediate transcriptional repression by unliganded thyroid hormone (TR) and retinoid acid receptors. The aptly named NR corepressor (NCoR) and silencing mediator for RAR and TR (SMRT) contain multiple repression domains that serve as docking platforms for recruitment of additional components in the corepressor complex including HDAC and mSin3. Later studies discovered that NCoR and SMRT can interact with additional NRs in the absence of hormone - including vitamin D receptors and PPARs – or in the presence of antagonists – such as ER, AR, glucocorticoid receptor, and progesterone receptor (reviewed in [117] and see section 2.1.3). These findings imply that NCoR and SMRT are specifically involved in active repression by NRs when they are not bound by agonist hormones. However, the ability to associate with NCoR and SMRT is also influenced by factors that can change the accessibility of corepressor docking sites on the NR. These include receptor isoform and heterodimer composition (i.e. for RAR), as well as DNA binding sequence and interactions with other transcription factors at the promoter [117].
The NR interaction domains located in the C-terminus of NCoR and SMRT consist of helical motifs known as corepressor NR (CoRNR) boxes with the consensus sequence L/I-X-X-I/V-I or LXXXI/LXXXI/L. Differences in affinity for NRs are affected by sequence variations in the CoRNR box motif itself, adjacent amino acids, and the corepressor binding surface of the receptor [117-118]. Interestingly, alternative splicing of SMRT generates isoforms with different affinities for different receptors [119]. In analogous to the coactivator NR box, the CoRNR box interacts with residues found in helices 3, 5 and 6 of the LBD hydrophobic groove [118]. However, as previously discussed, the ligand-dependent positioning of helix 12/AF-2 relative to the LBD determines whether coactivator or corepressor can bind. In the unliganded receptor, the extended conformation of helix 12 away from the LBD removes steric restraints that would otherwise prevent the CoRNR box from binding [120]. By contrast, repositioning of helix 12 close to the LBD in the presence of agonist switches the steric accessibility of the binding surface to accommodate the shorter coactivator NR box. Another study using corepressor peptide mimics indicated that helix 12 contains a CoRNR box that can also compete for corepressor binding [121]. This finding provides an alternative explanation for the poor interaction between AF-2 and NCoR/SMRT in the agonist-bound receptor conformation. More recently, however, new receptor interaction motifs were identified at the N-terminus of SMRT and NCoR, and surprisingly the motifs interact with the DNA binding domain of receptors such as ERa, underscoring the complexity of corepressor-receptor interaction [122].
3.2. RIP140 and LCoR – Repressors of agonist-bound NRs
Since the discovery of NCoR and SMRT, the number of identified corepressors has expanded to include several diverse proteins that are distinguished structurally and functionally in their mechanism of repression (reviewed in [120]). Most of these recently described corepressors possess LXXLL NR boxes and can thus associate with the AF-2 of agonist-bound NRs. A few, such as NSD1 and COPR1, contain both activation and repression domains, as well as distinct NR interaction domains that function in the absence or presence of ligand depending on the target receptor [120]. The majority of NR box-containing corepressors, such as RIP140 and LCoR, contain only repression domains and inhibit agonist-activated NRs in a LXXLL-dependent manner [123-124]. Although RIP140 was initially identified as a coactivator depending on the cell and promoter context [117], it is now generally considered a corepressor. Both RIP140 and LCoR are widely expressed in human tissue and have been found to interact with and negatively regulate a large number of ligand-activated NRs [124 -125]. Gene knockout studies in mice suggest that RIP140 plays crucial roles in energy homeostasis and female fertility [126-127].
A distinguishing feature in corepressors of agonist-bound NRs is their ability to repress transcription through HDAC-dependent and –independent mechanisms in a receptor-dependent manner. For RIP140 and NCoR, this property may be mediated in part by secondary corepressors C-terminal-binding protein (CtBP) 1 and 2, which associate with ancillary mediators of repressive histone modifications including histone deacetylases such as HDACs/mSin3 and histone methyl transferases such as G9A, Eu-HMT, and PcG/polycomb complex proteins [128-129]. Recruitment of HDAC6 to a central region in LCoR appears critical for its blocking of gene expression by agonist bound progesterone receptor and ERα [130-131]. The complexity and multiplicity of repressor domains present in a single corepressor protein indicates that corepressors may utilize diverse mechanisms to attenuate the transcriptional activation by NRs.
3.3. Clinical Significance of Corepressors as Therapeutic Targets
Mutations that alter specific NR interactions with corepressors such as NCoR and SMRT have been implicated in the pathogenesis of several forms of human endocrine disorders and malignancies. For example, such mutations in the isoforms of thyroid hormone receptors are initiated with the resistance to thyroid hormone (RTH) syndrome, hepatocellular carcinoma, renal carcinoma, and papillary thyroid cancer while mutations in PPAR-γ are associated with familial severe insulin resistance [117, 132-136]. These genetic lesions typically produce dominant-negative NRs that exhibit impaired release of corpressors in response to ligand and consequently interfere with the transcriptional activation by wild-type receptors.
On the other hand, changes in the level of specific corepressors or activity of HDACs that are recruited by corepressors have been implicated in inflammatory and malignant diseases [137-141]. The involvement of metastasis-associated (MTA) proteins in diverse transcriptional programs that regulate breast cancer progression indicates its potential value as a therapeutic target [142-144]. One promising study used small peptides to mimic the function of MTA1s, a short splice-variant of MTA1 with a unique NR box motif that sequesters ERα in the cytoplasm [145]. The MTA1s peptide containing the motif inhibited ER transactivation, estrogen-dependent proliferation, anchorage-independent growth and in vivo tumor progression of MCF7 cells [146]. Nonetheless, more in vivo studies will be needed to assess the therapeutic efficacy of targeting MTA and other corepressor proteins in tumors of various tissues. Ultimately, this type of investigation should also shed light on the critical roles of different corepressor proteins in cancer progression.
4. Concluding Remarks
Substantial research progress in NR coregulator function and mechanisms provided valuable insights into their contributions to disease development. This review focused on thus far relatively well-characterized representatives from both classes of coregulators and coincidentally, the evidence we present here attribute to their pathological and clinical relevance in various malignancies. However, as illustrated by the coactivator PGC-1α, some coregulators have established roles in physiological processes that are deregulated in other types of disease. Moreover, the widespread tissue distribution of various NR coactivators such as p160/SRC family members and corepressors NCoR and SMRT indicates that they are involved in regulating a diverse array of normal physiological functions as well. Indeed, tissue culture and animal models combined with human studies strongly suggest that altered expression and function of an expanding number of coregulators underlies the etiology of many pathological conditions involving metabolism, neurodegeneration, and hormonal disturbances.
While many clinical studies support the prognostic significance of coregulator expression in diseased tissues, studies that explore the potential of specific coactivators and corepressors as molecular targets are needed to expedite development of new drugs for treating or preventing hormone-related diseases. It is now evident that altered NR structure and function alone cannot explain the development or progression of relevant diseases. Thus, therapeutic strategies to target disease-promoting pathways with focus on modulating the expression or activity of specific coregulators can be viable and effective as well. However, as the function of coregulators is still being elucidated and new coregulators are being identified, targeting coregulators for new therapeutics represents both a new opportunity and enormous challenge. The latter lies in the complex gene networks regulated by the coregulators and also in their function as integrators for multiple, distinctive cell signaling pathways. Therefore, success of any targeting strategy will likely depend on deeper insights into the downstream pathways and gene networks that are regulated by specific coregulators, as well as the role played by post-translational modifications of chromatin and coregulators themselves. Nonetheless, given their crucial functions revealed, coregulators that possess well-defined functional motifs and/or enzymatic activities can be readily exploited for identification of compounds with therapeutic potential. The fact that some of the coregulators play important roles in hormone responsive and non-responsive diseases can make the effort in targeting them more rewarding.
Fig 1.
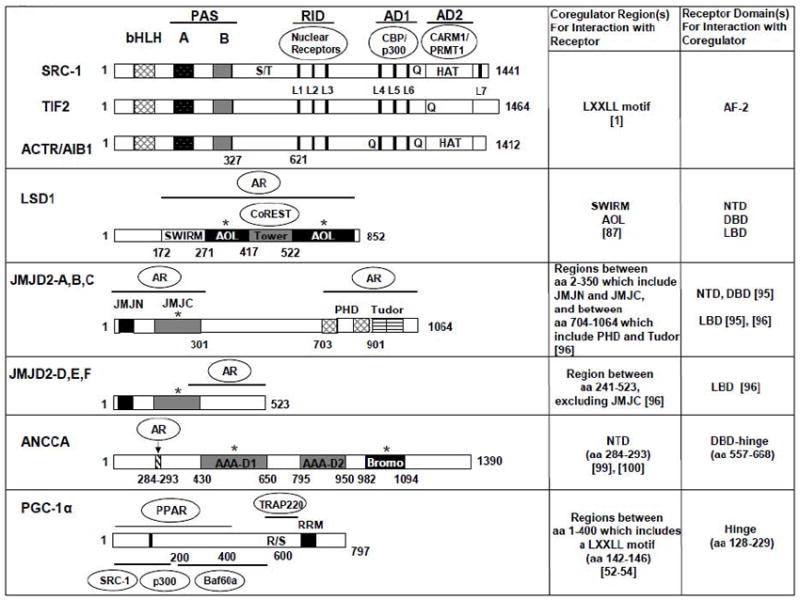
The functional domains of coactivators featured in the review and their involvement in interactions with nuclear receptors and secondary coregulators. The locations of conserved structural and functional domains for the full-length human coactivator proteins are indicated by filled or textured boxes and bars, with amino acid residues numbered. bHLH, basic helix-loop-helix; PAS, Per/ARNT/Sim domain; S/T, serine/threonine-rich regions; RID, receptor interaction domain; L1–L6 (L7), LXXLL alpha-helix motifs, indicated by the black vertical lines;Q, glutamine-rich regions; HAT, histone acetyltransferase domains; AD1 and AD2, transcriptional activation domains; AR (androgen receptor); AOL (amine oxidase-like); JMJ (Jumonji); PHD (plant homeodomain); AAA (ATPases associated with various cellular activities); R/S (arginine/serine-rich region); RRM (RNA recognition motif). Interaction partners for RID, AD1, and AD2 are outlined inside ovals in the schematic for the three p160 coactivators. For the remaining coactivators, the domains highlighted by the lines or arrow are involved in binding the indicated NR or secondary coactivator, and the corepressor CoREST binds the Tower domain in LSD1. TRAP220 is the PPARγ-interacting subunit of TRAP/DRIP/Mediator complex. Baf60a is a core subunit of the SWI/SNF complex. * indicates that the domain’s presence or activity is required for coactivation function. References in [] are included in the table specifying interactions for each NR-coactivator pair.
Fig 2.
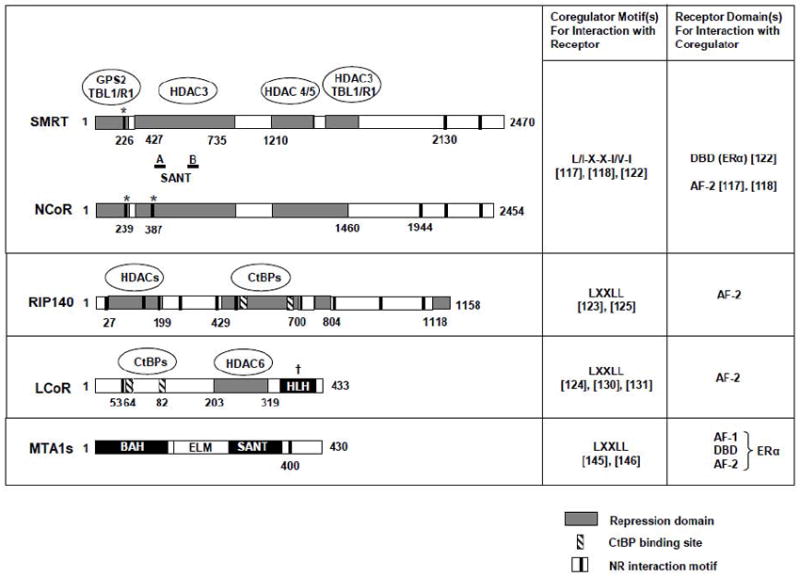
The functional domains of corepressors featured in the review and their involvement in interactions with nuclear receptors and secondary corepressors. The transcriptional repression and NR interaction domains are indicated by gray-filled bars and black vertical lines, respectively, with associated secondary corepressors outlined inside the ovals. Other structural domains are indicated by black or white-filled bars, with amino acid residues numbered. The location of conserved SANT domains A and B in NCoR and SMRT are marked by the horizontal lines. The approximate location of CtBP binding sites are indicated in striped vertical bars. With the exception of MTA1s, the schematics for all corepressors represent full-length isoforms. * indicates recently identified NR interaction domains that bind the DBD in ERα. † indicates a required domain for corepression function. SANT (SWI3, ADA2, N-CoR and TFIIIB DNA binding domain); HLH (helix loop helix); BAH (bromo-adjacent homology); ELM (Egl-27 and MTA1 homology). The consensus NR interaction motif sequences for each corepressor are reported on the right along with the NR domain(s) that they bind and relevant references in [].
Acknowledgments
This work was supported in part by NIH grants R01DK53528 (M.L. Privalsky), R01CA113860, R01CA134766 and R01DK060019 (H-W Chen), and DoD grant W81XWH-07-1-0312 (J. X Zou).
Abbreviations
- NR
nuclear hormone receptor
- LBD
ligand binding domain
- DBD
DNA binding domain
- SERM
selective estrogen receptor modulator
- SRC
steroid receptor coactivator
- ACTR
activator for receptors
- AIB1
amplified in breast cancer 1
- TIF2
transcriptional intermediary factor 2
- PGC-1
PPAR gamma coactivator-1
- HAT
histone acetyltransferase
- CARM1
coactivator-associated arginine methyltransferase 1
- PRMT
protein arginine methyltransferase
- ANCCA
AAA nuclear coregulator cancer-associated
- AAA
ATPases associated with various cellular activities
- SMRT
silencing mediator for RAR and TR
- NCoR
NR corepressor
- MTA
metastasis-associated protein
- RIP140
receptor-interacting protein 140
- LCoR
ligand-dependent corepressor
- JMJD
jumonji C domain-containing histone demethylase
- LSD1
lysine-specific histone demethylase 1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Glass CK, Rosenfeld MG. The coregulator exchange in transcriptional functions of nuclear receptors. Genes & Development. 2000;14:121–141. [PubMed] [Google Scholar]
- 2.Xu J, Li Q. Review of the in Vivo Functions of the p160 Steroid Receptor Coactivator Family. Mol Endocrinol. 2003;17:1681–1692. doi: 10.1210/me.2003-0116. [DOI] [PubMed] [Google Scholar]
- 3.Edwards DP. The Role of Coactivators and Corepressors in the Biology and Mechanism of Action of Steroid Hormone Receptors. Journal of Mammary Gland Biology and Neoplasia. 2000;5:307–324. doi: 10.1023/a:1009503029176. [DOI] [PubMed] [Google Scholar]
- 4.Shao W, Keeton EK, McDonnell DP, Brown M. Coactivator AIB1 links estrogen receptor transcriptional activity and stability. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:11599–11604. doi: 10.1073/pnas.0402997101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kushner PJ, et al. Estrogen receptor action through target genes with classical and alternative response elements. Pure Appl Chem. 2003;75:1757–1769. [Google Scholar]
- 6.Metivier R, Penot G, Flouriot G, Pakdel F. Synergism Between ERα Transactivation Function 1 (AF-1) and AF-2 Mediated by Steroid Receptor Coactivator Protein-1: Requirement for the AF-1 α-Helical Core and for a Direct Interaction Between the N- and C-Terminal Domains. Mol Endocrinol. 2001;15:1953–1970. doi: 10.1210/mend.15.11.0727. [DOI] [PubMed] [Google Scholar]
- 7.McInerney EM, Rose DW, Flynn SE, Westin S, Mullen T-M, Krones A, Inostroza J, Torchia J, Nolte RT, Assa-Munt N, Milburn MV, Glass CK, Rosenfeld MG. Determinants of coactivator LXXLL motif specificity in nuclear receptor transcriptional activation. Genes Dev. 1998;12:3357–3368. doi: 10.1101/gad.12.21.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Darimont BD, Wagner RL, Apriletti JW, Stallcup MR, Kushner PJ, Baxter JD, Fletterick RJ, Yamamoto KR. Structure and specificity of nuclear receptor-coactivator interactions. Genes Dev. 1998;12:3343–3356. doi: 10.1101/gad.12.21.3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doi M, Hirayama J, Sassone-Corsi P. Circadian Regulator CLOCK Is a Histone Acetyltransferase. 2006;125:497–508. doi: 10.1016/j.cell.2006.03.033. [DOI] [PubMed] [Google Scholar]
- 10.Moras D, Gronemeyer H. The nuclear receptor ligand-binding domain: structure and function. Current Opinion in Cell Biology. 1998;10:384–391. doi: 10.1016/s0955-0674(98)80015-x. [DOI] [PubMed] [Google Scholar]
- 11.Wurtz J-M, Bourguet W, Renaud J-P, Vivat V, Chambon P, Moras D, Gronemeyer H. A canonical structure for the ligand-binding domain of nuclear receptors. Nat Struct Mol Biol. 1996;3:87–94. doi: 10.1038/nsb0196-87. [DOI] [PubMed] [Google Scholar]
- 12.Hall JM, McDonnell DP, Korach KS. Allosteric Regulation of Estrogen Receptor Structure, Function, and Coactivator Recruitment by Different Estrogen Response Elements. Mol Endocrinol. 2002;16:469–486. doi: 10.1210/mend.16.3.0814. [DOI] [PubMed] [Google Scholar]
- 13.Klinge CM, Jernigan SC, Smith SL, Tyulmenkov VV, Kulakosky PC. Estrogen response element sequence impacts the conformation and transcriptional activity of estrogen receptor α. Molecular and Cellular Endocrinology. 2001;174:151–166. doi: 10.1016/s0303-7207(01)00382-3. [DOI] [PubMed] [Google Scholar]
- 14.Hsia EYC, Zou JX, Chen H-W. Progress in Molecular Biology and Translational Science. Elsevier Inc.; 2009. Chapter 8 The Roles and Action Mechanisms of p160/SRC Coactivators and the ANCCA Coregulator in Cancer; pp. 261–298. [DOI] [PubMed] [Google Scholar]
- 15.Harigopal M, Heymann J, Ghosh S, Anagnostou V, Camp R, Rimm D. Estrogen receptor co-activator (AIB1) protein expression by automated quantitative analysis (AQUA) in a breast cancer tissue microarray and association with patient outcome. Breast Cancer Research and Treatment. 2009;115:77–85. doi: 10.1007/s10549-008-0063-9. [DOI] [PubMed] [Google Scholar]
- 16.Dihge L, Bendahl P-O, Grabau D, Isola J, Lövgren K, Rydén L, Fernö M. Epidermal growth factor receptor (EGFR) and the estrogen receptor modulator amplified in breast cancer (AIB1) for predicting clinical outcome after adjuvant tamoxifen in breast cancer. Breast Cancer Research and Treatment. 2008;109:255–262. doi: 10.1007/s10549-007-9645-1. [DOI] [PubMed] [Google Scholar]
- 17.Myers E, Hill ADK, Kelly G, McDermott EW, O’Higgins NJ, Buggy Y, Young LS. Associations and Interactions between Ets-1 and Ets-2 and Coregulatory Proteins, SRC-1, AIB1, and NCoR in Breast Cancer. Clinical Cancer Research. 2005;11:2111–2122. doi: 10.1158/1078-0432.CCR-04-1192. [DOI] [PubMed] [Google Scholar]
- 18.Fleming FJ, Myers E, Kelly G, Crotty TB, McDermott EW, O’Higgins NJ, Hill ADK, Young LS. Expression of SRC-1, AIB1, and PEA3 in HER2 mediated endocrine resistant breast cancer; a predictive role for SRC-1. Journal of Clinical Pathology. 2004;57:1069–1074. doi: 10.1136/jcp.2004.016733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Osborne CK, Bardou V, Hopp TA, Chamness GC, Hilsenbeck SG, Fuqua SAW, Wong J, Allred DC, Clark GM, Schiff R. Role of the Estrogen Receptor Coactivator AIB1 (SRC-3) and HER-2/neu in Tamoxifen Resistance in Breast Cancer. J Natl Cancer Inst. 2003;95:353–361. doi: 10.1093/jnci/95.5.353. [DOI] [PubMed] [Google Scholar]
- 20.Gregory CW, He B, Johnson RT, Ford OH, Mohler JL, French FS, Wilson EM. A mechanism for androgen receptor-mediated prostate cancer recurrence after androgen deprivation therapy. Cancer Res. 2001;61:4315–4319. [PubMed] [Google Scholar]
- 21.Yan J, Erdem H, Li R, Cai Y, Ayala G, Ittmann M, Yu-Lee L-Y, Tsai SY, Tsai M-J. Steroid Receptor Coactivator-3/AIB1 Promotes Cell Migration and Invasiveness through Focal Adhesion Turnover and Matrix Metalloproteinase Expression. Cancer Res. 2008;68:5460–5468. doi: 10.1158/0008-5472.CAN-08-0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou H-J, Yan J, Luo W, Ayala G, Lin S-H, Erdem H, Ittmann M, Tsai SY, Tsai M-J. SRC-3 Is Required for Prostate Cancer Cell Proliferation and Survival. Cancer Res. 2005;65:7976–7983. doi: 10.1158/0008-5472.CAN-04-4076. [DOI] [PubMed] [Google Scholar]
- 23.Gnanapragasam VJ, Leung HY, Pulimood AS, Neal DE, Robson CN. Expression of RAC 3, a steroid hormone receptor co-activator in prostate cancer. Br J Cancer. 2001;85:1928–1936. doi: 10.1054/bjoc.2001.2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iwase H, Omoto Y, Toyama T, Yamashita H, Hara Y, Sugiura H, Zhang Z. Clinical Significance of AIB1 Expression in Human Breast Cancer. Breast Cancer Research and Treatment. 2003;80:339–345. doi: 10.1023/A:1024916126532. [DOI] [PubMed] [Google Scholar]
- 25.Alkner S, Bendahl P-O, Grabau D, Lovgren K, Stal O, Ryden L, Ferno M o.b.o.t.S. Swedish. South-East Swedish Breast Cancer Groups, AIB1 is a predictive factor for tamoxifen response in premenopausal women. Ann Oncol. 2010;21:238–244. doi: 10.1093/annonc/mdp293. [DOI] [PubMed] [Google Scholar]
- 26.Agoulnik IU, Vaid A, Nakka M, Alvarado M, Bingman WE, Erdem H, Frolov A, Smith CL, Ayala GE, Ittmann MM, Weigel NL. Androgens Modulate Expression of Transcription Intermediary Factor 2, an Androgen Receptor Coactivator whose Expression Level Correlates with Early Biochemical Recurrence in Prostate Cancer. Cancer Research. 2006;66:10594–10602. doi: 10.1158/0008-5472.CAN-06-1023. [DOI] [PubMed] [Google Scholar]
- 27.Louie MC, Zou JX, Rabinovich A, Chen H-W. ACTR/AIB1 Functions as an E2F1 Coactivator To Promote Breast Cancer Cell Proliferation and Antiestrogen Resistance. Mol Cell Biol. 2004;24:5157–5171. doi: 10.1128/MCB.24.12.5157-5171.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zou JX, Zhong Z, Shi X-B, Tepper CG, White RWd, Kung H-J, Chen H. ACTR/AIB1/SRC-3 and androgen receptor control prostate cancer cell proliferation and tumor growth through direct control of cell cycle genes. The Prostate. 2006;66:1474–1486. doi: 10.1002/pros.20477. [DOI] [PubMed] [Google Scholar]
- 29.Al-azawi D, Ilroy MM, Kelly G, Redmond AM, Bane FT, Cocchiglia S, Hill ADK, Young LS. Ets-2 and p160 proteins collaborate to regulate c-Myc in endocrine resistant breast cancer. Oncogene. 2007;27:3021–3031. doi: 10.1038/sj.onc.1210964. [DOI] [PubMed] [Google Scholar]
- 30.Gregory CW, Fei X, Ponguta LA, He B, Bill HM, French FS, Wilson EM. Epidermal Growth Factor Increases Coactivation of the Androgen Receptor in Recurrent Prostate Cancer. J Biol Chem. 2004;279:7119–7130. doi: 10.1074/jbc.M307649200. [DOI] [PubMed] [Google Scholar]
- 31.Ueda T, Mawji NR, Bruchovsky N, Sadar MD. Ligand-independent Activation of the Androgen Receptor by Interleukin-6 and the Role of Steroid Receptor Coactivator-1 in Prostate Cancer Cells. J Biol Chem. 2002;277:38087–38094. doi: 10.1074/jbc.M203313200. [DOI] [PubMed] [Google Scholar]
- 32.Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer. 2001;1:34–45. doi: 10.1038/35094009. [DOI] [PubMed] [Google Scholar]
- 33.Shou J, Massarweh S, Osborne CK, Wakeling AE, Ali S, Weiss H, Schiff R. Mechanisms of Tamoxifen Resistance: Increased Estrogen Receptor-HER2/neu Cross-Talk in ER/HER2-Positive Breast Cancer. J Natl Cancer Inst. 2004;96:926–935. doi: 10.1093/jnci/djh166. [DOI] [PubMed] [Google Scholar]
- 34.Massarweh S, Osborne CK, Creighton CJ, Qin L, Tsimelzon A, Huang S, Weiss H, Rimawi M, Schiff R. Tamoxifen Resistance in Breast Tumors Is Driven by Growth Factor Receptor Signaling with Repression of Classic Estrogen Receptor Genomic Function. Cancer Res. 2008;68:826–833. doi: 10.1158/0008-5472.CAN-07-2707. [DOI] [PubMed] [Google Scholar]
- 35.Su Q, Hu S, Gao H, Ma R, Yang Q, Pan Z, Wang T, Li F. Role of AIB1 for Tamoxifen Resistance in Estrogen Receptor-Positive Breast Cancer Cells. Oncology. 2008;75:159–168. doi: 10.1159/000159267. [DOI] [PubMed] [Google Scholar]
- 36.Zhao W, Zhang Q, Kang X, Jin S, Lou C. AIB1 is required for the acquisition of epithelial growth factor receptor-mediated tamoxifen resistance in breast cancer cells. Biochemical and Biophysical Research Communications. 2009;380:699–704. doi: 10.1016/j.bbrc.2009.01.155. [DOI] [PubMed] [Google Scholar]
- 37.Nettles KW, Greene GL. Ligand control of coregulator recruitment to nuclear receptors. Annual Review of Physiology. 2005;67:309–333. doi: 10.1146/annurev.physiol.66.032802.154710. [DOI] [PubMed] [Google Scholar]
- 38.Shang Y, Brown M. Molecular Determinants for the Tissue Specificity of SERMs. Science. 2002;295:2465–2468. doi: 10.1126/science.1068537. [DOI] [PubMed] [Google Scholar]
- 39.Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, Greene GL. The Structural Basis of Estrogen Receptor/Coactivator Recognition and the Antagonism of This Interaction by Tamoxifen. Cell. 1998;95:927–937. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 40.Chang C-Y, Norris JD, Gron H, Paige LA, Hamilton PT, Kenan DJ, Fowlkes D, McDonnell DP. Dissection of the LXXLL Nuclear Receptor-Coactivator Interaction Motif Using Combinatorial Peptide Libraries: Discovery of Peptide Antagonists of Estrogen Receptors α and β. Mol Cell Biol. 1999;19:8226–8239. doi: 10.1128/mcb.19.12.8226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rodriguez AL, Tamrazi A, Collins ML, Katzenellenbogen JA. Design, Synthesis, and in Vitro Biological Evaluation of Small Molecule Inhibitors of Estrogen Receptor α Coactivator Binding. Journal of Medicinal Chemistry. 2003;47:600–611. doi: 10.1021/jm030404c. [DOI] [PubMed] [Google Scholar]
- 42.Shao D, Berrodin TJ, Manas E, Hauze D, Powers R, Bapat A, Gonder D, Winneker RC, Frail DE. Identification of novel estrogen receptor α antagonists. The Journal of Steroid Biochemistry and Molecular Biology. 2004;88:351–360. doi: 10.1016/j.jsbmb.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 43.Zhou H-B, Collins ML, Gunther JR, Comninos JS, Katzenellenbogen JA. Bicyclo[2.2.2]octanes: Close structural mimics of the nuclear receptor-binding motif of steroid receptor coactivators. Bioorganic & Medicinal Chemistry Letters. 2007;17:4118–4122. doi: 10.1016/j.bmcl.2007.05.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Becerril J, Hamilton Andrew D. Helix Mimetics as Inhibitors of the Interaction of the Estrogen Receptor with Coactivator Peptides13. Angewandte Chemie International Edition. 2007;46:4471–4473. doi: 10.1002/anie.200700657. [DOI] [PubMed] [Google Scholar]
- 45.Gunther JR, Moore TW, Collins ML, Katzenellenbogen JA. Amphipathic Benzenes Are Designed Inhibitors of the Estrogen Receptor α/Steroid Receptor Coactivator Interaction. ACS Chemical Biology. 2008;3:282–286. doi: 10.1021/cb800056r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Parent AA, Gunther JR, Katzenellenbogen JA. Blocking Estrogen Signaling After the Hormone: Pyrimidine-Core Inhibitors of Estrogen Receptor-Coactivator Binding. Journal of Medicinal Chemistry. 2008;51:6512–6530. doi: 10.1021/jm800698b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.LaFrate AL, Gunther JR, Carlson KE, Katzenellenbogen JA. Synthesis and biological evaluation of guanylhydrazone coactivator binding inhibitors for the estrogen receptor. Bioorganic & Medicinal Chemistry. 2008;16:10075–10084. doi: 10.1016/j.bmc.2008.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang LH, Yang XY, Zhang X, An P, Kim H-J, Huang J, Clarke R, Osborne CK, Inman JK, Appella E, Farrar WL. Disruption of estrogen receptor DNA-binding domain and related intramolecular communication restores tamoxifen sensitivity in resistant breast cancer. Cancer Cell. 2006;10:487–499. doi: 10.1016/j.ccr.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 49.Redmond AM, Bane FT, Stafford AT, McIlroy M, Dillon MF, Crotty TB, Hill AD, Young LS. Coassociation of Estrogen Receptor and p160 Proteins Predicts Resistance to Endocrine Treatment; SRC-1 is an Independent Predictor of Breast Cancer Recurrence. Clinical Cancer Research. 2009;15:2098–2106. doi: 10.1158/1078-0432.CCR-08-1649. [DOI] [PubMed] [Google Scholar]
- 50.Arnold LA, Estebanez-Perpina E, Togashi M, Jouravel N, Shelat A, McReynolds AC, Mar E, Nguyen P, Baxter JD, Fletterick RJ, Webb P, Guy RK. Discovery of Small Molecule Inhibitors of the Interaction of the Thyroid Hormone Receptor with Transcriptional Coregulators. Journal of Biological Chemistry. 2005;280:43048–43055. doi: 10.1074/jbc.M506693200. [DOI] [PubMed] [Google Scholar]
- 51.Gunther JR, Parent AA, Katzenellenbogen JA. Alternative Inhibition of Androgen Receptor Signaling: Peptidomimetic Pyrimidines As Direct Androgen Receptor/Coactivator Disruptors. ACS Chemical Biology. 2009;4:435–440. doi: 10.1021/cb900043e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Puigserver P, Adelmant G, Wu Z, Fan M, Xu J, O’Malley B, Spiegelman BM. Activation of PPAR Coactivator-1 Through Transcription Factor Docking. Science. 1999;286:1368–1371. doi: 10.1126/science.286.5443.1368. [DOI] [PubMed] [Google Scholar]
- 53.Wallberg AE, Yamamura S, Malik S, Spiegelman BM, Roeder RG. Coordination of p300-Mediated Chromatin Remodeling and TRAP/Mediator Function through Coactivator PGC-1α. Molecular Cell. 2003;12:1137–1149. doi: 10.1016/s1097-2765(03)00391-5. [DOI] [PubMed] [Google Scholar]
- 54.Li S, Liu C, Li N, Hao T, Han T, Hill DE, Vidal M, Lin JD. Genome-wide Coactivation Analysis of PGC-1α Identifies BAF60a as a Regulator of Hepatic Lipid Metabolism. Cell Metabolism. 2008;8:105–117. doi: 10.1016/j.cmet.2008.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lin JD. Minireview: The PGC-1 Coactivator Networks: Chromatin-Remodeling and Mitochondrial Energy Metabolism. Mol Endocrinol. 2009;23:2–10. doi: 10.1210/me.2008-0344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A Cold-Inducible Coactivator of Nuclear Receptors Linked to Adaptive Thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- 57.Vega RB, Huss JM, Kelly DP. The Coactivator PGC-1 Cooperates with Peroxisome Proliferator-Activated Receptor α in Transcriptional Control of Nuclear Genes Encoding Mitochondrial Fatty Acid Oxidation Enzymes. Mol Cell Biol. 2000;20:1868–1876. doi: 10.1128/mcb.20.5.1868-1876.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huss JM, Torra IP, Staels B, Giguere V, Kelly DP. Estrogen-Related Receptor α Directs Peroxisome Proliferator-Activated Receptor α Signaling in the Transcriptional Control of Energy Metabolism in Cardiac and Skeletal Muscle. Mol Cell Biol. 2004;24:9079–9091. doi: 10.1128/MCB.24.20.9079-9091.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lin J, Wu H, Tarr PT, Zhang C-Y, Wu Z, Boss O, Michael LF, Puigserver P, Isotani E, Olson EN, Lowell BB, Bassel-Duby R, Spiegelman BM. Transcriptional co-activator PGC-1α drives the formation of slow-twitch muscle fibres. Nature. 2002;418:797–801. doi: 10.1038/nature00904. [DOI] [PubMed] [Google Scholar]
- 60.Leone TC, Lehman JJ, Finck BN, Schaeffer PJ, Wende AR, Boudina S, Courtois M, Wozniak DF, Sambandam N, Bernal-Mizrachi C, Chen Z, Holloszy JO, Medeiros DM, Schmidt RE, Saffitz JE, Abel ED, Semenkovich CF, Kelly DP. PGC-1α Deficiency Causes Multi-System Energy Metabolic Derangements: Muscle Dysfunction, Abnormal Weight Control and Hepatic Steatosis. PLoS Biol. 2005;3:e101. doi: 10.1371/journal.pbio.0030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Arany Z, Novikov M, Chin S, Ma Y, Rosenzweig A, Spiegelman BM. Transverse aortic constriction leads to accelerated heart failure in mice lacking PPAR-γ coactivator 1α. Proceedings of the National Academy of Sciences. 2006;103:10086–10091. doi: 10.1073/pnas.0603615103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yoon JC, Puigserver P, Chen G, Donovan J, Wu Z, Rhee J, Adelmant G, Stafford J, Kahn CR, Granner DK, Newgard CB, Spiegelman BM. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature. 2001;413:131–138. doi: 10.1038/35093050. [DOI] [PubMed] [Google Scholar]
- 63.Michael LF, Wu Z, Cheatham RB, Puigserver P, Adelmant G, Lehman JJ, Kelly DP, Spiegelman BM. Restoration of insulin-sensitive glucose transporter (GLUT4) gene expression in muscle cells by the transcriptional coactivator PGC-1. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:3820–3825. doi: 10.1073/pnas.061035098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, Miyazaki Y, Kohane I, Costello M, Saccone R, Landaker EJ, Goldfine AB, Mun E, DeFronzo R, Finlayson J, Kahn CR, Mandarino LJ. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:8466–8471. doi: 10.1073/pnas.1032913100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hammarstedt A, Jansson PA, Wesslau C, Yang X, Smith U. Reduced expression of PGC-1 and insulin-signaling molecules in adipose tissue is associated with insulin resistance. Biochemical and Biophysical Research Communications. 2003;301:578–582. doi: 10.1016/s0006-291x(03)00014-7. [DOI] [PubMed] [Google Scholar]
- 66.Semple RK, Crowley VC, Sewter CP, Laudes M, Christodoulides C, Considine RV, Vidal-Puig A, O’Rahilly S. Expression of the thermogenic nuclear hormone receptor coactivator PGC-1α is reduced in the adipose tissue of morbidly obese subjects. Int J Obes Relat Metab Disord. 2003;28:176–179. doi: 10.1038/sj.ijo.0802482. [DOI] [PubMed] [Google Scholar]
- 67.Mootha VK, Lindgren CM, Eriksson K-F, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D, Groop LC. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 68.Koo S-H, Satoh H, Herzig S, Lee C-H, Hedrick S, Kulkarni R, Evans RM, Olefsky J, Montminy M. PGC-1 promotes insulin resistance in liver through PPAR-α-dependent induction of TRB-3. Nat Med. 2004;10:530–534. doi: 10.1038/nm1044. [DOI] [PubMed] [Google Scholar]
- 69.Wu Z, Boss O. Targeting PGC-1α to control energy homeostasis. Expert Opinion on Therapeutic Targets. 2007;11:1329–1338. doi: 10.1517/14728222.11.10.1329. [DOI] [PubMed] [Google Scholar]
- 70.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1α and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 71.Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, Geny B, Laakso M, Puigserver P, Auwerx J. Resveratrol Improves Mitochondrial Function and Protects against Metabolic Disease by Activating SIRT1 and PGC-1α. Cell. 2006;127:1109–1122. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 72.Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, Prabhu VV, Allard JS, Lopez-Lluch G, Lewis K, Pistell PJ, Poosala S, Becker KG, Boss O, Gwinn D, Wang M, Ramaswamy S, Fishbein KW, Spencer RG, Lakatta EG, Le Couteur D, Shaw RJ, Navas P, Puigserver P, Ingram DK, de Cabo R, Sinclair DA. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444:337–342. doi: 10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Beher D, Wu J, Cumine S, Kim KW, Lu S-C, Atangan L, Wang M. Resveratrol is Not a Direct Activator of SIRT1 Enzyme Activity. Chemical Biology & Drug Design. 2009;74:619–624. doi: 10.1111/j.1747-0285.2009.00901.x. [DOI] [PubMed] [Google Scholar]
- 74.Palsamy P, Subramanian S. Resveratrol, a natural phytoalexin, normalizes hyperglycemia in streptozotocin-nicotinamide induced experimental diabetic rats. Biomedicine & Pharmacotherapy. 2008;62:598–605. doi: 10.1016/j.biopha.2008.06.037. [DOI] [PubMed] [Google Scholar]
- 75.Palsamy P, Subramanian S. Modulatory effects of resveratrol on attenuating the key enzymes activities of carbohydrate metabolism in streptozotocin-nicotinamide-induced diabetic rats. Chemico-Biological Interactions. 2009;179:356–362. doi: 10.1016/j.cbi.2008.11.008. [DOI] [PubMed] [Google Scholar]
- 76.Surani MA, Hayashi K, Hajkova P. Genetic and Epigenetic Regulators of Pluripotency. Cell. 2007;128:747–762. doi: 10.1016/j.cell.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 77.Collado M, Blasco MA, Serrano M. Cellular Senescence in Cancer and Aging. Cell. 2007;130:223–233. doi: 10.1016/j.cell.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 78.Foster SL, Medzhitov R. Gene-specific control of the TLR-induced inflammatory response. Clinical Immunology. 2009;130:7–15. doi: 10.1016/j.clim.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jones PA, Baylin SB. The Epigenomics of Cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kampranis SC, Tsichlis PN, George FVW, George K. Advances in Cancer Research. Academic Press; 2009. Chapter 4 Histone Demethylases and Cancer; pp. 103–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bannister AJ, Schneider R, Kouzarides T. Histone Methylation: Dynamic or Static? Cell. 2002;109:801–806. doi: 10.1016/s0092-8674(02)00798-5. [DOI] [PubMed] [Google Scholar]
- 82.Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y. Histone Demethylation Mediated by the Nuclear Amine Oxidase Homolog LSD1. Cell. 2004;119:941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 83.Shi Y-J, Matson C, Lan F, Iwase S, Baba T, Shi Y. Regulation of LSD1 Histone Demethylase Activity by Its Associated Factors. Molecular Cell. 2005;19:857–864. doi: 10.1016/j.molcel.2005.08.027. [DOI] [PubMed] [Google Scholar]
- 84.Kouzarides T. Chromatin Modifications and Their Function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 85.Shilatifard A. Chromatin Modifications by Methylation and Ubiquitination: Implications in the Regulation of Gene Expression. Annual Review of Biochemistry. 2006;75:243–269. doi: 10.1146/annurev.biochem.75.103004.142422. [DOI] [PubMed] [Google Scholar]
- 86.Eissenberg JC, Shilatifard A. Leaving a mark: the many footprints of the elongating RNA polymerase II. Current Opinion in Genetics & Development. 2006;16:184–190. doi: 10.1016/j.gde.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 87.Metzger E, Wissmann M, Yin N, Muller JM, Schneider R, Peters AHFM, Gunther T, Buettner R, Schule R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. 2005;437:436–439. doi: 10.1038/nature04020. [DOI] [PubMed] [Google Scholar]
- 88.Perillo B, Ombra MN, Bertoni A, Cuozzo C, Sacchetti S, Sasso A, Chiariotti L, Malorni A, Abbondanza C, Avvedimento EV. DNA Oxidation as Triggered by H3K9me2 Demethylation Drives Estrogen-Induced Gene Expression. Science. 2008;319:202–206. doi: 10.1126/science.1147674. [DOI] [PubMed] [Google Scholar]
- 89.Huang Y, Greene E, Murray Stewart T, Goodwin AC, Baylin SB, Woster PM, Casero RA. Inhibition of lysine-specific demethylase 1 by polyamine analogues results in reexpression of aberrantly silenced genes. Proceedings of the National Academy of Sciences. 2007;104:8023–8028. doi: 10.1073/pnas.0700720104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lee MG, Wynder C, Schmidt DM, McCafferty DG, Shiekhattar R. Histone H3 Lysine 4 Demethylation Is a Target of Nonselective Antidepressive Medications. Chemistry & Biology. 2006;13:563–567. doi: 10.1016/j.chembiol.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 91.Zhu Q, Liu C, Ge Z, Fang X, Zhang X, Straat K, Bjorkholm M, Xu D. Lysine-Specific Demethylase 1 (LSD1) Is Required for the Transcriptional Repression of the Telomerase Reverse Transcriptase (hTERT) Gene. PLoS ONE. 2008;3:e1446. doi: 10.1371/journal.pone.0001446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Scoumanne A, Chen X. The Lysine-specific Demethylase 1 Is Required for Cell Proliferation in Both p53-dependent and -independent Manners. Journal of Biological Chemistry. 2007;282:15471–15475. doi: 10.1074/jbc.M701023200. [DOI] [PubMed] [Google Scholar]
- 93.Huang J, Sengupta R, Espejo AB, Lee MG, Dorsey JA, Richter M, Opravil S, Shiekhattar R, Bedford MT, Jenuwein T, Berger SL. p53 is regulated by the lysine demethylase LSD1. Nature. 2007;449:105–108. doi: 10.1038/nature06092. [DOI] [PubMed] [Google Scholar]
- 94.Kahl P, Gullotti L, Heukamp LC, Wolf S, Friedrichs N, Vorreuther R, Solleder G, Bastian PJ, Ellinger J, Metzger E, Schule R, Buettner R. Androgen Receptor Coactivators Lysine-Specific Histone Demethylase 1 and Four and a Half LIM Domain Protein 2 Predict Risk of Prostate Cancer Recurrence. Cancer Res. 2006;66:11341–11347. doi: 10.1158/0008-5472.CAN-06-1570. [DOI] [PubMed] [Google Scholar]
- 95.Wissmann M, Yin N, Muller JM, Greschik H, Fodor BD, Jenuwein T, Vogler C, Schneider R, Gunther T, Buettner R, Metzger E, Schule R. Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression. Nat Cell Biol. 2007;9:347–353. doi: 10.1038/ncb1546. [DOI] [PubMed] [Google Scholar]
- 96.Shin S, Janknecht R. Activation of androgen receptor by histone demethylases JMJD2A and JMJD2D. Biochemical and Biophysical Research Communications. 2007;359:742–746. doi: 10.1016/j.bbrc.2007.05.179. [DOI] [PubMed] [Google Scholar]
- 97.Xiang Y, Zhu Z, Han G, Ye X, Xu B, Peng Z, Ma Y, Yu Y, Lin H, Chen AP, Chen CD. JARID1B is a histone H3 lysine 4 demethylase up-regulated in prostate cancer. Proceedings of the National Academy of Sciences. 2007;104:19226–19231. doi: 10.1073/pnas.0700735104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yamane K, Toumazou C, Tsukada Y-I, Erdjument-Bromage H, Tempst P, Wong J, Zhang Y. JHDM2A, a JmjC-Containing H3K9 Demethylase, Facilitates Transcription Activation by Androgen Receptor. Cell. 2006;125:483–495. doi: 10.1016/j.cell.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 99.Zou JX, Revenko AS, Li LB, Gemo AT, Chen H-W. ANCCA, an estrogen-regulated AAA+ ATPase coactivator for ERα, is required for coregulator occupancy and chromatin modification. Proceedings of the National Academy of Sciences. 2007;104:18067–18072. doi: 10.1073/pnas.0705814104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zou JX, Guo L, Revenko AS, Tepper CG, Gemo AT, Kung H-J, Chen H-W. Androgen-Induced Coactivator ANCCA Mediates Specific Androgen Receptor Signaling in Prostate Cancer. Cancer Res. 2009;69:3339–3346. doi: 10.1158/0008-5472.CAN-08-3440. [DOI] [PubMed] [Google Scholar]
- 101.Ciro M, Prosperini E, Quarto M, Grazini U, Walfridsson J, McBlane F, Nucifero P, Pacchiana G, Capra M, Christensen J, Helin K. ATAD2 Is a Novel Cofactor for MYC, Overexpressed and Amplified in Aggressive Tumors. Cancer Res. 2009;69:8491–8498. doi: 10.1158/0008-5472.CAN-09-2131. [DOI] [PubMed] [Google Scholar]
- 102.Hanson PI, Whiteheart SW. AAA+ proteins: have engine, will work. Nat Rev Mol Cell Biol. 2005;6:519–529. doi: 10.1038/nrm1684. [DOI] [PubMed] [Google Scholar]
- 103.Indiani C, O’Donnell M. The replication clamp-loading machine at work in the three domains of life. Nat Rev Mol Cell Biol. 2006;7:751–761. doi: 10.1038/nrm2022. [DOI] [PubMed] [Google Scholar]
- 104.Erzberger JP, Berger JM. Evolutionary relationships and structural mechanisms of AAA+ proteins. Annual Review of Biophysics and Biomolecular Structure. 2006;35:93–114. doi: 10.1146/annurev.biophys.35.040405.101933. [DOI] [PubMed] [Google Scholar]
- 105.Burgess SA, Walker ML, Sakakibara H, Knight PJ, Oiwa K. Dynein structure and power stroke. Nature. 2003;421:715–718. doi: 10.1038/nature01377. [DOI] [PubMed] [Google Scholar]
- 106.Siddiqui SM, Sauer RT, Baker TA. Role of the processing pore of the ClpX AAA+ ATPase in the recognition and engagement of specific protein substrates. Genes & Development. 2004;18:369–374. doi: 10.1101/gad.1170304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Schlieker C, Weibezahn J, Patzelt H, Tessarz P, Strub C, Zeth K, Erbse A, Schneider-Mergener J, Chin JW, Schultz PG, Bukau B, Mogk A. Substrate recognition by the AAA+ chaperone ClpB. Nat Struct Mol Biol. 2004;11:607–615. doi: 10.1038/nsmb787. [DOI] [PubMed] [Google Scholar]
- 108.Martin A, Baker TA, Sauer RT. Pore loops of the AAA+ ClpX machine grip substrates to drive translocation and unfolding. Nat Struct Mol Biol. 2008;15:1147–1151. doi: 10.1038/nsmb.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hsia EYC, Kalashnikova EV, Revenko AS, Zou JX, Borowsky AD, Chen H-W. Deregulated E2F and the AAA+ Coregulator ANCCA Drive Proto-Oncogene ACTR/AIB1 Overexpression in Breast Cancer. Molecular Cancer Research. 2010;8:183–193. doi: 10.1158/1541-7786.MCR-09-0095. [DOI] [PubMed] [Google Scholar]
- 110.Wang Y, Klijn JGM, Zhang Y, Sieuwerts AM, Look MP, Yang F, Talantov D, Timmermans M, Meijer-van Gelder ME, Yu J, Jatkoe T, Berns EMJJ, Atkins D, Foekens JA. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. The Lancet. 2005;365:671–679. doi: 10.1016/S0140-6736(05)17947-1. [DOI] [PubMed] [Google Scholar]
- 111.Teschendorff A, Naderi A, Barbosa-Morais N, Pinder S, Ellis I, Aparicio S, Brenton J, Caldas C. A consensus prognostic gene expression classifier for ER positive breast cancer. Genome Biology. 2006;7:R101. doi: 10.1186/gb-2006-7-10-r101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ma X-J, Salunga R, Tuggle JT, Gaudet J, Enright E, McQuary P, Payette T, Pistone M, Stecker K, Zhang BM, Zhou Y-X, Varnholt H, Smith B, Gadd M, Chatfield E, Kessler J, Baer TM, Erlander MG, Sgroi DC. Gene expression profiles of human breast cancer progression. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:5974–5979. doi: 10.1073/pnas.0931261100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.van ’t Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AAM, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT, Schreiber GJ, Kerkhoven RM, Roberts C, Linsley PS, Bernards R, Friend SH. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 114.Pollack JR, Sorlie T, Perou CM, Rees CA, Jeffrey SS, Lonning PE, Tibshirani R, Botstein D, Borresen-Dale A-L, Brown PO. Microarray analysis reveals a major direct role of DNA copy number alteration in the transcriptional program of human breast tumors. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:12963–12968. doi: 10.1073/pnas.162471999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Marmorstein R, Berger SL. Structure and function of bromodomains in chromatin-regulating complexes. Gene. 2001;272:1–9. doi: 10.1016/s0378-1119(01)00519-4. [DOI] [PubMed] [Google Scholar]
- 116.Ogura T, Whiteheart SW, Wilkinson AJ. Conserved arginine residues implicated in ATP hydrolysis, nucleotide-sensing, and inter-subunit interactions in AAA and AAA+ ATPases. Journal of Structural Biology. 2004;146:106–112. doi: 10.1016/j.jsb.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 117.Privalsky ML. The Role of Corepressors in Transcriptional Regulation by Nuclear Hormone Receptors. Annual Review of Physiology. 2004;66:315–360. doi: 10.1146/annurev.physiol.66.032802.155556. [DOI] [PubMed] [Google Scholar]
- 118.Perissi V, Staszewski LM, McInerney EM, Kurokawa R, Krones A, Rose DW, Lambert MH, Milburn MV, Glass CK, Rosenfeld MG. Molecular determinants of nuclear receptor-corepressor interaction. Genes & Development. 1999;13:3198–3208. doi: 10.1101/gad.13.24.3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Goodson ML, Jonas BA, Privalsky ML. Alternative mRNA Splicing of SMRT Creates Functional Diversity by Generating Corepressor Isoforms with Different Affinities for Different Nuclear Receptors. Journal of Biological Chemistry. 2005;280:7493–7503. doi: 10.1074/jbc.M411514200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gurevich I, Flores AM, Aneskievich BJ. Corepressors of agonist-bound nuclear receptors. Toxicology and Applied Pharmacology. 2007;223:288–298. doi: 10.1016/j.taap.2007.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Heldring N, Pawson T, McDonnell D, Treuter E, Gustafsson J-Ãk, Pike ACW. Structural Insights into Corepressor Recognition by Antagonist-bound Estrogen Receptors. Journal of Biological Chemistry. 2007;282:10449–10455. doi: 10.1074/jbc.M611424200. [DOI] [PubMed] [Google Scholar]
- 122.Varlakhanova N, Snyder C, Jose S, Hahm JB, Privalsky ML. Estrogen Receptors Recruit SMRT and N-CoR Corepressors through Newly Recognized Contacts between the Corepressor N Terminus and the Receptor DNA Binding Domain. Mol Cell Biol. 2010;30:1434–1445. doi: 10.1128/MCB.01002-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lee C-H, Chinpaisal C, Wei L-N. Cloning and Characterization of Mouse RIP140, a Corepressor for Nuclear Orphan Receptor TR2. Mol Cell Biol. 1998;18:6745–6755. doi: 10.1128/mcb.18.11.6745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Fernandes I, Bastien Y, Wai T, Nygard K, Lin R, Cormier O, Lee HS, Eng F, Bertos NR, Pelletier N, Mader S, Han VKM, Yang X-J, White JH. Ligand-Dependent Nuclear Receptor Corepressor LCoR Functions by Histone Deacetylase-Dependent and -Independent Mechanisms. Molecular Cell. 2003;11:139–150. doi: 10.1016/s1097-2765(03)00014-5. [DOI] [PubMed] [Google Scholar]
- 125.Augereau P, Badia E, Balaguer P, Carascossa S, Castet A, Jalaguier S, Cavaillès V. Negative regulation of hormone signaling by RIP140. The Journal of Steroid Biochemistry and Molecular Biology. 2006;102:51–59. doi: 10.1016/j.jsbmb.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 126.Leonardsson G, Steel JH, Christian M, Pocock V, Milligan S, Bell J, So P-W, Medina-Gomez G, Vidal-Puig A, White R, Parker MG. Nuclear receptor corepressor RIP140 regulates fat accumulation. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:8437–8442. doi: 10.1073/pnas.0401013101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.White R, Leonardsson G, Rosewell I, Ann Jacobs M, Milligan S, Parker M. The nuclear receptor co-repressor Nrip1 (RIP140) is essential for female fertility. Nat Med. 2000;6:1368–1374. doi: 10.1038/82183. [DOI] [PubMed] [Google Scholar]
- 128.Shi Y, Sawada J-I, Sui G, Affar EB, Whetstine JR, Lan F, Ogawa H, Po-Shan Luke M, Nakatani Y, Shi Y. Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature. 2003;422:735–738. doi: 10.1038/nature01550. [DOI] [PubMed] [Google Scholar]
- 129.Chinnadurai G. CtBP, an Unconventional Transcriptional Corepressor in Development and Oncogenesis. Molecular Cell. 2002;9:213–224. doi: 10.1016/s1097-2765(02)00443-4. [DOI] [PubMed] [Google Scholar]
- 130.Palijan A, Fernandes I, Bastien Y, Tang L, Verway M, Kourelis M, Tavera-Mendoza LE, Li Z, Bourdeau V, Mader S, Yang XJ, White JH. Function of Histone Deacetylase 6 as a Cofactor of Nuclear Receptor Coregulator LCoR. Journal of Biological Chemistry. 2009;284:30264–30274. doi: 10.1074/jbc.M109.045526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Palijan A, Fernandes I, Verway M, Kourelis M, Bastien Y, Tavera-Mendoza LE, Sacheli A, Bourdeau V, Mader S, White JH. Ligand-dependent Corepressor LCoR Is an Attenuator of Progesterone-regulated Gene Expression. Journal of Biological Chemistry. 2009;284:30275–30287. doi: 10.1074/jbc.M109.051201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lin K-H, Shieh H-Y, Chen S-L, Hsu H-C. Expression of mutant thyroid hormone nuclear receptors in human hepatocellular carcinoma cells. Molecular Carcinogenesis. 1999;26:53–61. doi: 10.1002/(sici)1098-2744(199909)26:1<53::aid-mc7>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 133.Lin K-H, Zhu X-G, Hsu H-C, Chen S-L, Shieh H-Y, Chen S-T, McPhie P, Cheng S-Y. Dominant Negative Activity of Mutant Thyroid Hormone α1 Receptors from Patients with Hepatocellular Carcinoma. Endocrinology. 1997;138:5308–5315. doi: 10.1210/endo.138.12.5625. [DOI] [PubMed] [Google Scholar]
- 134.Kamiya Y, Puzianowska-Kuznicka M, McPhie P, Nauman J, Cheng S-y, Nauman A. Expression of mutant thyroid hormone nuclear receptors is associated with human renal clear cell carcinoma. Carcinogenesis. 2002;23:25–33. doi: 10.1093/carcin/23.1.25. [DOI] [PubMed] [Google Scholar]
- 135.Puzianowska-Kuznicka M, Krystyniak A, Madej S-Y, Cheng J. Nauman, Functionally Impaired TR Mutants Are Present in Thyroid Papillary Cancer. J Clin Endocrinol Metab. 2002;87:1120–1128. doi: 10.1210/jcem.87.3.8296. [DOI] [PubMed] [Google Scholar]
- 136.Rosen MD, Privalsky ML. Thyroid Hormone Receptor Mutations Found in Renal Clear Cell Carcinomas Alter Corepressor Release and Reveal Helix 12 as Key Determinant of Corepressor Specificity. Mol Endocrinol. 2009;23:1183–1192. doi: 10.1210/me.2009-0126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Thakur MK, Paramanik V. Role of Steroid Hormone Coregulators in Health and Disease. Hormone Research in Paediatrics. 2009;71:194–200. doi: 10.1159/000201107. [DOI] [PubMed] [Google Scholar]
- 138.Kristensen LS, Nielsen HM, Hansen LL. Epigenetics and cancer treatment. European Journal of Pharmacology. 2009;625:131–142. doi: 10.1016/j.ejphar.2009.10.011. [DOI] [PubMed] [Google Scholar]
- 139.Lane AA, Chabner BA. Histone Deacetylase Inhibitors in Cancer Therapy. J Clin Oncol. 2009;27:5459–5468. doi: 10.1200/JCO.2009.22.1291. [DOI] [PubMed] [Google Scholar]
- 140.Mroz RM, Noparlik J, Chyczewska E, Braszko JJ, Holownia A. Molecular basis of chronic inflammation in lung diseases: new therapeutic approach. J Physiol Pharmacol. 2007;58:453–460. [PubMed] [Google Scholar]
- 141.Marwick JA, Ito K, Adcock IM, Kirkham PA. Oxidative stress and steroid resistance in asthma and COPD: pharmacological manipulation of HDAC-2 as a therapeutic strategy. Expert Opinion on Therapeutic Targets. 2007;11:745–755. doi: 10.1517/14728222.11.6.745. [DOI] [PubMed] [Google Scholar]
- 142.Singh R, Kumar R. MTA Family of Transcriptional Metaregulators in Mammary Gland Morphogenesis and Breast Cancer. Journal of Mammary Gland Biology and Neoplasia. 2007;12:115–125. doi: 10.1007/s10911-007-9043-7. [DOI] [PubMed] [Google Scholar]
- 143.Toh Y, Nicolson G. The role of the MTA family and their encoded proteins in human cancers: molecular functions and clinical implications. Clinical and Experimental Metastasis. 2009;26:215–227. doi: 10.1007/s10585-008-9233-8. [DOI] [PubMed] [Google Scholar]
- 144.Manavathi B, Singh K, Kumar R. MTA family of coregulators in nuclear receptor biology and pathology. Nucl Recept Signal. 2007;5:e010. doi: 10.1621/nrs.05010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kumar R, Wang R-A, Mazumdar A, Talukder AH, Mandal M, Yang Z, Bagheri-Yarmand R, Sahin A, Hortobagyi G, Adam L, Barnes CJ, Vadlamudi RK. A naturally occurring MTA1 variant sequesters oestrogen receptor-α in the cytoplasm. Nature. 2002;418:654–657. doi: 10.1038/nature00889. [DOI] [PubMed] [Google Scholar]
- 146.Singh RR, Kaluarachchi K, Chen M, Rayala SK, Balasenthil S, Ma J, Kumar R. Solution Structure and Antiestrogenic Activity of the Unique C-terminal, NR-box Motif-containing Region of MTA1s. Journal of Biological Chemistry. 2006;281:25612–25621. doi: 10.1074/jbc.M604444200. [DOI] [PubMed] [Google Scholar]