

Abstract
An analysis of Sox9 binding profiles in developing chondrocytes identified marked enrichment of an AP-1-like motif. Here, we have explored the functional interplay between Sox9 and AP-1 in mammalian chondrocyte development. Among AP-1 family members, Jun and Fosl2 were highly expressed within prehypertrophic and early hypertrophic chondrocytes. Chromatin immunoprecipitation followed by DNA sequencing (ChIP-seq) showed a striking overlap in Jun- and Sox9-bound regions throughout the chondrocyte genome, reflecting direct binding of each factor to the same enhancers and a potential for protein-protein interactions within AP-1- and Sox9-containing complexes. In vitro reporter analysis indicated that direct co-binding of Sox9 and AP-1 at target motifs promoted gene activity. By contrast, where only one factor can engage its DNA target, the presence of the other factor suppresses target activation consistent with protein-protein interactions attenuating transcription. Analysis of prehypertrophic chondrocyte removal of Sox9 confirmed the requirement of Sox9 for hypertrophic chondrocyte development, and in vitro and ex vivo analyses showed that AP-1 promotes chondrocyte hypertrophy. Sox9 and Jun co-bound and co-activated a Col10a1 enhancer in Sox9 and AP-1 motif-dependent manners consistent with their combined action promoting hypertrophic gene expression. Together, the data support a model in which AP-1 family members contribute to Sox9 action in the transition of chondrocytes to the hypertrophic program.
KEY WORDS: Chondrocyte development, Hypertrophy, Sox9, Transcriptional program, Mouse
Summary: Genome-wide interaction analyses and mechanistic studies demonstrate that Sox9 and AP-1 family members collaborate to regulate the hypertrophic chondrocyte transition during skeletal development in mice.
INTRODUCTION
The SRY-box (Sox) transcriptional regulator Sox9 is a central regulator of endochondral skeletal development (reviewed by Akiyama and Lefebvre, 2011; Lefebvre and Dvir-Ginzberg, 2016). Initial Sox9 expression in condensing mesenchymal cells precedes active chondrogenesis (Bi et al., 1999). Subsequently, Sox9 expression is maintained in proliferating immature chondrocytes and post-mitotic prehypertrophic chondrocytes (Akiyama et al., 2005; Dy et al., 2012). Sox9 transcription terminates in early hypertrophic chondrocytes (Dy et al., 2012). However, Sox9 protein persists through hypertrophic development: only terminal hypertrophic chondrocytes at the hypertrophic-osteoblast interface of the bone shaft lack Sox9 (Dy et al., 2012).
Genetic analysis in the mouse has shown that Sox9 is necessary for establishing chondrocytes in the cranial, axial and appendicular skeleton (Akiyama et al., 2002; Bi et al., 2001; Mori-Akiyama et al., 2003). Moreover, Sox9 is sufficient to initiate chondrogenic programs when activated in mesenchymal stem cells (Ikeda et al., 2004), embryonic stem cells (Ikeda et al., 2004) and human dermal fibroblasts (Ikeda et al., 2004; Ohba et al., 2015; Outani et al., 2013). Inactivating mutations in human SOX9 result in campomelic dysplasia, an autosomal dominant disorder characterized by hypoplasia of the endochondral skeleton and bowing of skeletal elements (Foster, 1996; Giordano et al., 2001; Kwok et al., 1995). Molecular studies indicate that Sox9 promotes expression of a broad spectrum of cartilage matrix components, promotes cell division and cell survival, and inhibits chondrocytes from adopting an alternative pathway of osteoblast development (reviewed by Lefebvre and Dvir-Ginzberg, 2016).
Sox9 directs chondrogenic programs in concert with other transcriptional components, notably the related Sox members Sox5 and Sox6. The combinatorial interaction of Sox5, Sox6 and Sox9 at Acan and Col2a1 enhancers promotes the expression of these key genes within mitotic chondrocytes (reviewed by Akiyama and Lefebvre, 2011; Lefebvre, 2002). Although genome-wide binding analysis of Sox9 and Sox6 indicates that they co-regulate genomic targets within proliferating chondrocytes (Liu and Lefebvre, 2015), Sox action in hypertrophic chondrocyte development is less clear.
Genetic removal of Sox9 broadly within both proliferating and prehypertrophic chondrocytes prevents hypertrophic progression. In the absence of Sox9, chondrocytes switch fate generating ectopic osteoblasts (Dy et al., 2012). Runx2, a key transcriptional regulator promoting hypertrophic chondrocyte development, is expressed in mitotic and postmitotic chondrocytes of the growth plate where Runx2 has been proposed to counter Sox9's transactivation of early chondrocyte targets such as Col2a1 (Cheng and Genever, 2010; Zhou et al., 2006). Sox9 has also been reported to interact with Mef2 to activate Col10a1, a matrix-encoding gene specifically expressed by hypertrophic chondrocytes (Dy et al., 2012). Furthermore, other reports suggest that a Gli-Sox9 interaction at a distal enhancer of Col10a1 suppresses Col10a1 expression in proliferating chondrocytes (Leung et al., 2011). Together, these studies suggest that Sox9 actions on chondrocyte programs are temporally and spatially regulated by interplay with a variety of additional transcriptional regulators.
Direct analysis of DNA binding has been particularly insightful in distinguishing conflicting regulatory models. Recently, we performed a genome-scale analysis comparing Sox9-DNA interactions in somite-derived and neural crest-derived chondrocytes (Ohba et al., 2015). Analysis of motif recovery identified a highly significant enrichment of an activator protein-1 (AP-1) motif, suggesting co-integration of AP-1 factor engagement into a Sox9-driven chondrocyte regulatory network. AP-1 family members have been linked to osteoblast and osteoclast regulation (reviewed by Wagner, 2002; Wagner and Eferl, 2005) and joint formation (Kan and Tabin, 2013) where Sox9 is either absent or present only at low levels. However, Fosl2 mutants show a reduction in the hypertrophic chondrocyte zone and in mineralized hypertrophic cartilage matrix (Karreth et al., 2004) whereas Jun removal delays chondrocyte hypertrophy specifically in the baso-occipital bone (Behrens et al., 2003).
Here, we explored Sox9, Jun and Fosl2 interactions in the developing mouse skeleton. Jun and Fosl2 are specifically expressed in prehypertrophic and early hypertrophic chondrocytes where endogenous Sox9 levels are maximal. ChIP-seq analysis shows extensive overlap between Jun- and Sox9-bound genomic target regions in mouse primary rib chondrocytes. DNA association likely reflects both direct Jun DNA binding through AP-1 motifs, and indirect protein-protein association with Sox9 mediated by the DNA-binding region of Jun. In vivo and in vitro functional assays suggest that AP-1 and Sox9 co-regulatory pathways promote the chondrocyte transition to post-mitotic hypertrophic chondrocytes and activation of the hypertrophic chondrocyte marker gene Col10a1. These findings highlight the cooperative interplay of transcriptional programs underpinning development of the mammalian skeleton.
RESULTS
Jun and Fosl2 are specifically expressed in prehypertrophic chondrocytes
Our recent ChIP-seq analysis of Sox9 action in mammalian chondrocytes identified a striking enrichment of an AP-1 factor binding motif (TGAG/CTCA), in addition to the expected Sox9 dimer and monomer motifs (Ohba et al., 2015), suggesting that AP-1 family members might act in conjunction with Sox9 to regulate the chondrogenic program.
To address potential AP-1 action, we performed RNA-seq on neonatal rib cartilage to examine the expression of all AP-1 family members that are known to interact with the recovered AP-1 motif: Jun (previously known as c-Jun), Junb, Jund, Fos (cFos), Fosb, Fosl1 (Fra1) and Fosl2 (Fra2). Several members of the AP-1 family showed significant rib chondrocyte expression through this analysis (Table S1). Given that AP-1 factors interchangeably form functional homodimer and heterodimer protein complexes (reviewed by Eferl and Wagner, 2003), the data raised the possibility of considerable heterogeneity in endogenous AP-1 complexes. Among the expressed family members, Jun and Fosl2 show strong expression within hypertrophic chondrocytes (see below) and both are linked to chondrocyte development (Behrens et al., 2003; Hess et al., 2003; Kan and Tabin, 2013; Karreth et al., 2004); these two AP-1 family members were selected for further study.
In situ hybridization to limb and rib skeletal elements with Jun and Fosl2 probes showed strong specific expression of both factors within prehypertrophic and early stage hypertrophic chondrocytes where Col10a1 is first activated (Fig. 1A-D′; Fig. S1A). Immunostaining showed high levels of Jun and Sox9 within Ki67−Sp7+ prehypertrophic chondrocytes extending into the initial domain of Col10a1+ hypertrophic chondrocytes (Fig. 1E-I′; Fig. S1B). These data suggest a potential interplay between Sox9 and AP-1 members at this key transition point in cartilage development.
Fig. 1.
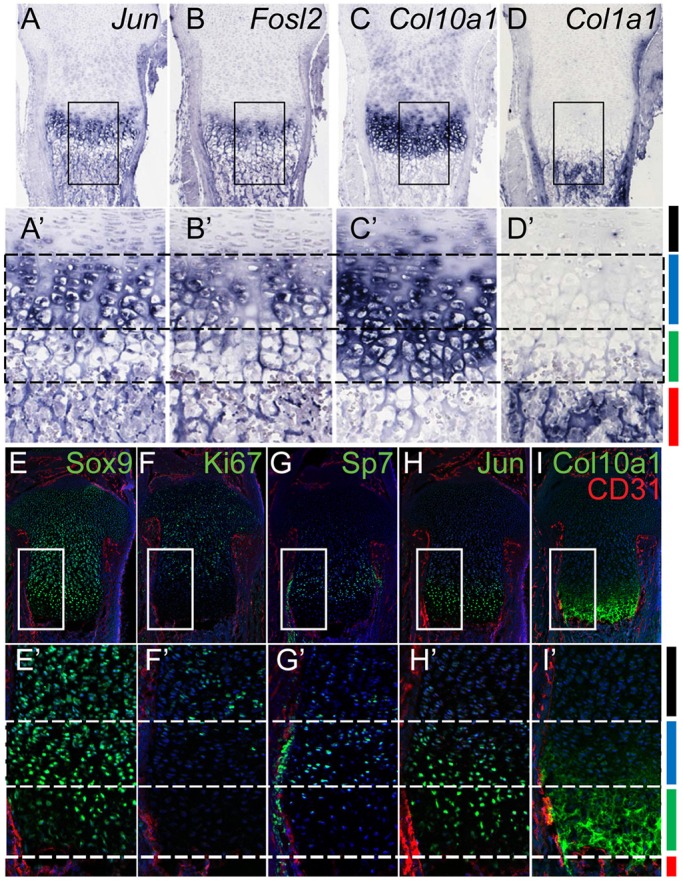
Jun and Fosl2 are specifically expressed in prehypertrophic and early hypertrophic chondrocytes. (A-D′) In situ hybridization comparing expression of Jun, Fosl2, Col10a1 and Col1a1 in the growth plate of the P1 mouse tibia. Boxed regions are magnified in A′-D′. (E-I′) Immunostaining of E17.5 mouse tibia sections to detect Sox9 (E), Jun (H), the proliferative cell marker Ki67 (F), the prehypertrophic chondrocyte marker Sp7 (G) and the hypertrophic chondrocyte marker Col10a1 (I). CD31 (red) demarcates the vasculature, and nuclei are visualized by DAPI staining (blue). Boxed regions appear as higher magnification images in E′-I′. Color bars on the right of D′ and I′ indicate distinct skeletal zones: red, bone-forming ossification region; green, mature hypertrophic chondrocytes; blue, prehypertrophic chondrocytes; black, columnar chondrocytes.
Co-occupancy of the chondrocyte genome by Sox9 and Jun
To address directly AP-1 action within this chondrocyte population, we performed ChIP-seq with anti-Jun antibody on rib chromatin preparations previously used to define Sox9 interactions in vivo (Ohba et al., 2015). The populations used in both this study and the previous one (Ohba et al., 2015) include proliferating and prehypertrophic chondrocytes, but not mature hypertrophic chondrocytes. A Cisgenome analysis (Ji et al., 2008; Jiang et al., 2010) of the ChIP-seq data predicted a large number of Jun interaction sites: 99,162 peaks were significantly enriched above the input control. Motif recovery by MEME-ChIP (Bailey et al., 2009) and Cisgenome predicted a significantly enriched AP-1 consensus binding motif within the dataset (Fig. 2A; Table S2). An AP-1 motif was centered within Jun ChIP-seq peaks (Fig. S2A) and was present within a large percentage of recovered peaks (67.6%: 66,986 of 99,162) consistent with direct engagement of AP-1 complexes containing a Jun component. In summary, the general features of the dataset point to a comprehensive, highly confident set of genomic interactions for Jun within developing chondrocytes.
Fig. 2.

Co-occupation of Jun and Sox9 at putative cis-regulatory modules of chondrocyte target genes. (A) Motif recovery by MEME-ChIP program showing enriched motif logo within top 500 Jun ChIP-seq peaks. Black bars under each motif logo indicate significance of recovery expressed as −log10 of E value. (B) Venn diagram for Sox9 and Jun ChIP-seq peak intersection. The P-value indicates the significance in the overlap of Sox9 ChIP-seq peaks with Jun ChIP-seq peaks. (C) Circos correlation visualization of Sox9 and Jun ChIP-seq peaks. Individual Sox9, Jun and p300 ChIP-seq peaks were clustered based on their genomic coordinates onto reference mouse genome mm9. Clustered peaks were merged to show higher order clustering of ChIP-seq peaks. Super-enhancer associated genes are labeled (Ohba et al., 2015). Prominent extracellular matrix protein-encoding genes expressed in cartilage are highlighted in red. See also Fig. S3. (D) Heatmap of sequence reads at genomic loci around Sox9 ChIP-seq peaks. Signal intensity of Sox9 and Jun ChIP-seq were plotted ±2 kb from Sox9 ChIP-seq peak centers and ranked with Sox9 ChIP-seq peak ranking (Ohba et al., 2015). (E) Scatter plot of Sox9 and Jun ChIP-seq signal at Sox9 ChIP-seq peaks. x-axis and y-axis represent Sox9 and Jun ChIP-seq signals, respectively, at Sox9 ChIP-seq peaks. ChIP-seq signals are shown as log2 of sequence reads per peak per 10 million reads. (F) A screenshot of Sox9 Jun and H3K4Me2 ChIP-seq signal at Col10a1 locus. Asterisk highlights the Col10a1 hypertrophic enhancer module verified in published transgenic studies (Chambers et al., 2002; Gebhard et al., 2004; Leung et al., 2011; Li et al., 2011; Riemer et al., 2002; Zheng et al., 2009), which was explored in this study. (G) Intersection of Jun ChIP-seq peaks with Sox9 ChIP-seq peaks. x-axis indicates ranking of Jun ChIP-seq peaks determined by Cisgenome peak calling. y-axis indicates frequency of Jun ChIP-seq peaks intersected with Sox9 ChIP-seq peaks where each bin samples one hundred peaks. (H) Comparison of Sox9 and Jun ChIP-seq signals with ChIP-seq examining various histone modifications and general transcriptional components within Jun-Sox9 shared peak regions and Jun-only peak regions (±1 kb from peak center). Overlap of H3K4Me2, p300, H3K27Ac, RNAPolII demarcate active enhancer modules, whereas H3K4Me3 demarcates active promoters. Experimental samples were compared with matched controls to show specificity of the ChIP-seq signal. ChIP-seq signals are displayed as sequence reads per peak, per five nucleotides of every 10 million reads. (I) GREAT GO analysis of biological processes (red), mouse phenotype (green) and MGI expression (blue) in Jun-Sox9 shared peak regions. (J) Venn diagram of the intersection of nearest gene neighbors to Jun and Sox9 ChIP-seq peaks.
Next, we investigated the overlap of Jun ChIP-seq peaks with Sox9 Class II ChIP-seq peaks identified in our previous study (Ohba et al., 2015). The Class II peaks represent direct Sox9-bound cis-regulatory modules closely linked to the activation of chondrocyte-specific gene regulatory programs (Ohba et al., 2015). Surprisingly, the majority of Sox9 Class II peaks (81%: 16,895 of 20,862) overlapped with Jun peaks (Fig. 2B); multiple interaction sites were prominent around genes encoding matrix proteins that are expressed at high levels in chondrocytes (Fig. 2C; Fig. S3). Furthermore, we observed a striking correlation in the relative levels of engagement between Jun and Sox9 at Jun-Sox9 co-bound regions (referred to as ‘Jun-Sox9 shared peak regions’ hereafter) as was reflected by the number of DNA sequence reads for bound regions (Fig. 2D,E). Importantly, Sox9 and Jun both associate strongly around Col10a1, a direct target of Sox9 regulation expressed in hypertrophic chondrocytes (Fig. 2F). Binding around Col10a1 highlighted a previously identified hypertrophic DNA enhancer module 4 kb upstream of Col10a1 that is sufficient to direct hypertrophic chondrocyte-restricted reporter gene expression in vivo (Fig. 2F) (Chambers et al., 2002; Gebhard et al., 2004; Leung et al., 2011; Li et al., 2011; Riemer et al., 2002; Zheng et al., 2009).
Interestingly, a comparison of DNA regions associated with Jun alone (referred to as ‘Jun-only peak regions’ hereafter) with those co-associated with Jun and Sox9 (Jun-Sox9 shared peaks) revealed several distinguishing features that suggest the latter are the most relevant to chondrocyte development. First, Jun-Sox9 shared peak regions showed a higher number of Jun sequence reads per peak and higher peak ranking consistent with stronger and/or more frequent engagement of Jun at sites co-engaged with Sox9 than Jun alone (Fig. 2G; Fig. S2B). Second, although both of these groups show strong enrichment of an AP-1 motif centered in peak regions indicative of bona fide Jun interactions with DNA targets (Fig. S2A), a Sox9 motif is only enriched in Jun-Sox9 shared peak regions (Fig. S2A; Table S3). Furthermore, the strong centering of Jun and Sox9 motifs within ChIP-seq peaks suggests direct DNA engagement of both factors within some shared cis-regulatory elements but also the potential for indirect association through protein-protein interactions (see later). Third, comparison of Jun-binding data with our published analysis of active chondrocyte enhancer signatures (p300 binding, H3K4Me2 and H3K27 acetylation enrichment and RNA polymerase II engagement; Ohba et al., 2015) showed that active enhancer signatures predominated only within Jun-Sox9 shared peak regions (Fig. 2H). Together, these data lend additional weight to the biological relevance of the Jun and Sox9 component of the dataset in active transcriptional processes within chondrocytes. Fourth, the biological relevance of Jun-Sox9 shared peaks was underscored by GREAT gene ontology (GO) analysis associating bound regions with neighboring potential target genes (McLean et al., 2010). The Jun-Sox9 shared peak regions showed a highly significant association with skeletal GO terms; the top GO Biological Process term was ‘cartilage development’ and analysis of MGI expression terms predicted skeletal structures (Fig. 2I). Jun-only peaks also showed strong matrix organization GO term enrichment (Fig. S2C), suggesting that many of these identified weaker target sites around the same set of genes engaged by Sox9, a conclusion underscored by the observation that the predicted Sox9 target gene set is essentially entirely encompassed by the predicted Jun-target gene set (Fig. 2J). Given all the evidence here that Sox9 co-association identifies a chondrocyte-related Jun/AP-1 signature with the strongest feature of active enhancers, we focused our analysis on this component of the Jun dataset.
Jun and Sox9 interactions in the regulation of chondrocyte gene expression
To understand the molecular mechanisms underlying the interplay of Jun and Sox9 during chondrogenesis, we analyzed the distribution of AP-1 and Sox9 motifs in Jun-Sox9 shared peak regions. Compared with matched controls (position-matched random control regions bioinformatically calculated from the genome), we observed a strong, significant co-occurrence of Sox9 and AP-1 motifs: 39% in the whole dataset (Fig. 3A; P-value=1.8e-14) and 65% in the subset of peaks with a Sox9 dimer-binding motif (Fig. S4A; P-value<2.2e-16). Only 16% of predicted peaks failed to display a Sox9 or AP-1 binding site suggesting that direct DNA binding of Sox9, AP-1, or both factors, was the primary mode of Sox9 and Jun interaction at Jun-Sox9 shared peak regions (Fig. 3A). The Jun-Sox9 shared peak regions containing both Sox9 and AP-1 motifs showed a higher Sox9 ChIP-seq signal than those with only Sox9 motifs though each showed a comparable active enhancer signature (Fig. 3B; Fig. S4B). These data suggest an enhanced engagement of Sox9 when AP-1 was bound at adjacent DNA sites.
Fig. 3.

Physical interaction between AP-1 factors and Sox9. (A) Enrichment of motifs in Jun-Sox9 shared peak regions. Compared with matched controls, Jun-Sox9 shared peak regions are categorized into four groups by the presence or absence of each factor's binding motif as indicated by the different colors. The pie charts show the frequency of each peak category. (B) Comparison of Sox9 ChIP-seq signal at Jun-Sox9 shared peak regions containing both Sox9 motif and AP-1 motifs (Sox9+AP-1 Motif) with those containing a Sox9 motif but no AP-1 motif (Sox9 Motif Only). *P=1.2e-13. (C) Diagram of deletion mutants used to determine Sox9 and Jun interactions (D). (D) Co-immunoprecipitation of Sox9 and Jun from HEK293T cells using serial deletion mutants illustrated in C. Deletions that prevent co-immunoprecipitation are highlighted in red. (E,F) Co-engagement of Sox9 and Jun on the chondrocyte genome. Following ChIP with the anti-Sox9 antibody, reChIP was performed as indicated. reChIP DNA was analyzed by conventional PCR (E) and quantitative PCR (F). Red bars in screenshots of CisGenome browser indicate regions analyzed by reChIP: (1) Sox9-AP-1-shared peaks with Sox9 and AP-1 motifs around the Col2a1 gene (shared peak region, chr15:97856565-97857517; amplified region, chr15:97857031-97857184) and Col10a1 gene (shared peak region, chr10:34076082-34076958; amplified region, chr10:34076498-34076672), (2) Sox9-AP-1-shared peaks only with Sox9 motif around the Col2a1 gene (shared peak region, chr15:97855718-97856360; amplified region, chr15:97856092-97856262) and Col10a1 gene (shared peak region, chr10:34062583-34063545; amplified region, chr10:34062970-34063158), and (3) Sox9-only peak with Sox9 motif around the Col2a1 gene (peak region, chr15:97839111-97840087; amplified region, chr15:97839448-97839608). #, undetected. Error bars represent s.d.
Although these results are consistent with cooperative binding interactions between Sox9 and AP-1 complexes at respective target sites, a significant number of the shared regions have only an AP-1 or Sox9 site suggesting protein-protein association between Sox9 and AP-1 complexes (Fig. 3A). Consistent with this view, Jun and Fosl2 co-immunoprecipitated with Sox9 when DNA-templated interactions were removed by digestion with the DNA nuclease benzonase (Fig. S4C). To further identify potential protein-protein interaction domains between Sox9 and Jun, we performed co-immunoprecipitation using a series of deletions directed by the domain architecture of each factor (Fig. 3C). These studies indicated that the N-terminal region of Sox9 interacts with a region of Jun encompassing the DNA-binding domain and adjacent proline/glutamine-enriched stretch (Fig. 3D).
We confirmed the protein-DNA-mediated and protein-protein-mediated interaction between Sox9 and AP-1 on the chondrocyte genome by ChIP-reChIP assay using the same chromatin preparations as those used in Sox9 and Jun ChIP-seq (Fig. 3E,F); enrichment was detected in (1) Sox9-AP-1-shared peaks with Sox9 and AP-1 motifs and (2) Sox9-AP-1-shared peaks with only Sox9 motif, but not in a Sox9-only peak with Sox9 motif, with more enrichment detected in (1) than in (2). These data, when considered with those in Fig. 3B and Fig. S4B, suggest that co-occupation of Sox9 and Jun within a cis-regulatory module may be promoted by the co-occurrence of each factor's recognition motif.
To understand the interplay of direct (protein-DNA mediated) and indirect (protein-protein mediated) Sox9 and AP-1 engagement on transcription of target genes, we generated a series of luciferase reporter constructs combining different enhancer-motif configurations with a basal promoter element: 4x48-Col2a1 (Fig. 4A; four copies of a 48 bp enhancer from Col2a1 intron 1 that contains multiple Sox9 binding motifs but no AP-1 motifs; Kan et al., 2009); 3xSox9BS(Col9a1) (Fig. 4B; three copies of a 17-nucleotide Sox9 dimer-binding motif from the Col9a1 enhancer; Ohba et al., 2015); 7xAP-1BS (Fig. 4C; seven copies of the optimal AP-1 motif); and 3xSox9BS(Col9a1)+7xAP-1BS (Fig. 4D; an artificial enhancer element combining Sox9 and AP-1 regulatory modules above).
Fig. 4.

Functional interactions between AP-1 factors and Sox9. (A-D) Reporter assay in HEK293T cells examining activation of a basal reporter luciferase construct containing four copies of a 48 bp Col2a1 intron 1 enhancer module (4x48-Col2a1) (A), three copies of a 17-nucleotide sequence containing a Sox9-binding site in a Col9a1 enhancer [3xSox9BS(Col9a1)] (B), seven copies of the AP-1 binding motif (7xAP-1BS) (C), and a module with both the 3xSox9BS(Col9a1) and 7xAP-1BS sequence (D). y-axis indicates relative light unit (RLU) values normalizing firefly luciferase to an internal Renilla luciferase control. *P <0.05; ns, non-significant. Error bars represent s.d.
As expected, Sox9 activated the 4x48-Col2a1 and 3xSox9BS(Col9a1) reporters (Fig. 4A,B). However, Sox9-mediated activation was reduced on co-transfection with Jun and Fosl2 (Fig. 4A,B). By contrast, TAM67, a dominant-negative variant of Jun that retains DNA binding but has no transcriptional activation capability, specifically suppressed endogenous AP-1 function (Brown et al., 1993), but enhanced Sox9-mediated activation of the reporter, presumably reflecting TAM67 inhibition of endogenous AP-1 complexes (Fig. 4A,B; Fig. S4D). As expected, Jun alone, or together with Fosl2, promoted activity of the 7xAP-1BS reporter; however, their activity was suppressed by Sox9 (Fig. 4C). By contrast, maximal activation of the 3xSox9BS(Col9a1)+7xAP-1BS reporter required both Sox9 and AP-1 family members and reporter activity was reduced by co-transfection with TAM67 (Fig. 4D). Together, these data suggest that protein-protein association between Sox9 and AP-1 family members reduced the transactivation activity of either complex evident in the response of target enhancers where direct binding sites are only present for one Sox9 or AP-1 factor. However, an additive transcriptional activity occurs when the DNA target contains binding sites for both Sox9 and AP-1. Consequently, the overall effect of AP-1 members on Sox9 regulation of chondrocyte targets is likely to be complex, reflecting the distribution of each factor's direct binding sites within the regulatory genome and non-DNA-dependent co-interaction.
Sox9 and AP-1 positively regulate chondrocyte hypertrophy
To understand the functional significance of the interplay between Sox9 and AP-1 in chondrocyte differentiation, we utilized a drug, SR11302, that inhibits AP-1-mediated transcriptional activation (Fanjul et al., 1994). We confirmed first that SR11302 inhibited Jun-mediated activation of its target in a dose-dependent manner (Fig. S4E). Next, we analyzed the effects of SR11302 on the expression of a Col10a1 enhancer-driven mCherry reporter (Col10a1:mCherry) (Maye et al., 2011) in primary mouse transgenic chondrocytes. Col10a1 is expressed in conjunction with Jun and Fosl2 activation at the hypertrophic chondrocyte transition (Fig. 1). SR11302 downregulated Col10a1:mCherry reporter activity (Fig. 5A,B) and the endogenous levels of Col10a1 mRNA (Fig. 5C). In addition, SR11302 inhibited normal activation of an endogenous Col10a1:mCherry transgenic reporter in intact femur explants, reducing the resulting hypertrophic zone and restricting ossification (Chung et al., 2001) (Fig. 5D). Whereas the levels of Col2a1 (predominantly but not exclusively expressed in proliferating immature chondrocytes), Sox9, and a number of genes displaying similar expression levels within distinct chondrocyte cell types (Actb, Epyc and Ndufv2) were not significantly changed on SR11302 treatment, expression of prehypertrophic chondrocyte-enriched genes (Sp7, Mef2c and Ihh) was elevated (Fig. 5C; Fig. S4F). Collectively, these data support a model wherein AP-1 family engagement promotes a progressive hypertrophic chondrocyte program.
Fig. 5.

AP-1 induces chondrocyte hypertrophy. (A,B) Col10a1:mCherry transgene expression in chondrocytes cultured with the AP-1 inhibitor SR11302. Primary rib chondrocytes isolated from Col10a1:mCherry transgenic mice were cultured for 10 days with SR11302 at the indicated concentrations in the presence of an insulin-transferrin-selenium (ITS) supplement with phosphate to induce hypertrophy. mCherry from the Col10a1:mCherry transgene indicates chondrocyte hypertrophy. Representative pictures of mCherry signal and quantitative data for mCherry-positive cells (biological triplicates) are shown in A and B, respectively. *P<0.05. (C) Expression of chondrocyte differentiation marker genes in the culture shown in A. Relative mRNA expression levels are determined by reverse transcription-quantitative polymerase chain reaction (RT-qPCR). *P<0.05. (D) Chondrocyte hypertrophy in femurs cultured with SR11302. Femurs isolated from E12.5 Col10a1:mCherry transgenic mice were cultured with 1 μM SR11302 for 5 days. mCherry highlights the hypertrophic chondrocyte domain, brackets indicate distance from articular surface to onset of mCherry-expressing hypertrophic domain. Arrows indicate the Col10a1:mCherry-expressing hypertrophic domain. Asterisk indicates the ossification domain. (E) mRNA expression of chondrocyte differentiation markers in cultured chondrocytes overexpressing Fosl2 and Jun or TAM67. Primary mouse rib chondrocytes were infected with virus overexpressing Fosl2-P2A-Jun, TAM67 or GFP control and cultured for 3 days. Triplicate analysis of mRNA expression for the indicated genes analyzed by RT-qPCR. Primers for TAM67 detected not only TAM67 but also full-length Jun, which enabled assessment of the ectopic expression of each factor. Col2a1 and Sox9 are broadly active in mitotic chondrocytes; Sp7, Mef2c and Ihh in prehypertrophic chondrocytes; and Col10a1 and Mmp13 in hypertrophic chondrocytes. *P<0.05 compared with the GFP control group. (F) A Tol2 transposase construct was used to introduce transgenes to ectopically express AP-1 factors (Fosl2-P2A-Jun transgene) or TAM67 under the control of mouse Col2a1 intron 6 enhancer (Ohba et al., 2015), which has been shown to direct robust transgene expression within zebrafish chondrocytes during skeletal development (Ohba et al., 2015). (G,H) Altered endochondral ossification in the pharyngeal arch of the 7-day zebrafish larva following overexpression of Fosl2-P2A-Jun and TAM67. The expression level of introduced genes was examined by RT-qPCR (G). Representative images of Alcian Blue/Alizarin Red staining are shown in H. Arrowheads, mineralization in palatoquadrate; arrows, mineralization in ceratohyal arch; asterisk, mineralization in hyosymplectic cartilage. Inset in each panel of H shows a representative Alizarin signal within the ceratohyal arch. (I) The histochemical signal in H was quantified using ImageJ (n=7 for each treatment). *P<0.05 comparing transgenic with stage-matched control groups. Error bars represent s.d.
To test this model further, we co-expressed Fosl2 and Jun, or expressed TAM67 or a GFP control, in neonatal mouse primary chondrocytes. Ectopic expression of Fosl2 and Jun downregulated expression of markers of proliferative and prehypertrophic chondrocytes (Col2a1, Sox9, Sp7, Mef2c and Ihh), but upregulated Col10a1, an early indicator of hypertrophic development (Fig. 5E); expression of Mmp13, a marker of the most mature mineralizing hypertrophic chondrocytes that do not show either Jun or Fosl2 activity (Fig. 1A,A′,B,B′,H), was unaltered on the ectopic expression of Fosl2 and Jun (Fig. 5E). Ectopic expression of TAM67 exerted opposite effects on expression of these genes to those invoked by Fosl2 and Jun (Fig. 5E). Next, we examined the effect of ectopic expression of Fosl2 and Jun on the developing zebrafish skeleton using Tol2 transposase for transposon-mediated transgenesis in the zebrafish embryo (Fig. 5F). Ectopic expression resulted in premature mineralization of endochondral cartilage elements (Fig. 5G-I). By contrast, mineralization was delayed on ectopic expression of TAM67 (Fig. 5G-I). Together, these data support findings from earlier gain-of-function and dominant-negative approaches in vitro and in vivo.
To examine Sox9 function in the hypertrophic program directly, we used an Sp7 (also known as osterix)-driven CRE transgenic mouse strain (Rodda and McMahon, 2006) to remove Sox9 activity specifically at the prehypertrophic transition, contrasting with other studies that have removed Sox9 from the columnar chondrocytes (Dy et al., 2012; Ikegami et al., 2011). Prehypertrophic chondrocyte-specific removal of Sox9 led to an expansion of the prehypertrophic chondrocyte zone (Fig. S5A,B) and pronounced loss of Col10a1-expressing hypertrophic chondrocytes (Fig. S5C,D; see also S5C′,D′ for higher magnification), providing evidence in support of Sox9 action in promoting progressive development of hypertrophic chondrocytes. In this, Col10a1 appears to be one likely shared target of Sox9 and Fosl2/Jun regulation from our primary ChIP-seq data (Fig. 2F) with strong binding to an in vivo-verified hypertrophic enhancer (Fig. 6A), transfection studies examining transcriptional responses in transfected primary rib chondrocytes (Fig. 6B), and data reviewed earlier (Gebhard et al., 2004; Leung et al., 2011; Li et al., 2011; Riemer et al., 2002).
Fig. 6.

Sox9 and AP-1 cooperatively induce the expression of Col10a1. (A) Screenshot of Col10a1 genomic locus showing a conserved block (black trace) enriched for Sox9 (red) and Jun (blue) ChIP-seq signals and an H3K4Me2 enhancer mark (purple) compared with input controls (brown). Conservation plot indicates phastcons value (conservation among 30 vertebrate species). (B) The effect on Col10a1 expression in primary rib chondrocyte culture following viral overexpression of Sox9 with either Fosl2-P2A-Jun or TAM67. mRNA expression values were determined by RT-qPCR 3 days after viral infection. y-axis indicates relative expression value over Gapdh value, normalized to the control groups infected with GFP-expressing viruses. *P<0.05. (C) Reporter assay analysis in HEK293T cells of luciferase constructs carrying the conserved block of a Col10a1 enhancer containing seven predicted AP-1 binding motifs and two Sox9 binding motifs. Wild-type enhancer construct (pGL-Col10WT-enh) and constructs with mutations to all AP-1 binding sites (pGL-Col10AM-enh) or all Sox9 binding sites (pGL-Col10SM-enh) were transfected with Sox9 and/or Jun and Fosl2 expression constructs. Relative expression of firefly luciferase was normalized to a Renilla luciferase internal control. *P<0.05. See also Fig. S6. Error bars represent s.d.
To examine this possibility further, we tested the effects of Sox9 or Jun on reporter gene expression driven from the Sox9/Jun-bound Col10a1 enhancer (Fig. S6). Individually, each factor upregulated reporter activity, and reporter activity was further enhanced when both Sox9 and Jun were transfected together (Fig. 6C, pGL-Col10WT-enh). Consistent with the direct DNA-dependent interaction of each factor, Sox9 and AP-1 were no longer able to activate the reporter when their predicted binding sites were mutated (Fig. 6C, pGL-Col10SM-enh and pGL-Col10AM-enh). Collectively, these data suggest that Col10a1 is one key, direct target upregulated by Sox9 and AP-1 in hypertrophic chondrocyte development.
DISCUSSION
Our data argue for a complex interplay between Sox9 and at least two AP-1 family members, Jun and Fosl2, in chondrocyte development. Whereas Jun binds a large array of DNA regions independently of Sox9, the strongest Jun-bound regions closely overlap with the large majority of Sox9 DNA targets in developing rib chondrocytes. Two distinct modes of Sox9 and AP-1 factor co-engagement were observed with distinct transcriptional consequences. Sox9 can form protein-protein complexes with Jun and Fosl2. At enhancers where direct binding of only Sox9 or AP-1 complexes is possible, protein-protein interactions attenuate the direct binding factor's transcriptional activity. By contrast, when Sox9 and AP-1 engage together through their cognate DNA-target sequence, their combined actions elevate expression above levels induced by either factor alone. Given our recent findings (Ohba et al., 2015) and those of other groups (Bhandari et al., 2012; Liu and Lefebvre, 2015; Oh et al., 2014, 2010) that the transcription of Sox9-specific chondrocyte targets is governed by integrating information from multiple independent enhancer elements, the overall impact of AP-1 factor activation on any given Sox9/AP-1 target gene is not easy to predict as it will reflect the combined action of multiple binding activities within any given enhancer and the integration of information from multiple enhancers; the inhibitory action of protein-protein interactions could function to fine-tune expression of target genes. However, the balance of the evidence herein suggests that the combined action of AP-1 factor input promotes a progressive hypertrophic chondrocyte program with Sox9. In addition, AP-1 family members other that Jun and Fosl2 may be expressed in proliferating chondrocytes and participate in Sox9-mediated gene regulation at the mitotic phase, although this study has not addressed this possibility. Extensive studies have linked AP-1 family action to bone formation, bone modeling, joint formation and intervertebral disc development (see Introduction). However, there is only limited prior evidence suggesting AP-1 action in chondrogenesis. Ectopic expression of Jun and JunD in vitro in chicken sternum chondrocytes has been reported to inhibit hypertrophic gene expression (Kameda et al., 1997). By contrast, in vivo Fosl2 removal from mammalian chondrocytes is reported to reduce the hypertrophic chondrocyte zone and associated calcified matrix, consistent with a positive role in hypertrophic chondrocyte development (Karreth et al., 2004).
Though Jun mutants do display joint phenotypes we saw no obvious phenotype in the remainder of the neonatal skeleton (Kan and Tabin, 2013). By contrast, small molecule-mediated AP-1 factor inhibition provided evidence in favor of an AP-1 input enhancing hypertrophic development. However, the precise action of this small molecule inhibitor is unclear, as is the extent of AP-1 family member inhibition. RNA-seq analysis indicates multiple AP-1 family members are expressed in hypertrophic chondrocytes in addition to Jun and Fosl2 as studied here. The potential for significant redundancy among family members precludes establishing a clear genetic resolution to the full potential of AP-1 family regulatory action in chondrocytes.
In contrast to AP-1 factors, Sox9 has been extensively studied in early chondrogenenesis. Sox9 is absolutely required for specification of mitotic chondrocytes (reviewed by Lefebvre, 2002). However, genetic studies have been less clear on the role of Sox9 in later, post-mitotic, hypertrophic chondrocyte development. Early deletion of Sox9 from Prrx1- or Col2a1-expressing cells blocks initiation or ongoing chondrogenesis, respectively (Akiyama et al., 2002). Further, spatial and temporal refinement of Sox9 removal from chondrocytes has supported a continuing role in promoting the hypertrophic chondrocyte transition (Dy et al., 2012; Ikegami et al., 2011).
Interestingly, our data show that Sox9 nuclear levels are highest at the prehypertrophic boundary precisely where Jun and Fosl2 expression initiate. Further, Sox9 and AP-1 family members co-bind each other's cis-regulatory regions, suggesting that cross- and auto-regulatory interactions might contribute to their observed patterns of gene activity. Removing Sox9 activity at the point of the prehypertrophic transition in vivo demonstrated that Sox9 is essential for the cellular progression to hypertrophic chondrocytes, consistent with the published studies discussed above. Further, drug-mediated attenuation of AP-1 factor activity blocked hypertrophic chondrocyte development, suggesting that both Sox9 and AP-1 have positive roles in the hypertrophic component of the endochondral skeletal program. One prominent co-regulated target in our study is Col10a1: Col10a1 activity in primary chondrocytes in vitro depends directly on Sox9 and AP-1 factor engagement in agreement with transfection studies of AP-1 factors in cell culture (Gebhard et al., 2004) but contrasting with transgenic studies that show that loss of a single Sox9-binding site within the context of a Col10a1 enhancer element results in premature activation of reporter in proliferating chondrocytes (Leung et al., 2011). The differences between these studies most likely reflect the limitation of analysis of single binding events in single enhancer elements where the in vivo picture is multiple binding events in multiple enhancer elements.
Previous studies have argued that Gli factors facilitate Sox9 inhibition of Col10a1 expression in proliferating chondrocytes (Leung et al., 2011). Interestingly, hedgehog responsiveness, as indicated by upregulation of Ptch1 and Gli1 targets, is lost at the prehypertrophic chondrocyte boundary (St-Jacques et al., 1999). Thus, the hypertrophic transition may act as a regulatory switch point for Sox9 control of hypertrophic target gene activity removing negative interactions (Gli), and activating de novo AP-1 factors to promote hypertrophic gene activity with Sox9.
Parathyroid hormone-like hormone (Pthlh; also known as Pthrp) signaling through the Pthlh receptor is a key inhibitor of the hypertrophic chondrocyte program (reviewed by Kronenberg, 2003). Pthlh is reported to induce AP-1 factors and enhance their DNA-binding activity in vitro, and dominant-negative forms of Fos have been reported to abolish Pthlh-mediated suppression of chondrocyte maturation in culture (Ionescu et al., 2001). In addition, Pthlh specifically activates Sox9 by phosphorylating Sox9 at serine 181 through protein kinase A exclusively in prehypertrophic chondrocytes, although the role of phosphorylated Sox9 has not been well studied (Huang et al., 2001).
Collectively, our in vivo and in vitro studies provide several lines of evidence supporting a direct ongoing role for Sox9 in the early stages of the hypertrophic program. Further, the data suggest that the actions of Sox9 in promoting the hypertrophic phase are augmented by the co-regulatory input of AP-1 family members at enhancer modules, a regulatory mode initiated on chondrocyte commitment to the hypertrophic program.
These studies suggest that the cell type-specific actions of Sox9 and AP-1 family members are crucially linked to the timing of chondrocyte development. Our Sox9 ChIP-seq analysis of primary chondrocytes reflects a mixed population of chondrocytes at different stages of chondrogenesis. Consequently, the exact timing of Sox9 action on a given enhancer is unclear and will require a significant technological improvement to be able to address in vivo given the small number of cells present within any skeletal element. However, the close concordance of Jun engagement at DNA targets with Sox9 and prehypertrophic-specific expression of Jun suggest that Jun interactions follow those of Sox9. Interestingly, Jun binds to many more genomic regions than does Sox9, but these ‘Jun-only’-associated regions do not display an active enhancer signature.
In this view, Sox9 likely provides the chondrocyte-context to a Jun regulatory input in chondrogenesis. Jun binds to a broad array of enhancers, many of which, such as those regulating Col2a1, were active under Sox9 control at an earlier stage (Ohba et al., 2015), whereas others, such as Col10a1, switch on in hypertrophic cells. As most of the early-activated enhancers remain active in the early phase of hypertrophic chondrocyte development, Jun and Fosl2 complexes formed at the hypertrophic transition could provide an additional layer of regulatory input into these early-initiating enhancers while providing de novo activating input into Col10a1 enhancers.
Although most of the Jun-Sox9 shared peak regions can be accounted for by a direct interaction of at least one of these factors, a small number (16%) of them show no readily identifiable Sox9 or AP-1 motif. This may reflect degeneracy of target sites. Alternatively, these data may point to additional interactions with other transcriptional regulators. In line with this, we recovered additional motifs within the Jun ChIP-seq dataset that might reflect an indirect association of Jun to the chondrocyte genome through engagement with other transcriptional regulatory factors. Of note, our analysis of Sox9-binding sites recovered significant enrichment for Runx-, Nfat-, Ets- and AT-rich homeodomain protein-binding motifs (Ohba et al., 2015). Of these possible co-operating factors, Runx2 is known to play an essential role in hypertrophic chondrocyte development and is normally targeted by Jun (Ding et al., 2012; Li et al., 2011; Papachristou et al., 2005; Yoshida et al., 2004) and Runx2 is reported to upregulate both Col10a1 and Sox9 in the Col10a1 expression domain (Ding et al., 2012), suggesting an interacting, positive regulatory loop. However, counter evidence suggests Sox9 upregulates Bapx1 (Nkx3-2), a negative regulator of Runx2 (Yamashita et al., 2009). Further, protein-protein interactions have been documented between Sox9 and Runx2 that are reported to attenuate Sox9-mediated transcriptional activation of target genes, suggesting a complex regulatory interplay (Zhou et al., 2006). Future studies developing strategies to incorporate Runx2, Bapx1, Mef2c and other transcriptional regulators of the hypertrophic program will help to decipher the regulatory logic of this key developmental event in mammalian skeletogenesis.
MATERIALS AND METHODS
Ethics statement
All experiments in this study were carried out in strict accordance with the recommendation in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health, USA. The procedures were approved by the Institutional Animal Care and Use Committee of The University of Southern California (IACUC # 11892).
In situ hybridization
Cryosections of postnatal day 1 (P1) Swiss Webster mouse skeletal samples were subjected to in situ hybridization with digoxigenin-labeled riboprobes. See supplementary Materials and Methods for further details of sample preparation and riboprobes.
Chromatin immunoprecipitation (ChIP), ChIP-seq library construction, and reChIP
ChIP was performed on P1 mouse rib chondrocytes as previously described (Ohba et al., 2015), using specific antibodies against Jun. ChIP-seq libraries were constructed using ThruPLEX DNA-seq Kit (Rubicon Genomics, R400429). See supplementary Materials and Methods for further details of chromatin preparation, antibodies, and library construction.
ReChIP was performed by sequential precipitations of chromatin using specific antibodies against Sox9 and Jun. DNA was analyzed by conventional polymerase chain reaction (PCR) and quantitative PCR (qPCR). See supplementary Materials and Methods for further details of the reChIP procedure, antibodies, and PCR primers.
ChIP-seq data analysis
Sequencing was performed in the Epigenome Center of the University of Southern California. Data were analyzed as previously described (Ohba et al., 2015); alignment of sequence reads, peak calling, de novo motif analysis, and gene ontology analysis of genes associated with obtained peaks were performed using ELAND, CisGenome software (Ji et al., 2008; Jiang et al., 2010), MEME-ChIP (Bailey et al., 2009), and GREAT (McLean et al., 2010) and DAVID (Huang da et al., 2009), respectively. See supplementary Materials and Methods for further details of these analyses.
Expression profiling
Ribs were digested with collagenase D from P1 Col10a1:mCherry transgenic mice. Fluorescence-activated cell sorting was performed to isolate RNA from 100,000 mCherry-positive chondrocytes. Sequencing was performed as mentioned above. Sequence reads were mapped with TopHat2 and quantified with Partek software. See supplementary Materials and Methods for further details of RNA isolation and sequencing library construction.
Plasmid construction
Plasmids expressing genes of interest were constructed by cloning of cDNA into mammalian expression vectors; cDNA was obtained by a PCR-based strategy. Luciferase reporter constructs were generated by cloning of annealed oligonucleotides into luciferase reporter vectors. Retroviral vectors were based on the pMX vector platform, and viral particles were produced in plat-E cells. See supplementary Materials and Methods for further details of cloning procedures.
Co-immunoprecipitation (co-IP), SDS-PAGE and western blotting
Nuclear extracts of HEK293T cells transfected with plasmids were subjected to co-IP, followed by SDS-PAGE of precipitated materials and western blotting. See supplementary Materials and Methods for further details of nuclear extract preparation, co-IP reaction, immunostaining of blotted membranes, and image acquisition.
Mouse primary chondrocyte culture
Chondrocytes were obtained from ribs of P1 Swiss Webster mice or Col10a1:mCherry transgenic mice by enzymatic digestion and subjected to cultures. mRNA expression was analyzed by reverse transcription (RT)-qPCR. See supplementary Materials and Methods for further details of cell isolation, cultures and mRNA expression analysis.
Mouse femur explant culture
Femurs were obtained from embryonic day (E) 12.5 Col10a1:mCherry transgenic mice and cultured with 1 μM of SR11302 (R&D Systems, 2476) or in DMSO in α-MEM/10% fetal bovine serum with antibiotics (penicillin-streptomycin; Sigma-Aldrich, P4333). Culture medium was changed daily with fresh SR11302 or DMSO.
Zebrafish assays
For zebrafish overexpression, Fosl2-P2A-Jun or TAM67 cDNAs, and E1 Col2a1 enhancer-reporter constructs were cloned into a Tol2 construct containing a zebrafish gata2 basal promoter (Kwan et al., 2007). Forty nanograms of each overexpression construct was injected together with 50 ng of Tol2 transposase into 1-cell-stage fertilized zebrafish embryos. The skeletal pattern was visualized by Alcian Blue and Alizarin Red staining of developing zebrafish (Walker and Kimmel, 2007). The extent of mineralization was quantified by measuring the red (Alizarin Red) pixels using ImageJ analysis of skeletal images.
Luciferase reporter assays
Luciferase and effector constructs were transfected into HEK293T cells with FuGENE-6 transfection reagent (Promega, E2691). After 48 h, luciferase activities were measured with the Dual-Glo Luciferase Reporter Assay System (Promega, E2920). For transfection of cells cultured on 96-well culture plates, 100 ng of total DNA was mixed with 0.3 μl FuGENE-6 reagent in DMEM per well. In the DNA mix, 5 ng of a thymidine kinase promoter driving a Renilla control construct was mixed with 10 ng of the experimental firefly luciferase construct. Up to 5 ng of Sox9-expressing construct and up to equal amounts of Fosl2 construct and Jun construct were used as effectors (up to 70 ng combined). A GFP construct was added to make up each DNA transfection to 100 ng DNA per well.
Immunostaining
Cryosections of E16.5 Sp7Cre;Sox9+/+ or Sp7Cre;Sox9flox/flox limbs were subjected to immunostaining using specific antibodies, in combination with secondary antibodies. See supplementary Materials and Methods for further details of sample preparation, antibodies and staining procedures.
Acknowledgements
We thank Jill McMahon, Gohar Saribekyan, Lora Barsky, Bernadette Masinsin, Seth Ruffins, Riana Parvez and Charles Meyer Nicolet from the USC Histology Core, the USC Flow Cytometry Core at the USC Imaging Center, and the USC Epigenome Center for providing technical assistance. We thank Dr Anton Valouev for his bioinformatics analysis assistance and Drs Toshiyuki Ikeda and Taku Saito for experimental materials. We are also grateful to Dr Gage Crump and Crump laboratory members for help with zebrafish studies, and Drs Henry M. Kronenberg, Clifford J. Tabin and Ung-il Chung for their helpful inputs.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
All authors planned experiments, analyzed the data and contributed to the writing of the manuscript. ChIP-seq, expression profiling data analysis, in vitro and in vivo validation of transcription regulation, zebrafish studies and immunofluorescence expression assays were performed by X.H. ChIP-seq, protein-protein interaction studies and immunofluorescence experiments were performed by S.O. H.H. assisted in data analysis.
Funding
This work was supported by a grant from the National Institutes of Health [DK056246 to A.P.M.]; Grant-in-Aid for Scientific Research [23689079, 26713054, 15K15732, 24240069 and 26221311] from the Japan Society for the Promotion of Science Core-to-Core Program A. Advanced Research Networks; a Takeda Science Foundation Research Grant; the University of Tokyo Graduate Program for Leaders in Life Innovation. Deposited in PMC for release after 12 months.
Data availability
ChIP-seq and RNA-seq data are available in NCBI Gene Expression Omnibus (GEO) under accession number GSE73372 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE73372).
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.134502.supplemental
References
- Akiyama H. and Lefebvre V. (2011). Unraveling the transcriptional regulatory machinery in chondrogenesis. J. Bone Miner. Metab. 29, 390-395. 10.1007/s00774-011-0273-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama H., Chaboissier M.-C., Martin J. F., Schedl A. and de Crombrugghe B. (2002). The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev. 16, 2813-2828. 10.1101/gad.1017802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama H., Kim J.-E., Nakashima K., Balmes G., Iwai N., Deng J. M., Zhang Z., Martin J. F., Behringer R. R., Nakamura T. et al. (2005). Osteo-chondroprogenitor cells are derived from Sox9 expressing precursors. Proc. Natl. Acad. Sci. USA 102, 14665-14670. 10.1073/pnas.0504750102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey T. L., Boden M., Buske F. A., Frith M., Grant C. E., Clementi L., Ren J., Li W. W. and Noble W. S. (2009). MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37, W202-W208. 10.1093/nar/gkp335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens A., Haigh J., Mechta-Grigoriou F., Nagy A., Yaniv M. and Wagner E. F. (2003). Impaired intervertebral disc formation in the absence of Jun. Development 130, 103-109. 10.1242/dev.00186 [DOI] [PubMed] [Google Scholar]
- Bhandari R. K., Haque M. M. and Skinner M. K. (2012). Global genome analysis of the downstream binding targets of testis determining factor SRY and SOX9. PLoS ONE 7, e43380 10.1371/journal.pone.0043380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi W., Deng J. M., Zhang Z., Behringer R. R. and de Crombrugghe B. (1999). Sox9 is required for cartilage formation. Nat. Genet. 22, 85-89. 10.1038/44573 [DOI] [PubMed] [Google Scholar]
- Bi W., Huang W., Whitworth D. J., Deng J. M., Zhang Z., Behringer R. R. and de Crombrugghe B. (2001). Haploinsufficiency of Sox9 results in defective cartilage primordia and premature skeletal mineralization. Proc. Natl. Acad. Sci. USA 98, 6698-6703. 10.1073/pnas.111092198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown P. H., Alani R., Preis L. H., Szabo E. and Birrer M. J. (1993). Suppression of oncogene-induced transformation by a deletion mutant of c-jun. Oncogene 8, 877-886. [PubMed] [Google Scholar]
- Chambers D., Young D. A., Howard C., Thomas J. T., Boam D. S., Grant M. E., Wallis G. A. and Boot-Handford R. P. (2002). An enhancer complex confers both high-level and cell-specific expression of the human type X collagen gene. FEBS Lett. 531, 505-508. 10.1016/S0014-5793(02)03606-2 [DOI] [PubMed] [Google Scholar]
- Cheng A. and Genever P. G. (2010). SOX9 determines RUNX2 transactivity by directing intracellular degradation. J. Bone Miner. Res. 25, 2680-2689. 10.1002/jbmr.174 [DOI] [PubMed] [Google Scholar]
- Chung U.-I., Schipani E., McMahon A. P. and Kronenberg H. M. (2001). Indian hedgehog couples chondrogenesis to osteogenesis in endochondral bone development. J. Clin. Invest. 107, 295-304. 10.1172/JCI11706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding M., Lu Y., Abbassi S., Li F., Li X., Song Y., Geoffroy V., Im H.-J. and Zheng Q. (2012). Targeting Runx2 expression in hypertrophic chondrocytes impairs endochondral ossification during early skeletal development. J. Cell. Physiol. 227, 3446-3456. 10.1002/jcp.24045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dy P., Wang W., Bhattaram P., Wang Q., Wang L., Ballock R. T. and Lefebvre V. (2012). Sox9 directs hypertrophic maturation and blocks osteoblast differentiation of growth plate chondrocytes. Dev. Cell 22, 597-609. 10.1016/j.devcel.2011.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eferl R. and Wagner E. F. (2003). AP-1: a double-edged sword in tumorigenesis. Nat. Rev. Cancer 3, 859-868. 10.1038/nrc1209 [DOI] [PubMed] [Google Scholar]
- Fanjul A., Dawson M. I., Hobbs P. D., Jong L., Cameron J. F., Harlev E., Graupner G., Lu X.-P. and Pfahl M. (1994). A new class of retinoids with selective inhibition of AP-1 inhibits proliferation. Nature 372, 107-111. 10.1038/372107a0 [DOI] [PubMed] [Google Scholar]
- Foster J. W. (1996). Mutations in SOX9 cause both autosomal sex reversal and campomelic dysplasia. Acta Paediatr. Jpn. 38, 405-411. 10.1111/j.1442-200X.1996.tb03515.x [DOI] [PubMed] [Google Scholar]
- Gebhard S., Poschl E., Riemer S., Bauer E., Hattori T., Eberspaecher H., Zhang Z., Lefebvre V., de Crombrugghe B. and von der Mark K. (2004). A highly conserved enhancer in mammalian type X collagen genes drives high levels of tissue-specific expression in hypertrophic cartilage in vitro and in vivo. Matrix Biol. 23, 309-322. 10.1016/j.matbio.2004.05.010 [DOI] [PubMed] [Google Scholar]
- Giordano J., Prior H. M., Bamforth J. S. and Walter M. A. (2001). Genetic study of SOX9 in a case of campomelic dysplasia. Am. J. Med. Genet. 98, 176-181. [DOI] [PubMed] [Google Scholar]
- Hess J., Hartenstein B., Teurich S., Schmidt D., Schorpp-Kistner M. and Angel P. (2003). Defective endochondral ossification in mice with strongly compromised expression of JunB. J. Cell Sci. 116, 4587-4596. 10.1242/jcs.00772 [DOI] [PubMed] [Google Scholar]
- Huang W., Chung U.-I., Kronenberg H. M. and de Crombrugghe B. (2001). The chondrogenic transcription factor Sox9 is a target of signaling by the parathyroid hormone-related peptide in the growth plate of endochondral bones. Proc. Natl. Acad. Sci. USA 98, 160-165. 10.1073/pnas.98.1.160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda T., Kamekura S., Mabuchi A., Kou I., Seki S., Takato T., Nakamura K., Kawaguchi H., Ikegawa S. and Chung U.-I. (2004). The combination of SOX5, SOX6, and SOX9 (the SOX trio) provides signals sufficient for induction of permanent cartilage. Arthritis Rheum. 50, 3561-3573. 10.1002/art.20611 [DOI] [PubMed] [Google Scholar]
- Ikegami D., Akiyama H., Suzuki A., Nakamura T., Nakano T., Yoshikawa H. and Tsumaki N. (2011). Sox9 sustains chondrocyte survival and hypertrophy in part through Pik3ca-Akt pathways. Development 138, 1507-1519. 10.1242/dev.057802 [DOI] [PubMed] [Google Scholar]
- Ionescu A. M., Schwarz E. M., Vinson C., Puzas J. E., Rosier R., Reynolds P. R. and O'Keefe R. J. (2001). PTHrP modulates chondrocyte differentiation through AP-1 and CREB signaling. J. Biol. Chem. 276, 11639-11647. 10.1074/jbc.M006564200 [DOI] [PubMed] [Google Scholar]
- Ji H., Jiang H., Ma W., Johnson D. S., Myers R. M. and Wong W. H. (2008). An integrated software system for analyzing ChIP-chip and ChIP-seq data. Nat. Biotechnol. 26, 1293-1300. 10.1038/nbt.1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H., Wang F., Dyer N. P. and Wong W. H. (2010). CisGenome Browser: a flexible tool for genomic data visualization. Bioinformatics 26, 1781-1782. 10.1093/bioinformatics/btq286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kameda T., Watanabe H. and Iba H. (1997). C-Jun and JunD suppress maturation of chondrocytes. Cell Growth Differ. 8, 495-503. [PubMed] [Google Scholar]
- Kan A. and Tabin C. J. (2013). c-Jun is required for the specification of joint cell fates. Genes Dev. 27, 514-524. 10.1101/gad.209239.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan A., Ikeda T., Saito T., Yano F., Fukai A., Hojo H., Ogasawara T., Ogata N., Nakamura K., Chung U.-I. et al. (2009). Screening of chondrogenic factors with a real-time fluorescence-monitoring cell line ATDC5-C2ER: identification of sorting nexin 19 as a novel factor. Arthritis Rheum. 60, 3314-3323. 10.1002/art.24878 [DOI] [PubMed] [Google Scholar]
- Karreth F., Hoebertz A., Scheuch H., Eferl R. and Wagner E. F. (2004). The AP1 transcription factor Fra2 is required for efficient cartilage development. Development 131, 5717-5725. 10.1242/dev.01414 [DOI] [PubMed] [Google Scholar]
- Kronenberg H. M. (2003). Developmental regulation of the growth plate. Nature 423, 332-336. 10.1038/nature01657 [DOI] [PubMed] [Google Scholar]
- Kwok C., Weller P. A., Guioli S., Foster J. W., Mansour S., Zuffardi O., Punnett H. H., Dominguez-Steglich M. A., Brook J. D., Young I. D. et al. (1995). Mutations in SOX9, the gene responsible for Campomelic dysplasia and autosomal sex reversal. Am. J. Hum. Genet. 57, 1028-1036. [PMC free article] [PubMed] [Google Scholar]
- Lefebvre V. (2002). Toward understanding the functions of the two highly related Sox5 and Sox6 genes. J. Bone Miner. Metab. 20, 121-130. 10.1007/s007740200017 [DOI] [PubMed] [Google Scholar]
- Lefebvre V. and Dvir-Ginzberg M. (2016). SOX9 and the many facets of its regulation in the chondrocyte lineage. Connect. Tissue Res. (in press). 10.1080/03008207.2016.1183667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung V. Y. L., Gao B., Leung K. K. H., Melhado I. G., Wynn S. L., Au T. Y. K., Dung N. W. F., Lau J. Y. B., Mak A. C. Y., Chan D. et al. (2011). SOX9 governs differentiation stage-specific gene expression in growth plate chondrocytes via direct concomitant transactivation and repression. PLoS Genet. 7, e1002356 10.1371/journal.pgen.1002356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F., Lu Y., Ding M., Napierala D., Abbassi S., Chen Y., Duan X., Wang S., Lee B. and Zheng Q. (2011). Runx2 contributes to murine Col10a1 gene regulation through direct interaction with its cis-enhancer. J. Bone Miner. Res. 26, 2899-2910. 10.1002/jbmr.504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C.-F. and Lefebvre V. (2015). The transcription factors SOX9 and SOX5/SOX6 cooperate genome-wide through super-enhancers to drive chondrogenesis. Nucleic Acids Res. 43, 8183-8203. 10.1093/nar/gkv688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maye P., Fu Y., Butler D. L., Chokalingam K., Liu Y., Floret J., Stover M. L., Wenstrup R., Jiang X., Gooch C. et al. (2011). Generation and characterization of Col10a1-mcherry reporter mice. Genesis 49, 410-418. 10.1002/dvg.20733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean C. Y., Bristor D., Hiller M., Clarke S. L., Schaar B. T., Lowe C. B., Wenger A. M. and Bejerano G. (2010). GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 28, 495-501. 10.1038/nbt.1630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori-Akiyama Y., Akiyama H., Rowitch D. H. and de Crombrugghe B. (2003). Sox9 is required for determination of the chondrogenic cell lineage in the cranial neural crest. Proc. Natl. Acad. Sci. USA 100, 9360-9365. 10.1073/pnas.1631288100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh C.-D., Maity S. N., Lu J.-F., Zhang J., Liang S., Coustry F., de Crombrugghe B. and Yasuda H. (2010). Identification of SOX9 interaction sites in the genome of chondrocytes. PLoS ONE 5, e10113 10.1371/journal.pone.0010113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh C.-D., Lu Y., Liang S., Mori-Akiyama Y., Chen D., de Crombrugghe B. and Yasuda H. (2014). SOX9 regulates multiple genes in chondrocytes, including genes encoding ECM proteins, ECM modification enzymes, receptors, and transporters. PLoS ONE 9, e107577 10.1371/journal.pone.0107577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohba S., He X., Hojo H. and McMahon A. P. (2015). Distinct transcriptional programs underlie Sox9 regulation of the mammalian chondrocyte. Cell Rep. 12, 229-243. 10.1016/j.celrep.2015.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Outani H., Okada M., Yamashita A., Nakagawa K., Yoshikawa H. and Tsumaki N. (2013). Direct induction of chondrogenic cells from human dermal fibroblast culture by defined factors. PLoS ONE 8, e77365 10.1371/journal.pone.0077365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papachristou D. J., Papachristou G. I., Papaefthimiou O. A., Agnantis N. J., Basdra E. K. and Papavassiliou A. G. (2005). The MAPK-AP-1/-Runx2 signalling axes are implicated in chondrosarcoma pathobiology either independently or via up-regulation of VEGF. Histopathology 47, 565-574. 10.1111/j.1365-2559.2005.02266.x [DOI] [PubMed] [Google Scholar]
- Riemer S., Gebhard S., Beier F., Poschl E. and von der Mark K. (2002). Role of c-fos in the regulation of type X collagen gene expression by PTH and PTHrP: localization of a PTH/PTHrP-responsive region in the human COL10A1 enhancer. J. Cell Biochem. 86, 688-699. 10.1002/jcb.10260 [DOI] [PubMed] [Google Scholar]
- Rodda S. J. and McMahon A. P. (2006). Distinct roles for Hedgehog and canonical Wnt signaling in specification, differentiation and maintenance of osteoblast progenitors. Development 133, 3231-3244. 10.1242/dev.02480 [DOI] [PubMed] [Google Scholar]
- St-Jacques B., Hammerschmidt M. and McMahon A. P. (1999). Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 13, 2072-2086. 10.1101/gad.13.16.2072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner E. F. (2002). Functions of AP1 (Fos/Jun) in bone development. Ann. Rheum. Dis. 61 Suppl. 2, ii40-ii42. 10.1136/ard.61.suppl_2.ii40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner E. F. and Eferl R. (2005). Fos/AP-1 proteins in bone and the immune system. Immunol. Rev. 208, 126-140. 10.1111/j.0105-2896.2005.00332.x [DOI] [PubMed] [Google Scholar]
- Yamashita S., Andoh M., Ueno-Kudoh H., Sato T., Miyaki S. and Asahara H. (2009). Sox9 directly promotes Bapx1 gene expression to repress Runx2 in chondrocytes. Exp. Cell Res. 315, 2231-2240. 10.1016/j.yexcr.2009.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida C. A., Yamamoto H., Fujita T., Furuichi T., Ito K., Inoue K.-I., Yamana K., Zanma A., Takada K., Ito Y. et al. (2004). Runx2 and Runx3 are essential for chondrocyte maturation, and Runx2 regulates limb growth through induction of Indian hedgehog. Genes Dev. 18, 952-963. 10.1101/gad.1174704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Q., Keller B., Zhou G., Napierala D., Chen Y., Zabel B., Parker A. E. and Lee B. (2009). Localization of the cis-enhancer element for mouse type X collagen expression in hypertrophic chondrocytes in vivo. J. Bone Miner. Res. 24, 1022-1032. 10.1359/jbmr.081249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G., Zheng Q., Engin F., Munivez E., Chen Y., Sebald E., Krakow D. and Lee B. (2006). Dominance of SOX9 function over RUNX2 during skeletogenesis. Proc. Natl. Acad. Sci. USA 103, 19004-19009. 10.1073/pnas.0605170103 [DOI] [PMC free article] [PubMed] [Google Scholar]