

ABSTRACT
Leishmaniasis is a devastating disease that disfigures or kills nearly two million people each year. Establishment and persistence of infection by the obligate intracellular parasite Leishmania requires repeated uptake by macrophages and other phagocytes. Therefore, preventing uptake could be a novel therapeutic strategy for leishmaniasis. Amastigotes, the life cycle stage found in the human host, bind Fc receptors and enter macrophages primarily through immunoglobulin-mediated phagocytosis. However, the host machinery that mediates amastigote uptake is poorly understood. We have previously shown that the Arg (also known as Abl2) non-receptor tyrosine kinase facilitates L. amazonensis amastigote uptake by macrophages. Using small-molecule inhibitors and primary macrophages lacking specific Src family kinases, we now demonstrate that the Hck, Fgr and Lyn kinases are also necessary for amastigote uptake by macrophages. Src-mediated Arg activation is required for efficient uptake. Interestingly, the dual Arg and Src kinase inhibitor bosutinib, which is approved to treat cancer, not only decreases amastigote uptake, but also significantly reduces disease severity and parasite burden in Leishmania-infected mice. Our results suggest that leishmaniasis could potentially be treated with host-cell-active agents such as kinase inhibitors.
KEY WORDS: Leishmania, Phagocytosis, Kinase, Macrophage, Src, Abl
Summary: The Src kinases Hck, Fgr and Lyn, and the Abl kinase Arg control Leishmania uptake by macrophages and subsequent infection. Hence, kinase inhibitors could potentially be used to treat leishmaniasis.
INTRODUCTION
The parasite Leishmania causes visceral or cutaneous disease in over a million people every year. Drugs used to treat leishmaniasis have serious side effects, and parasites are developing resistance to them. The Leishmania life cycle has two main stages: promastigotes in sand flies, and amastigotes in the mammalian host. If an infected sandfly injects promastigotes into a host, the promastigotes must be engulfed by phagocytes to establish infection. Leishmania then differentiates within the phagolysosome into the amastigote. If amastigotes are found outside of this acidic compartment, they must be re-engulfed to persist in the host (Kane and Mosser, 2000).
Several macrophage surface protein receptors allow Leishmania uptake. Promastigotes interact with multiple receptors, like the complement receptor CR3 (Russell and Wright, 1988); binding is enhanced by complement component fragment C3bi opsonization mediated by lipophosphoglycan (LPG) (Mosser et al., 1992; Puentes et al., 1988). The FcR subclass FcγR, which is required for IgG-mediated phagocytosis, is primarily responsible for amastigote uptake (Guy and Belosevic, 1993; Kima et al., 2000; Woelbing et al., 2006), and IgG opsonization of amastigotes facilitates these interactions (Morehead et al., 2002). Leishmania receptor binding causes actin-rich phagocytic cups to engulf the parasite (Lodge and Descoteaux, 2008); however, the signaling process directing cup formation is not well understood.
The Abl family kinases Abl and Arg (also known as Abl1 and Abl2, respectively) translate signals from growth factor and adhesion receptors into cytoskeletal rearrangements (Bradley and Koleske, 2009). Receptor engagement stimulates these kinases to bind and phosphorylate Arp2/3 complex activators (Lapetina et al., 2009; Miller et al., 2010), yielding dynamic cell edge protrusions that resemble phagocytic intermediates. Abl and Arg also facilitate endocytosis (Jacob et al., 2009; Tanos and Pendergast, 2006, 2007), autophagy (Yogalingam and Pendergast, 2008), viral (Reeves et al., 2005, 2011; Swimm et al., 2010) and bacterial uptake (Burton et al., 2003; Elwell et al., 2008; Ly and Casanova, 2009; Napier et al., 2011), and IgG-mediated phagocytosis (Greuber and Pendergast, 2012). We have previously reported that Abl and Arg allow complementary non-redundant processes during phagocytosis and Leishmania uptake (Wetzel et al., 2012). Genetic loss of Arg prevents efficient IgG-mediated phagocytosis and amastigote uptake, whereas loss of Abl reduces C3bi-mediated phagocytosis and L. amazonensis promastigote uptake. In addition, by using the Abl and Arg inhibitor imatinib and assessing mice lacking Abl or Arg, we have shown that Abl family kinases mediate infection in murine cutaneous leishmaniasis (Wetzel et al., 2012).
Src family kinases (SFKs) are non-receptor tyrosine kinases regulated by cell surface receptors that play roles in cell morphogenesis. Src and Lyn directly bind the FcR (Wu et al., 2001), and macrophages lacking the SFKs Hck, Lyn and Fgr have substantial defects in IgG-mediated phagocytosis (Fitzer-Attas et al., 2000), and viral (Abram and Lowell, 2008; Bavagnoli et al., 2011; Cheng et al., 2015) and bacterial uptake (Hauck et al., 1998; Paul et al., 2008; Van Langendonck et al., 1998). SFKs phosphorylate and activate Arg (Mader et al., 2011; Plattner et al., 2004; Tanis et al., 2003), and this can be amplified by Arg autophosphorylation on a distinct regulatory site (Bradley and Koleske, 2009). However, whether and how SFKs facilitate the uptake of Leishmania is not clear. Of note, if SFKs and Arg both were to mediate amastigote uptake, either within the same pathway, or in different pathways, combining Arg and SFK inhibitors might show increased efficacy over Arg and Abl inhibitors for disrupting the disease course of leishmaniasis.
Here, we provide evidence that host SFKs activate Arg to facilitate immunoglobulin-mediated phagocytosis and L. amazonensis amastigote uptake. Using kinase inhibitors and macrophages lacking specific SFKs, we show that Hck, Fgr and Lyn also mediate efficient amastigote uptake. SFKs signal through Arg to facilitate this process. Finally, the combination Arg and SFK inhibitor bosutinib not only reduces amastigote uptake by macrophages but also significantly ameliorates disease severity in Leishmania-infected mice. These results suggest that leishmaniasis could be treated with drugs that inhibit these kinases or other host cell processes.
RESULTS
SFKs are required for efficient amastigote but not promastigote uptake
We and others have demonstrated that IgG-opsonized amastigote uptake occurs primarily through an FcRγ-mediated process, whereas C3bi-opsonized promastigote uptake occurs primarily through a CR3-mediated process (Carter et al., 2009; Kima et al., 2000; Mosser and Edelson, 1985; Russell and Wright, 1988; Ueno and Wilson, 2012). SFKs are known to facilitate immunoglobulin-mediated phagocytosis. To test whether they might mediate Leishmania uptake, we examined whether the SFK inhibitor SU6656 (which has an IC50 of 20–700 nM, depending on the specific SFK; Blake et al., 2000) affected the uptake of L. amazonensis promastigotes or amastigotes. We used two-color immunofluorescence (Wetzel et al., 2003) to distinguish adherent from internalized parasites and measured the phagocytic index (number of particles internalized per 100 cells) in the presence of SU6656 or DMSO. We found that SU6656 inhibited IgG-opsonized bead phagocytosis by bone-marrow-derived macrophages (BMDMs) (Fig. S1A) with an approximate IC50 of 2.5 μM; its CC50 (cytotoxic concentration necessary to cause death to 50% of viable cells) for BMDMs over the same incubation period was above our highest concentration of 10 μM. Treating BMDM with 2.5 μM SU6656 decreased the phagocytic index for IgG-opsonized beads but not C3bi-opsonized beads (Fig. 1A). 2.5 μM SU6656 also decreased the phagocytic index for IgG-opsonized amastigotes by 40±7% (mean±.s.e.m.) relative to controls, but did not affect the uptake of C3bi-opsonized promastigotes (Fig. 1B,C). Amastigotes bound to SU6656-treated BMDMs at levels indistinguishable from controls (Fig. 1D), indicating that decreased invasion did not simply result from reduced adhesion. Similar results were found when the murine macrophage-like cell line RAW 264.7 was used instead of BMDMs (Fig. 1E; Fig. S1B). Internalization defects were observed even over incubations of up to 2 h, demonstrating that treated BMDMs did not overcome defects in phagocytosis even if a substantial amount of time elapsed (Fig. S1C). Treating macrophages with another SFK inhibitor, PP2 [IC50 for Src=1.4 μM (Blake et al., 1999); CC50 is substantially above 20 μM (Beausejour et al., 2012)], also caused a defect in amastigote uptake (Fig. 1F).
Fig. 1.

SFKs are required for optimal IgG-mediated phagocytosis and amastigote uptake. Macrophages were treated with 2.5 μM SU6656 or DMSO for 2 h and incubated with C3bi- or IgG-coated beads (A) or L. amazonensis C3bi-coated promastigotes or IgG-coated amastigotes (B–F) for 30 min. Two-color immunofluorescence distinguished between intracellular (green) and extracellular (orange) beads or parasites. Nuclei are labeled with DAPI. (A) SU6656 decreases IgG-coated but not C3bi-coated bead uptake by BMDMs. Results are the mean±s.e.m. phagocytic index (PI) for BMDMs treated with 2.5 μM SU6656 normalized to the DMSO-treated phagocytic index (100%) for each experiment. (B) SU6656 decreases amastigote uptake but not promastigote uptake by BMDMs. Graph shows the normalized mean±s.e.m. SU6656-treated phagocytic index compared to the DMSO-treated phagocytic index. (C) Image of anti-P8 antibody IgG-opsonized amastigote uptake by BMDMs treated with DMSO (top) or SU6656 (bottom). Left panels, representative fields; right panels, enlarged view of boxed area. Scale bars: 10 μm (left); 5 μm (right). (D) SU6656 does not affect amastigote adhesion. Results are percentages of adhered amastigotes per 100 SU6656-treated BMDMs (adhesive index, AI) normalized to the DMSO-treated adhesive index from the experiment in B. (E) SU6656 decreases amastigote uptake by RAW 264.7 cells. Shown is the normalized mean±s.e.m. phagocytic index for SU6656-treated compared to DMSO-treated RAW 264.7 cells. (F) The SFK inhibitor PP2 decreases amastigote uptake. Shown is the normalized mean±s.e.m. phagocytic index for amastigote uptake by PP2-treated RAW 264.7 cells compared to DMSO-treated controls. *P<0.05; **P<0.01 (one-sample t-test); n=3 separate experiments.
Hck, Fgr and Lyn are required for amastigote uptake
Both SU6656 and PP2 inhibit other kinases besides SFKs, such as Aurora kinases (Arai et al., 2012; Bain et al., 2003). Therefore, it was unclear whether the effects of these drugs resulted from SFK inhibition. There are nine SFKs, eight of which are expressed in macrophages. Previous data suggests that three SFKs, namely Hck, Fgr, and Lyn, are especially important for immunoglobulin-mediated phagocytosis (Fitzer-Attas et al., 2000). Thus, we tested the role of these kinases in L. amazonensis amastigote uptake by isolating BMDMs from mice lacking Hck, Fgr and Lyn (Hu et al., 2004). We found that these macrophages were defective in amastigote uptake (Fig. 2A), which was decreased by 41±8% (mean±.s.e.m.) relative to controls. Internalization defects were also demonstrated for IgG-opsonized beads (Fig. 2B), as seen previously (Fitzer-Attas et al., 2000). No defects in the uptake of promastigotes (Fig. 2A) or C3bi-opsonized beads (Fig. 2B) were observed in Hck−/− Fgr−/− Lyn−/− BMDMs.
Fig. 2.

Hck, Fgr and Lyn facilitate IgG-mediated phagocytosis and amastigote uptake. (A) Hck−/− Fgr−/− Lyn−/− BMDMs exhibit defects in amastigote uptake. BMDMs were incubated with opsonized promastigotes and amastigotes as described in Fig. 1. Graph shows the mean±s.e.m. phagocytic index (PI) for promastigotes and amastigotes for Hck−/− Fgr−/− Lyn−/− BMDMs, normalized to WT BMDMs. (B) Hck−/− Fgr−/− Lyn−/− BMDMs show defects in IgG-mediated phagocytosis. BMDMs were incubated with C3bi- or IgG-coated beads as in Fig. 1. Shown is the mean±s.e.m. phagocytic index for C3bi- or IgG-coated beads for Hck−/− Fgr−/− Lyn−/− BMDMs, normalized to WT BMDMs. *P<0.05 (one-sample t-test); n=3 separate experiments.
Src family kinases lie upstream of Arg in a signaling pathway that governs amastigote uptake
Arg facilitates both immunoglobulin-mediated phagocytosis and amastigote uptake (Wetzel et al., 2012). In other biological systems, Arg functions downstream of SFKs (Bradley and Koleske, 2009; Mader et al., 2011; Tegtmeyer and Backert, 2011). To determine whether a SFK–Arg signaling pathway governed amastigote uptake, we first treated Hck−/− Fgr−/− Lyn−/− BMDMs with imatinib. The IC50 of imatinib for Abl is 600 nM (Buchdunger et al., 1995) and its CC50 is ≥20 μM for a 2-h incubation with RAW 264.7 cells; hence, 3.3 μM was selected as in Wetzel et al., 2012. Treating Hck−/− Fgr−/− Lyn−/− BMDMs with imatinib did not cause additional defects in IgG-mediated phagocytosis or amastigote uptake (Fig. 3A), suggesting that SFKs and Abl family kinases might lie in the same pathway. To determine whether Abl or Arg was responsible for SFK-mediated processes, we used wild-type (WT), Ablflox/flox LysM Cre+, Arg−/− or Arg−/−Ablflox/flox LysM Cre+ BMDMs (henceforth referred to as double knockout, dKO) (Fig. 3B). We found that Ablflox/flox LysM Cre+ and dKO BMDMs had defects in C3bi-mediated phagocytosis and promastigote uptake, whereas Arg−/− and dKO BMDMs had defects in IgG-mediated phagocytosis and amastigote uptake. SU6656 inhibited IgG-mediated phagocytosis and amastigote uptake in WT and Ablflox/flox LysM Cre+ BMDMs but not in Arg−/− or dKO BMDMs.
Fig. 3.

An SFK-Arg signaling pathway facilitates IgG-mediated phagocytosis and amastigote uptake. (A) Treating Hck−/− Fgr−/− Lyn−/− BMDMs with imatinib does not further decrease (left) IgG-mediated phagocytosis or (right) IgG-opsonized amastigote uptake. n=3 separate experiments. The left graph shows the mean±s.e.m. phagocytic index (PI) for IgG-coated beads for Hck−/− Fgr−/− Lyn−/− BMDMs incubated with imatinib or DMSO, compared to WT BMDMs incubated with imatinib or DMSO (the latter normalized to 100%). The right graph shows the mean±s.e.m. phagocytic index for amastigotes for Hck−/− Fgr−/− Lyn−/− BMDMs incubated with imatinib or DMSO, compared to WT BMDMs incubated with imatinib or DMSO (the latter normalized to 100%). (B) Effects of SU6656 on BMDMs lacking Abl or Arg. Graphs show the mean±s.e.m. phagocytic index for WT, Ablflox/flox LysM Cre+ (referred to as Abl−/−), Arg−/−, or Arg−/−Ablflox/flox LysM Cre+ (dKO) BMDMs incubated with DMSO or SU6656 and allowed to take up C3bi-coated beads, IgG-coated beads, C3bi-promastigotes or IgG-amastigotes. Data are normalized to WT DMSO-treated BMDMs (set at 100%). n=3 experiments. (C) The Arg activator DPH stimulates IgG-mediated phagocytosis. Shown is one representative experiment of two experiments demonstrating how increasing DPH doses affect the phagocytic index for IgG-coated beads. The maximally stimulating dose, 250 nM, was selected for the remaining experiments. (D) DPH rescues IgG-mediated phagocytosis (left) and amastigote uptake (right) in Hck−/− Fgr−/− Lyn−/− BMDMs. n=3 separate experiments. The left graph shows the mean±s.e.m. phagocytic index for IgG-coated beads incubated with WT BMDMs or Hck−/− Fgr−/− Lyn−/− BMDMs treated with DMSO or DPH. The right graph shows the mean±s.e.m. phagocytic index for IgG-amastigotes incubated with WT BMDMs or Hck−/− Fgr−/− Lyn−/− BMDMs treated with DMSO or DPH. (E) Effects of DPH on BMDMs lacking Abl or Arg. Graphs show the mean±s.e.m. phagocytic index for WT, Ablflox/flox LysM Cre+ (referred to as Abl−/−), Arg−/− or dKO BMDMs incubated with DMSO or DPH and allowed to take up C3bi-coated beads, IgG-coated beads, C3bi-promastigotes or IgG-amastigotes. Data are normalized to phagocytic index for DMSO-incubated WT BMDMs (100%) for each condition. n=4 experiments. For all categories, *P<0.05; **P<0.01; n.s., not significant compared with DMSO-treated WT BMDMs (ANOVA).
We then used the small-molecule Abl family kinase activator DPH (Yang et al., 2011) to test whether activating Arg stimulated phagocytosis and amastigote uptake. DPH is known to activate Arg (Simpson et al., 2015) and Abl, with an EC50 of 250–400 nM (Yang et al., 2011); its CC50 is ≥10 μM for BMDMs. DPH stimulated phagocytosis in a dose-dependent manner (Fig. 3C). At low doses (250 nM), DPH rescued the IgG-opsonized bead and amastigote (Fig. 3D) uptake defects seen in Hck−/− Fgr−/− Lyn−/− BMDMs, returning uptake to 106±6% and 113±8% (mean±s.e.m.) of controls, respectively. DPH had no effect on amastigote uptake by Arg−/− BMDMs, suggesting that the drug was specifically activating Arg (Fig. 3D). We then treated WT, Ablflox/flox LysM Cre+, Arg−/− and dKO BMDMs with DPH prior to phagocytosis or Leishmania uptake. DPH only increased C3bi-mediated phagocytosis and promastigote uptake by BMDMs containing Abl (e.g. WT and Arg−/− BMDMs), whereas it only increased IgG-mediated phagocytosis and amastigote uptake by BMDMs containing Arg (e.g. WT and Ablflox/flox LysM Cre+ BMDMs) (Fig. 3E). Taken together, these results are most consistent with a signaling pathway in which SFKs act upstream of, and activate Arg but not Abl, during IgG-mediated phagocytosis and amastigote uptake.
The SFK and Arg inhibitor bosutinib decreases Leishmania uptake by macrophages
We next reasoned that the dual specificity SFK and Arg kinase inhibitor bosutinib might more effectively inhibit amastigote uptake. Bosutinib was recently approved by the US Food and Drug Administration (FDA) for treatment of chronic myelogenous leukemia and other cancers (Rusconi et al., 2014), and it inhibits purified Arg (Golas et al., 2003) and SFKs with an IC50 of ∼1 nM (Boschelli et al., 2001; Remsing Rix et al., 2009). Low bosutinib doses were sufficient to decrease IgG-opsonized bead uptake (Fig. 4A), with an approximate IC50 of 0.5 μM to prevent uptake; its CC50 for the same incubation period as for BMDMs was >5 μM. We found that 1 μM bosutinib consistently inhibited BMDM and RAW 264.7 cell uptake of C3bi- or IgG-coated beads (Fig. 4B,C). It also inhibited Leishmania promastigote and amastigote uptake by BMDMs by 49±3% and 46±5% (mean±s.e.m.), respectively (Fig. 4D,E). Treating RAW 264.7 cells with bosutinib and exposing them to parasites yielded similar results (Fig. 4F).
Fig. 4.

The SFK, Abl and Arg inhibitor bosutinib decreases phagocytosis and Leishmania uptake. (A) Effects of bosutinib on IgG-mediated phagocytosis. After treatment with the listed bosutinib doses or 0.1% DMSO, BMDMs were incubated with IgG-coated beads. Shown is the phagocytic index (PI) for each condition normalized to DMSO treatment (100%). A representative experiment of two experiments is shown. (B,C) Bosutinib decreases C3bi- or IgG-coated bead uptake. Shown is the mean±s.e.m. phagocytic index for C3bi- or IgG-opsonized beads incubated with bosutinib-treated BMDMs (B) or RAW 264.7 cells (C), normalized to DMSO-treated cells. n=3 experiments. (D) Bosutinib decreases promastigote uptake. Images of C3bi-coated promastigote uptake by WT BMDMs treated with DMSO (top) or bosutinib (bottom) are shown. Left panels, wide-field images; right panels, enlarged views of the boxed areas. Scale bars: 20 μm (left); 10 μm (right). (E,F) Bosutinib decreases L. amazonensis promastigote and amastigote uptake. Shown is the mean±s.e.m. phagocytic index for C3bi-opsonized promastigotes and IgG-opsonized amastigotes incubated with bosutinib-treated BMDMs (E) or RAW 264.7 cells (F), normalized to DMSO-treated cells. n=3 experiments. *P<0.05; **P<0.01 (by one-sample t-test).
Amastigote exposure activates SFK signaling through Arg
Arg kinase activity is required for optimal IgG-mediated phagocytosis (Fig. 3; Greuber and Pendergast, 2012; Wetzel et al., 2012). Incubation of RAW 264.7 cells with opsonized L. amazonensis amastigotes induced a significant increase in phosphorylation of the Abl and Arg substrate CrkII (a splice variant encoded by the Crk gene; denoted pCrk) (Kain and Klemke, 2001). This amastigote-induced increase in pCrk was significantly reduced following treatment with the Abl and Arg inhibitor imatinib, the SFK inhibitor SU6656, and the dual SFK and Arg inhibitor bosutinib (Fig. 5A,B). To determine whether CrkII activation was mediated through Abl or Arg during IgG-mediated amastigote binding and uptake, we incubated WT, Ablflox/flox LysM Cre+, Arg−/− and dKO (Arg−/−Ablflox/flox LysM Cre+) BMDMs with amastigotes with or without DPH. We found that pCrk levels increased significantly in WT and Ablflox/flox LysM Cre+ BMDMs incubated with amastigotes, but not in amastigote-treated Arg−/− or dKO BMDMs, indicating that CrkII activation during amastigote uptake was specifically dependent upon Arg and not Abl (Fig. 5C,D).
Fig. 5.

SFKs and Arg phosphorylate downstream effectors during amastigote uptake. (A,B) Phosphorylation of the SFK, Abl and Arg substrate CrkII (pCrk) induced upon amastigote uptake is decreased in imatinib-, SU6656- or bosutinib-treated macrophages. RAW 264.7 cells were allowed to adhere to uncoated plates (−) or plates coated with anti-P8 antibody-opsonized amastigotes (+ amastigotes) for 15 min before processing for immunoblotting. (A) Representative immunoblot of pCrk (top) and total CrkII (bottom) in DMSO-treated RAW 264.7 cells [with (+) or without (–) amastigotes] and amastigote-stimulated imatinib-, SU6656- or bosutinib-treated RAW 264.7 cells. (B) Graph presents the relative pCrk levels, normalized to CrkII levels, among RAW 264.7 cell categories shown in A. Levels of pCrk are normalized to amastigote-exposed DMSO-treated RAW 264.7 cells (100%). n=3 experiments. *P<0.05 compared with amastigote-stimulated DMSO-treated RAW 264.7 cells (ANOVA). (C,D) pCrk is induced upon amastigote uptake, is increased by DPH and is Arg-dependent. BMDMs [WT, Ablflox/flox LysM Cre+ (Abl−/−), Arg−/− or dKO] were adhered to plates as above before processing for immunoblotting. (C) Representative immunoblot of pCrk (top) and total CrkII (bottom) in DMSO-treated BMDMs (− amastigotes), DMSO-, DPH- and bosutinib-treated WT BMDMs (+ amastigotes), and DMSO- or DPH-treated Abl−/−, Arg−/− or dKO BMDMs (all +amastigotes). (D) Graph shows relative pCrk levels, normalized to total Crk levels, among the categories of BMDMs shown in C. Levels of pCrk for each category are normalized to the level in amastigote-exposed DMSO-treated BMDMs. n=5 experiments. *P≤0.05; n.s., not significant compared with amastigote-stimulated DMSO-treated WT BMDMs (ANOVA).
SFK and dual SFK and Arg inhibitors decrease lesion size and parasite burden in a murine model of cutaneous leishmaniasis
Knowing that inhibiting SFK and Arg significantly decreased parasite uptake in cultured cells, we next determined whether SFK and Arg inhibitors decreased the manifestations of cutaneous leishmaniasis in mice. Our previous work demonstrated that the Abl and Arg inhibitor imatinib decreased lesion size and parasite burden in mice (Wetzel et al., 2012). To test the effects of SFK inhibition, we provided 2 mg/kg body weight of PP2 by intraperitoneal injection three times weekly thoughout the course of infection. We found that PP2 significantly decreased the average lesion size in mice infected with L. amazonensis (Fig. 6A), as well as the parasite burden (Fig. 6B) by >10 fold. To determine whether inhibiting both SFKs and Arg would also provide benefit, we provided 30 mg/kg body weight of bosutinib or vehicle (DMSO) in drinking water throughout the course of infection. We found that bosutinib significantly decreased the average lesion size in L.-amazonensis-infected mice (Fig. 6C). A higher dose of bosutinib (150 mg/kg body weight) did not cause additional decreases in lesion size (Fig. S2).
Fig. 6.

SFKs, Abl and Arg permit efficient infection in a mouse model of cutaneous leishmaniasis. (A) PP2-treated mice have smaller lesions than untreated mice. Four or five C57BL/6 mice per category were injected with 1×106 L. amazonensis promastigotes and given 2 mg/kg body weight of PP2 or DMSO by intraperitoneal injection three times weekly, starting 7 days before infection and continuing until the mice were euthanized. Two experiments were performed; shown is one experiment containing 5 mice per group. Results represent the mean±s.e.m. foot size increase over the uninfected foot (normalized to 1). *P<0.05 (ANOVA). (B) Lesions in PP2-treated mice contain fewer parasites than DMSO-treated mice (quantified by limiting dilution). Plotted is the mean±s.e.m. total lesion parasite burden in millions at the end of the experiment in A; in this example, there were 3.7×106±1.5×106 parasites in the DMSO-treated mice versus 179×103±77×103 parasites in the PP2-treated mice. *P=0.042 (two-tailed t-test). (C) Bosutinib-treated mice have smaller lesions than untreated mice. Four to eight C57BL/6 mice per category were infected as in A and treated with 30 mg/kg body weight/day of bosutinib or DMSO in their drinking water, starting 4 days before infection and continuing until the mice were euthanized. Three experiments were performed; shown is a representative experiment containing five mice per group. *P<0.05 (ANOVA). (D) Lesions in bosutinib-treated mice contain fewer parasites than DMSO-treated mice. Plotted is the parasite burden in millions at the end of the experiment in C; here there were 3.3×106±0.7×106 parasites in the DMSO-treated mice versus 61×103±21×103 parasites in the bosutinib-treated mice. **P=0.0023 (two-tailed t-test). (E) Relationship between FcR signaling, SFKs and Arg during Leishmania amastigote uptake. Upon FcRγ ligation by amastigotes, Hck, Fgr and Lyn are activated. These SFKs phosphorylate and activate Arg kinase, which phosphorylates and activates CrkII, leading to actin polymerization.
We first tested whether bosutinib impacted on lesions by directly affecting parasite growth or survival. 10 μM bosutinib caused no growth defects for promastigotes (Fig. S3A) or amastigotes (Fig. S3B). Parasites could transition from promastigotes to amastigotes when cultures were grown in bosutinib, either axenically (Fig. S3C) or in BMDMs (Fig. S3D). Finally, we found no survival defects in amastigotes within bosutinib-treated previously infected BMDMs (Fig. S3E).
The reduced lesion size in bosutinib-treated mice could potentially result from differences in the immune response to infection. Of note, mice lacking SFKs and Abl family kinases have significant immunological defects, including deficits in B cell and T cell development, proliferation and function, which could alter the course of cutaneous leishmaniasis (Colucci et al., 1999; Gu et al., 2007; Kovacs et al., 2014; Liberatore and Goff, 2009; Lin et al., 1994; Silberman et al., 2008; Zipfel et al., 2004). To determine whether the immunological effects of Abl, Arg or SFK inhibition were causing the healing seen in bosutinib-treated mice, we isolated draining lymph nodes from infected DMSO and bosutinib-treated mice and profiled cytokine secretion after stimulation with parasite lysate (Soong et al., 1996). Overall, there were decreased amounts of cytokines secreted in response to L. amazonensis by lymph nodes isolated from bosutinib-treated mice compared to control mice (Table 1, bosutinib); a change in the overall ratio of T-helper 1 to T-helper 2 cells (Th1/Th2) was not noted (Table S1). Bosutinib did not affect cytokine release in response to concanavalin A (Con A) stimulation in these same mice (Table 1, bosutinib), suggesting that this change was specific to L. amazonensis stimulation. Consequently, the lower response to Leishmania antigen stimulation might reflect the reduced parasite load rather than an effect of bosutinib on the ongoing host response to infection. To evaluate this point, we examined the direct in vitro effect of bosutinib on the response of lymph node cells to leishmanial antigens. Cytokine secretion did not decrease when infected draining lymph nodes isolated from DMSO-treated infected mice were treated with 0.1% DMSO or bosutinib during culture (Table S2), suggesting that bosutinib did not directly inhibit cytokine secretion. Cytokine secretion did not significantly change in lymph nodes isolated from imatinib-treated or PP2-treated mice (Table 1, imatinib, PP2), with a few exceptions. Both imatinib (i.e. Abl family kinase inhibition alone) and PP2 decreased IL-1β levels, but based on previous studies, this decrease would not be expected to cause healing (Lima-Junior et al., 2013). PP2 also caused lower IFN-γ secretion at high antigenic stimulation (the opposite to what would be expected if the change were contributing to healing in PP2-treated mice; Soong et al., 2012) and decreased IL-17 secretion with Leishmania antigenic stimulation.
Table 1.
Cytokine secretion in bosutinib-, imatinib- and PP2-treated mice.
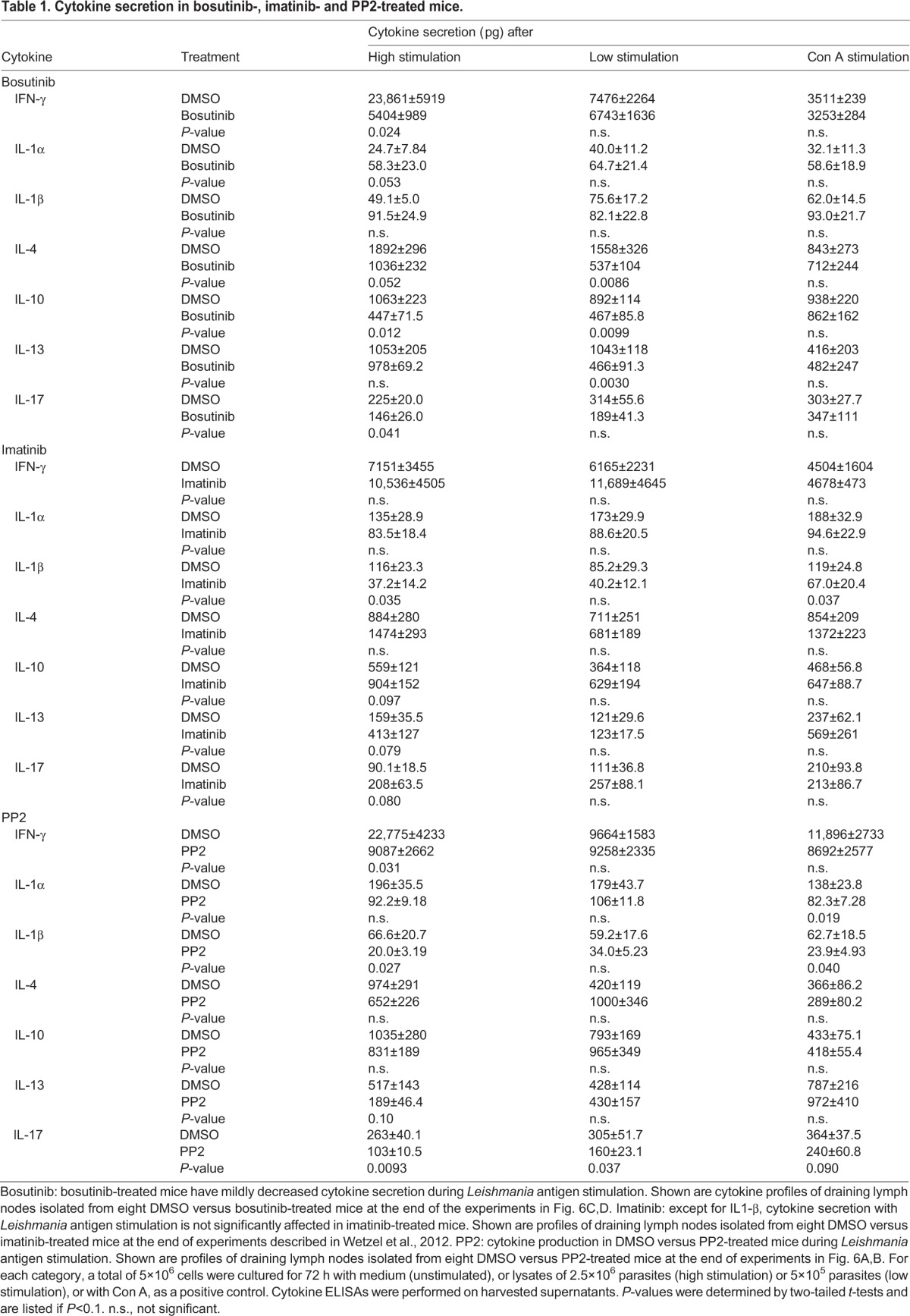
Cytokines that attract immune cells to infection sites, termed chemokines, could also affect the outcome of leishmaniasis. We next assessed whether Abl and Arg, SFK, or combined Abl and SFK inhibition affected chemokine secretion. The production of chemokines implicated in Leishmania pathogenesis [IP-10, MIP-1α (CCL3), MIP-2 (CXCL2), RANTES (CCL5), CXCL9 and MIP-1β (CCL4)] did not change with bosutinib or imatinib (Table S3A,B), suggesting that the Abl- and Arg-mediated differences in chemokines were not responsible for decreased lesions. PP2 caused decreased secretion of MIP-1α, MIP-2 and CXCL9 (Table S3C), but these changes are not known to decrease lesion size (Brandonisio et al., 2002; Lima-Junior et al., 2013; Muller et al., 2003; Oghumu et al., 2010; Ritter and Korner, 2002; Santiago et al., 2004).
Given that bosutinib caused a significant inhibition of parasite uptake, and uptake is required for parasite survival, we suspected that the decreased cytokine secretion in bosutinib-treated mice resulted from lower parasite burdens in these mice. Therefore, we determined the parasite burdens in DMSO- and bosutinib-treated mice at the end of the experiment (∼3 months of infection) by limiting dilution. We found that the number of parasites contained within the infected foot was far lower in bosutinib-treated mice than in DMSO-treated mice (Fig. 6D), with an average decrease of ∼50-fold. These results suggest that understanding the host cell processes required for parasite uptake could provide new candidates for treating leishmaniasis.
DISCUSSION
Previously, we have demonstrated that the host Arg non-receptor tyrosine kinase facilitates L. amazonensis amastigote uptake by macrophages (Wetzel et al., 2012). The results shown here are the first to demonstrate that SFKs, and specifically Hck, Fgr and Lyn, also facilitate amastigote uptake. SFKs activate Arg during efficient amastigote uptake. Finally, the small-molecule combination Arg and SFK inhibitor bosutinib decreases amastigote uptake by macrophages and significantly reduces disease severity and parasite burden in Leishmania-infected mice. In summary, host cell kinase inhibitors might provide novel exploratory candidates for the treatment of leishmaniasis.
Leishmania amastigotes isolated from lesions are coated with IgG and primarily bind to the FcR to stimulate uptake (Kima et al., 2000). Based on their role during IgG-mediated phagocytosis, one might predict a role for SFKs during Leishmania uptake. However, a previous in vivo study in mice lacking the SFK Fyn failed to demonstrate an effect of SFKs during Leishmania infection (Yamakami et al., 2001), in retrospect, likely due to redundant roles for multiple SFK members. Our work delineates specific roles for Hck, Fgr and Lyn for stimulating amastigote uptake, both based on pharmacological inhibition and BMDMs from mice lacking these three kinases.
Our work demonstrates that SFKs signal through Arg to facilitate amastigote uptake, as modeled in Fig. 6E. We do not observe additional deficits in IgG-mediated phagocytosis or amastigote uptake when Hck−/− Fgr−/− Lyn−/− BMDMs are treated with imatinib or when Arg−/− BMDMs are treated with SU6656, indicating that SFKs and Arg lie in the same signaling pathway, as previously shown in other systems (Mader et al., 2011; Plattner et al., 2004; Tanis et al., 2003). Another report has indicated that macrophages treated with both imatinib and SU6656 had IgG-mediated phagocytosis defects beyond those seen with either drug alone (Greuber and Pendergast, 2012), suggesting that SFKs, and Arg and Abl might partly signal through different pathways. These differences might have occurred if imatinib and SU6656 did not completely inhibit Abl, Arg and SFK activity, or if the kinases also provide a scaffolding function. In both cases, drug treatment alone might not fully prevent this IgG-mediated uptake pathway. Furthermore, using BMDMs lacking Abl or Arg, we directly demonstrate that a SFK–Arg pathway, and not a SFK–Abl pathway, is employed during IgG-mediated phagocytosis and amastigote uptake. These data are consistent with our previous report showing that Abl is required for C3bi-mediated phagocytosis and promastigote uptake, whereas Arg is required for IgG-mediated phagocytosis and amastigote uptake (Wetzel et al., 2012). We suspect that the differential usage of Abl and Arg during phagocytosis and Leishmania uptake is due to their different subcellular localization within macrophages.
We find that activating Arg in Hck−/− Fgr−/− Lyn−/− BMDMs with low or sub-EC50 doses (Yang et al., 2011) of DPH rescues uptake. Ours is the first demonstration that activating Abl family kinases facilitates phagocytosis. Interestingly, however, higher doses of DPH inhibit phagocytosis, suggesting that either there is a point where overly active Arg is deleterious, or that the drug affects other essential molecules at high concentrations. The action of SFKs and Arg allows phosphorylation of their downstream mediator CrkII during FcR engagement by amastigotes. We also find that DPH increases CrkII phosphorylation in an Arg-dependent manner, consistent with previous observations (Yang et al., 2011). Clearly, SFKs and Arg-independent mechanisms also contribute to IgG-mediated phagocytosis and amastigote uptake, given that inhibiting both kinases does not completely prevent these processes. Future studies will focus on uncovering novel signaling pathways permitting Leishmania uptake and further delineating the SFK–Arg signaling pathway that occurs.
As kinase inhibitors do not prevent 100% of activity in vivo, targeting multiple signaling molecules in the same pathway could, in theory, yield a greater effect than inhibiting a single kinase. Thus, we turned to the potent combination SFK, Abl and Arg inhibitor bosutinib. Despite its effects on multiple host cell kinases, there are sufficiently few side effects that bosutinib was approved by the FDA to treat chronic myelogenous leukemia (Rusconi et al., 2014). We find that bosutinib prevented C3bi- and IgG-mediated phagocytosis, as well as promastigote and amastigote uptake.
Our mouse studies also indicate that Leishmania survival and pathogenesis depend on SFKs, Abl and Arg. We propose that the smaller lesions seen in these kinase-inhibited mice result at least partly from uptake defects in macrophages. Consistent with this proposal, fewer parasites are contained in lesions in bosutinib-treated mice than in controls. Interestingly, bosutinib-treated mice had a larger decrease in parasite burden than we saw with imatinib (Wetzel et al., 2012) or PP2 therapy. Thus, our results imply, although do not show directly, that inhibiting multiple signaling proteins required for parasite uptake might improve results over inhibiting a single kinase alone.
Given that there are no direct effects of bosutinib treatment seen on parasite replication or survival within macrophages, the lesion size disparities in L. amazonensis infection could result from differences in cell entry and resulting parasite burden, or from differences in the host inflammatory and/or immune response (Soong et al., 1995). The murine immune response to leishmaniasis is complicated and is dependent on the parasite species and mouse strain. In general, Th1 responses are protective, and Th2 responses are deleterious to the host (Jones et al., 1998). For example, inhibiting phosphoinositide 3-kinase (PI3K)γ, which can signal though SFKs, Abl and Arg (Bradley and Koleske, 2009), affects Leishmania uptake and impairs the Th2 response (Cummings et al., 2012). Mice lacking SFKs have defects in macrophage recruitment (Park et al., 1999), the respiratory burst (Meng and Lowell, 1998), and B cell and T cell development and signaling (Lowell, 2011). Immunological defects in mice lacking Abl family kinases include defects in B cell and T cell development and signaling, among others; drugs affecting SFK and Abl and Arg also decrease immune cell proliferation (Gu et al., 2007; Huang et al., 2008; Liberatore and Goff, 2009; Silberman et al., 2008; Zipfel et al., 2004). All of these effects could contribute to the smaller lesions in Leishmania-infected bosutinib-treated mice.
We find that incubating cultured lymph nodes with bosutinib does not directly affect cytokine secretion. Cytokine profiling of lymph nodes isolated from infected bosutinib-treated mice demonstrated a decrease in many cytokines measured after Leishmania antigen stimulation, but not after Con A stimulation. These decreases are generally not statistically significant with Abl and Arg, or SFK inhibition alone, and none of the changes seen with single inhibition are known to facilitate healing (Ehrlich et al., 2014; Lima-Junior et al., 2013; Soong et al., 2012), with the possible exception of the decreased IL-17 secretion seen with PP2 (Sousa et al., 2014). One potential explanation for the bosutinib-induced cytokine decreases is that a lower parasite burden causes a diminished amount of cytokines to be secreted in response to L. amazonensis antigen. Another possibility is that Abl, Arg and SFKs promote an immune response to L. amazonensis, which is dampened by kinase inhibition, and does not reach statistical significance unless both families are inhibited. Unfortunately, it is difficult to distinguish between these possibilities in our system. Even in bosutinib-treated mice, the ratios of IFN-γ to TH2 cytokines do not improve, suggesting that the differences in lesion size and parasite burden are not fully explained by defective immune responses to Leishmania infection. However, decreases in IL-10, like those described here, could facilitate lesion healing and decrease parasite burden during L. amazonensis murine infection (Ji et al., 2005), contributing to the effects of bosutinib.
Chemokine profiling also does not reveal explanations for the smaller lesions seen with Abl and Arg, or SFK inhibitors. We examined levels of IP-10, MIP-1α, MIP-2, RANTES, CXCL9 and MIP-1β. Several changed upon PP2, but not bosutinib or imatinib treatment, and, based on previous studies, these changes would not be expected to facilitate healing (Brandonisio et al., 2002; Lima-Junior et al., 2013; Muller et al., 2003; Oghumu et al., 2010; Ritter and Korner, 2002; Santiago et al., 2004). Of note, PP2 inhibits multiple targets, not just SFKs (>50 targets were seen in Brandvold et al., 2012), and these off-target effects could contribute to its chemokine and cytokine profile. More studies of chemokine secretion after host manipulations like kinase inhibition might be beneficial for unraveling the pathogenesis of leishmaniasis.
Additionally, mice with defects in B cells or T cells can be disease resistant following L. amazonensis infection (Soong et al., 1997). Indeed, the diminished swelling that we see in bosutinib-treated mice might actually be less than what might have been expected from the parasite burden decrease alone. Using another Leishmania species might distinguish whether kinase-inhibition-induced immune deficiencies are diminishing the effect we would otherwise see from bosutinib.
In summary, we have shown that SFKs signal through Arg to facilitate IgG-mediated phagocytosis and Leishmania uptake. SFKs, and Abl and Arg also govern the subsequent pathogenesis of leishmaniasis in the mouse model. These results strongly suggest that improving our understanding of cell entry by Leishmania could provide novel treatment strategies for leishmaniasis. Furthermore, they support the concept that drugs that target conserved host cell processes, rather than the infectious agents themselves, could be used to treat a broad range of infectious diseases, including parasitic diseases like leishmaniasis. Thus far, we have focused on clinically available tyrosine kinase inhibitors with relatively benign side effect profiles. In contrast, the agents currently used to treat leishmaniasis have multiple toxic side effects, and resistance to these drugs is emerging. Host-cell-active agents could be combined with current antiparasitics, increasing their efficacy while lowering their toxicity. Future work will continue to explore the potential use of cell entry inhibitors for leishmaniasis.
MATERIALS AND METHODS
Mice
C57BL/6 mice were obtained from Jackson Labs (Bar Harbor, ME). Abl and Arg knockout mice [Ablflox/flox LysM Cre+, Arg−/−, and Arg−/−Ablflox/flox LysM Cre+ (termed dKO)] from a mixed background (C57BL/6X 129Sv/J) were backcrossed to C57BL/6 more than five times; littermates provided controls. Hck, Fgr and Lyn triple-knockout mice (Hck−/− Fgr−/− Lyn−/−) were as described in Hu et al. (2004). The Institutional Animal Care and Use Committees at Yale University and UT Southwestern approved all protocols.
Cell culture
RAW 264.7 cells (ATCC, Manassas, VA) were grown in Dulbeco's modified Eagle's medium (DMEM) with 10% heat-inactivated fetal bovine serum (FBS; Invitrogen, Grand Island, NY). For primary macrophage experiments, cells were harvested from femurs and tibias of WT, Ablflox/flox LysM Cre+, Arg−/−, dKO, and Hck−/− Fgr−/− Lyn−/− mice and differentiated into bone-marrow-derived macrophages (BMDMs), which was confirmed as in Wetzel et al. (2012). Cells were tested for Mycoplasma contamination (Uphoff and Drexler, 2013).
Parasite culture
L. amazonensis promastigotes (strain IFLA/BR/67/PH8, from Norma W. Andrews, University of Maryland, College Park, MD) were grown in Schneider's Drosophila medium with 10 μg/ml gentamicin and 15% heat-inactivated FBS at 24°C (Wetzel et al., 2012). For uptake experiments, promastigotes were incubated for 7 days to maximize infective metacyclics (i.e. those isolated through a step Percoll gradient; Sigma, St Louis, MO). Amastigotes were grown axenically (Wetzel et al., 2012). To assess for growth defects with bosutinib, medium containing 0.1% DMSO or 10 μM bosutinib (LC Laboratories, Woburn, MA) was used. Bosutinib remained in solution at neutral pH and has improved solubility at lower pH (Remsing Rix et al., 2009). Parasites were passed through mice to maintain virulence.
Phagocytosis assays
RAW 264.7 cells or BMDMs were plated at ∼50% confluence and incubated overnight in serum-free medium or M-CSF-starved medium, respectively. Except where indicated, for drug experiments with IgG-coated beads, coverslips were preincubated in medium containing 3.3 μM imatinib (LC Laboratories), 2.5 μM SU6656 (Sigma), 10 μM PP2 (Sigma), 1 μM bosutinib (LC Laboratories) or DMSO (0.1%) (Sigma) for 2 h. For C3bi-coated bead uptake, cells were preactivated with phorbol 12-myristate 13-acetate (PMA; Sigma), which added 0.1% DMSO to all PMA-treated cells. Experiments were conducted as described previously (Wetzel et al., 2012). Briefly, 2-μm latex yellow–green beads (Sigma) coated with human IgM (cat. no. I-8260, Sigma) were incubated in fresh mouse serum for C3bi opsonization or rabbit anti-IgM (cat. no. 270A, Sigma) for IgG opsonization (confirmed as described previously; Wetzel et al., 2012). Cells were incubated with 10–15 beads/cell for 30 min at 37°C, fixed with 3% formaldehyde for 15 min, blocked with 2% BSA without permeabilization, incubated with rabbit anti-human IgM plus Hoechst 33258 dye (Sigma) to visualize nuclei, then incubated with Alexa-Fluor-594-conjugated goat anti-rabbit-IgG secondary antibody (cat. no. A11034, Invitrogen). This two-color immunofluorescence assay (Wetzel et al., 2003) allowed distinguishing of internalized and external beads. Coverslips were visualized using a 40×1.0 NA aperture Nikon objective on a Nikon Eclipse TE2000-5 fluorescence microscope by a treatment-blinded observer. Images were acquired with a Qimaging Cooled Charge-coupled Device mono, 12-bit camera and Nikon Imaging software. At least ten randomly selected fields were visualized for a total of over 100 macrophages and beads per experiment. The mean phagocytic index (the number of particles internalized per 100 macrophages) for controls was set as the maximum (100%) value for each experiment, and those for experimental conditions were normalized to that value. Each experiment was performed three times and the mean±s.e.m. was calculated. A one-sample Student's t-test or two-way ANOVA was used to determine statistical significance.
Leishmania uptake assays
BMDMs or RAW 264.7 cells were plated and treated with inhibitors, DMSO or PMA as above. Metacyclic promastigotes were incubated in fresh mouse serum for C3bi opsonization, and BMDMs or RAW 264.7 cells were PMA-activated for 1 h. Amastigotes were coated with anti-P8-proteoglycolipid complex (monoclonal antibody IgG1; Pan and McMahon-Pratt, 1988). Coverslips with RAW cells or BMDMs were incubated with C3bi-opsonized promastigotes at a ratio of ten parasites to one cell or with IgG-opsonized amastigotes at two parasites to one cell for 30 min and fixed with 3% formaldehyde. External promastigotes were incubated with mouse anti-gp46 antibody; external amastigotes were incubated with mouse anti-P8 antibody as described previously (Wetzel et al., 2012). All were incubated with Alexa-Fluor-568-conjugated donkey anti-mouse-IgG secondary antibody (cat. no. A10037, Invitrogen). After permeabilization, parasites were re-incubated with mouse anti-gp46 (promastigotes) or mouse anti-P8 (amastigotes) antibodies and Alexa-Fluor-488-conjugated donkey anti-mouse-IgG secondary antibody (cat. no. A21202, Invitrogen) and DAPI (Sigma). At least ten random fields were selected containing at least 100 parasites and macrophages per experiment; each experiment was performed three times. The mean±s.e.m. of biological replicates is shown. Images for analysis were collected as above. The representative images shown in Figs 1 and 4 were visualized with a Zeiss LSM 700 confocal microscope with a Zeiss 40×1.3 NA EC Plan-Neofluar oil objective (promastigotes) or a Zeiss 63×1.4 NA Plan-Apochromat objective (amastigotes), acquired with a Zeiss Axiocam 506 mono, 6 megapixel monochrome camera and Zeiss Imaging software (Zeiss, Jena, Germany), and linearly processed in Adobe Photoshop (Adobe Systems, San Jose, CA). A one-sample Student's t-test or two-way ANOVA was used to determine statistical significance.
Intra-macrophage assays
For assays examining promastigote–amastigote transition within macrophages or intracellular survival, 10 L. amazonensis promastigotes were added per WT BMDM cell (starved of G-CSF overnight) and plated in triplicate wells at 50% confluence. After 4 h, the BMDMs were washed 5× with PBS, and DMSO or 2 μM bosutinib in medium containing G-CSF was added. The number of parasites per 100 BMDMs was counted at 24 h to assess amastigote transition or 72 h to assess survival.
Immunoblotting
To measure phosphorylation of the downstream SFK, Abl and Arg effector CrkII (42 kDa), starved RAW 264.7 cells or WT, Ablflox/flox LysM Cre+, Arg−/− or dKO BMDMs were added for 15 min to uncoated dishes or dishes precoated for 1 h with anti-P8 antibody (Pan and McMahon-Pratt, 1988) IgG-opsonized amastigotes. Cells were lysed with a buffer of 20 mM Tris-HCl pH 7.2, 2 mM EDTA, 150 mM NaCl and 1% Triton X-100 that contained protease and phosphatase inhibitors. For representative images, equivalent protein amounts were loaded on 10% SDS-PAGE gels, transferred to nitrocellulose membranes, and probed with antibodies against phosphorylated CrkII (pCrkII) (Y221, cat. no. 3491S, Cell Signaling, Beverly, MA) at 1:1500, or CrkII (cat. no. sc-9004, Santa Cruz Biotechnology, Dallas, TX) at 1:500. For analysis, relative amounts of pCrkII were compared with Image J analysis software and were normalized to CrkII (membranes were stripped of pCrkII and reprobed for CrkII, as in Wetzel et al., 2012). Imaging was performed by using a phosphorimager (ImageQuant LAS 4000, GE). Two-way ANOVA was used to determine statistical significance.
Murine infections
Four to eight female C57BL/6 mice per group were infected at between 6 and 8 weeks of age. Power calculations were performed as in Wetzel et al., 2012. 1×106 metacyclic promastigotes in PBS were injected subcutaneously in the dorsal right hind foot. Imatinib experiments were performed as described previously (Wetzel et al., 2012). For PP2-treated mice, two independent experiments were performed. Mice were provided 2 mg/kg body weight of PP2 or 5% DMSO (the diluent) by intraperitoneal injection three times a week, starting a week before infection and continuing through until the mice were euthanized. For bosutinib-treated mice, three independent experiments were conducted. Mice were provided 30 mg/kg body weight/day of bosutinib or <0.5% DMSO (the diluent) in drinking water starting 4 days before infection and continuing through until the mice were euthanized. Bottle weight was obtained to monitor water intake. Lesion size was monitored with calipers weekly by an investigator blinded to condition, and the infected:uninfected size ratio was calculated (Champsi and McMahon-Pratt, 1988). The control and drug-treated groups were compared to swelling at time 0 by ANOVA. Parasite burdens in lesions were determined upon experimental termination between experimental weeks 12 and 18 by limiting dilution (Soong et al., 1997).
Lymph nodes from infected DMSO versus drug-treated mice were harvested for cytokine and chemokine profiling (Soong et al., 1997, 1995, 1996). Cells were plated and stimulated with promastigote lysates or Con A (5 µg/ml; Sigma) (Wetzel et al., 2012). Supernatants were obtained after 72 h. The cytokines IL-4, IL-10, IL-13, IL-17 or IFN-γ were assessed by ELISA (BD Biosciences, San Jose, CA). Background levels were assessed using unstimulated cell supernatants. Levels of the cytokines and chemokines IL-1β, IL-1α, IP-10, MIP-1α (CCL3), MIP-2 (CXCL2), RANTES (CCL5), and MIP-1β (CCL4) were profiled by using the custom multiplex Luminex Platform (R&D Systems, Inc, Minneapolis, MN) through the UT Southwestern Microarray Core as described previously (Gonzalez-Fajardo et al., 2015; Navas et al., 2014). CXCL9 levels were assessed by ELISA (R&D Systems, Inc).
Acknowledgements
We thank Norma W. Andrews for providing L. amazonensis strain IFLA/BR/67/PH8 and the Koleske and McMahon-Pratt laboratories for helpful discussions. We thank Margaret A. Phillips and her laboratory for the use of equipment and reagents, James J. Collins for confocal microscope use, the parasitology group at UT Southwestern for valuable discussions, Indu Raman at the UT Southwestern Microarray Core for assistance with cytokine and chemokine profiling, and Emily T. Mamula, Imran Ullah, Hanspeter Niederstrasser, Meghan E. Kerrisk and Margaret A. Cooper for technical and experimental assistance.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
D.M.W. designed and performed experiments, analyzed data and wrote the manuscript. E.L.R. performed experiments and collected and analyzed data. S.L. assisted with the Hck−/− Fgr−/− Lyn−/− mouse studies and manuscript preparation. D.M.-P. and A.J.K. participated in experimental design and data analysis, discussed results and assisted with manuscript preparation.
Funding
This work was supported by the National Institutes of Health [grant numbers NRSA F32 AI094905 and K08 AI103036 to D.M.W.; R01 CA122142 to S.L.; R01 AI093775 to D.M.-P.; and R01 CA133346, R01 GM100411, and R01 NS089662 to A.J.K.]; and funds from the University of Texas Southwestern Medical Center, Department of Pediatrics (to D.M.W. and E.L.R.). Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.185595.supplemental
References
- Abram C. L. and Lowell C. A. (2008). The diverse functions of Src family kinases in macrophages. Front. Biosci. 13, 4426-4450. 10.2741/3015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai R., Tsuda M., Watanabe T., Ose T., Obuse C., Maenaka K., Minami A. and Ohba Y. (2012). Simultaneous inhibition of Src and Aurora kinases by SU6656 induces therapeutic synergy in human synovial sarcoma growth, invasion and angiogenesis in vivo. Eur. J. Cancer 48, 2417-2430. 10.1016/j.ejca.2011.12.028 [DOI] [PubMed] [Google Scholar]
- Bain J., McLauchlan H., Elliott M. and Cohen P. (2003). The specificities of protein kinase inhibitors: an update. Biochem. J. 371, 199-204. 10.1042/bj20021535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bavagnoli L., Dundon W. G., Garbelli A., Zecchin B., Milani A., Parakkal G., Baldanti F., Paolucci S., Volmer R., Tu Y. et al. (2011). The PDZ-ligand and Src-homology type 3 domains of epidemic avian influenza virus NS1 protein modulate human Src kinase activity during viral infection. PLoS ONE 6, e27789 10.1371/journal.pone.0027789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beausejour M., Noel D., Thibodeau S., Bouchard V., Harnois C., Beaulieu J.-F., Demers M.-J. and Vachon P. H. (2012). Integrin/Fak/Src-mediated regulation of cell survival and anoikis in human intestinal epithelial crypt cells: selective engagement and roles of PI3-K isoform complexes. Apoptosis 17, 566-578. 10.1007/s10495-012-0713-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake R. A., Garcia-Paramio P., Parker P. J. and Courtneidge S. A. (1999). Src promotes PKCdelta degradation. Cell Growth Differ. 10, 231-241. [PubMed] [Google Scholar]
- Blake R. A., Broome M. A., Liu X., Wu J., Gishizky M., Sun L. and Courtneidge S. A. (2000). SU6656, a selective src family kinase inhibitor, used to probe growth factor signaling. Mol. Cell. Biol. 20, 9018-9027. 10.1128/MCB.20.23.9018-9027.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boschelli D. H., Ye F., Wang Y. D., Dutia M., Johnson S. L., Wu B., Miller K., Powell D. W., Yaczko D., Young M. et al. (2001). Optimization of 4-phenylamino-3-quinolinecarbonitriles as potent inhibitors of Src kinase activity. J. Med. Chem. 44, 3965-3977. 10.1021/jm0102250 [DOI] [PubMed] [Google Scholar]
- Bradley W. D. and Koleske A. J. (2009). Regulation of cell migration and morphogenesis by Abl-family kinases: emerging mechanisms and physiological contexts. J. Cell Sci. 122, 3441-3454. 10.1242/jcs.039859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandonisio O., Panaro M. A., Fumarola I., Sisto M., Leogrande D., Acquafredda A., Spinelli R. and Mitolo V. (2002). Macrophage chemotactic protein-1 and macrophage inflammatory protein-1 alpha induce nitric oxide release and enhance parasite killing in Leishmania infantum-infected human macrophages. Clin. Exp. Med. 2, 125-129. 10.1007/s102380200017 [DOI] [PubMed] [Google Scholar]
- Brandvold K. R., Steffey M. E., Fox C. C. and Soellner M. B. (2012). Development of a highly selective c-Src kinase inhibitor. ACS Chem. Biol. 7, 1393-1398. 10.1021/cb300172e [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchdunger E., Zimmermann J., Mett H., Meyer T., Muller M., Regenass U. and Lydon N. B. (1995). Selective inhibition of the platelet-derived growth factor signal transduction pathway by a protein-tyrosine kinase inhibitor of the 2-phenylaminopyrimidine class. Proc. Natl. Acad. Sci. USA 92, 2558-2562. 10.1073/pnas.92.7.2558 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Burton E. A., Plattner R. and Pendergast A. M. (2003). Abl tyrosine kinases are required for infection by Shigella flexneri. EMBO J. 22, 5471-5479. 10.1093/emboj/cdg512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter C. R., Whitcomb J. P., Campbell J. A., Mukbel R. M. and McDowell M. A. (2009). Complement receptor 3 deficiency influences lesion progression during Leishmania major infection in BALB/c mice. Infect. Immun. 77, 5668-5675. 10.1128/iai.00802-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champsi J. and McMahon-Pratt D. (1988). Membrane glycoprotein M-2 protects against Leishmania amazonensis infection. Infect. Immun. 56, 3272-3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C.-Y., Huang W.-R., Chi P.-I., Chiu H.-C. and Liu H.-J. (2015). Cell entry of bovine ephemeral fever virus requires activation of Src-JNK-AP1 and PI3K-Akt-NF-kappaB pathways as well as Cox-2-mediated PGE2 /EP receptor signalling to enhance clathrin-mediated virus endocytosis. Cell. Microbiol. 17, 967-987. 10.1111/cmi.12414 [DOI] [PubMed] [Google Scholar]
- Colucci F., Soudais C., Rosmaraki E., Vanes L., Tybulewicz V. L. and Di Santo J. P. (1999). Dissecting NK cell development using a novel alymphoid mouse model: investigating the role of the c-abl proto-oncogene in murine NK cell differentiation. J. Immunol. 162, 2761-2765. [PubMed] [Google Scholar]
- Cummings H. E., Barbi J., Reville P., Oghumu S., Zorko N., Sarkar A., Keiser T. L., Lu B., Ruckle T., Varikuti S. et al. (2012). Critical role for phosphoinositide 3-kinase gamma in parasite invasion and disease progression of cutaneous leishmaniasis. Proc. Natl. Acad. Sci. USA 109, 1251-1256. 10.1073/pnas.1110339109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich A., Castilho T. M., Goldsmith-Pestana K., Chae W.-J., Bothwell A. L. M., Sparwasser T. and McMahon-Pratt D. (2014). The immunotherapeutic role of regulatory T cells in Leishmania (Viannia) panamensis infection. J. Immunol. 193, 2961-2970. 10.4049/jimmunol.1400728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elwell C. A., Ceesay A., Kim J. H., Kalman D. and Engel J. N. (2008). RNA interference screen identifies Abl kinase and PDGFR signaling in Chlamydia trachomatis entry. PLoS Pathog. 4, e1000021 10.1371/journal.ppat.1000021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzer-Attas C. J., Lowry M., Crowley M. T., Finn A. J., Meng F., DeFranco A. L. and Lowell C. A. (2000). Fcgamma receptor-mediated phagocytosis in macrophages lacking the Src family tyrosine kinases Hck, Fgr, and Lyn. J. Exp. Med. 191, 669-682. 10.1084/jem.191.4.669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golas J. M., Arndt K., Etienne C., Lucas J., Nardin D., Gibbons J., Frost P., Ye F., Boschelli D. H. and Boschelli F. (2003). SKI-606, a 4-anilino-3-quinolinecarbonitrile dual inhibitor of Src and Abl kinases, is a potent antiproliferative agent against chronic myelogenous leukemia cells in culture and causes regression of K562xenografts in nude mice. Cancer Res. 63, 375-381. [PubMed] [Google Scholar]
- Gonzalez-Fajardo L., Fernandez O. L., McMahon-Pratt D. and Saravia N. G. (2015). Ex vivo host and parasite response to antileishmanial drugs and immunomodulators. PLoS Negl. Trop. Dis. 9, e0003820 10.1371/journal.pntd.0003820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greuber E. K. and Pendergast A. M. (2012). Abl family kinases regulate FcgammaR-mediated phagocytosis in murine macrophages. J. Immunol. 189, 5382-5392. 10.4049/jimmunol.1200974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J. J., Zhang N., He Y.-W., Koleske A. J. and Pendergast A. M. (2007). Defective T cell development and function in the absence of Abelson kinases. J. Immunol. 179, 7334-7343. 10.4049/jimmunol.179.11.7334 [DOI] [PubMed] [Google Scholar]
- Guy R. and Belosevic M. (1993). Comparison of receptors required for entry of Leishmania major amastigotes into macrophages. Infect. Immun. 61, 1553-1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauck C. R., Meyer T. F., Lang F. and Gulbins E. (1998). CD66-mediated phagocytosis of Opa52 Neisseria gonorrhoeae requires a Src-like tyrosine kinase- and Rac1-dependent signalling pathway. EMBO J. 17, 443-454. 10.1093/emboj/17.2.443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y., Liu Y., Pelletier S., Buchdunger E., Warmuth M., Fabbro D., Hallek M., Van Etten R. A. and Li S. (2004). Requirement of Src kinases Lyn, Hck and Fgr for BCR-ABL1-induced B-lymphoblastic leukemia but not chronic myeloid leukemia. Nat. Genet. 36, 453-461. 10.1038/ng1343 [DOI] [PubMed] [Google Scholar]
- Huang Y., Comiskey E. O., Dupree R. S., Li S., Koleske A. J. and Burkhardt J. K. (2008). The c-Abl tyrosine kinase regulates actin remodeling at the immune synapse. Blood 112, 111-119. 10.1182/blood-2007-10-118232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob M., Todd L. A., Majumdar R. S., Li Y., Yamamoto K.-I. and Pure E. (2009). Endogenous cAbl regulates receptor endocytosis. Cell. Signal. 21, 1308-1316. 10.1016/j.cellsig.2009.03.016 [DOI] [PubMed] [Google Scholar]
- Ji J., Masterson J., Sun J. and Soong L. (2005). CD4+CD25+ regulatory T cells restrain pathogenic responses during Leishmania amazonensis infection. J. Immunol. 174, 7147-7153. 10.4049/jimmunol.174.11.7147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones D. E., Elloso M. M. and Scott P. (1998). Host susceptibility factors to cutaneous leishmaniasis. Front. Biosci. 3, D1171-D1180. 10.2741/A353 [DOI] [PubMed] [Google Scholar]
- Kain K. H. and Klemke R. L. (2001). Inhibition of cell migration by Abl family tyrosine kinases through uncoupling of Crk-CAS complexes. J. Biol. Chem. 276, 16185-16192. 10.1074/jbc.M100095200 [DOI] [PubMed] [Google Scholar]
- Kane M. M. and Mosser D. M. (2000). Leishmania parasites and their ploys to disrupt macrophage activation. Curr. Opin. Hematol. 7, 26-31. 10.1097/00062752-200001000-00006 [DOI] [PubMed] [Google Scholar]
- Kima P. E., Constant S. L., Hannum L., Colmenares M., Lee K. S., Haberman A. M., Shlomchik M. J. and McMahon-Pratt D. (2000). Internalization of Leishmania mexicana complex amastigotes via the Fc receptor is required to sustain infection in murine cutaneous leishmaniasis. J. Exp. Med. 191, 1063-1068. 10.1084/jem.191.6.1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs M., Nemeth T., Jakus Z., Sitaru C., Simon E., Futosi K., Botz B., Helyes Z., Lowell C. A. and Mocsai A. (2014). The Src family kinases Hck, Fgr, and Lyn are critical for the generation of the in vivo inflammatory environment without a direct role in leukocyte recruitment. J. Exp. Med. 211, 1993-2011. 10.1084/jem.20132496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapetina S., Mader C. C., Machida K., Mayer B. J. and Koleske A. J. (2009). Arg interacts with cortactin to promote adhesion-dependent cell edge protrusion. J. Cell Biol. 185, 503-519. 10.1083/jcb.200809085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberatore R. A. and Goff S. P. (2009). c-Abl-deficient mice exhibit reduced numbers of peritoneal B-1 cells and defects in BCR-induced B cell activation. Int. Immunol. 21, 403-414. 10.1093/intimm/dxp006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima-Junior D. S., Costa D. L., Carregaro V., Cunha L. D., Silva A. L. N., Mineo T. W. P., Gutierrez F. R. S., Bellio M., Bortoluci K. R., Flavell R. A. et al. (2013). Inflammasome-derived IL-1beta production induces nitric oxide-mediated resistance to Leishmania. Nat. Med. 19, 909-915. 10.1038/nm.3221 [DOI] [PubMed] [Google Scholar]
- Lin C. -T., Shen Z., Boros P. and Unkeless J. C. (1994). Fc receptor-mediated signal transduction. J. Clin. Immunol. 14, 1-13. 10.1007/BF01541170 [DOI] [PubMed] [Google Scholar]
- Lodge R. and Descoteaux A. (2008). Leishmania invasion and phagosome biogenesis. Subcell. Biochem. 47, 174-181. 10.1007/978-0-387-78267-6_14 [DOI] [PubMed] [Google Scholar]
- Lowell C. A. (2011). Src-family and Syk kinases in activating and inhibitory pathways in innate immune cells: signaling cross talk. Cold Spring Harb. Perspect. Biol. 3, a002352 10.1101/cshperspect.a002352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly K. T. and Casanova J. E. (2009). Abelson tyrosine kinase facilitates Salmonella enterica serovar Typhimurium entry into epithelial cells. Infect. Immun. 77, 60-69. 10.1128/IAI.00639-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mader C. C., Oser M., Magalhaes M. A. O., Bravo-Cordero J. J., Condeelis J., Koleske A. J. and Gil-Henn H. (2011). An EGFR-Src-Arg-cortactin pathway mediates functional maturation of invadopodia and breast cancer cell invasion. Cancer Res. 71, 1730-1741. 10.1158/0008-5472.CAN-10-1432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng F. and Lowell C. A. (1998). A beta 1 integrin signaling pathway involving Src-family kinases, Cbl and PI-3 kinase is required for macrophage spreading and migration. EMBO J. 17, 4391-4403. 10.1093/emboj/17.15.4391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M. M., Lapetina S., MacGrath S. M., Sfakianos M. K., Pollard T. D. and Koleske A. J. (2010). Regulation of actin polymerization and adhesion-dependent cell edge protrusion by the Abl-related gene (Arg) tyrosine kinase and N-WASp. Biochemistry 49, 2227-2234. 10.1021/bi901721u [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morehead J., Coppens I. and Andrews N. W. (2002). Opsonization modulates Rac-1 activation during cell entry by Leishmania amazonensis. Infect. Immun. 70, 4571-4580. 10.1128/IAI.70.8.4571-4580.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosser D. and Edelson P. (1985). The mouse macrophage receptor for C3bi (CR3) is a major mechanism in the phagocytosis of Leishmania promastigotes. J. Immunol. 135, 2785-2789. [PubMed] [Google Scholar]
- Mosser D., Springer T. and Diamond M. (1992). Leishmania promastigotes require opsonic complement to bind to the human leukocyte integrin Mac-1 (CD11b/CD18). J. Cell Biol. 116, 511-520. 10.1083/jcb.116.2.511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller K., Bischof S., Sommer F., Lohoff M., Solbach W. and Laskay T. (2003). Differential production of macrophage inflammatory protein 1gamma (MIP-1gamma), lymphotactin, and MIP-2 by CD4(+) Th subsets polarized in vitro and in vivo. Infect. Immun. 71, 6178-6183. 10.1128/IAI.71.11.6178-6183.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napier R. J., Rafi W., Cheruvu M., Powell K. R., Zaunbrecher M. A., Bornmann W., Salgame P., Shinnick T. M. and Kalman D. (2011). Imatinib-sensitive tyrosine kinases regulate mycobacterial pathogenesis and represent therapeutic targets against tuberculosis. Cell Host Microbe 10, 475-485. 10.1016/j.chom.2011.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navas A., Vargas D. A., Freudzon M., McMahon-Pratt D., Saravia N. G. and Gomez M. A. (2014). Chronicity of dermal leishmaniasis caused by Leishmania panamensis is associated with parasite-mediated induction of chemokine gene expression. Infect. Immun. 82, 2872-2880. 10.1128/IAI.01133-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oghumu S., Lezama-Davila C. M., Isaac-Marquez A. P. and Satoskar A. R. (2010). Role of chemokines in regulation of immunity against leishmaniasis. Exp. Parasitol. 126, 389-396. 10.1016/j.exppara.2010.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan A. and McMahon-Pratt D. (1988). Monoclonal antibodies specific for the amastigote stage of Leishmania pifanoi. I. Characterization of antigens associated with stage- and species-specific determinants. J. Immunol. 140, 2406-2414. [PubMed] [Google Scholar]
- Park R. K., Erdreich-Epstein A., Liu M., Izadi K. D. and Durden D. L. (1999). High affinity IgG receptor activation of Src family kinases is required for modulation of the Shc-Grb2-Sos complex and the downstream activation of the nicotinamide adenine dinucleotide phosphate (reduced) oxidase. J. Immunol. 163, 6023-6034. [PubMed] [Google Scholar]
- Paul R., Obermaier B., Van Ziffle J., Angele B., Pfister H.-W., Lowell C. A. and Koedel U. (2008). Myeloid Src kinases regulate phagocytosis and oxidative burst in pneumococcal meningitis by activating NADPH oxidase. J. Leukoc. Biol. 84, 1141-1150. 10.1189/jlb.0208118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plattner R., Koleske A. J., Kazlauskas A. and Pendergast A. M. (2004). Bidirectional signaling links the Abelson kinases to the platelet-derived growth factor receptor. Mol. Cell. Biol. 24, 2573-2583. 10.1128/MCB.24.6.2573-2583.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puentes S. M., Sacks D. L., da Silva R. P. and Joiner K. A. (1988). Complement binding by two developmental stages of Leishmania major promastigotes varying in expression of a surface lipophosphoglycan. J. Exp. Med. 167, 887-902. 10.1084/jem.167.3.887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves P. M., Bommarius B., Lebeis S., McNulty S., Christensen J., Swimm A., Chahroudi A., Chavan R., Feinberg M. B., Veach D. et al. (2005). Disabling poxvirus pathogenesis by inhibition of Abl-family tyrosine kinases. Nat. Med. 11, 731-739. 10.1038/nm1265 [DOI] [PubMed] [Google Scholar]
- Reeves P. M., Smith S. K., Olson V. A., Thorne S. H., Bornmann W., Damon I. K. and Kalman D. (2011). Variola and monkeypox viruses utilize conserved mechanisms of virion motility and release that depend on Abl and Src family tyrosine kinases. J. Virol. 85, 21-31. 10.1128/JVI.01814-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remsing Rix L. L., Rix U., Colinge J., Hantschel O., Bennett K. L., Stranzl T., Muller A., Baumgartner C., Valent P., Augustin M. et al. (2009). Global target profile of the kinase inhibitor bosutinib in primary chronic myeloid leukemia cells. Leukemia 23, 477-485. 10.1038/leu.2008.334 [DOI] [PubMed] [Google Scholar]
- Ritter U. and Korner H. (2002). Divergent expression of inflammatory dermal chemokines in cutaneous leishmaniasis. Parasite Immunol. 24, 295-301. 10.1046/j.1365-3024.2002.00467.x [DOI] [PubMed] [Google Scholar]
- Rusconi F., Piazza R., Vagge E. and Gambacorti-Passerini C. (2014). Bosutinib: a review of preclinical and clinical studies in chronic myelogenous leukemia. Expert Opin. Pharmacother. 15, 701-710. 10.1517/14656566.2014.882898 [DOI] [PubMed] [Google Scholar]
- Russell D. G. and Wright S. D. (1988). Complement receptor type 3 (CR3) binds to an Arg-Gly-Asp-containing region of the major surface glycoprotein, gp63, of Leishmania promastigotes. J. Exp. Med. 168, 279-292. 10.1084/jem.168.1.279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago H. D. C., Oliveira C. F., Santiago L., Ferraz F. O., de Souza D. D. G., de-Freitas L. A. R., Afonso L. C. C., Teixeira M. M., Gazzinelli R. T. and Vieira L. Q. (2004). Involvement of the chemokine RANTES (CCL5) in resistance to experimental infection with Leishmania major. Infect. Immun. 72, 4918-4923. 10.1128/IAI.72.8.4918-4923.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson M. A., Bradley W. D., Harburger D., Parsons M., Calderwood D. A. and Koleske A. J. (2015). Direct interactions with the integrin β1 cytoplasmic tail activate the Abl2/Arg kinase. J. Biol. Chem. 290, 8360-8372. 10.1074/jbc.M115.638874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silberman I., Sionov R. V., Zuckerman V., Haupt S., Goldberg Z., Strasser A., Ben-Sasson Z. S., Baniyash M., Koleske A. J. and Haupt Y. (2008). T cell survival and function requires the c-Abl tyrosine kinase. Cell Cycle 7, 3847-3857. 10.4161/cc.7.24.7267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soong L., Duboise S., Kima P. and McMahon-Pratt D. (1995). Leishmania pifanoi amastigote antigens protect mice against cutaneous leishmaniasis. Infect. Immun 63, 3559-3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soong L., Xu J.-C., Grewal I. S., Kima P., Sun J., Longley B. J., Ruddle N. H., McMahon-Pratt D. and Flavell R. A. (1996). Disruption of CD40-CD40 ligand interactions results in an enhanced susceptibility to Leishmania amazonensis infection. Immunity 4, 263-273. 10.1016/S1074-7613(00)80434-3 [DOI] [PubMed] [Google Scholar]
- Soong L., Chang C., Sun J., Longley B. J., Ruddle N., Flavell R. and McMahon-Pratt D. (1997). Role of CD4+ T cells in pathogenesis associated with Leishmania amazonensis infection. J. Immunol. 158, 5374-5383. [PubMed] [Google Scholar]
- Soong L., Henard C. A. and Melby P. C. (2012). Immunopathogenesis of non-healing American cutaneous leishmaniasis and progressive visceral leishmaniasis. Semin. Immunopathol. 34, 735-751. 10.1007/s00281-012-0350-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa L. M. A., Carneiro M. B. H., Resende M. E., Martins L. S., dos Santos L. M., Vaz L. G., Mello P. S., Mosser D. M., Oliveira M. A. P. and Vieira L. Q. (2014). Neutrophils have a protective role during early stages of Leishmania amazonensis infection in BALB/c mice. Parasite Immunol. 36, 13-31. 10.1111/pim.12078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swimm A. I., Bornmann W., Jiang M., Imperiale M. J., Lukacher A. E. and Kalman D. (2010). Abl family tyrosine kinases regulate sialylated ganglioside receptors for polyomavirus. J. Virol. 84, 4243-4251. 10.1128/JVI.00129-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanis K. Q., Veach D., Duewel H. S., Bornmann W. G. and Koleske A. J. (2003). Two distinct phosphorylation pathways have additive effects on Abl family kinase activation. Mol. Cell. Biol. 23, 3884-3896. 10.1128/MCB.23.11.3884-3896.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanos B. and Pendergast A. M. (2006). Abl tyrosine kinase regulates endocytosis of the epidermal growth factor receptor. J. Biol. Chem. 281, 32714-32723. 10.1074/jbc.M603126200 [DOI] [PubMed] [Google Scholar]
- Tanos B. and Pendergast A. M. (2007). Abi-1 forms an epidermal growth factor-inducible complex with Cbl: role in receptor endocytosis. Cell. Signal. 19, 1602-1609. 10.1016/j.cellsig.2007.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tegtmeyer N. and Backert S. (2011). Role of Abl and Src family kinases in actin-cytoskeletal rearrangements induced by the Helicobacter pylori CagA protein. Eur. J. Cell Biol. 90, 880-890. 10.1016/j.ejcb.2010.11.006 [DOI] [PubMed] [Google Scholar]
- Ueno N. and Wilson M. E. (2012). Receptor-mediated phagocytosis of Leishmania: implications for intracellular survival. Trends Parasitol. 28, 335-344. 10.1016/j.pt.2012.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uphoff C. C. and Drexler H. G. (2013). Eradication of mycoplasma contaminations. Methods Mol Biol. 946, 15-26. 10.1007/978-1-62703-128-8_2 [DOI] [PubMed] [Google Scholar]
- Van Langendonck N., Velge P. and Bottreau E. (1998). Host cell protein tyrosine kinases are activated during the entry of Listeria monocytogenes. Possible role of pp60c-src family protein kinases. FEMS Microbiol. Lett. 162, 169-176. 10.1016/S0378-1097(98)00111-6 [DOI] [PubMed] [Google Scholar]
- Wetzel D. M., Hakansson S., Hu K., Roos D. and Sibley L. D. (2003). Actin filament polymerization regulates gliding motility by apicomplexan parasites. Mol. Biol. Cell 14, 396-406. 10.1091/mbc.E02-08-0458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetzel D. M., McMahon-Pratt D. and Koleske A. J. (2012). The Abl and Arg kinases mediate distinct modes of phagocytosis and are required for maximal Leishmania infection. Mol. Cell. Biol. 32, 3176-3186. 10.1128/mcb.00086-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woelbing F., Kostka S. L., Moelle K., Belkaid Y., Sunderkoetter C., Verbeek S., Waisman A., Nigg A. P., Knop J., Udey M. C. et al. (2006). Uptake of Leishmania major by dendritic cells is mediated by Fcgamma receptors and facilitates acquisition of protective immunity. J. Exp. Med. 203, 177-188. 10.1084/jem.20052288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y., Suzuki-Inoue K., Satoh K., Asazuma N., Yatomi Y., Berndt M. C. and Ozaki Y. (2001). Role of Fc receptor gamma-chain in platelet glycoprotein Ib-mediated signaling. Blood 97, 3836-3845. 10.1182/blood.V97.12.3836 [DOI] [PubMed] [Google Scholar]
- Yamakami K., Akao S., Wakabayashi K., Tadakuma T. and Yoshizawa N. (2001). Mice lacking protein tyrosine kinase fyn develop a T helper-type 1 response and resist Leishmania major infection. Environ. Health Prev. Med. 6, 132-135. 10.1007/BF02897960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Campobasso N., Biju M. P., Fisher K., Pan X.-Q., Cottom J., Galbraith S., Ho T., Zhang H., Hong X. et al. (2011). Discovery and characterization of a cell-permeable, small-molecule c-Abl kinase activator that binds to the myristoyl binding site. Chem. Biol. 18, 177-186. 10.1016/j.chembiol.2010.12.013 [DOI] [PubMed] [Google Scholar]
- Yogalingam G. and Pendergast A. M. (2008). Abl kinases regulate autophagy by promoting the trafficking and function of lysosomal components. J. Biol. Chem. 283, 35941-35953. 10.1074/jbc.M804543200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipfel P. A., Zhang W., Quiroz M. and Pendergast A. M. (2004). Requirement for Abl kinases in T cell receptor signaling. Curr. Biol. 14, 1222-1231. 10.1016/j.cub.2004.07.021 [DOI] [PubMed] [Google Scholar]