

Abstract
Myocardin-related transcription factors (MRTFs) play a central role in the regulation of actin expression and cytoskeletal dynamics. Stimuli that promote actin polymerization allow for shuttling of MRTFs to the nucleus where they activate serum response factor (SRF), a regulator of actin and other cytoskeletal protein genes. SRF is an essential regulator of skeletal muscle differentiation and numerous components of the muscle sarcomere, but the potential involvement of MRTFs in skeletal muscle development has not been examined. We explored the role of MRTFs in muscle development in vivo by generating mutant mice harboring a skeletal muscle-specific deletion of MRTF-B and a global deletion of MRTF-A. These double knockout (dKO) mice were able to form sarcomeres during embryogenesis. However, the sarcomeres were abnormally small and disorganized, causing skeletal muscle hypoplasia and perinatal lethality. Transcriptome analysis demonstrated dramatic dysregulation of actin genes in MRTF dKO mice, highlighting the importance of MRTFs in actin cycling and myofibrillogenesis. MRTFs were also shown to be necessary for the survival of skeletal myoblasts and for the efficient formation of intact myotubes. Our findings reveal a central role for MRTFs in sarcomere formation during skeletal muscle development and point to the potential involvement of these transcriptional co-activators in skeletal myopathies.
KEY WORDS: Myogenesis, MRTF, SRF, Actin cycling, Myopathy
Summary: MRTFs are regulators of actin cycling and cytoskeletal gene expression. Loss of MRTFs in the mouse skeletal muscle causes muscle hypoplasia due to defects in sarcomere formation.
INTRODUCTION
Myocardin-related transcription factors A and B (MRTF-A and MRTF-B; also known as MKL1/MAL and MKL2) are crucial regulators of actin cycling and cytoskeletal dynamics (Olson and Nordheim, 2010). These transcriptional co-activators function by binding to serum response factor (SRF), a versatile transcription factor belonging to the MADS-box family of transcription factors (Cen et al., 2003; Miano, 2003; Norman et al., 1988; Wang et al., 2002). In turn, SRF acts as a central regulator of cytoskeletal gene expression, by binding its consensus sequence, known as the CArG box [CC(A/T)6GG], a motif frequently associated with genes involved in actin turnover and cytoskeletal dynamics (Posern and Treisman, 2006; Treisman, 1986, 1992). SRF is required for the expression of cytoskeletal genes and for myogenic differentiation (Croissant et al., 1996; Wei et al., 1998).
SRF is essential for mesoderm development during embryogenesis and the loss of SRF leads to embryonic lethality due to defects in gastrulation (Arsenian et al., 1998). Skeletal muscle-specific deletion of SRF causes severe muscle hypoplasia. In these mutant mice, muscle fibers are present, but are unable to undergo hypertrophy, leading to perinatal lethality (Li et al., 2005b). Furthermore, conditional deletion of SRF in adult mice using a tamoxifen-inducible Cre recombinase leads to progressive loss of muscle mass and sarcopenia (Lahoute et al., 2008).
Although the function of SRF in skeletal muscle development and maintenance has been studied extensively, less is known about the role of MRTFs in skeletal muscle development. MRTF-A and MRTF-B are ubiquitously expressed transcriptional co-activators that have been implicated in myogenic differentiation, based on siRNA knockdown studies in cultured muscle cells (Selvaraj and Prywes, 2003). Global deletion of MRTF-A does not affect viability or cause any overt disease phenotypes, apart from defects in mammary myoepithelial cell maintenance (Li et al., 2006; Sun et al., 2006). By contrast, loss of MRTF-B causes embryonic lethality due to defects in branchial arch and neural crest development (Li et al., 2005a). Neither MRTF-A nor MRTF-B null mice demonstrate any skeletal muscle pathology.
Myocardin-related transcription factors are upregulated during muscle injury and regeneration (Mokalled et al., 2012). Another member of the MRTF family, MEF2 activating motif and SAP domain containing transcriptional regulator (MASTR), cooperates with MyoD (also known as MYOD1) to turn on the myogenic program during skeletal muscle regeneration (Creemers et al., 2006; Meadows et al., 2008). MASTR and MRTF-A are required for skeletal muscle regeneration in adult mice. Indeed, global or satellite cell-specific genetic deletion of MASTR leads to impairment in skeletal muscle regeneration, which is augmented in MASTR; MRTF-A double-null animals (Mokalled et al., 2012).
In addition to modulating SRF-dependent transcription, MRTFs influence actin cycling at the protein level, by acting as ‘actin sensors’ in the cytoplasm, through their ability to bind actin via their RPEL domains (Miralles et al., 2003). Actin continuously cycles between two states in the cytoplasm: monomeric or globular (G) actin, and polymerized or filamentous (F) actin. When G-actin is in abundance in the cytoplasm, MRTFs are bound by monomeric actin and sequestered in the cytoplasm, preventing their translocation into the nucleus and activation of SRF-dependent genes encoding actin and other cytoskeletal components (Olson and Nordheim, 2010). Thus, MRTFs can regulate actin cycling in the cell at both the transcriptional and post-translational levels. Several actin-binding proteins contribute to this dynamic turnover process to maintain the balance between F- and G-actin. Previously, we and others showed that the actin-binding protein leiomodin 3 (LMOD3) promotes the polymerization of actin in skeletal muscle and is required for muscle development (Cenik et al., 2015; Yuen et al., 2014). The Lmod3 promoter contains a CArG box, and Lmod3 gene expression is strongly regulated by MRTFs. Furthermore, MRTF-A and -B levels were reduced in Lmod3 null mice, indicating that dysregulation of cellular actin dynamics can directly influence MRTF signaling (Cenik et al., 2015).
To investigate further the potential roles of the MRTF/SRF pathway in muscle development, we generated mice lacking both MRTF-A and -B in skeletal muscle. The severe lethal phenotype of these mice reveals essential and redundant roles of MRTF-A and -B in the control of sarcomerogenesis and muscle cell survival and points to the potential involvement of MRTFs in various skeletal myopathies.
RESULTS
MRTF dKO mice fail to thrive and have defects in muscle development
In order to characterize the role of MRTFs in muscle development and function, we utilized previously generated mouse lines with a global deletion of MRTF-A and a floxed allele of MRTF-B (Li et al., 2006; Mokalled et al., 2010). These mice were crossed with two skeletal muscle-specific Cre lines, myogenin-Cre (Cheng et al., 1993) and HSA-Cre (Miniou et al., 1999), in order to generate mice harboring a deletion of MRTF-B in skeletal muscle in the background of a global deletion of MRTF-A (Fig. S1A). Myogenin-Cre and HSA-Cre both display pan-myofiber-specific expression restricted to the skeletal muscle lineage beginning at embryonic days (E) 8 and 9, respectively. Mice with Cre deletion of MRTF-B and global deletion of MRTF-A are henceforth referred to as double knockout (dKO) mice. Cre-positive MRTF-A+/−; MRTF-Bflox/flox females were bred to MRTF-A−/−; MRTF-Bflox/flox males in order to generate MRTF dKO mice. The HSA-Cre- and myogenin-Cre-positive dKO lines are respectively referred to as HdKO and MdKO mice. Loss of MRTF-A and MRTF-B expression was confirmed by quantitative real-time PCR (RT-qPCR) analysis (Fig. 1, data shown for HdKO mice). Whole hindlimb tissue from E17.5 mice, which contained subcutaneous tissue and bone as well as muscle, was used for this analysis. By RT-qPCR analysis, the MRTF-A transcript was reduced by 80%. The presence of MRTF-A transcript in dKO muscle is expected based on the MRTF-A targeting strategy and is consistent with previously published reports (Costello et al., 2015; Li et al., 2006; McDonald et al., 2015). The original targeting strategy for generating the MRTF-A null mice led to the splicing of exon 8 to exon 12 and production of a transcript with an in-frame fusion. This transcript translates into a non-functional truncated protein lacking the SRF binding domain (Li et al., 2006). The partial reduction in MRTF-B mRNA was attributed to the mixed cellular content of the muscle tissue.
Fig. 1.
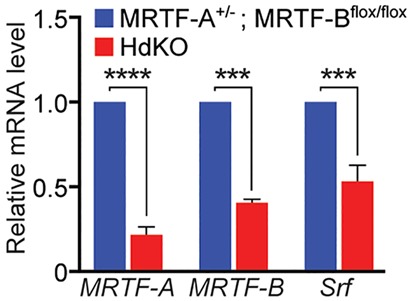
MRTF/SRF gene expression in MRTF dKO mice. MRTF-A, MRTF-B and SRF transcript expression in hindlimb muscle of E17.5 control (MRTF-A+/−; MRTF-Bflox/flox) and HdKO mice as measured by RT-qPCR. Experiments were performed in triplicate with three biological replicates for each genotype, and expression was normalized to 18S rRNA. All data are shown as mean±s.e.m. The two-tailed Mann–Whitney U test was used for pairwise comparisons between groups. ***P<0.001, ****P<0.0001.
MRTF dKO mice were not born at Mendelian ratios, with a significantly lower number of dKO pups being observed compared with the expected numbers (Table 1). Both HdKO and MdKO mice displayed perinatal lethality, with over 90% of the animals dying within a few hours of birth.
Table 1.
Expected and observed numbers of MRTF dKO mice at birth following intercrosses of Cre-positive MRTF-A+/−; MRTF-Bflox/flox females to MRTF-A−/−; MRTF-Bflox/flox males

HdKO mice that survived to birth were able to move, but were smaller in size and had a relatively flaccid appearance compared with their healthy littermates (Fig. 2A). Among the six HdKO mice that were born, only one survived to postnatal day (P) 3 (Fig. 2A, right). The remaining mice died at P1, their viability ranging between 1 and 12 h after birth.
Fig. 2.
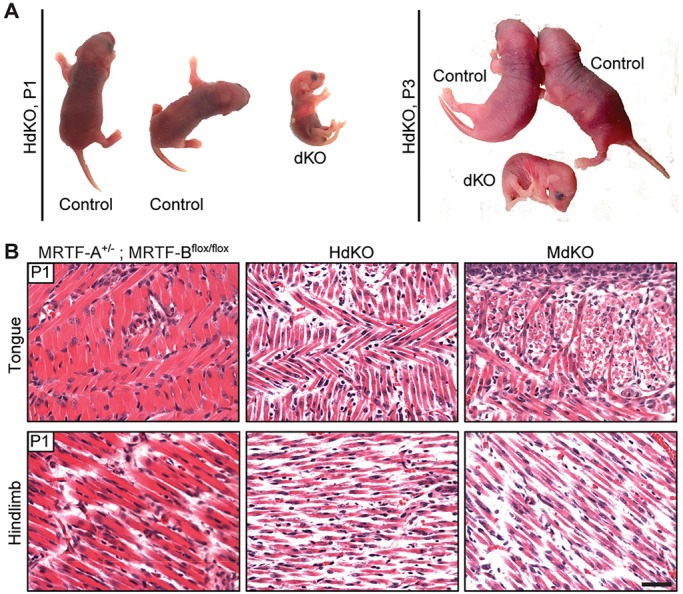
Failure to thrive and muscle hypoplasia phenotype in MRTF dKO mice. (A) HdKO mice at P1 and P3 show failure to thrive and reduced muscle mass. Control: MRTF-A+/−; MRTF-Bflox/flox. (B) H&E staining of coronal sections of tongue and transverse sections of hindlimb demonstrate reduced fiber size in dKO mice, compared with control (MRTF-A+/−; MRTF-Bflox/flox). Scale bar: 40 μm.
The myogenin-Cre line displayed a more severe phenotype; none of the MdKO mice survived past P1. All animals that were born alive died within 12 h of birth. A subset of MdKO neonates was able to feed and breathe. However, the alveoli were smaller, indicating respiratory distress as a possible reason for mortality. Other MdKO mice that were discovered dead at P1 had no milk spots and their alveoli were not expanded (Fig. S1B), suggesting that they were stillborn. In the HdKO line, fewer in utero deaths occurred compared with the MdKO, and only one dead pup was observed to have non-patent alveoli and no milk spot (Fig. S1B). Together, these results pointed towards a more severe phenotype in the MdKO line compared with the HdKO line.
Skeletal muscle tissues were harvested from surviving and freshly dead MdKO and HdKO mice and histological analysis was performed. Severe skeletal muscle hypoplasia was observed in all muscle types examined (Fig. 2B). To investigate possible MRTF dosage effects, Cre-positive MRTF-A+/−; MRTF-Bflox/flox animals, as well as Cre-negative MRTF-A+/−; MRTF-Bflox/flox and Cre-negative MRTF-A−/−; MRTF-Bflox/flox were also assessed for skeletal muscle defects. No skeletal muscle abnormalities were observed by conventional histology in these animals (Fig. S1C).
The variability in time of lethality in dKO mice indicated the possibility of a difference in penetrance of the phenotype, with mice that have lower penetrance showing a higher likelihood of survival to birth. This would lead to an inherently biased selection of healthier mice when characterizing the phenotype. Therefore, we also evaluated mice at E10.5, E12.5, E15.5 and E17.5 to determine whether the dKO animals displayed the same defects in muscle development at earlier stages of development (Fig. 3). Consistent with our findings in P1 mice, the dKO embryos also showed clear muscle defects with visibly thinner diaphragms and smaller myofibers (Fig. 3B), as early as E12.5. Myofiber membranes were visualized by wheat germ agglutinin staining (Fig. 4A) and the cross-sectional area of myofibers was measured. Quantification of these measurements showed reduction of myofiber size for all muscle groups that were analyzed (Fig. 4B). Nuclei numbers per myofiber showed no significant change (Fig. S2).
Fig. 3.
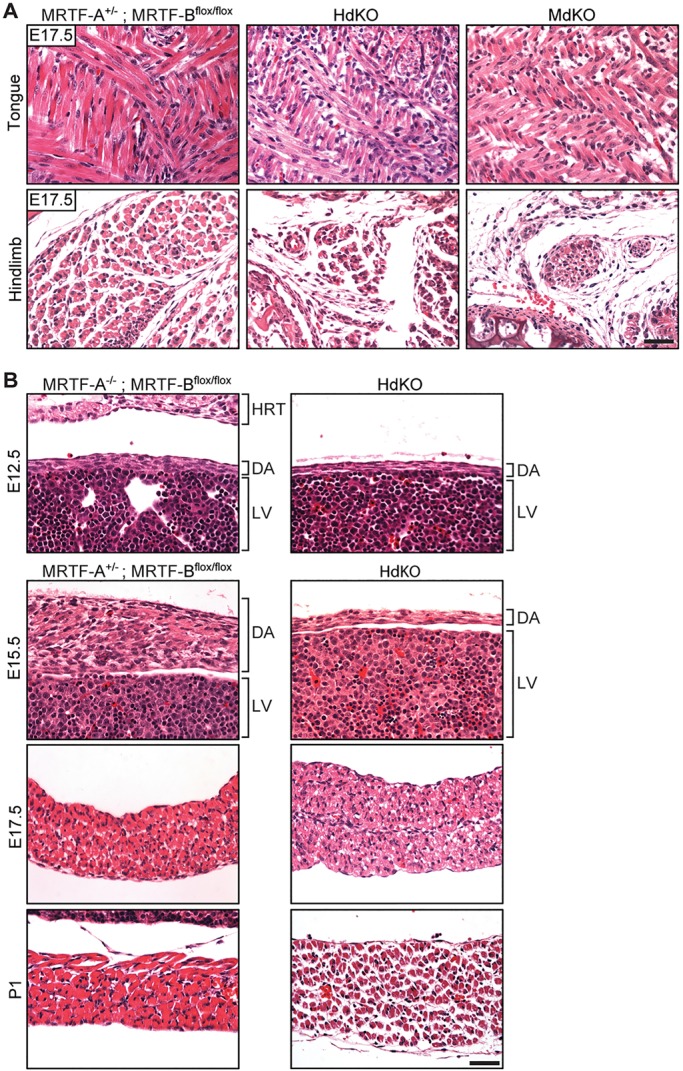
Hypoplasia of myofibers is observed in embryonic muscle tissue of MRTF dKO animals. (A) H&E staining of coronal sections of tongue and transverse sections of hindlimb from E17.5 control (MRTF-A+/−; MRTF-Bflox/flox) and dKO embryos shows a reduction in myofiber size in the dKO tissue. (B) H&E staining of WT and MRTF dKO diaphragm at E12.5, E15.5, E17.5 and P1 shows myofiber hypotrophy at all ages that becomes more pronounced with age. DA, diaphragm; HRT, heart; LV, liver. Scale bars: 40 μm.
Fig. 4.
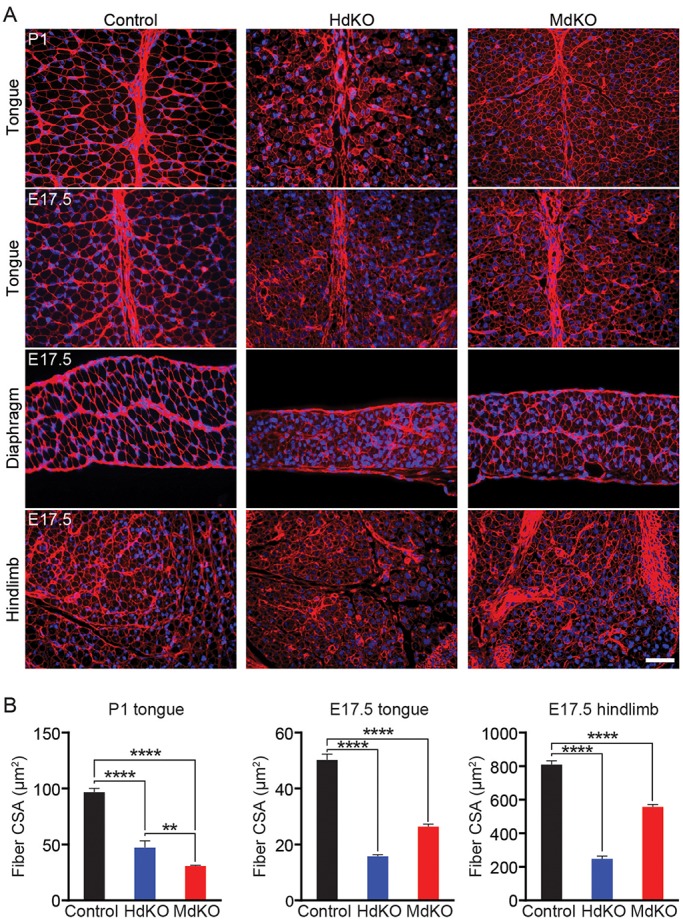
MRTF dKO mice have reduced myofiber cross-sectional areas. (A) Wheat germ agglutinin (WGA) staining of muscle membranes in tongue, diaphragm and hindlimb muscles of E17.5 and P1 mice show reduction of myofiber size in MdKO and HdKO mice compared with control (MRTF-A+/−; MRTF-Bflox/flox) littermates. WGA (red), DAPI (blue). Scale bar: 40 μm. (B) Quantification of images in A. Five images per sample were acquired and analyzed, from three mice per genotype. All data are shown as mean±s.e.m. Comparisons between groups were conducted using the unpaired t-test; two-tailed P-values are shown. **P<0.01, ****P<0.0001. CSA, cross-sectional area.
MRTF dKO mice display severe sarcomere defects
To determine whether sarcomere integrity or organization was affected in the MRTF dKO mice, we performed transmission electron microscopy on E17.5 hindlimb tissue of both HdKO and MdKO animals (Fig. 5). The sarcomeres observed in the HdKO and MdKO mice were sparse and smaller than normal. At higher magnification, the HdKO mice showed abnormal sarcomeres, with regions in the myofibers showing what appeared to be silhouettes of sarcomeres that did not mature correctly (Fig. 5). MdKO mice also showed this defect; although the general architecture of the sarcomere was present, the ultrastructural components of the organized thick and thin filaments, such as the A, I and M lines, were not distinctly visible. This indicates that although mice harboring the genetic deletion of MRTFs do not survive long enough to develop well-defined pathologies of the muscle, MRTFs are nevertheless required for the full development of the sarcomere in vivo.
Fig. 5.
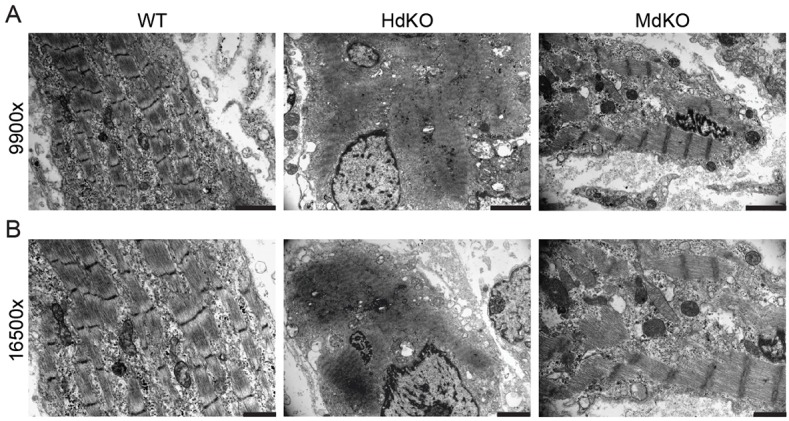
Electron microscopy reveals sarcomere defects in MRTF dKO muscles. (A,B) Transmission electron microscopic images of hindlimb tissue from E17.5 WT (MRTF-A+/+; MRTF-B+/+) and MRTF dKO mice showing a decrease in size and number of sarcomeres and impairment in sarcomere formation. Magnification values are shown on the left. Scale bars: 1 μm.
Dysregulation of genes involved in actin cycling and cytoskeletal development in MRTF dKO mice
To understand the molecular mechanisms underlying the failure of MRTF dKO mice to thrive, we performed RNA-seq analysis on hindlimb muscle collected from E17.5 HdKO animals. A total of 148 genes were found to be dysregulated at the mRNA level (Fig. 6A), with 111 genes downregulated and 37 upregulated (for a full list of these genes, refer to Table S2). Gene ontology (GO) analysis showed that pathways involved in cytoskeletal organization were highly dysregulated in the MRTF dKO mice (Fig. 6B; Fig. S3B). Many genes in these categories are known downstream targets of MRTF/SRF signaling (Esnault et al., 2014). Targets from RNA-seq were validated by RT-qPCR analysis (Fig. 6D).
Fig. 6.

MRTF dKO mice demonstrate a dysregulation in cytoskeletal genes. (A) Heat map showing the differentially expressed genes in muscle tissue from WT (MRTF-A+/+; MRTF-B+/+) and MRTF dKO mice at E17.5. (B,C) Gene ontology analysis (B) and ingenuity pathway analysis (IPA) (C) of differentially expressed genes identified top pathways involved in cytoskeletal development and actin cycling. (D) RT-qPCR analysis was performed on E17.5 hindlimb HdKO and WT muscle tissue to validate selected RNA-seq hits and selected MRTF/SRF target genes. Experiments were performed in triplicate with three biological replicates and expression was normalized to 18S rRNA. All data are shown as mean±s.e.m. The Kruskal–Wallis test was used for multiple comparisons, followed by Dunn's post-test for pairwise comparisons. ***P<0.001, ****P<0.0001.
Because the MRTF pathway functions through the binding of the MRTF/SRF complex to the CArG box motif, we investigated whether any of our RNA-seq hits were known transcriptional targets of MRTF/SRF. Previously, Esnault and colleagues identified MRTF/SRF target genes by ChIP-seq analysis in cultured fibroblasts, which include but are not limited to actin filament genes, genes involved in cellular motility and adhesion, and transcriptional machinery regulators (Esnault et al., 2014). Comparison of our hits from RNA-seq with these MRTF ChIP-seq datasets revealed that the overlapping targets mainly included actin cycling-associated genes (Fig. S3C), providing additional evidence that the deletion of MRTFs in vivo disrupts the actin-dependent transcriptional circuit required for muscle development. This is substantiated by the observation of a decrease in the mRNA expression of Lmod3, which encodes a known actin-binding protein that is a target of MRTFs (Fig. 6D).
MRTF dKO myoblasts have reduced survival and defects in myotube formation
To investigate further the mechanistic basis of the muscle pathology observed in the dKO mice, we isolated primary myoblasts from Cre-negative MRTF-A−/−; MRTF-Bflox/flox embryos at E17.5, as previously described (Millay et al., 2013). Cre recombinase was introduced by adenoviral infection to delete MRTF-B in the cells and efficiency of recombination was confirmed by genotyping for the MRTF-B floxed and null alleles (Fig. 7A). Immunocytochemistry with an antibody against phospho-histone H3, a marker of mitosis, demonstrated a marked decrease in the proliferation of dKO myoblasts compared with MRTF-A−/−; MRTF-Bflox/flox (referred to as control) myoblasts (Fig. 7B,C). Furthermore, there was an increase in the apoptosis marker cleaved caspase 3 (Fig. 7D), indicating an overall decrease in survival of the dKO cells. Upon induction of differentiation, dKO myofibers were observed to form myotubes, as shown by fast myosin (MY-32) staining (Fig. 7D). However, these myotubes were shorter, thinner and more disorganized compared with their control counterparts (Fig. 7D). Interestingly, the myotubes positive for cleaved caspase 3 did not display or were weakly positive for MY-32 staining and did not fuse with other MY-32-positive cells (Fig. 7D, arrowheads). In summary, these results demonstrate that MRTFs are necessary for myoblast survival and myotube formation in vitro.
Fig. 7.
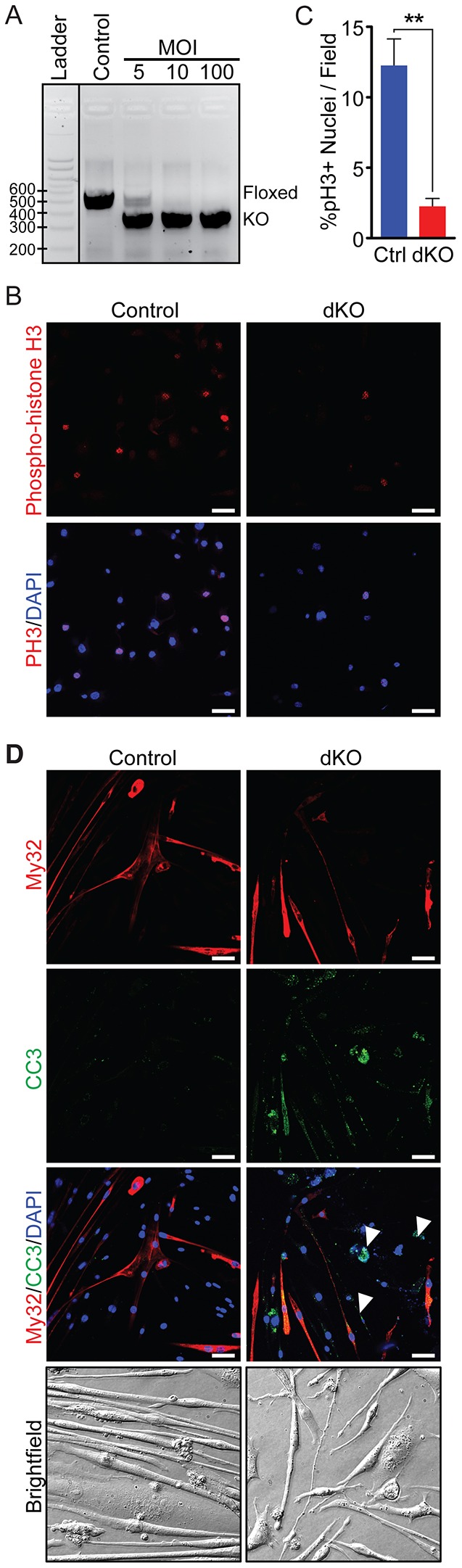
Loss of MRTFs leads to decreased myoblast survival and defects in myotube formation. (A) DNA gel demonstrating the loss of the floxed MRTF-B allele and the presence of the knockout MRTF-B allele upon induction with adenoviral Cre recombinase (first lane: adeno-CMV-null; last three lanes: adeno-CMV-iCre). Samples were run on a 2% TAE gel. MOI, multiplicity of infection. (B) Phospho-histone H3 (pH3) analysis of control (MRTF-A−/−; MRTF-Bflox/flox) and dKO (MRTF-A−/−; MRTF-B−/−) myoblasts demonstrates a decrease in proliferation in dKO myoblasts. (C) Quantification of images in panel B. All data are shown as mean±s.e.m. The two-tailed Mann–Whitney U test was used for pairwise comparisons between groups. **P<0.01. (D) Cleaved caspase-3 and MY-32 staining of control and dKO myotubes demonstrates an increase in apoptosis and myotube defects in dKO cultures. Scale bars: 40 μm.
DISCUSSION
In the present study, we demonstrate that MRTFs are necessary for skeletal muscle development in vivo and in vitro. The absence of MRTF-A and -B in skeletal muscle results in perinatal death accompanied by small myofibers and incomplete sarcomere formation. These results highlight the importance of the MRTF/SRF pathway as a central regulator of several muscle-specific genes (Cenik et al., 2015; Garg et al., 2014; Kuwahara et al., 2007; Meadows et al., 2008).
The contractile function of skeletal muscle relies on a large collection of contractile proteins and regulators of actin dynamics. Not only do MRTFs regulate a majority of the genes encoding these proteins, they also associate directly with monomeric G-actin to influence actin cycling (Olson and Nordheim, 2010). When bound to G-actin, MRTFs are prevented from associating with SRF, whereas in the unbound state they associate with SRF to drive expression of actin and sarcomeric proteins. Thus, the MRTF/SRF axis has a direct impact on the structural integrity of the components of the sarcomere. Moreover, these muscle-specific genes thought to signal through MRTF/SRF are important regulators of actin cycling. In the case of the muscle-specific protein LMOD3, MRTF regulates actin turnover by binding actin directly (Cenik et al., 2015). Other MRTF targets, such as MASTR, contribute to myogenesis by recruiting myogenic transcription factors, such as MyoD (Meadows et al., 2008).
We have recently shown that MRTFs are also required for the development and maintenance of cardiac muscle (Mokalled et al., 2015). Deletion of MRTF-A and -B in the heart led to postnatal lethality due to sarcomere disarray in the heart. Interestingly, the cardiac MRTF dKO mice demonstrated better survival than mice with skeletal muscle MRTF deletion: 75% of the mice died within the first 2 weeks of birth. This improved survival can possibly be attributed to compensation by myocardin, another member of the MRTF transcription factor family, which is required for cardiac development (Huang et al., 2009; Wang et al., 2001). Myocardin expression is restricted to the heart, and it is considered to be one of the primary regulators of SRF activity (Wang et al., 2001). There is no skeletal muscle-specific equivalent of myocardin, thus leading to a more severe phenotype in the muscle-specific MRTF dKO. Regardless, variability in phenotype penetrance and time of lethality was observed, which could be attributed to the mixed background of the MRTF dKO mice.
The MRTF/SRF axis is not only crucial for skeletal muscle and cardiac development, but also important for several other biological processes, such as the development of the hematopoietic system (Costello et al., 2015), thymocyte development (Costello et al., 2010; Mylona et al., 2011), glucose and lipid metabolism (McDonald et al., 2015; Sward et al., 2016), and neuronal development and motility (Kalita et al., 2012; Knoll, 2010).
The myogenic regulatory factors (MRFs) MYF5, MyoD, myogenin and MRF4 (MYF6) function as master regulators of myofibrillogenesis (reviewed by Berkes and Tapscott, 2005; Sweetman, 2012) and are also known to act as co-activators of SRF (Puri and Sartorelli, 2000). Genetic deletions of Mrf4, MyoD or Myf5 alone do not produce overt defects in sarcomere formation in mice (Braun et al., 1992; Rawls et al., 1998; Rudnicki et al., 1992), whereas genetic deletion of myogenin causes perinatal lethality due to a complete lack of myoblast fusion and myofiber formation (Hasty et al., 1993; Nabeshima et al., 1993). MyoD has also been shown to associate directly with SRF and to serve as a co-activator of SRF-dependent muscle genes (L'Honore et al., 2003). Interestingly, the MRTF dKO phenotype is more severe than the phenotypes of mice with single deletions of MyoD, Myf5 or Mrf4. The MTRF dKO phenotype is instead similar to that of the SRF deletion, indicating a central role for the SRF/MRTF axis in muscle development (Li et al., 2005b).
The RNA-seq analysis of MRTF dKO muscle revealed extreme dysregulation of genes involved in sarcomere formation and function. Not only were genes coding for actins and myosins downregulated in these animals, but also other components of the cytoskeletal machinery, as well as direct transcriptional targets of MRTF/SRF and several members of the Rho GTPase family, were found to be downregulated. Rho GTPases are effector proteins of cell motility signaling, which promote actin polymerization by activating several pathways including the ROCK-LiM-cofilin pathway (Cotton and Claing, 2009; Jaffe and Hall, 2005). Finally, the dysregulation of genes encoding proteins involved in cell-cell junctions, such as junctophilins and dysferlin, indicate that loss of MRTFs not only disrupts cell motility, but also intercellular interactions.
Myoblasts lacking MRTF-A and -B showed impaired proliferation and an increase in apoptosis, indicating that MRTFs are necessary for survival of muscle cells. The differentiation and fusion of myoblasts into functional myotubes, which is necessary for the formation of functional myofibers, was not completely blocked by the loss of MRTFs. In dKO myoblasts, we observed no change in expression of myomaker (TMEM8C), a key trigger for myoblast fusion (Millay et al., 2013). However, MRTF dKO myotubes were structurally impaired, demonstrating a marked decrease in myotube size and disorganization of existing myotubes. This was partially attributable to the decrease in survival, as the cells undergoing apoptosis were unable to contribute to myotube formation. In addition, given our prior demonstration of the importance of sarcomere components for myotube elongation and growth (Johnson et al., 2013), we speculate that dysregulation of these types of SRF targets in MRTF dKO myoblasts disrupts the formation of multinucleated myotubes.
Several pathologies of skeletal muscle, including severe congenital myopathies such as nemaline myopathy, are characterized by sarcomeric disarray caused by abnormalities in actin regulation (de Rezende Pinto et al., 2015; Romero et al., 2013). Interestingly, unlike the genetic deletion of MRTF/SRF targets, such as LMOD3, loss of MRTFs in skeletal muscle did not cause nemaline myopathy. This suggests that although MRTF/SRF target genes play a greater role in sarcomere maintenance and disease states, the MRTF/SRF signaling cascade itself has a more fundamental role in maintaining cytoskeletal architecture, and therefore directly impacts development. Given the evidence that MRTFs are required for the hypertrophy of skeletal muscle during development (Collard et al., 2014; Lamon et al., 2009; Mokalled et al., 2012), it would be interesting to investigate whether the in vivo overexpression of MRTFs can rescue diseases with degenerative and skeletal muscle atrophy phenotypes. Overall, our findings provide further insight into the mechanistic basis of muscle development and identify MRTFs as potential therapeutic targets for skeletal muscle diseases associated with muscle wasting and hypoplasia.
MATERIALS AND METHODS
Generation of mouse lines
MRTF-A−/− and MRTF-Bflox/flox were previously generated as described (Li et al., 2005a; Mokalled et al., 2010) and crossed to myogenin and HSA-Cre animals in order to obtain dKO animals. As female MRTF-A null mice do not lactate, the breeding required the use of MRTF-A+/−; MRTF-Bflox/flox females. Cre-negative MRTF-A+/−; MRTF-Bflox/flox and/or Cre-negative MRTF-A−/−; MRTF-Bflox/flox animals were used as controls for all experiments, with the exception of the RNA-seq analysis, for which wild-type animals of the same genetic background were used. All experimental procedures involving animals in this study were reviewed and approved by the Institutional Animal Care and Use Committee of the University of Texas (UT) Southwestern Medical Center. See supplementary Materials and Methods for details of genotyping.
Histology and immunohistochemistry
All tissues were fixed in 4% paraformaldehyde, followed by paraffin embedding and sectioning, followed by routine Hematoxylin and Eosin (H&E) staining as previously described (Cenik et al., 2015). Wheat germ agglutinin (WGA) and DAPI staining was performed using Alexa Fluor 555-conjugated WGA (Molecular Probes, W32464) as described previously (Liu et al., 2011). Imaging of H&E-stained slides and immunostained slides was carried out on a Leica DM2000 upright microscope. Myofiber sizes were calculated using ImageJ software (NIH) for area threshold and particle analysis.
Electron microscopy analysis
For electron microscopy analysis, samples were prepared for electron microscopy as previously described (Cenik et al., 2015), and images were acquired using a FEI Tecnai G2 Spirit electron microscope equipped with an LaB6 source Gatan CCD camera and operated at 120 kV.
RNA extraction and RT-qPCR analysis
Total RNA was extracted from animal tissue using the TRIzol reagent (Thermo Fisher Scientific), as per manufacturer's instructions. RT-qPCR analysis for validation of MRTF-A and MRTF-B takeout was performed as previously described (Mokalled et al., 2015). Validation of RNA-seq hits was performed using TaqMan probes (for a full list of probes, see Table S1). See supplementary Materials and Methods for further details.
RNA-seq analysis of MRTF dKO tissue
E17.5 embryos were dissected and hindlimb muscle was removed by separating the muscle and connective tissue of the de-skinned limb from the bone, followed by snap freezing of the tissue with liquid nitrogen. The frozen tissue was thawed in TRIzol reagent, and hand-homogenized by mini-pestle, followed by homogenization with a 20G needle. Following chloroform extraction, samples were loaded on phase-lock gel tubes (Eppendorf, Heavy 2.0 ml) and centrifuged. The supernatant from this extraction was used for RNA extraction using the RNeasy Mini Kit (Qiagen) as per manufacturer's instructions. RNA quality was verified by the Agilent 2100 Bioanalyzer and RNA-seq was performed using Illumina HiSeq 2500 by the UT Southwestern Medical Center Genomics and Microarray Core Facility. Data analysis was performed by the UT Southwestern Department of Clinical Sciences. For detailed information on the bioinformatic analysis of RNA-seq data, refer to the supplementary Materials and Methods.
Isolation and culturing of primary embryonic myoblasts
Myoblasts were isolated from MRTF-A−/−; MRTF-Bflox/flox females bred to males of the same genotype, at E17.5, as previously described (Millay et al., 2013). Following enrichment for myoblasts, experiments were performed using pure myoblast populations plated on laminin-coated cell culture dishes. MRTF-B was deleted by infecting cells with a pre-packaged adenoviral Cre recombinase (CMV-iCre, Vector Biolabs); control cells were concurrently infected with a null adenovirus counterpart (CMV-null Vector Biolabs). Following titration of adenoviral particles in order to determine the optimal multiplicity of infection (MOI), experiments were conducted following 48 h of infection at a MOI of 100.
Immunocytochemistry
Primary myoblasts were cultured in growth media (Ham's F-10, 20% fetal bovine serum, 1% penicillin-streptomycin) for 48 h after infection with adenoviruses. Following this, cells were either fixed with 4% paraformaldehyde and stained with an antibody against phospho-histone H3 (Ser10; 06-570, Millipore), or differentiation was induced by serum starvation (Dulbecco's modified Eagle medium, 2% horse serum). Induced cells were allowed to differentiate for 48 h, after which they were fixed and stained with MY-32 (Sigma) and cleaved caspase 3 (Cell Signaling). All antibodies were used at 1:100 and immunofluorescence staining was performed as previously described (Kutluk Cenik et al., 2013; Millay et al., 2013). Cells in 35 mm tissue culture dishes were visualized on a Leica DM2000 upright epifluorescent microscope and a Zeiss LSM780 upright confocal microscope.
Statistics
All data are presented as the mean value or percentage change±s.e.m. Comparisons between two datasets were made using the Mann–Whitney U test for nonparametric datasets and the two-tailed Student's t-test for parametric datasets. The Kruskal–Wallis test was used for non-parametric analysis of multiple groups, and Dunn's post-test was used for pairwise comparisons. A P-value of less than 0.05 was considered statistically significant. Statistical significance is indicated as follows: *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
Acknowledgements
We thank Rebecca Jackson, Robyn Leidel, Phoebe Doss, Anza Darehshouri and Kate Luby-Phelps of the UT Southwestern Electron Microscopy Core Facility for their assistance and advice. We would also like to thank Abhjit Bughde and Kate Luby-Phelps of the UT Southwestern Live Cell Imaging Core Facility for assistance with confocal microscopy (K.L.P. and the LCIF are funded by the NIH, grant S10 RR029731-01). We are grateful to José Cabrera for assistance with graphics.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
B.K.C., N.L., E.N.O. and R.B.-D. developed the concepts and performed data analysis; B.K.C., S.B. and N.L. performed experiments; B.C. performed bioinformatics analysis; and B.K.C., N.L., E.N.O. and R.B.-D. prepared and edited the manuscript.
Funding
This work was supported by grants from the National Institutes of Health (NIH) [HL-077439, DK-099653, U01-HL-100401, AR-067294, HD-087351 and HL-130253]; and the Robert A. Welch Foundation [1-0025, to E.N.O.]. N.L. is supported by a Beginning Grant-in-Aid (BGIA) from the American Heart Association (AHA) [13BGIA17150004]. B.K.C. is supported by a T32 NIH Pharmacological Sciences training grant [5T32GM007062-40]. Deposited in PMC for release after 12 months.
Data availability
RNA-seq data are available at Gene Expression Omnibus under accession number GSE84063 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE84063).
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.135855.supplemental
References
- Arsenian S., Weinhold B., Oelgeschläger M., Rüther U. and Nordheim A. (1998). Serum response factor is essential for mesoderm formation during mouse embryogenesis. EMBO J. 17, 6289-6299. 10.1093/emboj/17.21.6289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkes C. A. and Tapscott S. J. (2005). MyoD and the transcriptional control of myogenesis. Semin. Cell Dev. Biol. 16, 585-595. 10.1016/j.semcdb.2005.07.006 [DOI] [PubMed] [Google Scholar]
- Braun T., Rudnicki M. A., Arnold H.-H. and Jaenisch R. (1992). Targeted inactivation of the muscle regulatory gene Myf-5 results in abnormal rib development and perinatal death. Cell 71, 369-382. 10.1016/0092-8674(92)90507-9 [DOI] [PubMed] [Google Scholar]
- Cen B., Selvaraj A., Burgess R. C., Hitzler J. K., Ma Z., Morris S. W. and Prywes R. (2003). Megakaryoblastic leukemia 1, a potent transcriptional coactivator for serum response factor (SRF), is required for serum induction of SRF target genes. Mol. Cell. Biol. 23, 6597-6608. 10.1128/MCB.23.18.6597-6608.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenik B. K., Garg A., McAnally J. R., Shelton J. M., Richardson J. A., Bassel-Duby R., Olson E. N. and Liu N. (2015). Severe myopathy in mice lacking the MEF2/SRF-dependent gene leiomodin-3. J. Clin. Invest. 125, 1569-1578. 10.1172/JCI80115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng T. C., Wallace M. C., Merlie J. P. and Olson E. N. (1993). Separable regulatory elements governing myogenin transcription in mouse embryogenesis. Science 261, 215-218. 10.1126/science.8392225 [DOI] [PubMed] [Google Scholar]
- Collard L., Herledan G., Pincini A., Guerci A., Randrianarison-Huetz V. and Sotiropoulos A. (2014). Nuclear actin and myocardin-related transcription factors control disuse muscle atrophy through regulation of Srf activity. J. Cell Sci. 127, 5157-5163. 10.1242/jcs.155911 [DOI] [PubMed] [Google Scholar]
- Costello P., Nicolas R., Willoughby J., Wasylyk B., Nordheim A. and Treisman R. (2010). Ternary complex factors SAP-1 and Elk-1, but not net, are functionally equivalent in thymocyte development. J. Immunol. 185, 1082-1092. 10.4049/jimmunol.1000472 [DOI] [PubMed] [Google Scholar]
- Costello P., Sargent M., Maurice D., Esnault C., Foster K., Anjos-Afonso F. and Treisman R. (2015). MRTF-SRF signaling is required for seeding of HSC/Ps in bone marrow during development. Blood 125, 1244-1255. 10.1182/blood-2014-08-595603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotton M. and Claing A. (2009). G protein-coupled receptors stimulation and the control of cell migration. Cell. Signal. 21, 1045-1053. 10.1016/j.cellsig.2009.02.008 [DOI] [PubMed] [Google Scholar]
- Creemers E. E., Sutherland L. B., Oh J., Barbosa A. C. and Olson E. N. (2006). Coactivation of MEF2 by the SAP domain proteins myocardin and MASTR. Mol. Cell 23, 83-96. 10.1016/j.molcel.2006.05.026 [DOI] [PubMed] [Google Scholar]
- Croissant J. D., Kim J.-H., Eichele G., Goering L., Lough J., Prywes R. and Schwartz R. J. (1996). Avian serum response factor expression restricted primarily to muscle cell lineages is required for alpha-actin gene transcription. Dev. Biol. 177, 250-264. 10.1006/dbio.1996.0160 [DOI] [PubMed] [Google Scholar]
- de Rezende Pinto W. B., de Souza P. V. S. and Oliveira A. S. B. (2015). Normal muscle structure, growth, development, and regeneration. Curr. Rev. Musculoskelet. Med. 8, 176-181. 10.1007/s12178-015-9267-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esnault C., Stewart A., Gualdrini F., East P., Horswell S., Matthews N. and Treisman R. (2014). Rho-actin signaling to the MRTF coactivators dominates the immediate transcriptional response to serum in fibroblasts. Genes Dev. 28, 943-958. 10.1101/gad.239327.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg A., O'Rourke J., Long C., Doering J., Ravenscroft G., Bezprozvannaya S., Nelson B. R., Beetz N., Li L., Chen S. et al. (2014). KLHL40 deficiency destabilizes thin filament proteins and promotes nemaline myopathy. J. Clin. Invest. 124, 3529-3539. 10.1172/JCI74994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasty P., Bradley A., Morris J. H., Edmondson D. G., Venuti J. M., Olson E. N. and Klein W. H. (1993). Muscle deficiency and neonatal death in mice with a targeted mutation in the myogenin gene. Nature 364, 501-506. 10.1038/364501a0 [DOI] [PubMed] [Google Scholar]
- Huang J., Min Lu M., Cheng L., Yuan L.-J., Zhu X., Stout A. L., Chen M., Li J. and Parmacek M. S. (2009). Myocardin is required for cardiomyocyte survival and maintenance of heart function. Proc. Natl. Acad. Sci. USA 106, 18734-18739. 10.1073/pnas.0910749106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe A. B. and Hall A. (2005). Rho GTPases: biochemistry and biology. Annu. Rev. Cell Dev. Biol. 21, 247-269. 10.1146/annurev.cellbio.21.020604.150721 [DOI] [PubMed] [Google Scholar]
- Johnson A. N., Mokalled M. H., Valera J. M., Poss K. D. and Olson E. N. (2013). Post-transcriptional regulation of myotube elongation and myogenesis by Hoi Polloi. Development 140, 3645-3656. 10.1242/dev.095596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalita K., Kuzniewska B. and Kaczmarek L. (2012). MKLs: co-factors of serum response factor (SRF) in neuronal responses. Int. J. Biochem. Cell Biol. 44, 1444-1447. 10.1016/j.biocel.2012.05.008 [DOI] [PubMed] [Google Scholar]
- Knöll B. (2010). Actin-mediated gene expression in neurons: the MRTF-SRF connection. Biol. Chem. 391, 591-597. 10.1515/bc.2010.061 [DOI] [PubMed] [Google Scholar]
- Kutluk Cenik B., Ostapoff K. T., Gerber D. E. and Brekken R. A. (2013). BIBF 1120 (nintedanib), a triple angiokinase inhibitor, induces hypoxia but not EMT and blocks progression of preclinical models of lung and pancreatic cancer. Mol. Cancer Ther. 12, 992-1001. 10.1158/1535-7163.MCT-12-0995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwahara K., Teg Pipes G. C., McAnally J., Richardson J. A., Hill J. A., Bassel-Duby R. and Olson E. N. (2007). Modulation of adverse cardiac remodeling by STARS, a mediator of MEF2 signaling and SRF activity. J. Clin. Invest. 117, 1324-1334. 10.1172/JCI31240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahoute C., Sotiropoulos A., Favier M., Guillet-Deniau I., Charvet C., Ferry A., Butler-Browne G., Metzger D., Tuil D. and Daegelen D. (2008). Premature aging in skeletal muscle lacking serum response factor. PLoS ONE 3, e3910 10.1371/journal.pone.0003910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamon S., Wallace M. A., Léger B. and Russell A. P. (2009). Regulation of STARS and its downstream targets suggest a novel pathway involved in human skeletal muscle hypertrophy and atrophy. J. Physiol. 587, 1795-1803. 10.1113/jphysiol.2009.168674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- L'Honore A., Lamb N. J., Vandromme M., Turowski P., Carnac G. and Fernandez A. (2003). MyoD distal regulatory region contains an SRF binding CArG element required for MyoD expression in skeletal myoblasts and during muscle regeneration. Mol. Biol. Cell 14, 2151-2162. 10.1091/mbc.E02-07-0451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Zhu X., Chen M., Cheng L., Zhou D., Lu M. M., Du K., Epstein J. A. and Parmacek M. S. (2005a). Myocardin-related transcription factor B is required in cardiac neural crest for smooth muscle differentiation and cardiovascular development. Proc. Natl. Acad. Sci. USA 102, 8916-8921. 10.1073/pnas.0503741102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S., Czubryt M. P., McAnally J., Bassel-Duby R., Richardson J. A., Wiebel F. F., Nordheim A. and Olson E. N. (2005b). Requirement for serum response factor for skeletal muscle growth and maturation revealed by tissue-specific gene deletion in mice. Proc. Natl. Acad. Sci. USA 102, 1082-1087. 10.1073/pnas.0409103102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S., Chang S., Qi X., Richardson J. A. and Olson E. N. (2006). Requirement of a myocardin-related transcription factor for development of mammary myoepithelial cells. Mol. Cell. Biol. 26, 5797-5808. 10.1128/MCB.00211-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N., Bezprozvannaya S., Shelton J. M., Frisard M. I., Hulver M. W., McMillan R. P., Wu Y., Voelker K. A., Grange R. W., Richardson J. A. et al. (2011). Mice lacking microRNA 133a develop dynamin 2-dependent centronuclear myopathy. J. Clin. Invest. 121, 3258-3268. 10.1172/JCI46267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald M. E., Li C., Bian H., Smith B. D., Layne M. D. and Farmer S. R. (2015). Myocardin-related transcription factor A regulates conversion of progenitors to beige adipocytes. Cell 160, 105-118. 10.1016/j.cell.2014.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meadows S. M., Warkman A. S., Salanga M. C., Small E. M. and Krieg P. A. (2008). The myocardin-related transcription factor, MASTR, cooperates with MyoD to activate skeletal muscle gene expression. Proc. Natl. Acad. Sci. USA 105, 1545-1550. 10.1073/pnas.0703918105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miano J. M. (2003). Serum response factor: toggling between disparate programs of gene expression. J. Mol. Cell. Cardiol. 35, 577-593. 10.1016/S0022-2828(03)00110-X [DOI] [PubMed] [Google Scholar]
- Millay D. P., O'Rourke J. R., Sutherland L. B., Bezprozvannaya S., Shelton J. M., Bassel-Duby R. and Olson E. N. (2013). Myomaker is a membrane activator of myoblast fusion and muscle formation. Nature 499, 301-305. 10.1038/nature12343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miniou P., Tiziano D., Frugier T., Roblot N., Le Meur M. and Melki J. (1999). Gene targeting restricted to mouse striated muscle lineage. Nucleic Acids Res. 27, e27 10.1093/nar/27.19.e27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miralles F., Posern G., Zaromytidou A.-I. and Treisman R. (2003). Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell 113, 329-342. 10.1016/S0092-8674(03)00278-2 [DOI] [PubMed] [Google Scholar]
- Mokalled M. H., Johnson A., Kim Y., Oh J. and Olson E. N. (2010). Myocardin-related transcription factors regulate the Cdk5/Pctaire1 kinase cascade to control neurite outgrowth, neuronal migration and brain development. Development 137, 2365-2374. 10.1242/dev.047605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokalled M. H., Johnson A. N., Creemers E. E. and Olson E. N. (2012). MASTR directs MyoD-dependent satellite cell differentiation during skeletal muscle regeneration. Genes Dev. 26, 190-202. 10.1101/gad.179663.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokalled M. H., Carroll K. J., Cenik B. K., Chen B., Liu N., Olson E. N. and Bassel-Duby R. (2015). Myocardin-related transcription factors are required for cardiac development and function. Dev. Biol. 406, 109-116. 10.1016/j.ydbio.2015.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mylona A., Nicolas R., Maurice D., Sargent M., Tuil D., Daegelen D., Treisman R. and Costello P. (2011). The essential function for serum response factor in T-cell development reflects its specific coupling to extracellular signal-regulated kinase signaling. Mol. Cell. Biol. 31, 267-276. 10.1128/MCB.01058-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabeshima Y., Hanaoka K., Hayasaka M., Esumi E., Li S., Nonaka I. and Nabeshima Y.-I. (1993). Myogenin gene disruption results in perinatal lethality because of severe muscle defect. Nature 364, 532-535. 10.1038/364532a0 [DOI] [PubMed] [Google Scholar]
- Norman C., Runswick M., Pollock R. and Treisman R. (1988). Isolation and properties of cDNA clones encoding SRF, a transcription factor that binds to the c-fos serum response element. Cell 55, 989-1003. 10.1016/0092-8674(88)90244-9 [DOI] [PubMed] [Google Scholar]
- Olson E. N. and Nordheim A. (2010). Linking actin dynamics and gene transcription to drive cellular motile functions. Nat. Rev. Mol. Cell Biol. 11, 353-365. 10.1038/nrm2890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posern G. and Treisman R. (2006). Actin’ together: serum response factor, its cofactors and the link to signal transduction. Trends Cell Biol. 16, 588-596. 10.1016/j.tcb.2006.09.008 [DOI] [PubMed] [Google Scholar]
- Puri P. L. and Sartorelli V. (2000). Regulation of muscle regulatory factors by DNA-binding, interacting proteins, and post-transcriptional modifications. J. Cell. Physiol. 185, 155-173. [DOI] [PubMed] [Google Scholar]
- Rawls A., Valdez M. R., Zhang W., Richardson J., Klein W. H. and Olson E. N. (1998). Overlapping functions of the myogenic bHLH genes MRF4 and MyoD revealed in double mutant mice. Development 125, 2349-2358. [DOI] [PubMed] [Google Scholar]
- Romero N. B., Sandaradura S. A. and Clarke N. F. (2013). Recent advances in nemaline myopathy. Curr. Opin. Neurol. 26, 519-526. 10.1097/WCO.0b013e328364d681 [DOI] [PubMed] [Google Scholar]
- Rudnicki M. A., Braun T., Hinuma S. and Jaenisch R. (1992). Inactivation of MyoD in mice leads to up-regulation of the myogenic HLH gene Myf-5 and results in apparently normal muscle development. Cell 71, 383-390. 10.1016/0092-8674(92)90508-A [DOI] [PubMed] [Google Scholar]
- Selvaraj A. and Prywes R. (2003). Megakaryoblastic leukemia-1/2, a transcriptional co-activator of serum response factor, is required for skeletal myogenic differentiation. J. Biol. Chem. 278, 41977-41987. 10.1074/jbc.M305679200 [DOI] [PubMed] [Google Scholar]
- Sun Y., Boyd K., Xu W., Ma J., Jackson C. W., Fu A., Shillingford J. M., Robinson G. W., Hennighausen L., Hitzler J. K. et al. (2006). Acute myeloid leukemia-associated Mkl1 (Mrtf-a) is a key regulator of mammary gland function. Mol. Cell. Biol. 26, 5809-5826. 10.1128/MCB.00024-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sward K., Stenkula K. G., Rippe C., Alajbegovic A., Gomez M. F. and Albinsson S. (2016). Emerging roles of the myocardin family of proteins in lipid and glucose metabolism. J. Physiol. (in press). 10.1113/jp271913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweetman D. (2012). The myogenic regulatory factors: critical determinants of muscle identity in development, growth and regeneration In Skeletal Muscle: From Myogenesis to Clinical Relations (ed. J. Cseri), pp. 31-48. Rijeka: InTech.
- Treisman R. (1986). Identification of a protein-binding site that mediates transcriptional response of the c-fos gene to serum factors. Cell 46, 567-574. 10.1016/0092-8674(86)90882-2 [DOI] [PubMed] [Google Scholar]
- Treisman R. (1992). The serum response element. Trends Biochem. Sci. 17, 423-426. 10.1016/0968-0004(92)90013-Y [DOI] [PubMed] [Google Scholar]
- Wang D., Chang P. S., Wang Z., Sutherland L., Richardson J. A., Small E., Krieg P. A. and Olson E. N. (2001). Activation of cardiac gene expression by myocardin, a transcriptional cofactor for serum response factor. Cell 105, 851-862. 10.1016/S0092-8674(01)00404-4 [DOI] [PubMed] [Google Scholar]
- Wang D. Z., Li S., Hockemeyer D., Sutherland L., Wang Z., Schratt G., Richardson J. A., Nordheim A. and Olson E. N. (2002). Potentiation of serum response factor activity by a family of myocardin-related transcription factors. Proc. Natl. Acad. Sci. USA 99, 14855-14860. 10.1073/pnas.222561499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L., Zhou W., Croissant J. D., Johansen F. E., Prywes R., Balasubramanyam A. and Schwartz R. J. (1998). RhoA signaling via serum response factor plays an obligatory role in myogenic differentiation. J. Biol. Chem. 273, 30287-30294. 10.1074/jbc.273.46.30287 [DOI] [PubMed] [Google Scholar]
- Yuen M., Sandaradura S. A., Dowling J. J., Kostyukova A. S., Moroz N., Quinlan K. G., Lehtokari V. L., Ravenscroft G., Todd E. J., Ceyhan-Birsoy O. et al. (2014). Leiomodin-3 dysfunction results in thin filament disorganization and nemaline myopathy. J. Clin. Invest. 124, 4693-4708. 10.1172/JCI75199 [DOI] [PMC free article] [PubMed] [Google Scholar]