

Abstract
Infection with the protozoan Leishmania infantum can lead to asymptomatic infection and protective immunity, or to the progressive and potentially fatal disease visceral leishmaniasis (VL). Published studies show host genetic background determines in part whether infected individuals will develop a symptomatic or asymptomatic outcome. The purpose of the current study was to fine map chromosome regions previously linked with risk for symptomatic (chromosome 9) or asymptomatic (chromosomes 15 and 19) manifestations of L. infantum infection. We conducted a family-based genetic study of VL and asymptomatic infection (detected by a DTH skin test) with a final post quality control sample of 961 individuals with full genotype and phenotype information from highly endemic neighborhoods of northeast Brazil. A total of 5485 SNPs under the linkage peaks on chromosomes 9, 15 and 19 were genotyped. No strong SNP associations were observed for the DTH phenotype. The most significant associations with the VL phenotype were with SNP rs1470217 (p = 5.9e−05; pcorrected = 0.057) on chromosome 9, and with SNP rs8107014 (p = 1.4e−05; pcorrected = 0.013) on chromosome 19. SNP rs1470217 is situated in a 180 kb intergenic region between TMEM215 (Transmembrane protein 215) and APTX (Aprataxin). SNP rs8107014 lies in the intron between exons 26 and 27 of a 34 exon transcript (ENST00000204005) of LTBP4, (Latent transforming growth factor-beta-binding protein 4a). The latter supports growing evidence that the transforming growth factor-beta pathway is important in the immunopathogenesis of VL.
Keywords: Visceral leishmaniasis, Fine mapping, Linkage regions, Tropical disease, Genetic risk factors
1. Introduction
Visceral leishmaniasis (VL) is a debilitating parasitic disease of humans caused by protozoa belonging to the Leishmania donovani complex. Symptomatic VL is a severe progressive infection which can be fatal even with treatment. Despite its potential severity, 80–90% of individuals infected with the causative parasites harbor either sub-clinical or asymptomatic infection (Blackwell et al., 2009). The hypothesis that human genetic variants also influence susceptibility to both VL and a positive DTH response is supported by segregation analyses in Brazilian populations (Feitosa et al., 1999; Peacock et al., 2001). Efforts to identify the specific genes conferring susceptibility have inspired candidate gene (reviewed Blackwell, 2010; Blackwell et al., 2009), as well as genome-wide linkage (Bucheton et al., 2003; Jamieson et al., 2007; Jeronimo et al., 2007a; Miller et al., 2007) and association (Fakiola et al., 2013) studies. Previously we carried out a genome-wide linkage study (Jeronimo et al., 2007a) that identified a region of putative linkage to symptomatic VL on human chromosome 9, with further regions on chromosomes 15 and 19 identified as carrying loci regulating asymptomatic disease as measured by a delayed type hypersensitivity (DTH) skin test response to crude leishmanial antigen. The purpose of the current study was to fine map these chromosome regions using high density single nucleotide polymorphism (SNP) genotyping and association analyses. The results support growing evidence that the transforming growth factor-beta pathway is important in the immunopathogenesis of VL.
2. Materials and methods
2.1. Subject sample and phenotype
Details of the study site in Natal, Rio Grande do Norte, Brazil, enrollment of subjects, and clinical phenotyping are described in full in our previous genome-wide linkage (Jeronimo et al., 2007a) and candidate gene (Jeronimo et al., 2007b) studies. Briefly, criteria for diagnosis of VL were a clinical presentation with hepatosplenomegaly, fever, cachexia and pancytopenia, positive parasitologic diagnosis (positive bone marrow aspirate, positive serology), and response to treatment. As before (Jeronimo et al., 2007a; Jeronimo et al., 2007b), the cutoff for a positive Montenegro test for Leishmania antigen was ≥5 mm of induration. The study was approved by the institutional review boards of the Universidade Federal do Rio Grande do Norte (numbers 19–01 and 21–01); the Comissão Nacional de Ética em Pesquisa (CONEP numbers 4581 and 4575); the University of Iowa; Johns Hopkins University; the University of Virginia; and the National Human Genome Research Institute, National Institutes of Health. Written consent was obtained from adults and from parents or guardians of minors <18 years of age, and written assent was obtained from minors 12–17 years of age.
2.2. Numbers of subjects
DNA for genotyping was available for 1200 individuals (49% male; 51% female), who all contributed to calculation of allele frequencies and linkage disequilibrium (LD) blocks. Full phenotype data was available for 961 genotyped individuals (145 VL; 421 DTH+; 395 DTH−). The study sample comprised 49% males and 51% females.
2.3. SNP selection and genotyping
SNPs (N = 6026) were selected to cover three regions of putative linkage in our prior study (Jeronimo et al., 2007a). Based on our knowledge of admixture in the region of northeast Brazil (Ettinger et al., 2009), tagging SNPs (minor allele frequency > 0.05) were selected from LD blocks using the CEU and YRI populations in HapMap (Table S1). SNP selection was based on >1 SNP per LD block with r2 > 0.8. SNPs between LD blocks were included to ensure coverage. The median distance between the 5485 post quality control (cf. below) SNPs was 10.2 kb. Genotyping was performed by the Center for Inherited Diseases Research at Johns Hopkins University, Baltimore, MD, USA, using the Illumina Infinium genoptyping platform. SNPs with median p < 0.001 for deviation from Hardy-Weinberg equilibrium (Wigginton et al., 2005) across unrelated individuals were removed. PEDSTATS (Wigginton and Abecasis, 2005) and MERLIN (Abecasis et al., 2002) software were used to remove Mendelian errors and unlikely genotypes (unlikely recombination events). Individuals or SNPs with >2% inconsistent calls or errors were removed from the analysis. Nuclear families with >5% errors were also excluded. After quality control, 5485 of the original 6026 SNPs were retained in the analysis. The call rate for SNPs among genotyped individuals was 99.91% after quality control.
2.4. Association analyses
Family-based association tests for qualitative traits (VL or DTH positive results) were conducted on all 5485 SNPs using the LAMP software package (Li et al., 2005). The population prevalence for DTH+ was set at 0.7, and at 0.5 for VL, based on observed prevalence in the study population. A modified Bonferroni threshold for significance was calculated to take account of the number of LD blocks identified using a conservative method (Gabriel et al., 2002) implemented in the Haploview program (Barrett et al., 2005). There were 289 blocks on chromosome 9, 355 blocks on chromosome 15 and 317 blocks on chromosome 19. Considering the total of 961 LD blocks, a threshold of p = 5.2e−05 (i.e. p = 0.05/961) was required to achieve significance at α = 0.05. Individual corrected p-values were nominal p-values multiplied by 961 LD blocks. In addition, p-values were calculated separately for each region from permutation tests, using a set of 1000 simulated populations generated with the MERLIN software simulation feature. Simulated data sets maintain the same allele frequencies and missing data points as the original study population. Plots of associations were generated with the Locuszoom software package (Pruim et al., 2010).
3. Results
3.1. Allelic associations
Table S2 lists the most significant associations (uncorrected p < 0.001) between each phenotype and markers in linkage regions.
On chromosome 9, SNP rs1470217 had a nominal p value of p = 5.9e−05 (empirical simulation p = 0.089; Bonferroni corrected p = 0.057) for association with a VL outcome (Table 1, Fig. 1, Table S2), with the A allele as the risk allele for VL. Despite the fact that SNP rs1470217 was selected as part of LD block 120 on chromosome 9 (see Table S1), this SNP association was not well supported by other SNPs in strong LD (Fig. 2A) in our study population. Therefore, deeper coverage of SNPs may be required to validate the association in this region. SNP rs1470217 is situated in a 180 kb intergenic region between TMEM215 (Transmembrane protein 215) and APTX (Aprataxin) (Fig. 2A). No significant associations were detected between the DTH phenotype and SNPs on chromosomes 9 (Fig. 1).
Table 1.
Locations and allele frequencies of the SNPs most highly associated with the VL phenotype. The number of VL affected individuals for each genotype is shown to provide insight into the possible effect of each allele. p-Values provided are: pun for uncorrected, pes for empirical simulation corrected, pcorr for modified Bonferroni correction (i.e. uncorrected p-value multiplied by 961 LD blocks). The overall threshold for significance taking account of the number of LD blocks is p = 5.2e−05 (i.e. p = 0.05/961 LD blocks) for an α = 0.05.
| Associated SNP | Position relative to nearest genes | Population allele frequencies | Genotype frequencies | Total (affected) per genotype | p-Values |
|---|---|---|---|---|---|
| rs1470217 | 115 kb downstream of TMEM215 | A = 0.678 | GG = 0.13 | GG = 107 (13) | pun = 5.9e−05 |
| Chr 9p21.1 | 69 kb downstream of APTX | G = 0.322 | AG = 0.45 | AG = 299 (78) | pes = 0.089 |
| Intergenic | AA = 0.42 | AA = 259 (48) | pcorr = 0.057 | ||
| rs8107014 | LTBP4 | C = 0.539 | CC = 0.34 | CC = 254 (48) | pun = 1.4e−05 |
| Chr 19q13.2 | Intronic | T = 0.461 | CT = 0.5 | CT = 331 (63) | pes = 0.022 |
| TT = 0.16 | TT = 80 (28) | pcorr = 0.013 |
Fig. 1.
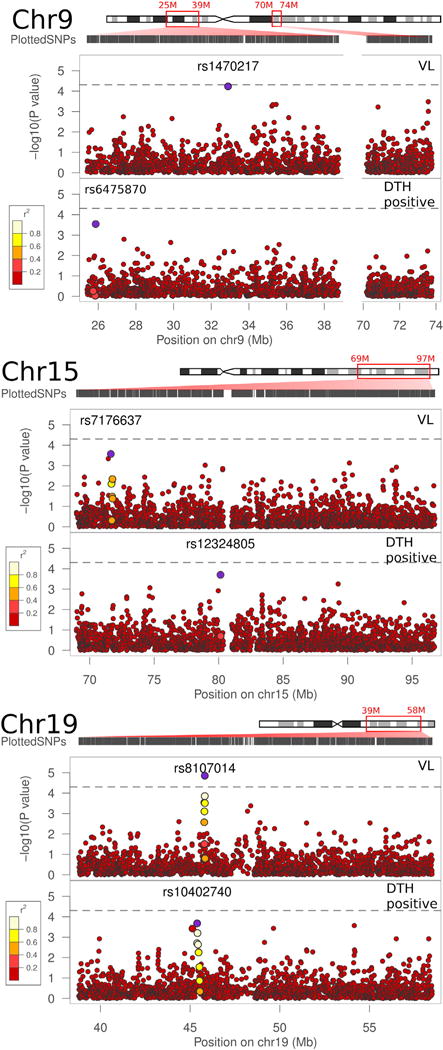
Allelic association results in linkage follow-up regions. Linkage follow-up regions on chromosomes 9 (A), 15 (B), and 19 (C) are indicated by the red boxes on the chromosome illustrations. The distribution of plotted SNPs across the region of interest is shown under the illustration of each chromosome. Three panels for each chromosome show the p-values for associations of SNPs with the VL and DTH positive traits (LAMP analysis). The large blue points represent the strongest association for that trait within the region of interest. The LD structure between the SNP with the best p-value and surrounding SNPs is indicated by a color gradient on other large points, with white being in strong LD, and red being in weak LD. The dashed line represents an adjusted Bonferroni threshold of significance at a p = 4.97e−05 cutoff. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Fig. 2.
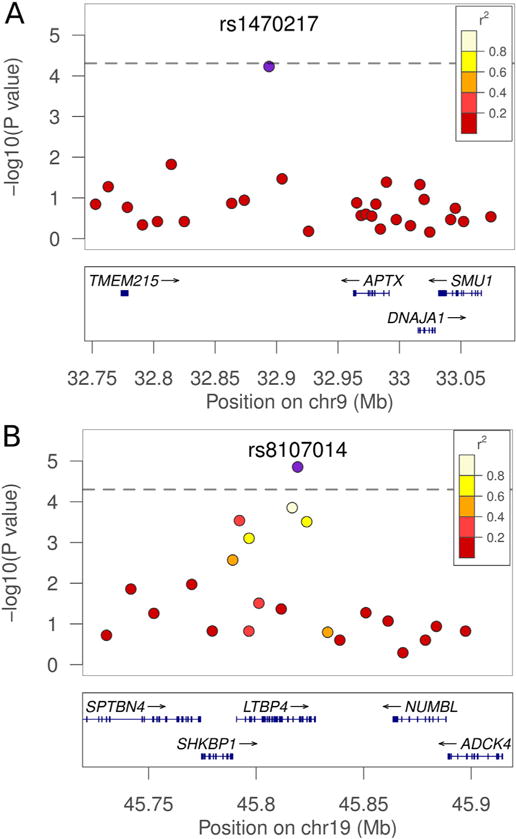
Regions of highest association with VL in linkage follow-up regions. Associations between the SNPs on chromosomes 9 (A) and 19 (B) are shown in detail. On chromosome 9, rs1470217 is indicated as the blue plot point. The distribution of SNPs covering 350 kb is shown above. On chromosome 19, rs8107014 is shown with 200 kb around the region. The blue points represent the strongest association for the VL trait within the region of interest. The LD structure between the SNP with the best p-value and surrounding SNPs is indicated by a color gradient on flanking SNPs, with white being in strong LD and red being in weak LD. The dashed line represents an adjusted Bonferroni threshold of significance at a p = 5.2e−05 cutoff (i.e. p = 0.05/961 LD blocks). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
On chromosome 15, no associations were observed for the VL or DTH phenotypes that withstood Bonferroni correction or permutation tests (Fig. 1, Table S2).
On chromosome 19, the most significant association (nominal p = 1.4e-05; empirical simulation p = 0.022; Bonferroni corrected p = 0.013) was observed between the VL phenotype and SNP rs8107014 (Table 1, Fig. 1, Table S2), with the minor T allele as the risk allele for VL. This SNP is located within the intron between the 26th and 27th exons of a 34 exon transcript (ENST00000204005) of LTBP4 (Latent transforming growth factor-beta-binding protein 4) (Fig. 2B), though shorter splice variants are also annotated. SNP rs8107014 has been previously reported to be associated with gene expression of LTBP4 in peripheral blood monocytes (Zeller et al., 2010), and has been recorded as part of an eQTL region in HaploRegv4.1 (http://www.broadinstitute.org/mammals/haploreg/detail_v4.1.php?query=&id=rs8107014). There were no significant associations of these SNPs with the DTH+phenotype. The SNP rs10402740 on chromosome 19 showing weak association (nominal p = 2.1e−04; Bonferroni corrected p = 0.202) with the DTH positive phenotype (Fig. 1, Table S2) is located 426 kb upstream of rs8107014, more proximal to the centromere, and is not in LD with rs8107014 (r2 = 0.005).
4. Discussion
The current study was designed to fine map regions under linkage peaks previously shown by us (Jeronimo et al., 2007a) to contain putative regions of linkage to VL susceptibility (chromosome 9), or to asymptomatic infection as determined by DTH skin test reactivity to leishmanial antigen (chromosomes 15 and 19). Although these regions were not highlighted in a recent large-scale genome-wide association of the VL phenotype in India and Brazil (Fakiola et al., 2013), the linkage peaks were considered worthy of follow-up as the only genome-wide linkage study in which the asymptomatic DTH skin test phenotype had been studied (Jeronimo et al., 2007a). In the event, the only associations that withstood correction for multiple testing were for the symptomatic VL phenotype, with high density SNP mapping pinpointing a region on Chromosome 9 intergenic between TMEM215 and APTX, and SNPs within LTBP4 on chromosome 19, as the peaks of association with VL. TMEM215 encodes a protein annotated as Transmembrane protein 215, with little known about its function other than having putative interactions with PDZ domains (Luck et al., 2011). APTX encodes Aprataxin, a member of the histidine triad superfamily, members of which have nucleotide binding and diadenosine polyphosphate hydrolase activities (Date et al., 2001). Deleterious mutations in APTX are associated with ataxia-ocular apraxia (Date et al., 2001), and Aprataxin is known to function together with other molecules to protect the genome against oxidative damage (Harris et al., 2009). At present there are no obvious functional links between either of these genes and VL pathogenesis. On the other hand, the more robust association observed for a group of intronic SNPs (top SNP rs8107014) at LTBP4 on chromosome 19 is of some interest in relation to prior knowledge of the role of the transforming growth factor beta (TGF-β) pathway in VL disease (Gantt et al., 2003; Gomes et al., 2000). For example, TGF-β is important in VL both as a suppressor of T cell responses (Gantt et al., 2003; Gomes et al., 2000) and potentially as an activator of T helper 17 development (Gantt et al., 2003). In addition, we have previously demonstrated genetic associations between SNPs at TGFBI on chromosome 5q31.1 and DTH responses to leishmanial antigen in our study population (Jeronimo et al., 2007b). TGFBI encodes the protein keratoepithelin, which is upregulated by TGF-β, and is expressed in skin epithelial cells where it could modulate the DTH phenotype. LTBP4 binds to inactive TGF-β complexed with latency associated peptide both intracellularly and upon release from cells (Oklo and Hesketh, 2000). The LTBP4 complex remains in extracellular tissues until activated via a number of mechanisms that alters its physiochemical characteristics, causing it to release its cargo. Further functional studies will be required to determine the possible role of LTBP4 in contributing to TGF-β regulation of VL pathogenesis. Our findings are also consistent with our prior reports of association between cutaneous leishmaniasis and genes in, or affecting, the TGF-β pathway (Castellucci et al., 2012; Castellucci et al., 2011), lending support to the broader importance of this pathway in pathogenesis of leishmaniasis.
Supplementary Material
Acknowledgments
This work was supported by NIH grant R01 AI076233 (JMB and MEW), AI045540 (MEW) and AI067874 (MEW, JMB). Data were collected under the Tropical Medicine Research Center grant P50 AI-30639 (SMBJ, MEW, JMB). Partial support was received from Merit Review grants from the Department of Veterans’ Affairs (1i01BX001983 and 5I01BX000536; MEW). The work was performed in part during the tenure of J.L.W. on NIH training grants T32 GM008629 and T32 GM082729. The authors are grateful to Anne Kwitek, Ph.D. and to Jeffrey Murray, M.D. for their helpful advice and discussions regarding genetic data analysis.
Footnotes
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.meegid.2016.05.005.
Competing interests
The authors declare they have no competing interests.
Author contributions
JLW performed statistical genetics analyses and drafted the manuscript. PD designed the fine mapping strategy, performed and advised on statistical genetics analyses. ELN participated in subject entry. GRM participated in subject entry and sample processing in Brazil. DRM participated in subject entry and sample processing in Brazil. HGL entered subjects, participated in data and sample collection and data gathering. MF advised on statistical genetics analyses, and revised the manuscript. JMB advised on statistical genetics analyses, and revised the manuscript. SMBJ and MEW oversaw the study design. SMBJ initiated subject entry, data collection and sample processing, supervised field site activities, was responsible for accuracy of data entry, and revised the manuscript. MEW supervised statistical genetics analyses and drafted the manuscript with JLW.
Contributor Information
Jason L. Weirather, Email: jason-weirather@uiowa.edu.
Priya Duggal, Email: pduggal@jhsph.edu.
Eliana L. Nascimento, Email: eltomaz@gmail.com, etomaz@cb.ufrn.br.
Gloria R. Monteiro, Email: gloriag74@hotmail.com.
Daniella R. Martins, Email: daniellamartins@cb.ufrn.br.
Henio G. Lacerda, Email: heniolacerda@ufrnet.br.
Michaela Fakiola, Email: mf300@cam.ac.uk.
Jenefer M. Blackwell, Email: jenefer.blackwell@telethonkids.org.au.
Selma M.B. Jeronimo, Email: smbj@cb.ufrn.br.
References
- Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin–rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- Blackwell JM. Chapter 35: Immunogenetics of host response to parasites in humans. In: Kaufmann SHE, Rouse B, Sacks D, editors. Immunology of Infectious Diseases. ASM Publications; Washington: 2010. pp. 483–490. [Google Scholar]
- Blackwell JM, Fakiola M, Ibrahim ME, Jamieson SE, Jeronimo SB, Miller EN, Mishra A, Mohamed HS, Peacock CS, Raju M, Sundar S, Wilson ME. Genetics and visceral leishmaniasis: of mice and man. Parasite Immunol. 2009;31:254–266. doi: 10.1111/j.1365-3024.2009.01102.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucheton B, Abel L, El-Safi S, Kheir MM, Pavek S, Lemainque A, Dessein AJ. A major susceptibility locus on chromosome 22q12 plays a critical role in the control of kala-azar. Am J Hum Genet. 2003;73:1052–1060. doi: 10.1086/379084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellucci L, Jamieson SE, Miller EN, de Almeida LF, Oliveira J, Magalhaes A, Guimaraes LH, Lessa M, Lago E, de Jesus AR, Carvalho EM, Blackwell JM. FLI1 polymorphism affects susceptibility to cutaneous leishmaniasis in Brazil. Genes Immun. 2011;12:589–594. doi: 10.1038/gene.2011.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellucci L, Jamieson SE, Almeida L, Oliveira J, Guimaraes LH, Lessa M, Fakiola M, Jesus AR, Nancy Miller E, Carvalho EM, Blackwell JM. Wound healing genes and susceptibility to cutaneous leishmaniasis in Brazil. Infect Genet Evol. 2012;12:1102–1110. doi: 10.1016/j.meegid.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Date H, Onodera O, Tanaka H, Iwabuchi K, Uekawa K, Igarashi S, Koike R, Hiroi T, Yuasa T, Awaya Y, Sakai T, Takahashi T, Nagatomo H, Sekijima Y, Kawachi I, Takiyama Y, Nishizawa M, Fukuhara N, Saito K, Sugano S, Tsuji S. Early-onset ataxia with ocular motor apraxia and hypoalbuminemia is caused by mutations in a new HIT superfamily gene. Nat Genet. 2001;29:184–188. doi: 10.1038/ng1001-184. [DOI] [PubMed] [Google Scholar]
- Ettinger NA, Duggal P, Braz RF, Nascimento ET, Beaty TH, Jeronimo SM, Pearson RD, Blackwell JM, Moreno L, Wilson ME. Genetic admixture in Brazilians exposed to infection with Leishmania chagasi. Ann Hum Genet. 2009;73:304–313. doi: 10.1111/j.1469-1809.2009.00510.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakiola M, Strange A, Cordell HJ, Miller EN, Pirinen M, Su Z, Mishra A, Mehrotra S, Monteiro GR, Band G, Bellenguez C, Dronov S, Edkins S, Freeman C, Giannoulatou E, Gray E, Hunt SE, Lacerda HG, Langford C, Pearson R, Pontes NN, Rai M, Singh SP, Smith L, Sousa O, Vukcevic D, Bramon E, Brown MA, Casas JP, Corvin A, Duncanson A, Jankowski J, Markus HS, Mathew CG, Palmer CN, Plomin R, Rautanen A, Sawcer SJ, Trembath RC, Viswanathan AC, Wood NW, Wilson ME, Deloukas P, Peltonen L, Christiansen F, Witt C, Jeronimo SM, Sundar S, Spencer CC, Blackwell JM, Donnelly P. Common variants in the HLA-DRB1-HLA-DQA1 HLA class II region are associated with susceptibility to visceral leishmaniasis. Nat Genet. 2013;45:208–213. doi: 10.1038/ng.2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feitosa MF, Axevedo E, Lima AM, Krieger H. Genetic causes involved in Leishmania chagasi infection in northeastern Brazil. Genet Mol Biol. 1999;22:1–5. [Google Scholar]
- Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, Higgins J, DeFelice M, Lochner A, Faggart M, Liu-Cordero SN, Rotimi C, Adeyemo A, Cooper R, Ward R, Lander ES, Daly MJ, Altshuler D. The structure of haplotype blocks in the human genome. Science. 2002;296:2225–2229. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- Gantt KR, Schultz-Cherry S, Rodriguez N, Jeronimo SM, Nascimento ET, Goldman TL, Recker TJ, Miller MA, Wilson ME. Activation of TGF-beta by Leishmania chagasi: importance for parasite survival in macrophages. J Immunol. 2003;170:2613–2620. doi: 10.4049/jimmunol.170.5.2613. [DOI] [PubMed] [Google Scholar]
- Gomes NA, Gattass CR, Barreto-De-Souza V, Wilson ME, DosReis GA. TGF-beta mediates CTLA-4 suppression of cellular immunity in murine kalaazar. J Immunol. 2000;164:2001–2008. doi: 10.4049/jimmunol.164.4.2001. [DOI] [PubMed] [Google Scholar]
- Harris JL, Jakob B, Taucher-Scholz G, Dianov GL, Becherel OJ, Lavin MF. Aprataxin, poly-ADP ribose polymerase 1 (PARP-1) and apurinic endonuclease 1 (APE1) function together to protect the genome against oxidative damage. Hum Mol Genet. 2009;18:4102–4117. doi: 10.1093/hmg/ddp359. [DOI] [PubMed] [Google Scholar]
- Jamieson SE, Miller EN, Peacock CS, Fakiola M, Wilson ME, Bales-Holst A, Shaw MA, Silveira F, Shaw JJ, Jeronimo SM, Blackwell JM. Genome-wide scan for visceral leishmaniasis susceptibility genes in Brazil. Genes Immun. 2007;8:84–90. doi: 10.1038/sj.gene.6364357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeronimo SM, Duggal P, Ettinger NA, Nascimento ET, Monteiro GR, Cabral AP, Pontes NN, Lacerda HG, Queiroz PV, Gomes CE, Pearson RD, Blackwell JM, Beaty TH, Wilson ME. Genetic predisposition to self-curing infection with the protozoan Leishmania chagasi: a genomewide scan. J Infect Dis. 2007a;196:1261–1269. doi: 10.1086/521682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeronimo SM, Holst AK, Jamieson SE, Francis R, Martins DR, Bezerra FL, Ettinger NA, Nascimento ET, Monteiro GR, Lacerda HG, Miller EN, Cordell HJ, Duggal P, Beaty TH, Blackwell JM, Wilson ME. Genes at human chromosome 5q31.1 regulate delayed-type hypersensitivity responses associated with Leishmania chagasi infection. Genes Immun. 2007b;8:539–551. doi: 10.1038/sj.gene.6364422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Boehnke M, Abecasis GR. Joint modeling of linkage and association: identifying SNPs responsible for a linkage signal. Am J Hum Genet. 2005;76:934–949. doi: 10.1086/430277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luck K, Fournane S, Kieffer B, Masson M, Nomine Y, Trave G. Putting into practice domain-linear motif interaction predictions for exploration of protein networks. PLoS One. 2011;6:e25376. doi: 10.1371/journal.pone.0025376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller EN, Fadl M, Mohamed HS, El Zein A, Jamieson SE, Cordell HJ, Peacock CS, Fakiola M, Raju M, Khalil EA, El Hassan AM, Ibrahim ME, Blackwell JM. Y chromosome lineage- and village-specific genes on chromosomes 1p22 and 6q27 that control visceral leishmaniasis in the Sudan. PLoS Genet. 2007;3:679–688. doi: 10.1371/journal.pgen.0030071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oklo R, Hesketh R. Review article: the latent transforming growth factor beta binding protein (LTBP) family. Biochem J. 2000;352:601–610. [PMC free article] [PubMed] [Google Scholar]
- Peacock CS, Collins A, Shaw MA, Silveira F, Costa J, Coste CH, Nascimento MD, Siddiqui R, Shaw JJ, Blackwell JM. Genetic epidemiology of visceral leishmaniasis in northeastern Brazil. Genet Epidemiol. 2001;20:383–396. doi: 10.1002/gepi.8. [DOI] [PubMed] [Google Scholar]
- Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, Boehnke M, Abecasis GR, Willer CJ. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26:2336–2337. doi: 10.1093/bioinformatics/btq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigginton JE, Abecasis GR. PEDSTATS: descriptive statistics, graphics and quality assessment for gene mapping data. Bioinformatics. 2005;21:3445–3447. doi: 10.1093/bioinformatics/bti529. [DOI] [PubMed] [Google Scholar]
- Wigginton JE, Cutler DJ, Abecasis GR. A note on exact tests of Hardy-Weinberg equilibrium. Am J Hum Genet. 2005;76:887–893. doi: 10.1086/429864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeller T, Wild P, Szymczak S, Rotival M, Schillert A, Castagne R, Maouche S, Germain M, Lackner K, Rossmann H, Eleftheriadis M, Sinning CR, Schnabel RB, Lubos E, Mennerich D, Rust W, Perret C, Proust C, Nicaud V, Loscalzo J, Hübner N, Tregouet D, Münzel T, Ziegler A, Tiret L, Blankenberg S, Cambien F. Genetics and beyond? The transcriptome of human monocytes and disease susceptibility. PLoS One. 2010;5:e10693. doi: 10.1371/journal.pone.0010693. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.