

Abstract
With 5-year survival rates below 5%, small cell lung carcinoma (SCLC) has very poor prognosis and requires improved therapies. Despite an excellent overall response to first-line therapy, relapses are frequent and further treatments are disappointing. The goal of the study was to improve second-line therapy of SCLC.
The effect of chemotherapeutic agents was evaluated in cell lines (apoptosis, reactive oxygen species, and RNA and protein expression) and in mouse models (tumour development).
We demonstrate here that valproic acid, a histone deacetylase inhibitor, improves the efficacy of a second-line regimen (vindesine, doxorubicin and cyclophosphamide) in SCLC cells and in mouse models.
Transcriptomic profiling integrating microRNA and mRNA data identifies key signalling pathways in the response of SCLC cells to valproic acid, opening new prospects for improved therapies.
Short abstract
Valproic acid improves second-line regimen of SCLC response in preclinical models http://ow.ly/Rsyd8
Introduction
Lung cancer is the leading cause of cancer-related death worldwide. The outcome of small cell lung carcinoma (SCLC) patients is the poorest of any histological subtype, with 5-year survival rates of <25% and <5% for limited- and extensive-stage disease, respectively [1]. Despite overall first-line response rates ranging between 60% and 80% (extensive), and 80% and 90% (limited), most tumours relapse. The prognosis remains very poor, with median survival rates of only 8–13 months (extensive) and 14–20 months (limited) [2]. Although significant efforts to develop new therapeutic strategies have been made during the last decade, results are still disappointing [2–5]. Future improvements in outcomes will require clarification of the molecular basis of this disease [1].
Epigenetic errors contribute to the initiation, progression and response to therapy of cancer (reviewed by Barnes et al. [6] and Petta et al. [7]). We and others previously proposed a working hypothesis postulating that histone deacetylase (HDAC) inhibitors induce antitumor activity by reversing epigenetic errors [8–11]. In particular, valproic acid (VPA) is an inhibitor of HDACs displaying appropriate pharmacokinetic properties, and yielding only moderate toxicity that is acceptable in the context of an anticancer treatment [12–14]. By modulating a broad range of activities, including proliferation, apoptosis and differentiation, VPA has antitumoural properties in several cancers, including SCLC [15–21].
Although there is no standard second-line therapy for SCLC, possible treatments most often comprise a combination of three chemotherapeutic agents: a DNA crosslinking agent (e.g. cyclophosphamide), an inhibitor of topoisomerase II (e.g. doxorubicin) and a mitotic spindle poison (e.g. vindesine) (here referred to collectively as “VAC”). With the aim of improving the treatment of extensive SCLC, we evaluated the capacity of VPA to increase the anticancer effect of the VAC regimen in cell cultures and in xenograft mouse models. The mechanisms involved in chemotherapeutic response to VPA were then studied by transcriptomic analyses.
Materials and methods
Cell culture conditions
Human SCLC cell lines (H146, H526 and H69) were purchased from the ATCC (Manassas, VA, USA) and cultivated as detailed previously [19]. Cells were incubated with VPA (Sigma-Aldrich, Diegem, Belgium), mafosfamide (Baxter, Braine-l'Alleud, Belgium), cyclophosphamide (Baxter), doxorubicin (Pfizer, Elsene, Belgium) and vindesine (Lilly, Brussels, Belgium), alone or in combination. Since cyclophosphamide needs to be activated in vivo by the hepatic metabolism, its active form, mafosfamide, was used for in vitro experiments. Optimal drug concentrations were determined by MTS viability assays.
Detection of apoptosis
Apoptosis was quantified by flow cytometry after ethanol fixation and propidium iodide incorporation, as outlined previously [22]. A synergy index was calculated using the formula:
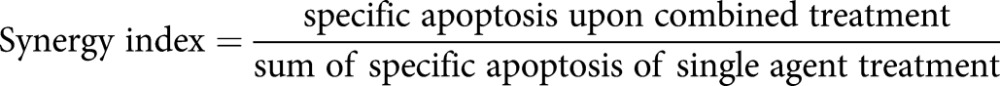 |
The percentage of specific apoptosis was determined using the formula:
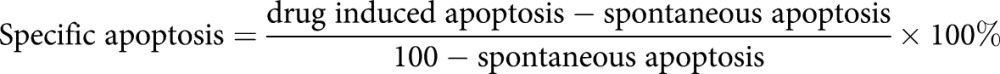 |
When the synergy index was >1, 1 or <1, the effects were defined as synergistic, additive or antagonistic, respectively.
To assess the role of caspases in apoptotic pathways, 5×105 cells were incubated with or without: 20 µM Z-Val-Ala-Asp(OMe)-CH2F (Becton Dickinson, Erembodegem, Belgium), a total pan-caspase inhibitor; 20 µM negative control (Z-FA-fmk) (Becton Dickinson); 40 µM Z-Ile-Glu(OMe)-Thr-Asp(OMe)-CH2F (Calbiochem, Overijse, Belgium), a caspase-8 specific inhibitor; or 40 µM Z-Leu-Glu(OMe)-His-Asp(OMe)-CH2F (Calbiochem), caspase 9 specific inhibitor; all compounds being diluted in dimethylsulfoxide.
Quantification of reactive oxygen species
Reactive oxygen species (ROS) were detected using 5,6-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate acetyl ester (CM-H2DCFDA; InVitrogen, Ghent, Belgium). After 30 min of pre-incubation with 5 µM CM-H2DCFDA, the different drugs were added alone or in combination. After 24 h of culture, SCLC cell lines (5×105 cells per mL in 24-well plates) were harvested, washed with PBS and analysed by flow cytometry (FACS Aria; Becton Dickinson). ROS production was quantified using the fluorescence intensity of chloromethyldichlorofluorescein. 10 000 events were collected and analysed with the FACS Diva software (Becton Dickinson). Cells were also treated with 100 µM hydrogen peroxide or 10 mM N-acetylcysteine (Calbiochem), a free-radical scavenger, as positive and negative controls, respectively.
Immunoblot analysis
Protein expression levels and intracellular translocations were assessed using cytosolic and nucleic buffers, and standard protocols for western blotting as detailed previously [19]. The antibodies used were: anti-acetylated histone H3 (Upstate, Overijse, Belgium), anti-actin, anti-Erk (both Sigma-Aldrich), anti-Bax, anti-Bcl-2 (both Dako Cytomation, Heverlee, Belgium), anti-Bid, anti-cytochrome c (Becton Dickinson), anti-BclxL, anti-phospho-Erk, anti-caspase 8, anti-caspase 9 (Cell Signaling, Leiden, the Netherlands), anti-γH2AX and anti-VDAC1 (Abcam, Cambridge, UK).
Evaluation of regimen efficacy in severe combine immunodeficiency mice
The Institutional Animal Care and Usage Committee of the University of Pennsylvania (Philadelphia, PA, USA) and the University of Liege (Liege, Belgium) approved all animal protocols in compliance with the Guide for the Care and Use of Laboratory Animals, according to the Declaration of Helsinki. The severe combined immunodeficiency (SCID) mice (BALB/c HanHsd-Prkdc; Jackson Laboratories, Sacramento, CA, USA) or NOD/SCID mice received a standard research diet throughout the experiment. H146 and H69 cells (2×106), embedded in 50% Matrigel Basement Membrane Matrix High Concentration (BD Biosciences, Erembodegem, Belgium), were implanted subcutaneously into the flanks of 7-week-old female SCID mice. When tumours reached a volume of 300–400 mm³, mice were administered with daily intraperitoneal injections of VPA (400 mg·kg−1·day−1) or PBS as a control. 3 days after the first VPA administration, intraperitoneal injections of cyclophosphamide (40 mg·kg−1), vindesine (0.5 mg·kg−1) and doxorubicin (3 mg·kg−1) were performed (at days 14 and 17, and days 8, 13, 25 and 34 for H69 and H146, respectively). The schedule of drugs injections was first determined in preliminary dose response experiments. Tumour volumes were calculated twice a week using the formula:
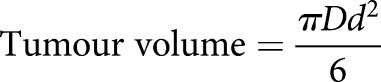 |
where D is the largest diameter and d the smallest. Groups of at least six mice were tested under each experimental condition.
RNA extraction
Cell lines were cultivated for 4 h in presence of 1 mM VPA. RNA was then extracted after two washes in PBS and lysis Trizol RNA Isolation Solution (Ambion, Ghent, Belgium), according to the manufacturer's protocol. For microarray analysis, a second separation between the aqueous and organic phases was performed on Phase Lock Gel (5 Prime, Leuven, Belgium) to optimise RNA recovery and purity. All the extracted RNAs were assessed for quantity and purity using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Temse, Belgium) and stored at −80°C until the amplification step.
Microarray analysis
The details of the microarray analysis have been described previously [19]. Briefly, reference RNAs were obtained by pooling equal amounts of total RNAs from three different SCLC cell lines (H146, H526 and H69). RNAs were reverse-transcribed with Moloney murine leukaemia virus reverse transcriptase using a polydeoxythymidine primer and amplified by PCR. The cDNAs were transcribed into copy RNAs (cRNAs) transcribed and labelled with Cy3 (reference) or Cy5 (sample) dyes using the Low Input RNA Linear Amplification Plus kit (Agilent Technologies, Diegem, Belgium). After quality checking, the labelled cRNAs and RNAs spiked in were hybridised on an Agilent oligonucleotide microarray (two-colour Whole Human Genome 4×44K arrays) and analysed according to manufacturer's protocol (GE2_v5_95_Feb07; Feature Extraction software, version 9.5.3.1). The statistical analyses were performed with Genespring GX, version 7.3.1 (Agilent Technologies). Additional normalisation steps were performed (per spot: division by the control signal; per array: normalisation to the 50th centile; per gene: normalisation to the median). Transcripts that were not present in at least one sample were excluded from additional analyses. Welch's t-test was used to assess the statistical significance, excluding genes whose expression varied by a factor inferior to 2 across the sample set of interest. The threshold for statistical significance after Benjamini's correction for multiple testing was 0.05.
Real-time reverse transcription PCR
mRNA levels were analysed by real-time quantitative reverse transcription PCR (qRT-PCR) as described previously [19], using the primers in table 1.
TABLE 1.
Real-time quantitative reverse transcription PCR primers
| Target | Forward primer | Reverse primer |
| BAG3 | 5′-ACAACAGCCGCACCACTAC-3′ | 5′-GAGCACAGGAATGGGAATGT-3′ |
| BBC3 | 5′-CCTGGAGGGTCCTGTACAATCT-3′ | 5′-GCACCTAATTGGGCTCCATCT-3′ |
| C17orf59 | 5′-AGGGATGTTGCTCTGCTTGT-3′ | 5′-AGGGATGTTGCTCTGCTTGT-3′ |
| C18orf4 | 5′-TGGAACCCACAATCACAAGA-3′ | 5′-CTTGAGTGCCCAAACCAAAT-3′ |
| C4orf24 | 5′-GCGTTTTTGGTGTTGCTGT-3′ | 5′-TTTCCGCGAATGATGAAAGT-3′ |
| C6orf204 | 5′-GAGGACGGACGTGAGGAGT-3′ | 5′-CAGGTAGCCATGCTGATGAA-3′ |
| C6ST | 5′-AGACCTGATTCCCTGTGGTG-3′ | 5′-TGCCTTTTCTTAGGGGTGTG-3′ |
| C20orf29 | 5′-TCTTCGGTGCTGAGAAGTCA-3′ | 5′-CTGGAAAGGGTTCTCTGCTG-3′ |
| DLH2 | 5′-GCACATGGGTTCCTACCAGT-3′ | 5′-TCCTTCTCAGGCTCGTTGTT-3′ |
| EFL4 | 5′-TTCTCTCCTCGACTGTGAAGC-3′ | 5′-CGTAGTGGGGGCAGACAAT-3′ |
| EPH2 | 5′- GACCCTTATGTTGCCTTTCG-3′ | 5′-TCACCACCATAGTCTCCCAAA-3′ |
| FOXO1A | 5′-AAGAGCGTGCCCTACTTCAA-3′ | 5′-CACCCTCTGGATTGAGCATC-3′ |
| FZD7 | 5′-CGACGCTCTTTACCGTTCTC-3′ | 5′-GCCATGCCGAAGAAGTAGAG-3′ |
| GPCR37 | 5′-GCATCTCCAACTCCCTCTTG-3′ | 5′-GTGGTGACTCCCAGAGAAGC-3′ |
| HEY1 | 5′-CGAGGTGGAGAAGGAGAGTG-3′ | 5′-CTGGGTACCAGCCTTCTCAG-3′ |
| HPRT | 5′-GGTCAGGCAGTATAATCCAAAG-3′ | 5′-AAGGGCATATCCTACAACAAAC-3′ |
| HSC40 | 5′-GCCTTGAAGTACCACCCAGA-3′ | 5′-CAAAGAAGGAGGCAAAGGTG-3′ |
| IER5 | 5′-CAGCATCTTCGGTTCCAGTT-3′ | 5′-TCCAGGGGTTCATGTCTCTC-3′ |
| RAB43 | 5′-GGACGAGCAGTACGATTTCC-3′ | 5′-GCGGTAGTAGCTCTGGGTGA-3′ |
| SLIT | 5′-GGCCCAGCAATTAACAAAAA-3′ | 5′-AAGGGTGGATATGTGGTGGA-3′ |
| TNFRSF19 | 5′-GCTGAACGGAACTCTCCAAC-3′ | 5′-CCGAAGCCACATTCCTTAGA-3′ |
| WIPI1 | 5′-GCCTGGTGGTGGTAGTCAGT-3′ | 5′-TCCAGGAGGGTCTTCAACAG-3′ |
MicroRNA analysis
MicroRNA (miRNA) expression was determined in triplicates using the TaqMan Low Density Array method (DNAVision, Gosselies, Belgium). Fluorescence data (Bioanalyzer 2100 Nano chip; Agilent Technologies) were normalised by means of two endogenous controls (RNU44 and RNU48). Results were expressed as fold change (2−(mean(ΔCT(VAC+VPA))–mean(ΔCT(VAC)), where CT is the cycle threshold). The Benjamini and Hochberg method was used for multiple testing corrections.
Statistical analysis
All cell culture experiments were performed at least three times and data are presented as mean±sd. Statistical significance was calculated using Student's t-test and data were considered statistically significant, very statistically significant or highly statistically significant when p<0.05, p<0.01 and p<0.001, respectively.
Results
VPA increases apoptosis in SCLC cells treated with VAC in vitro and in vivo
To evaluate the synergism between VPA and the second-line regimen VAC, VPA, at a concentration achievable in patients (1 mM) [23], was combined with therapeutically relevant doses of mafosfamide (10 µM), doxorubicin (0.3 µM) and vindesine (20 nM). After 24 h of culture, the combination of VPA and VAC significantly increased apoptotic rates in three SCLC cell lines (H146, H69 and H526; p<0.01 by Student's t-test) (figure 1). In fact, the effect of VPA on the combined VAC treatment was synergistic (synergy index of 2.9, 2.4 and 1.6 in H146, H69 and H526 cells, respectively).
FIGURE 1.
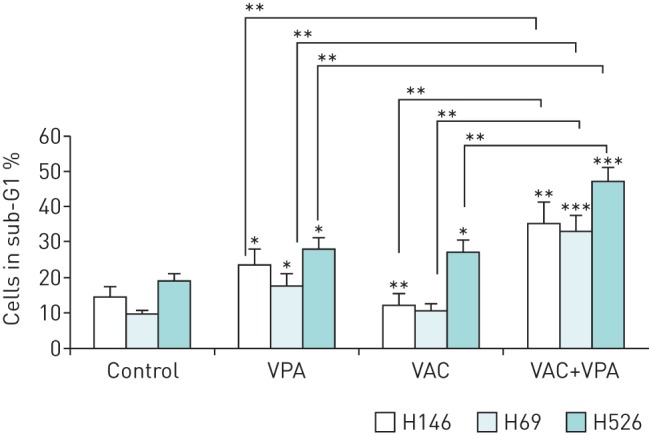
Valproic acid (VPA) synergises with VAC (mafosfamide, doxorubicin and vindesine) to induce apoptosis in small cell lung carcinoma (SCLC) cell lines. Three SCLC cell lines (H146, H526 and H69) were cultivated during 24 h with or without VPA (1 mM) in combination with mafosfamide (10 µM), vindesine (20 nM) and doxorubicin (0.3 µM). Apoptosis rates were determined by flow cytometry after ethanol fixation and propidium iodide staining. Cells in sub-G1 phase represent apoptotic cells with fragmented DNA. The percentages of cells undergoing apoptosis are presented as mean±sd of three independent experiments. For clarity, Student's t-tests between conditions and their associated control are indicated above individual conditions. *: p<0.05; **: p<0.01; ***: p<0.001.
We next determined the involvement of specific caspases using pharmacological inhibitors of caspases 3, 8 and 9 (figure 2). To facilitate comparison between treatments, control results (Z-FA-fmk) were arbitrarily normalised to 1. All three inhibitors significantly reduced apoptosis of SCLC cells, indicating that the mechanism is caspase-dependent, and involves both extrinsic and intrinsic pathways.
FIGURE 2.
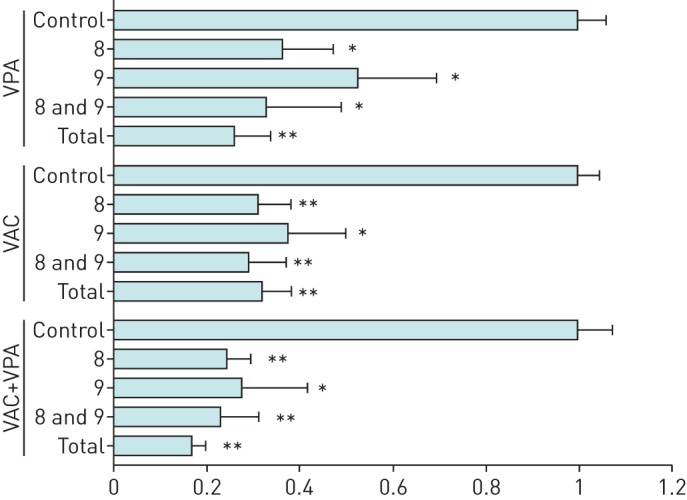
Apoptosis induced by valproic acid (VPA) and VAC (mafosfamide, doxorubicin and vindesine) is dependent on caspases 8 and 9. Different caspase inhibitors were added to cell cultures for 2 h: a total pan-caspase inhibitor (Z-Val-Ala-Asp(OMe)-CH2F, 20 µM); a negative control (Z-FA-fmk, 20 µM); a caspase 8 inhibitor (Z-Ile-Glu(OMe)-Thr-Asp(OMe)-CH2F, 40 µM); a caspase 9 inhibitor (Z-Leu-Glu(OMe)-His-Asp(OMe)-CH2F, 40 µM); or a combination of the caspase 8 and 9 inhibitors (40 µM each). Cells were then cultivated for 48 h with or without VPA (1 mM) in combination with mafosfamide (10 µM), vindesine (20 nM) and doxorubicin (0.3 µM). Cells in sub-G1 phase were considered to be apoptotic. The percentages of cells undergoing apoptosis from three independent experiments were normalised to the Z-FA-fmk control, arbitrarily set to 1. Results are shown for the H526 cell line. Similar results were obtained with H146 and H69. Data are presented as mean–sd. *: p<0.05 compared to Z-FA-fmk control by paired Student's t-test; **:p<0.01 compared to Z-FA-fmk control by paired Student's t-test.
To evaluate the role of reactive oxygen species in the apoptotic process, intracellular ROS levels were monitored by flow cytometry in H146, H69 and H526 cells cultivated for 24 h in presence of VPA and/or VAC. VPA, but not VAC, significantly increased ROS production in all three cell lines (p<0.05) (figure 3a, c and e). Despite significantly decreasing ROS levels, the free-radical scavenger N-acetylcysteine was not sufficient to inhibit apoptosis (figure 3b, d and f).
FIGURE 3.

Valproic acid (VPA)-induced apoptosis is independent of reactive oxygen species (ROS) production. Three small cell lung cancer cell lines (H146 (a and b), H526 (c and d) and H69 (e and f)) were incubated 1 h with or without 10 mM N-acetylcysteine (NAC), a ROS scavenger. Then, the cells were cultivated for 24 h with or without VPA (1 mM) in combination with mafosfamide (10 µM), vindesine (20 nM) and doxorubicin (0.3 µM). Prior to the addition of the drugs, cell cultures were split into two fractions. The first was incubated for 1 h in presence of 5 µM 5,6-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate acetyl ester at 37°C in order to detect the ROS levels by flow cytometry. 500 µM hydrogen peroxide was used as positive control for ROS production. a, c and e) Data are presented as mean±sd fluorescence intensity of chloromethyldichlorofluorescein related to the level obtained in control. b, d and f) The remaining cell fraction was used to determine the rates of apoptosis by flow cytometry after ethanol fixation and propidium iodide staining. Data are represented as mean±sd of three independent experiments. For clarity, statistical tests between conditions and their associated control are indicated above individual conditions. *: p<0.05 by paired Student's t-test; **: p<0.01 by paired Student's t-test.
We next evaluated expression of a series of proteins involved in the cell cycle and apoptosis (figure 4). Western blot analysis demonstrated that VPA-induced apoptosis involved hyperacetylation of histone H3 and phosphorylation of Erk1/2. Double-stranded DNA breaks in the presence of VPA were revealed by γH2AX blotting. VPA created an imbalance between pro- and anti-apoptotic modulators: a decrease in cytoplasmic Bcl-2 and BclxL; cleavage of Bid (into t-Bid); processing of caspase 8 and caspase 9; the appearance of a cleaved form of Bax in the mitochondria; and release of cytochrome c into the cytoplasm.
FIGURE 4.
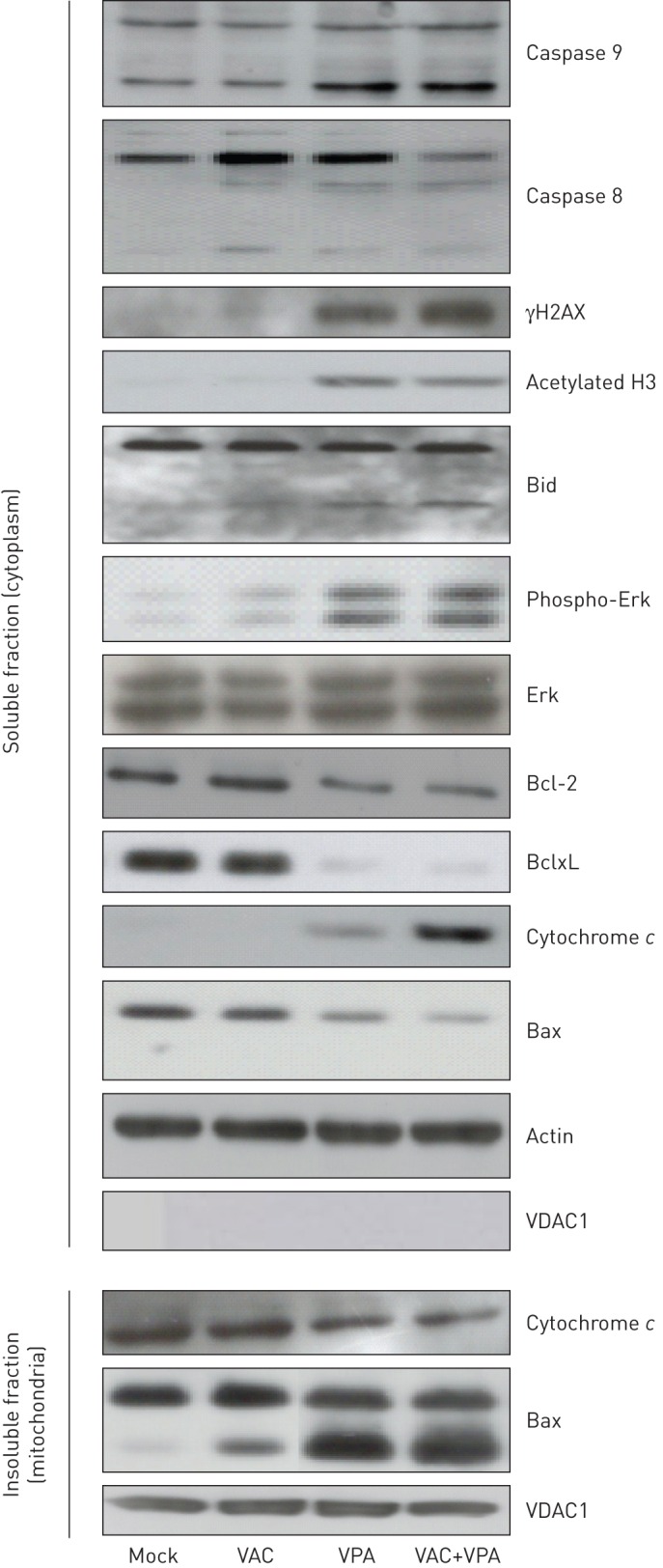
Protein expression profiling of small cell lung carcinoma treated with valproic acid (VPA) and VAC (mafosfamide, doxorubicin and vindesine). Cell lines were cultivated for 24 h with or without VPA (1 mM) in combination with mafosfamide (10 µM), vindesine (20 nM) and doxorubicin (0.3 µM). After culture, the cytosolic and mitochondrial fractions were analysed by Western blotting with the indicated antibodies. Protein concentrations were normalised according to actin levels. The purity of mitochondrial and cytosolic fractions was assessed by expression of VDAC1. Results are shown for the H146 cell line. Similar results were obtained with H69 and H526 cell lines. As expected for a histone deacetylase inhibitor, VPA induced hyper- acetylation of histone H3 when added alone or in combination with VAC. In the presence of VPA, the proapoptotic protein Bid was cleaved, generating the processed and active t-Bid isoform (lower band), and cytochrome c was released from the mitochondria into the cytoplasm (compare upper and lower panels). Under these conditions, Bax was concentrated into mitochondria, generating a cleaved 18-kDa isoform (lower band), and expression of BclxL was strongly reduced, while there was only slightly reduction of the level of Bcl-2. VPA induced phosphorylation of H2AX (γH2AX) and Erk1/2 independently of VAC treatment.
Based on the proapoptotic synergy between VPA and VAC in SCLC cells, we investigated the ability of VPA to improve antitumor efficacy in mouse models. Therefore, SCLC cells (H69 and H146) were injected subcutaneously into the flanks of SCID mice, and the mice were treated with VPA, cyclophophamide (40 mg·kg−1), doxorubicin (3 mg·kg−1) and vindesine (0.5 mg·kg−1), as described in the materials and methods section. Although partial responses were observed with VPA or VAC, tumours eventually relapsed (figure 5). In contrast, tumour growth was restricted in both SCID mouse xenograft models when VPA was combined with VAC (p<0.001 and p<0.05 by Student's t-test, for H69 and H146, respectively).
FIGURE 5.

Valproic acid (VPA) prevents small cell lung carcinoma (SCLC) tumour growth in combination with cyclophosphamide, vindesine and doxorubicin. Two human SCLC cell lines (H69 (a) and H146 (b)) were injected subcutaneously into severe combined immunodeficiency mice in 50% matrigel. Groups of at least six mice were tested in each experimental condition. When tumours reached a volume of 300–400 mm³, mice were administered with daily intraperitoneal injections of VPA (400 mg·kg−1·day−1) or PBS as a control. 3 days after the first VPA administration, intraperitoneal injections of cyclophosphamide (40 mg·kg−1), vindesin (0.5 mg·kg−1) and doxorubicin (3 mg·kg−1) were performed (at days 14 and 17, and days 8, 13, 25 and 34 for H69 and H146, respectively (arrows)). The tumour volume (in cubic millimetres) is presented as means±sd and was calculated at regular intervals of time. There is a statistical significant difference according to the Student's t-test: **: p<0.01 for VAC+VPA versus VAC only treatments.
We conclude that, in two preclinical models of SCLC, VPA improves the efficacy of VAC.
VPA modulates key cellular pathways in SCLC, including cell death and tumour invasion
To better characterise the molecular mechanisms involved, we analysed the transcriptome of H526 cells treated for 4 h with VPA and/or VAC using Agilent microarrays. Bioinformatic analyses revealed a list of genes that were significantly up- or down-regulated by a factor >2 in presence of VPA. Among these, 138 were also specifically identified in cells treated with VPA+VAC compared to VAC (online supplementary material). Expression of the most significantly modulated genes was confirmed by qRT-PCR in three SCLC cell lines (table 2).
TABLE 2.
The 21 most significantly modulated genes were selected from microarray analysis comparing treatment with valproic acid plus VAC versus VAC alone
| Name | Common name | Genbank accession number | H69 | H526 | H146 |
| BAG3 | BAG3 | NM_004281 | 2.3 | 2.2 | 2.0 |
| BBC3 | PUMA | NM_014417 | 2.8 | 5.2 | 2.3 |
| C17orf59 | PRO2472 | NM_017622 | 1.8 | 3.1 | 3.0 |
| C18orf4 | C18orf4 | NM_032160 | 13.8 | 4.2 | 1.3 |
| C20orf29 | FLJ11168 | NM_018347 | 0.3 | 0.4 | 3.6 |
| C4orf24 | C4orf24 | NM_152618 | 10.8 | 4.6 | 2.6 |
| C6orf204 | C6orf204 | BC045657 | 2.0 | 2.0 | 1.6 |
| C6ST | C6ST | NM_004267 | 1.4 | 5.7 | 4.2 |
| DLH2 | TES1 | NM_004405 | 9.1 | 4.1 | 1.0 |
| EFL4 | EFL4 | NM_005227 | 8.7 | 5.4 | 14.3 |
| EPH2 | DKFZP566F2124 | NM_015630 | 0.4 | 0.3 | 3.8 |
| FOXO1A | FOXO1A | NM_002015 | 4.6 | 3.4 | 67.2 |
| FZD7 | FzD7 | NM_003507 | 3.2 | 17.9 | 7.5 |
| GPCR37 | PAELR | NM_005302 | 2.1 | 3.6 | 20.0 |
| HEY1 | HERP2 | NM_012258 | 0.7 | 2.4 | 1.9 |
| HSC40 | HSC40 | NM_012266 | 2.0 | 3.4 | 24.9 |
| IER5 | SBBI48 | NM_016545 | 1.5 | 1.9 | 4.4 |
| RAB43 | RAB41 | NM_198490 | 10.5 | 5.3 | 6.1 |
| SLIT | LRRC12 | NM_052910 | 3.3 | 3.4 | 1.8 |
| TNFRSF19 | TROY | NM_148957 | 1.1 | 2.0 | 3.5 |
| WIPI1 | WIPI49 | NM_017983 | 1.7 | 2.7 | 1.0 |
Data are fold changes obtained by quantitiative revserse transcription PCR of transcripts isolated from three small cell lung carcinoma cell lines (H69, H526 and H146). VAC: mafosfamide, doxorubicin and vindesine.
With 54 genes involved, cell death was the top scored biological function affected, as revealed by Ingenuity (Qiagen, Venlo, the Netherlands) (figure 6). The network related to this function included several genes involved in apoptosis: BBC3 (Puma, a pro-apoptotic member of the Bcl2 family), TNFRSF19 and TNFRSF12A (upstream effectors of apoptosis). To extend this transcriptomic analysis, we next analysed expression of miRNAs [24, 25]. After correction for multiple testing, only two miRNAs were significantly modulated when adding VPA, both of them being involved in tumour invasion: miR-589 (fold increase of 441 with VPA, corrected p-value <0.01) and miR-575 (fold decrease of 0.018, corrected p-value <0.05) (table 3).
FIGURE 6.
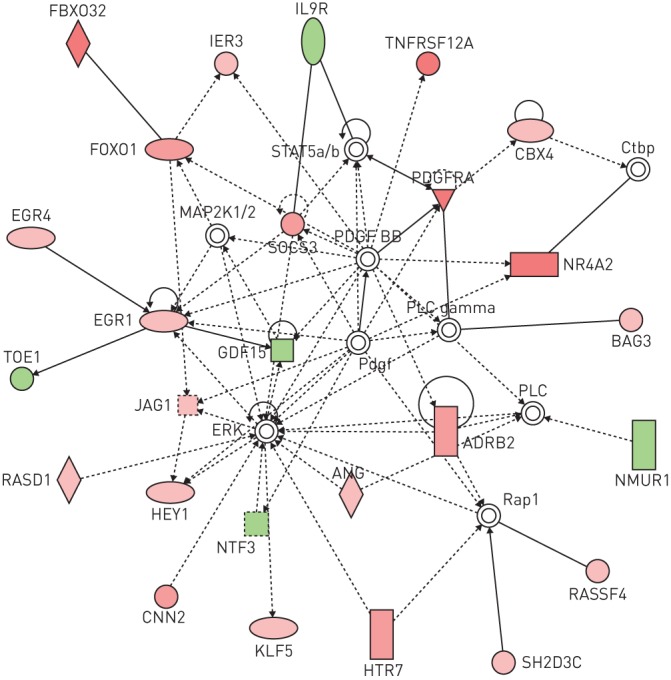
Valproic acid (VPA) modulates expression of genes involved in cell death pathways. Genes transcriptionally modified by VPA extrated from the online supplementary material were analysed with the Ingenuity software. The most significantly affected molecular and cellular function was cell death (p<3.89×10−0.5, with 54 genes modulated: ADRB2, ALX3, ARC, BAG3, BBC3, BCL6, CNN2, CYP26B1, DLL1, DLX2, DUSP10, EGR1, EGR4, EPHA2, FBXO32, FOXO1, GAS2, GDF15, GFI1, GJB2, GPC3, GPR37, HEY1, IER3, IL9R, JAG1, KLF2, KLF5, LATS2, MERTK, MITF, NKX3-2, NR4A2, NTF3, NUAK2, PAX6, PDGFRA, PPARA, RASD1, RASSF4, RUNX1, RUNX1T1, S100A10, SGK1, SIRT4, SOCS3, SSTR2, TNFRSF19, TNFRSF12A, TSLP, USE1, VIPR2, WNT11 and ZFP36). Red and green indicate up- and down-regulated genes, respectively.
TABLE 3.
MicroRNAs modulated by valproic acid
| TaqMan target | miRBase number | Fold change | p-value by t-test | Corrected p-value |
| hsa-miR-589 | hsa-miR-589* | 441.31735 | 0.000015 | 0.005305 |
| hsa-miR-575 | hsa-miR-575 | 0.01787 | 0.000175 | 0.031945 |
| hsa-miR-200a* | hsa-miR-200a* | 4.23666 | 0.001085 | 0.112437 |
| hsa-miR-345 | NA | 5.41671 | 0.001232 | 0.112437 |
| hsa-miR-124a | hsa-miR-124 | 0.27756 | 0.002318 | 0.120889 |
| hsa-miR-16 | hsa-miR-16 | 0.38224 | 0.001808 | 0.120889 |
| hsa-miR-616 | hsa-miR-616* | 0.06316 | 0.002145 | 0.120889 |
| hsa-miR-182* | hsa-miR-182* | 0.28658 | 0.003472 | 0.154357 |
| hsa-miR-376a* | NA | 0.04461 | 0.004229 | 0.154357 |
| hsa-miR-130b | hsa-miR-130b | 0.46074 | 0.005376 | 0.165957 |
| hsa-miR-9 | hsa-miR-9 | 3.51174 | 0.005456 | 0.165957 |
| hsa-let-7a | hsa-let-7a | 3.52777 | 0.008200 | 0.201666 |
| hsa-miR-106b | hsa-miR-106b | 0.49053 | 0.008288 | 0.201666 |
| hsa-miR-142-3p | hsa-miR-142-3p | 4.82212 | 0.007863 | 0.201666 |
| hsa-miR-200c | hsa-miR-200c | 2.30733 | 0.010624 | 0.212291 |
| hsa-miR-29c | hsa-miR-29c | 0.43967 | 0.011632 | 0.212291 |
| hsa-miR-564 | hsa-miR-564 | 4.74989 | 0.011571 | 0.212291 |
| hsa-miR-9* | hsa-miR-9* | 0.43287 | 0.010819 | 0.212291 |
| hsa-miR-99b | hsa-miR-99b | 0.49626 | 0.011479 | 0.212291 |
| hsa-miR-324-5p | hsa-miR-324-5p | 0.49551 | 0.014315 | 0.237493 |
| hsa-let-7g | hsa-let-7g | 2.60556 | 0.017417 | 0.264876 |
| hsa-miR-181b | hsa-miR-181b | 2.16330 | 0.019021 | 0.272956 |
| hsa-miR-594 | NA | 0.40264 | 0.019443 | 0.272956 |
| hsa-let-7d | hsa-let-7d | 2.82868 | 0.022429 | 0.303208 |
| hsa-miR-572 | hsa-miR-572 | 0.30744 | 0.029794 | 0.362498 |
| hsa-miR-335 | hsa-miR-335 | 0.38050 | 0.038849 | 0.386740 |
| hsa-miR-565 | NA | 0.40463 | 0.044928 | 0.395449 |
| hsa-miR-99a | hsa-miR-99a | 0.45290 | 0.049659 | 0.421521 |
Expression of microRNAs was determined by TaqMan Low Density Array in H526 cells treated with valproic acid+VAC or VAC alone. For fold changes <0.5 or >2, the uncorrected p-value was <0.05 (common microRNAs when normalised to RNU44 and RNU48). NA: not applicable; VAC: mafosfamide, doxorubicin and vindesine.
Discussion
The preclinical evidence provided in this report indicates that VPA synergistically increases the apoptotic rates of three different cell lines in response to a second-line therapy for SCLC (figure 1). The mechanism is caspase-dependent, and involves both extrinsic and intrinsic pathways (figure 2). VPA increases the level of ROS, and modifies the balance between pro- and antiapoptotic modulators (figures 3 and 4). The new regimen of VPA+VAC efficiently reduces tumour growth in SCID mouse models engrafted with human SCLC cells (figure 5). This evidence spurred the European Lung Cancer Work Party to launch a phase I/II clinical trial to assess the combination of VPA with VAC in patients presenting with relapsing or refractory SCLC (registered at www.elcwp.org with identifier number 01081). Results presented in the accompanying article by Berghmans et al. [26] indicate that the new regimen induces a significant response in relapsing/refractory SCLC patients. However, since the treatment does not translate into a significant progression-free survival, it is not recommended for second-line therapy.
Our transcriptomic data provide some evidence to better understand the mechanisms involved and to give insights for novel regimen. VPA modulates key genes modulating pathways linked to cell cycle and apoptosis. Among these, Puma (BBC3, Bcl-2 binding component 3), TNFRSF12A (TWEAK/Fn14), sodium/potassium ATPase and HEY1 (a mediator of Notch signalling) are significantly upregulated by VPA. In particular, the sodium/potassium ATPase is involved in sensitivity to platinium [27] and could be a major factor involved in VPA efficacy. Puma is able to interact with Bax and BclxL, promoting translocation of Bax to the mitochondria and competitive binding to BclxL, impeding the latter's ability to inhibit Bax proapoptotic activity [28]. Parallel analyses of miRNA expression identified two particular miRNAs whose expression is tightly correlated with VPA treatment. When the VAC+VPA regimen is compared to VAC alone, miR-589 is increased 441-fold while miR-575 expression is reduced to 0.018. miRNA-589 modulates epithelial–mesenchymal transition in human mesothelial cells [29] while miR-575 promotes growth and invasion of lung cancer cells [30]. Causal involvement of these microRNAs in treatment efficacy could be further investigated by antagomiRs and miRNA mimics.
VPA also regulates expression of modulators of DNA damage response such as Bcl-6, which inhibits TP53 and modulates DNA damage-induced apoptotic responses [31]. Most important is the concomitant modulation of several genes involved in the Wnt pathway, including FZD2, FZD5, FZD7, FZD10 and WNT11. In this context, our preliminary data show that VPA activates TCF/LEF transcription factors in SCLC cells (data not shown). Furthermore, two general Wnt inhibitors (quercetin and ethacrynic acid) impair VPA-induced apoptosis. At first glance, involvement of Wnt in the therapeutic response may appear surprising, since this pathway has been directly correlated with tumorigenicity. For example, overexpression of FZD7 was associated with aberrant activation of the Wnt canonical pathway in oesophagus and colorectal cancers [32, 33]. However, the complexity of the Wnt pathways is clearly less well-understood or at least heterogenous in lung cancer [34]. In fact, expression of WNT7A, which targets FZD7, has been shown to be downregulated in lung cancer [35]. However, SCLC is characterised by overexpression of Wnt inhibitors such as NLK, Sox11 and TCF-4 [36]. In contrast, a tumour suppressor role of the Wnt pathway in SCLC has also been reported [37]. In this perspective, our results indicate that Wnt signalling is associated with a chemotherapeutic response. Similarly, there is a relationship between hyperinduced canonical Wnt activity and enhanced apoptosis in HDAC inhibitor-treated colorectal tumour cells [38].
In conclusion, this report provides preclinical evidence for the use of VPA in second-line therapy of SCLC and opens new prospects for improvement using Wnt activators [39, 40].
Acknowledgements
We are grateful to Jean-Paul Sculier and Thierry Berghmans (Department of Intensive Care Unit and Thoracic Oncology, Institut Jules Bordet, Centre des Tumeurs de l'Université Libre de Bruxelles, Brussels, Belgium) for their collaboration in setting up the clinical trial reported in their companion article to this one [26]. We thank the GIGA technology platforms.
Footnotes
This article has supplementary material available from openres.ersjournals.com
Support statement: The work was supported by the Fonds National de la Recherche Scientifique (FNRS), the Télévie, the Interuniversity Attraction Poles programme BELVIR initiated by the Belgian Science Policy Office, the Action de Recherche Concertée Glyvir of the Communauté française de Belgique, the Belgian Foundation against Cancer, the Sixth Research Framework Programme of the European Union (project INCA LSHC-CT-2005-018704), the Neoangio excellence programme and the Partenariat Public Privé INCA of the Direction générale des Technologies, de la Recherche et de l'Energie/DG06 of the Walloon government, the Centre anticancéreux près ULg, the Synbiofor and Agricultureislife projects of GxABT, the ULg Fonds Spéciaux pour la Recherche, the Plan Cancer of the Service Public Fédéral. L. Willems (research director) is member of the FNRS. Funding information for this article has been deposited with FundRef.
Conflict of interest: Disclosures can be found alongside this article at openres.ersjournals.com
References
- 1.Amini A, Byers LA, Welsh JW, et al. Progress in the management of limited-stage small cell lung cancer. Cancer 2014; 120: 790–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Demedts IK, Vermaelen KY, van Meerbeeck JP. Treatment of extensive-stage small cell lung carcinoma: current status and future prospects. Eur Respir J 2010; 35: 202–215. [DOI] [PubMed] [Google Scholar]
- 3.Oze I, Hotta K, Kiura K, et al. Twenty-seven years of phase III trials for patients with extensive disease small-cell lung cancer: disappointing results. PLoS One 2009; 4: e7835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rossi A, Maione P, Palazzolo G, et al. New targeted therapies and small-cell lung cancer. Clin Lung Cancer 2008; 9: 271–279. [DOI] [PubMed] [Google Scholar]
- 5.Osterlind K. Chemotherapy in small cell lung cancer. Eur Respir J 2001; 18: 1026–1043. [DOI] [PubMed] [Google Scholar]
- 6.Barnes PJ, Adcock IM, Ito K. Histone acetylation and deacetylation: importance in inflammatory lung diseases. Eur Respir J 2005; 25: 552–563. [DOI] [PubMed] [Google Scholar]
- 7.Petta V, Gkiozos I, Strimpakos A, et al. Histones and lung cancer: are the histone deacetylases a promising therapeutic target? Cancer Chemother Pharmacol 2013; 72: 935–952. [DOI] [PubMed] [Google Scholar]
- 8.Sasaki H, Moriyama S, Nakashima Y, et al. Histone deacetylase 1 mRNA expression in lung cancer. Lung Cancer 2004; 46: 171–178. [DOI] [PubMed] [Google Scholar]
- 9.Schrump DS, Fischette MR, Nguyen DM, et al. Clinical and molecular responses in lung cancer patients receiving Romidepsin. Clin Cancer Res 2008; 14: 188–198. [DOI] [PubMed] [Google Scholar]
- 10.Leclercq S, Gueugnon F, Boutin B, et al. A 5-aza-2´-deoxycytidine/valproate combination induces cytotoxic T-cell response against mesothelioma. Eur Respir J 2011; 38: 1105–1116. [DOI] [PubMed] [Google Scholar]
- 11.Scherpereel A, Berghmans T, Lafitte JJ, et al. Valproate-doxorubicin: promising therapy for progressing mesothelioma. A phase II study. Eur Respir J 2011; 37: 129–135. [DOI] [PubMed] [Google Scholar]
- 12.Chapman A, Keane PE, Meldrum BS, et al. Mechanism of anticonvulsant action of valproate. Prog Neurobiol 1982; 19: 315–359. [DOI] [PubMed] [Google Scholar]
- 13.Cinatl J Jr, Cinatl J, Scholz M, et al. Antitumor activity of sodium valproate in cultures of human neuroblastoma cells. Anticancer Drugs 1996; 7: 766–773. [DOI] [PubMed] [Google Scholar]
- 14.DeVane CL. Pharmacokinetics, drug interactions, and tolerability of valproate. Psychopharmacol Bull 2003; 37: Suppl. 2, 25–42. [PubMed] [Google Scholar]
- 15.Bouzar AB, Boxus M, Defoiche J, et al. Valproate synergizes with purine nucleoside analogues to induce apoptosis of B-chronic lymphocytic leukaemia cells. Br J Haematol 2009; 144: 41–52. [DOI] [PubMed] [Google Scholar]
- 16.Crisanti MC, Wallace AF, Kapoor V, et al. The HDAC inhibitor panobinostat (LBH589) inhibits mesothelioma and lung cancer cells in vitro and in vivo with particular efficacy for small cell lung cancer. Mol Cancer Ther 2009; 8: 2221–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duenas-Gonzalez A, Candelaria M, Perez-Plascencia C, et al. Valproic acid as epigenetic cancer drug: preclinical, clinical and transcriptional effects on solid tumors. Cancer Treat Rev 2008; 34: 206–222. [DOI] [PubMed] [Google Scholar]
- 18.Gottlicher M, Minucci S, Zhu P, et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J 2001; 20: 6969–6978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hubaux R, Vandermeers F, Crisanti MC, et al. Preclinical evidence for a beneficial impact of valproate on the response of small cell lung cancer to first-line chemotherapy. Eur J Cancer 2010; 46: 1724–1734. [DOI] [PubMed] [Google Scholar]
- 20.Vandermeers F, Hubert P, Delvenne P, et al. Valproate, in combination with pemetrexed and cisplatin, provides additional efficacy to the treatment of malignant mesothelioma. Clin Cancer Res 2009; 15: 2818–2828. [DOI] [PubMed] [Google Scholar]
- 21.Achachi A, Florins A, Gillet N, et al. Valproate activates bovine leukemia virus gene expression, triggers apoptosis, and induces leukemia/lymphoma regression in vivo. Proc Natl Acad Sci USA 2005; 102: 10309–10314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hubaux R, Thu KL, Coe BP, et al. EZH2 promotes E2F-driven SCLC tumorigenesis through modulation of apoptosis and cell-cycle regulation. J Thorac Oncol 2013; 8: 1102–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Spiller HA, Krenzelok EP, Klein-Schwartz W, et al. Multicenter case series of valproic acid ingestion: serum concentrations and toxicity. J Toxicol Clin Toxicol 2000; 38: 755–760. [DOI] [PubMed] [Google Scholar]
- 24.Scott GK, Mattie MD, Berger CE, et al. Rapid alteration of microRNA levels by histone deacetylase inhibition. Cancer Res 2006; 66: 1277–1281. [DOI] [PubMed] [Google Scholar]
- 25.Suzuki H, Maruyama R, Yamamoto E, et al. Epigenetic alteration and microRNA dysregulation in cancer. Front Genet 2013; 4: 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berghmans T, Lafitte J-J, Scherpereel A, et al. VAC chemotherapy with valproic acid for refractory/relapsing small cell lung cancer: a phase II study. ERJ Open Res 2015; 1: 00029-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stewart DJ. Tumor and host factors that may limit efficacy of chemotherapy in non-small cell and small cell lung cancer. Crit Rev Oncol Hematol 2010; 75: 173–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Y, Xing D, Liu L. PUMA promotes Bax translocation by both directly interacting with Bax and by competitive binding to Bcl-X L during UV-induced apoptosis. Mol Biol Cell 2009; 20: 3077–3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang K, Zhang H, Zhou X, et al. miRNA589 regulates epithelial-mesenchymal transition in human peritoneal mesothelial cells. J Biomed Biotechnol 2012; 2012: 673096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang H, Yan C, Shi X, et al. MicroRNA-575 targets BLID to promote growth and invasion of non-small cell lung cancer cells. FEBS Lett 2015; 589: 805–811. [DOI] [PubMed] [Google Scholar]
- 31.Phan RT, Dalla-Favera R. The BCL6 proto-oncogene suppresses p53 expression in germinal-centre B cells. Nature 2004; 432: 635–639. [DOI] [PubMed] [Google Scholar]
- 32.Tanaka S, Akiyoshi T, Mori M, et al. A novel frizzled gene identified in human esophageal carcinoma mediates APC/β-catenin signals. Proc Natl Acad Sci USA 1998; 95: 10164–10169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ueno K, Hiura M, Suehiro Y, et al. Frizzled-7 as a potential therapeutic target in colorectal cancer. Neoplasia 2008; 10: 697–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hubaux R, Thu KL, Lam WL. Re: the Wnt signaling pathway in non-small cell lung cancer. J Natl Cancer Inst 2014; 106. [DOI] [PubMed] [Google Scholar]
- 35.Calvo R, West J, Franklin W, et al. Altered HOX and WNT7A expression in human lung cancer. Proc Natl Acad Sci USA 2000; 97: 12776–12781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coe BP, Lockwood WW, Girard L, et al. Differential disruption of cell cycle pathways in small cell and non-small cell lung cancer. Br J Cancer 2006; 94: 1927–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park K-S, Lim J, Singh A, et al. The tumor suppressor role of Wnt/β-catenin pathway in small cell lung cancer. Am J Respir Crit Care Med 2012; 185: A5539. [Google Scholar]
- 38.Bordonaro M, Lazarova DL, Sartorelli AC. Hyperinduction of Wnt activity: a new paradigm for the treatment of colorectal cancer? Oncol Res 2008; 17: 1–9. [DOI] [PubMed] [Google Scholar]
- 39.Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer 2013; 13: 11–26. [DOI] [PubMed] [Google Scholar]
- 40.Cai Y, Mohseny AB, Karperien M, et al. Inactive Wnt/β-catenin pathway in conventional high-grade osteosarcoma. J Pathol 2010; 220: 24–33. [DOI] [PubMed] [Google Scholar]