

Abstract
In recent years, the notion that ovarian carcinoma results from ovulation-induced inflammation of the fallopian tube epithelial cells (FTECs) has gained evidence. However, the mechanistic pathway for this process has not been revealed yet. In the current study, we propose the mutator protein activation-induced cytidine deaminase (AID) as a link between ovulation-induced inflammation in FTECs and genotoxic damage leading to ovarian carcinogenesis. We show that AID, previously shown to be functional only in B lymphocytes, is expressed in FTECs under physiological conditions, and is induced in vitro upon ovulatory-like stimulation and in vivo in carcinoma-associated FTECs. We also report that AID activity results in epigenetic, genetic and genomic damage in FTECs. Overall, our data provides new insights into the etiology of ovarian carcinogenesis and may set the ground for innovative approaches aimed at prevention and early detection.
Introduction
Undoubtedly, cancer is a disease of somatic mutations, genomic instability, and epigenetic abnormalities. The mechanisms behind these phenomena remain elusive in most malignancies. Multitude of ovulation cycles has been proposed as a trigger of ovarian cancer [1], [2], [3]. The normal physiological process of ovulation consists of both inflammation and hormonal fluctuations mediated by cytokines, chemokines and steroidal hormones present in the follicular fluid (FF) [4], [5]. Inflammation is a common pathway that drives carcinogenesis in several organ systems [6], [7], whereas hormonal signaling pathways play a role in tumor progression, once genetic aberrations have been initiated. In the past decade it has become clear that most gynecological serous papillary carcinomas originate in the fallopian tube fimbria [8], [9], turning the research focus to the fallopian tube epithelial cells (FTECs) rather than the ovarian surface epithelium. To that aim we have previously developed an ex vivo model system of FTECs [10] which can be genetically modified [11], immortalized [12] and incubated with human follicular fluid to simulate the effect of ovulation [13].
Endogenous cytidine deaminases (AID and apolipoprotein B mRNA editing enzyme, catalytic polypeptide (APOBECs)) attract much attention in recent literature as possibly responsible for both point mutations and chromosomal rearrangements in multiple cancer types. Activation-induced cytidine deaminase (AID, the product of the gene AICDA) normally mediates somatic hyper-mutation and class-switch recombination in immunoglobulin genes [14]. It preferentially acts on cytidine in the consensus sequence of WRCY or WRC (W = A/T, R = A/G, Y = C/T) [15], on single-stranded DNA in tissue-specifically transcribed genes. AID targets co-localize with regions of histone H3K27 acetylation and super-enhancer activity [16], [17]. The deamination reaction results in uracil residues within the DNA sequence, triggering the base-excision repair enzyme uracil-DNA-glycosylase (UNG) to form apurinic/apyrimidinic (AP) sites. When replication occurs before repair has been completed with high fidelity, C:G → T:A mutations are incorporated [18]. In cases where trans-lesion DNA polymerases are absent, the stalled replication fork collapses, resulting in double-strand DNA breaks and chromosomal rearrangements [14], [19], [20]. In addition, as deamination of 5-methylcytidine (5 mC) results in the formation of thymidine, AID has been implicated in global genomic hypomethylation [21], [22], [23].
AID is expressed mainly in B-lymphocytes, where it also has non-immunoglobulin targets, including genes that are tightly related to lymphomagenesis [24], [25]. Its activity in epithelial tissues has been previously described, and shown to be induced by inflammatory stimuli as well as estradiol [26], [27]. As mentioned, both are components of ovulation. AID has been implicated in inflammation-associated malignancies [28], [29], such as Helicobacter pylori–induced gastric adenocarcinoma [30], [31], HBV-related hepatocellular carcinoma [32], [33] and cholangiocarcinoma [34], and also in breast carcinoma [35], and malignant melanoma [36]. AID is up-regulated as early as in esophageal intraepithelial squamous carcinoma, and persists in invasive carcinoma [37]. Its role in gynecological malignancies has not been studied, and no model has been developed to study the function of AID in early tumorigenesis.
The apolipoprotein B mRNA editing enzyme, catalytic polypeptide (APOBEC), is a family of deaminase enzymes involved in restriction of retroviral and mobile retroelement sequences. APOBEC3 has been subject to remarkable evolutionary expansion in primates. The preferred context of most APOBEC3s activity is of 5′ thymidine preceding the mutated cytidine [38]. The mutational signature of APOBEC3-family members has been observed in multiple malignancies [39], [40] including breast cancer [41], [42], [43]. They act on single-stranded DNA and have a typical tendency to cluster resulting in catastrophic regional hyper-mutation, termed ‘kataegis’ [40], [44]. Specifically, APOBEC3B (A3B) is overexpressed in several tumor types, including ovarian carcinoma [45].
In the basis of the current work lies the hypothesis that endogenous physiological stimuli related to ovulation may drive genetic and genomic aberrations that lead to serous carcinogenesis. We show that upon exposure of the fallopian tube fimbria epithelium to hormonal and inflammatory stimuli, AID is induced. Its activity leads to genomic and epigenetic modifications, which may play a causal role in ovarian carcinogenesis. In the current study, we also report increased AID expression in FTECs adjacent to established tumors in vivo. These observations suggest a generalized ‘mutagenic field effect’ in the fimbria priming it to develop serous carcinoma. We therefore propose that AID is a mechanistic link between ovulation-induced inflammation and early stages of serous carcinogenesis.
Materials and Methods
Cell Culture
Fresh, grossly normal fallopian tube fimbriae, removed from patients undergoing gynecological surgery for indications other than ovarian cancer, were allocated by the Chaim Sheba Medical Center Institutional Tissue Bank upon approval of the institutional ethics committee. Tissues were dissociated as previously described [13], and the harvested cells were > 95% pure (repeatedly verified with PAX8 immunostaining). Overall, more than 30 specimens from different donors were utilized for this research. Immortalized FTECs (iFTECs) were kindly provided by the Drapkin lab, Dana-Farber Cancer Institute, Boston, MA, USA [12]. AID overexpressing FTECs (AID-OE FTECs) were produced by retroviral infection of iFTECs with pMSCVgfp::AID, a gift from Dr. Nina Papavasiliou (Addgene plasmid # 15925, [46]). Control cells were produced by retroviral infection of iFTECS with MSCV-IRES-GFP, a gift from Dr. Tannishtha Reya (Addgene plasmid # 20672).
Immunohistochemistry, Immunofluorescence and Immunoblotting
Archival tissues were retrieved from the Department of Pathology at the Chaim Sheba Medical Center with the appropriate ethical committee approvals. We constructed tissue microarrays (TMAs) of ~ 30 representative cases (in duplicates) of normal fimbriae of 4 clinical conditions and of high-grade serous ovarian carcinoma tumors (HGSOC, see Results section). All slides were simultaneously stained and scored according to the signal intensity and distribution, ranging from negative (no staining or faint staining in < 10% of cells), to weak (faint cytoplasmic staining in > 10% of cells), moderate (intermediate cytoplasmic staining of > 10% of cells, or strong cytoplasmic staining of 10–50% of cells) and strong (strong cytoplasmic staining of > 50% of cells). AID expression scores are shown in Supplementary Figure 1. Immunostaining was performed with anti-AID rabbit polyclonal antibody (Abcam, Burlingame, CA, USA at 1:150 dilution for 1 h at RT) and anti-p53 mouse monoclonal antibody (clone DO1, EMD-Millipore, Billerica, MA, USA at 1:70 dilution for 1 h at RT).
Following 2% paraformaldehyde fixation (Electron Microscopy Sciences, Hatfield, PA, USA) FTECs and AID-OE FTECs were immunostained with anti-γH2A.X (Millipore, Billerica, MA, USA at 1:1000 dilution, 4°C, ON), followed by incubation with a fluorescent secondary antibody. Cell positivity was defined as having at least 5 specific nuclear foci.
Anti-AID Rabbit monoclonal antibody (Cell Signaling, Danvers, MA, USA at 1:120 dilution, 4°C, ON) was used for Western blot.
RNA Extraction and qRT-PCR
Total RNA was extracted using QIAzol reagent (Qiagen, Valencia, CA, USA) followed by RNeasy clean-up kit (Qiagen) according to manufacturer's protocol. 1 μg of total RNA was used for reverse transcription using high capacity cDNA reverse transcription kit (Applied Biosystems, Grand Island, NY, USA). mRNA concentrations were assessed using FastStart Universal SYBR Green Master (ROX) (Roche, Indianapolis, IN, USA) with the following primers: AID: 5′- GGGAACCCCAACCTCAGTCT, 5′-CCTTGCGGTCCTCACAGAAG, A3B: 5′-TTGAAAACGAACCCATCCTC, 5′-AGGGGGTCCAGGATACAAAC, B2M: 5′-TTCTGGCCTGGAGGCTATC, 5′- TCAGGAAATTTGACTTTCCATTC.
FF Treatment and Drug Intervention
FF was obtained from 16 women undergoing oocyte retrieval (for reasons other than hormonal infertility) who provided written informed consent, following approval by the institutional ethics committee. It was processed as described before and pooled to overcome batch effects [13]. Serum-supplemented culture medium was used as control for FF, hence an equivalent concentration of 1% Ultroser G (PALL Life Sciences, Cergy-Saint-Christophe, France) was added to the FF [13]. β-estradiol (Sigma Aldrich, St. Louis, MO, USA) was used at a final concentration of 10 nM for either 4 or 24 h. Fulvestrant (Sigma Aldrich, St. Louis, MO, USA) was used at a final concentration of 100 nM. Cells were exposed to the antagonist 24 h before exposure to FF pool and throughout 4 h of incubation with FF. TNFα (Sigma Aldrich, St. Louis, MO, USA) was used at a final concentration of 5 ng/ml for 4 h. Infliximab (Janssen Biotech, Titusville, NJ, USA) was used at a final concentration of 10 μg/ml for 4 h, and was mixed with the FF pool 1 h prior to the application on the cells, in order to achieve complete TNFα blockade.
Fluorescence Based In Vitro Deamination Assay
The protocol was adopted and modified from Leonard et al. and is illustrated in Figure 5A [45]. Untreated control iFTECs, AID-OE FTECs, or iFTECs incubated with FF pool were lysed in 25 mM HEPES pH 7.4, 150 mM NaCl, 1 mM MgCl2, 1 mM ZnCl2, 1 mM EDTA, 0.5% Triton X-100, 10% Glycerol, 1:100 protease inhibitor Cocktail (Roche Pharmaceuticals, Indianapolis, IN, USA). For each sample, 550 μg total protein was incubated for 2 h, 37°C in 50 mM Tris HCl pH 7.4, 10 mM EDTA containing 0.02 unit uracil-DNA glycosylase (UNG, New England Biolabs, Ipswich, MA, USA), 2 μg RNase A (Qiagen, Valencia, CA, USA), and 200 pmol of either an experimental AID motif-containing dual-fluorescent probe (5′[Cy3] TATTATACTAATGGATTTAT [Cy5]) or control (A3B-motif) fluorescent probe (5′[Cy3] TATTATTCCGATGGATTTAT [Cy5]). 4 N NaOH was added for 30 minutes, at 37°C, and then neutralized with 4 N HCl in 2 M Tris HCl. The 5′ Cy3 and 3′ Cy5 labeling yields fluorescence resonance energy transfer (FRET) as long as the probe is intact. Deamination of the C nucleotide resulted in uracil, the subsequent formation of an AP site by UNG and break of the probe by NaOH. Hence, AID activity leads to loss of the FRET signal and gain of Cy3 fluorescent signal. Fluorescence was detected with excitation at 550 nm and emission at 570 nm using Gemini Microplate Reader (Molecular Devices, Sunnyvale, CA, USA). Fluorescent signal detected for the AID motif-containing probe was normalized by the fluorescent signal obtained for the control probe, and the ratio between treated cells and control cells was calculated.
Figure 5.

AID is functional following overexpression, as well as in FF-treated iFTECs. (A) Schematic illustration of FRET-based in vitro deamination assay. (B) FRET-based in vitro deamination assay using FF pool treated iFTECs (n = 4, P = .014) as well as AID–OE FTECs (n = 2, P = .06) as compared to untreated controls. Cells were incubated with either AID consensus probe or a control probe for normalization. (C) Total 5 mC levels in FF pool-treated FTECs (n = 4, Wilcoxon test P = 0.02), FF pool-treated iFTECs (n = 4, P = .02), or AID-OE FTECs (n = 2, P = .05). (D) Amount of AP sites in FF pool-treated iFTECs, as compared to untreated controls, immediately after 4-h incubation with FF pool (n = 2, t test P = .19), or after additional 4-h incubation with normal medium (n = 2, t test P = .006).
Quantification of DNA Damage
We used MethylFlash methylated DNA 5 mC quantification kit (Epigentek, Farmingdale, NY, USA) to examine 5 mC levels in DNA of AID-OE FTECs, FTECs and iFTECs extracted 20 h following 4-h incubation with FF pool.
The number of AP sites was measured using OxiSelect Oxidative DNA Damage Quantitation Kit (Cell Biolabs, San Diego, CA, USA) in AID-OE FTECs and iFTECs following 4-h exposure to FF pool.
Microarray Analysis
Microarray experiments were performed on AID-OE FTECs and control cells. For expression profiling total RNA was profiled using GeneChip PrimeView Human Gene Expression Array (Affymetrix, Santa Clara, CA, USA), according to the manufacturer's standard protocols. To study global copy number variations, genomic DNA was extracted using ZR-Duet DNA/RNA MiniPrep Kit (Zymo Research, Irvine, CA, USA), and examined using CytoScan HD Array Kit (Affymetrix, Santa Clara, CA, USA), according to the manufacturer's standard protocols. The microarray .CEL files were deposited in GEO.
Bioinformatic Analysis
293 ovarian cancer and matched normal whole exome datasets were retrieved from The Cancer Genome Atlas (TCGA) database [47]. The muTect tool [48] was used to identify somatic point mutations with at least 25 × coverage in both the normal and cancer samples. To filter out only high confidence mutations the ‘t_lod_fstar’ parameter was set to a threshold of ≥ 50. For each mutation, the compatibility with the AID motif was scored. We then implemented a previously published pipeline for detection of mutagenesis patterns [39]. Briefly, the pipeline includes two main stages: Mutations' clustering and detection of enriched mutagenesis pattern in clustered mutations. We clustered the mutations as described and calculated P-value for each cluster using negative binomial distribution. For clusters of C:G mutations, enrichment for AID/APOBEC motifs was calculated. The enrichment was calculated as the ratio between the numbers of mutations with each motif to the cluster context.
Statistical Analysis
Data are presented as mean value ± standard error. For RT-PCR experiments, statistical significance (P < .05) was assessed by either t test or by Wilcoxon signed-rank test as indicated. Statistically significant results are highlighted with ‘*’.
Results
AID is Expressed in Fallopian Tube Fimbria Epithelial Cells
To test whether AID is expressed in normal human fallopian tube fimbriae, we immunostained TMAs of grossly normal fimbriae of patients undergoing salpingectomy for the following indications: (A) HGSOC, (B) risk-reducing bilateral salpingo-oophorectomy (RRBSO) in BRCA mutation carriers, (C) leiomyoma (condition not expected to affect the FTECs) or (D) ectopic pregnancy (non-malignant pathology of the tube). AID is indeed expressed in human FTECs, while not in tubal stromal cells (Figure 1). AID is localized mainly in the cytoplasm of the epithelial cells, as observed in B lymphocytes, and in accordance with previous literature [49]. Age did not significantly correlate with AID staining score in any of the 4 groups, or when all cases were analyzed together (Pearson's test, r = 0.41).
Figure 1.

AID is expressed in FTECs. Histological sections of fallopian tubes were immuno-stained with either AID antibody (A, B, D, E, F) or p53 antibody (C). Tissues were derived from patients undergoing salpingectomy for the following indications: HGSOC (A, n = 28), RRBSO in BRCA gene mutation carriers (B, n = 24), leiomyoma (E, n = 27) or ectopic pregnancy (F, n = 29). Boxes represent × 2 enlargement of the marked area. (C, D) Incidental serous tubal intraepithelial carcinoma (STIC) diagnosed in RRBSO specimen from a BRCA mutation carrier. Consecutive slides were stained for p53 to highlight the lesion (C), and for AID (D). The signal intensity for all cases is graphically summarized (G).
The highest expression of AID is seen in normal fallopian tube sections derived from HGSOC patient (Figure 1, A, G), with 78% of the patients staining positive to some degree. Serous tubal intraepithelial carcinoma (STIC) precursor lesions can be highlighted within the fimbria of HGSOC patient by strong staining for p53 (nuclear stain, Figure 1C), whereas the adjacent normal epithelium is p53-negative. Interestingly, whereas p53-positive cells are weakly positive for AID, a much stronger AID signal was detected in surrounding p53-negative cells (Figure 1D).
FTECs of BRCA mutation carriers who underwent RRBSO showed AID positivity in 33% of the cases (Figure 1, B, G). In most of the cases examined, the signal intensity score was low.
52% of fimbriae of patients who underwent salpingectomy due to uterine leiomyoma, who we referred to as controls, had weak to moderate AID staining (Figure 1, E, G). Conversely, only 3.5% of the fimbria resected due to ectopic pregnancy were positive of AID with weak intensity score (Figure 1, F, G). These differences probably reflect less ovulation cycles experienced until resection, as the median ages of the ectopic pregnancy, leiomyoma and RRBSO groups were 33 (range: 20–45) vs. 52 (range: 38–67) and 43 years (range: 35–66), respectively.
Next, we wanted to characterize AID expression in HGSOC tumors (n = 27). The intensity of AID staining varied considerably: 18.2% of the tumors showed high AID signal, 24.2% of the tumors were scored as moderate, 30.3% were scored as weak and 27.3% were negative for AID (Supp. Figure 1). Our data shows greater degree of AID positivity than previously suggested by the TCGA transcriptomic data showing AID mRNA up-regulation in only 3.8–6.4% of the HGSOC analyzed (cBioPortal analysis [50], [51]).
AID is Up-Regulated in Inflammatory Conditions
To study the effect of inflammation, we looked at AID expression in cases of acute or chronic salpingitis (Figure 2, Supp Figure 2, n = 7). As demonstrated in Figure 2A, chronic inflammatory infiltrate co-localizes with intense AID staining in FTECs. It also coincides with abundant TP53-positive cells (Figure 2B), indicating genotoxic stress, TP53 activation or possible initiation of precursor lesions. These results may suggest a causal association between AID expression and the abundant TP53-positive pre-malignant lesions in the fallopian tube (‘p53 signatures’) [9].
Figure 2.

AID is up-regulated in inflammatory conditions. Histological sections of inflamed fallopian tubes were immuno-stained with either AID antibody (A) or p53 antibody (B). Chronic inflammation co-localized with extensive AID and p53 staining. Arrows indicate cells stained positively for both antibodies.
Exposure of Fallopian Tube Derived Cells to Follicular Fluid Induces AID Expression
We hypothesized that exposure of normal human FTECs to FF, depicting natural ovulation stimuli, up-regulates AID expression. As we reported previously [13], incubation of FTECs with FF does not compromise their viability and proliferation. Following 4-h incubation with FF obtained from different IVF patients, the level of AID mRNA vs. untreated control was induced by 1.2-fold to 3.6-fold (Figure 3A). Interestingly, a pool from 6 different FF samples resulted in more pronounced, 4.1-fold AID mRNA induction, suggesting a synergistic effect of several FF components. A significant induction of AID by FF pool was seen as early as after 45 minutes of treatment (Figure 3B, n = 5, Wilcoxon test P = .007), and was maximal following 4-h incubation with FF pool (n = 7, P = .003). Interestingly, transcription down-regulation was seen after 24-h treatment. Similarly, up-regulation of AID protein levels following 4-h stimulation with FF pool is demonstrated by immunostaining (Figure 3C, n = 3) as well as by western blot analysis (Figure 3, D, E).
Figure 3.

Treatment of FTECs with FF induces AID expression. (A) AID transcription induction following 4 h incubation with individual FF and pooled FF. (B) Time course of AID induction following FF pool stimulation from 15 min to 24 h (15 min n = 3, Wilcoxon test, P = .64, 45 min n = 5, P = .007, 2 h n = 4, P = .28, 4 h n = 7, P = .003, 24 h n = 3, P = .06). (C) Increased AID protein expression in the cytoplasm of FTECs following stimulation with FF pool for 4 h as observed by immunohistochemistry (× 20 magnification (top) and × 40 magnification (bottom)). Increased AID protein expression following incubation with FF pool as observed by western blot analysis in FTECs (D), and in iFTECs (E).
Previous work showed A3B up-regulation in ovarian tumors as compared to normal ovarian tissues [45]. RT-PCR results indicate that while the baseline amount of A3B mRNA in FTECs was ~ 16-fold higher than that of AID mRNA, no induction of A3B expression was detected following 15-min to 24-h treatment with FF pool (Supp. Figure 3).
Estradiol and TNFα Redundantly Contribute to FF-Mediated AID Induction
Human FF contains factors responsible for oocyte maturation and ovulation, including steroid hormones, growth factors, cytokines and proteolytic enzymes [52]. Ex vivo induction of AID by estradiol was previously shown in mouse ovaries [27]. Incubation of iFTECs with 10 nM β-estradiol (E2) for 24 h resulted in 4.8-fold AID mRNA induction (Figure 4, n = 4, Wilcoxon test P = .021). Pretreatment with 100 nM fulvestrant, an estrogen receptor antagonist, for 24 h completely repressed the induction of AID by E2 (1.08-fold induction compared to control, Figure 4, n = 3). To test whether E2 is the major component responsible for AID elevation, we pretreated iFTECs with fulvestrant as before, for 24 h prior to incubation with FF pool. Fulvestrant alone and FF pool alone served as controls. Surprisingly, treatment with fulvestrant did not significantly decrease FF-mediated AID induction (2.4- vs. 2.8-fold increase in AID mRNA respectively, Figure 4, n = 3).
Figure 4.
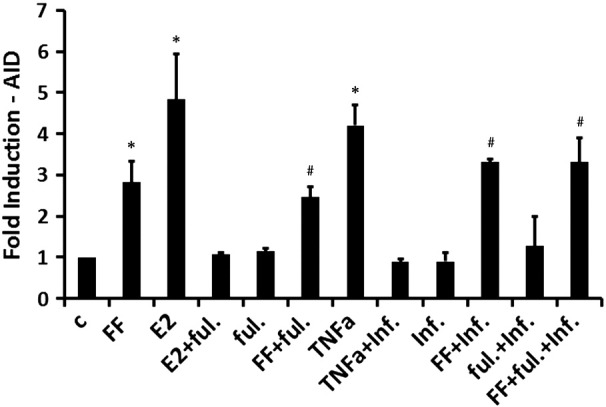
Blockade of either TNFα, or estradiol signaling does not inhibit FF induced AID mRNA induction. Induced AID expression in FTECs following 4-h incubation of iFTECs with either control medium (c), FF pool (n = 4, Wilcoxon test P = .021), or TNFα (5 ng/ml, n = 4, P = .021), as well as 24-h incubation with 10 nM β-Estradiol (E2, n = 4, P = .021). Blockade of TNFα by infliximab (inf., 10 μg/ml, n = 3), of E2 by fulvestrant (ful., 100 nM, n = 3), or of both factors together (n = 3) did not attenuate FF induced AID transcription (P > .05). Controls were carried out by incubation of the cells with either fulvestrant (n = 3), infliximab (n = 3), a combination of fulvestrant and infliximab (n = 3), a combination of E2 and fulvestrant (n = 3) or a combination of TNFα and infliximab (n = 3). Results are presented as fold induction compared to normal control medium. # represents data that is significantly different from control, but insignificantly different from FF-treated cells.
The pro-inflammatory factor TNFα was previously reported to be a component of human FF [52]. Stimulation of iFTECs with 5 ng/ml TNFα resulted in 4.2-fold AID mRNA induction (Figure 4, n = 4, Wilcoxon test P = .021). 1-h pre-incubation of TNFα with 10 μg/ml infliximab, an anti-TNFα monoclonal antibody, completely blocked this effect of TNFα (0.9-fold induction compared to control, n = 3). However, 1-h incubation of FF pool with 10 μg/ml infliximab, did not attenuate the observed increase in AID mRNA in comparison to FF pool alone (3.3- vs. 2.8-fold, Figure 4, n = 3). Lastly, incubation with both fulvestrant and infliximab did not significantly decrease the AID-inducing effect of FF (3.3-fold increase, Figure 4, n = 3). These results suggest that whereas E2 and TNFα are potent AID inducers neither one is essential, and other FF components may possibly compensate for their neutralization.
AID is Functional in FF-Treated FTECs
To verify the functionality of AID in FTECs, we performed an in vitro FRET-based deamination assay (Figure 5A). We measured 37% increase in Cy3 fluorescent signal in FF pool-treated iFTECs as compared to untreated control (Figure 5B, n = 4, t test P = .01). We repeated the experiment using stable AID-OE FTECs, which exhibit more robust and durable effects of AID activation. Following overexpression of AID, cells were viable and did not show significant alterations in cell cycle distribution, anchorage independent growth, or proliferation rate as compared to control (empty vector, data not shown). Using this system, we measured 20% increase in Cy3 fluorescent signal as compared to control (Figure 5B, n = 2, P = .06). These results reflect specific deamination of the only cytidine in the oligonucleotide probe by AID and directly indicate that AID deamination activity is increased in iFTECs following stimulation with FF pool, as well as in stably AID-overexpressing cells.
AID Activity Induces a Decrease in Overall DNA Methylation
As AID deamination results in the conversion of methylated cytidine to thymidine and the repair mechanisms do not restore the methylation pattern, we used DNA methylation in FTECs as surrogate for AID activity.
Following short-term incubation with FF pool (4 h), the global amount of 5 mC was reduced by 28% and 21% in iFTECs (n = 4, Wilcoxon test P = .02) and FTECs (n = 4, P = .02), respectively (Figure 5C). Similarly, We observed a 50% reduction in 5 mC levels in AID-OE FTECs as compared to control (Figure 5C, n = 2, P = .05). These results indicate that AID is active in FTECs under conditions similar to physiological ovulation, and that the induced changes in global DNA methylation are cumulative.
It is noteworthy that AID overexpression in FTECs results in a modest transcriptional change. Using an oligonucleotide microarray, at a twofold change cut-off, only 145 genes were up-regulated and 24 genes were down-regulated (Supp. Table 2). This observation is consistent with the fact that AID is not expected to directly affect transcriptional programs.
AID Induces the Formation of AP Sites in iFTECs
We compared the rate of AP sites in the DNA of iFTECs following exposure to FF pool. Though not significant, we observed a 26% increase in the amount of AP sites (from 93.4/105-bases to 117.8/105-bases (Figure 5D, n = 2, t test P = .19)). The effect was more prominent when DNA extraction was delayed by 4 h, supposedly accounting for prolonged AID activity and kinetics of repair processes, leading to 36% increase in the amount of AP sites (136/105-bases, n = 2, P = .006). However, the rate of AP sites in DNA extracted from stable AID-OE FTECs was not significantly greater than in control cells (data not shown). Unlike deamination of 5 mC, which leads to irreversible loss of methylation, the formation of AP sites is a transient event, which undergoes repair before replication, hence is not cumulative.
AID Activity Induces DNA Damage in FTECs
We immunostained AID-OE FTECs, as compared to control cells, for phospho-Histone H2A.X (γH2A.X) to quantitate dsDNA breaks (Figure 6, A, B). We observed a 50% increase in γH2A.X positive cells (t test, P = .007). This data is in accordance with our previous report showing a similar 70% increase in γH2A.X positive cells following transient exposure of FTECs to FF pool [13].
Figure 6.

AID activity induces DNA damage in FTECs. (A, B) A 1.5-fold increase in γH2AX staining in AID-OE FTECs (n = 157), vs. control cells (n = 170) (t test, P = .007, scale-1 μm Red- γH2AX, Blue-DAPI).
To further characterize the DNA damage caused by AID, we analyzed global chromosomal rearrangements in AID-OE FTECs as compared to control cells using Affymetrix CytoScan microarrays. We revealed 149 unique losses and 148 unique gains in AID-OE FTECs, compared to 96 losses and 69 gains in control cells. The copy number gains correspond to 1.16% of the genome in AID-OE FTECs vs. 0.31% in the control cells, while the percentage of the genome which is covered by copy number losses is 0.90% and 0.92% in AID-OE and control FTECs.
AID Motif Mutagenic Pattern in HGSOC
We analyzed whole-exome sequencing data of 293 HGSOC samples from TCGA for evidence for AID-related mutagenesis pattern, as previously reported [40], [44]. Applying the bioinformatic methods described by Roberts et al. [39] to clustered mutations only, we were unable to detect AID-motif (WRC) enrichment. We did, however, find 1.82- and 2.39-fold enrichment of A3B-related TC and TCW motifs respectively (Supp. Figure 4). A statistically significant enrichment for A3B motif was seen in only 1 out of 293 tumors analyzed. Both results are in agreement with Roberts et al., in which only 3 out of 317 ovarian tumors showed enrichment for the A3B signal [39].
A total of 33,859 somatic mutations were detected in the 293 whole-exome datasets, more than 99% were not clustered. Of these mutations, 32.5% were C:G → T:A transition, which is the most prevalent mutational outcome of AID function [18] (Table 1). Supplementary Table 1 shows the proportion of mutations with AID-motif out of the total number of mutations detected in each of the tumors analyzed. The rate of the AID motif in the human exome (WRCY or RGYW) was calculated by us as 6.6%, while the average rate of AID-motif mutations in the 293 tumors was 8.2% (Fisher's Exact Test P = 4.52e-219). Interestingly, in 7% (21/293 tumors) the rate of AID-motif mutations was more than 2-fold higher than expected by random.
Table 1.
Distribution of Somatic Mismatch Mutations in HGSOC Tumors. 293 Whole-Exome Data Sets Were Retrieved From TCGA and the Type of Mismatch Mutation (MM) Was Quantified. About One Third are C:G → T:A Transitions
| MM type | Number | Percent |
|---|---|---|
| C > A | 2613 | 9.294 |
| G > A | 4532 | 16.12 |
| T > A | 1401 | 4.983 |
| A > C | 967 | 3.439 |
| G > C | 2389 | 8.497 |
| T > C | 2143 | 7.622 |
| A > G | 2114 | 7.519 |
| C > G | 2342 | 8.33 |
| T > G | 995 | 3.539 |
| A > T | 1428 | 5.079 |
| C > T | 4602 | 16.368 |
| G > T | 2589 | 9.209 |
| Total | 28115 | 100 |
Discussion
It is widely accepted that recurrent somatic mutations confer proliferative advantage to the tumor. Nevertheless, it is still unclear why specific genes and chromosomal “hotspots” are particularly susceptible to mutagenesis in multiple different types of tissues. Significant progress has been made in the past few years in identifying mutation patterns and defining the concept of ‘kataegis’, clustered hypermutations [40], [44]. Assuming the existence of an endogenous mutagenic mechanism, AID/APOBEC deaminases have been suggested as responsible for these catastrophic events [53]. Aside from genetic predisposition to genomic instability delivered by BRCA1/2 mutations, very little is known about the molecular drivers of mutations and genomic instability during serous papillary carcinogesis. Over 40 years ago Fathalla hypothesized that ovulation causes repeated inflammatory damage followed by cyclic repair of the ovarian epithelium, culminating in malignant transformation [2]. While the same hypothesis can be easily applied to the fimbria as the tissue-of-origin, no mechanistic sequence of events has been delineated yet. In the current study, we investigate the role of AID in HGSOC using three complementary approaches: In the first part of this research, we show that AID expression is up-regulated in vivo under pro-carcinogenic circumstances and in inflammatory conditions in the tissue-of-origin, whereas it is not uniformly highly expressed in advanced HGSOCs, suggesting a significant role during the initial steps of the tumorigenic process. In the second part, we show that AID plays a key role in vitro in instigating genotoxic damage in the cell-of-origin of HGSOC—the FTEC [9]. To do that, we uniquely model the effects of normal ovulation, epitomized in this research in the transient exposure of FTECs to contents of human FF. Under these unique experimental conditions we observed a significant, tightly-regulated induction of AID, along with an increase in dsDNA breaks, AP sites, and genomic rearrangements, and reduction in global DNA methylation. We have showed that various FF components, including E2 and TNFα are capable of inducing AID transcription, but none is indispensable. The kinetics of AID induction by FF is likely to be the sum of several factors, most are yet unknown. Finally, in the third part of the research we present a bioinformatic analysis performed on a large set of HGSOC sequencing data showing enrichment for somatic mutations that occur within an AID motif.
Perhaps the most intriguing finding of this work, is that, overall, a single, transient exposure of FTEC cells to FF, mimicking the physiological process of normal ovulation, results in robust genotoxic effect. It is reasonable to speculate that most of the DNA damage is subsequently resolved both in vitro and in vivo, while a fraction accumulates and exacerbates with repeat inflammatory insults, resulting in malignancy. To that end we used AID-OE FTECs and found that global hypomethylation and chromosomal rearrangements were accentuated, representing cumulative permanent genetic and epigenetic aberrations. We also show that unlike dsDNA damage, AP sites did not accumulate over time in AID-OE cells, suggesting more efficient base excision repair mechanisms than that of dsDNA breaks, previously shown to be inherently delayed in FTECs [10].
In vivo, generalized up-regulation is seen in tissue sections of fimbria of patients with established HGSOC and salpingitis. Previous literature indicates that the chronically inflamed colonic mucosa of patients with inflammatory bowel disease undergoes a ‘field change’ of cancer-associated molecular alterations anteceding the appearance of dysplasia [54]. Similarly, our data shows that AID is robustly expressed throughout the epithelium of the fimbria under chronic inflammatory conditions, accompanied by p53 positivity, and even more so in regions adjacent to pre-cancerous lesions, suggesting causal relations. For the first time, we show that AID is active in the epithelium of the müllerian tract in both benign and malignant conditions. AID-expressing perturbed FTECs are hence primed for malignant transformation, after accruing sufficient DNA damage.
As mentioned before, A3B had been associated with epithelial carcinogenesis, including ovarian cancer (in which only 3 out of 317 tumors showed enrichment for the A3B signal) [39]. Using the same computational approach, we performed an independent analysis of somatic mutations in advanced HGSOC tumors from the TCGA, in search for enrichment for clusters of point mutations associated with AID motif (WRCY). Nevertheless, our results did not support such conclusion, in line with previous data, failing to show that AID causes mutation clustering. To the best of our knowledge there are no publications demonstrating global enrichment for AID motifs based on whole-exome sequencing. Any existing data is confined to preselect cancer-related genes [25], [55]. A single research that looked at AID motif in whole-exome sequencing of gastric carcinoma, alluded to an unconventional AID motif [30]. It is conceivable that while AID does not directly form clustered mutations, AID-mediated dsDNA breaks lead to secondary activation of break-induced replication and incorporation of random clustered mutations (without preferential AID-motif) which mask the overall enrichment for AID motif [44]. This notion is supported by our observations regarding increased level of DNA breaks and chromosomal rearrangements in AID-OE FTECs, as well as by vast body of data regarding the role of AID in class-switch recombination and chromosomal translocations [20], [24]. We do, however, show that almost one third of the somatic point mutations in HGSOC are C:G → T:A transitions, and that most HGSOC tumors have above-random frequency of AID-motif mutations.
In conclusion, our research suggests the engagement of AID as the ‘missing link’ between ovulation-induced inflammation, DNA damage, and carcinogenesis of the fallopian tube epithelium. Obviously, the process of tumorigenesis extends over many years and cannot be attributed to a single factor. We highlight a mechanism by which the repeated physiological process of ovulation may be considered an endogenous carcinogen leading to HGSOC, necessitating further research in order to better define at-risk populations and improve early-detection or chemo-prevention in these women.
Acknowledgements
This research was supported by research grants from the Israel Cancer Research Fund Clinical Research Career Development Award, the Israeli Ministry of Health Chief Scientist Research Grant, The Israel Cancer Association, and the Chaim Sheba Medical Center Dr. Pinchas Bornstein Talpiot Medical Leadership Program.
We thank the teams of the Department of Gynecologic Oncology, the immunohistochemistry lab at the Department of Pathology, and the Institutional Tissue Banks, all at the Chaim Sheba Medical Center, Ramat Gan, Israel.
Footnotes
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.neo.2015.12.003.
Appendix A. Supplementary data
Supplementary materials
References
- 1.Purdie DM, Bain CJ, Siskind V, Webb PM, Green AC. Ovulation and risk of epithelial ovarian cancer. Int J Cancer. 2003;104:228–232. doi: 10.1002/ijc.10927. [DOI] [PubMed] [Google Scholar]
- 2.Fathalla MF. Incessant ovulation—a factor in ovarian neoplasia? Lancet. 1971;2:163. doi: 10.1016/s0140-6736(71)92335-x. [DOI] [PubMed] [Google Scholar]
- 3.Fleming JS, Beaugie CR, Haviv I, Chenevix-Trench G, Tan OL. Incessant ovulation, inflammation and epithelial ovarian carcinogenesis: revisiting old hypotheses. Mol Cell Endocrinol. 2006;247:4–21. doi: 10.1016/j.mce.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 4.Ambekar AS, Nirujogi RS, Srikanth SM, Chavan S, Kelkar DS, Hinduja I, Zaveri K, Prasad TS, Harsha HC, Pandey A. Proteomic analysis of human follicular fluid: A new perspective towards understanding folliculogenesis. J Proteomics. 2013;87:68–77. doi: 10.1016/j.jprot.2013.05.017. [DOI] [PubMed] [Google Scholar]
- 5.Buscher U, Chen FC, Kentenich H, Schmiady H. Cytokines in the follicular fluid of stimulated and non-stimulated human ovaries; is ovulation a suppressed inflammatory reaction? Hum Reprod. 1999;14:162–166. doi: 10.1093/humrep/14.1.162. [DOI] [PubMed] [Google Scholar]
- 6.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Salvador S, Gilks B, Kobel M, Huntsman D, Rosen B, Miller D. The fallopian tube: primary site of most pelvic high-grade serous carcinomas. Int J Gynecol Cancer. 2009;19:58–64. doi: 10.1111/IGC.0b013e318199009c. [DOI] [PubMed] [Google Scholar]
- 9.Levanon K, Crum C, Drapkin R. New insights into the pathogenesis of serous ovarian cancer and its clinical impact. J Clin Oncol. 2008;26:5284–5293. doi: 10.1200/JCO.2008.18.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Levanon K, Ng V, Piao HY, Zhang Y, Chang MC, Roh MH, Kindelberger DW, Hirsch MS, Crum CP, Marto JA. Primary ex vivo cultures of human fallopian tube epithelium as a model for serous ovarian carcinogenesis. Oncogene. 2009;29:1103–1113. doi: 10.1038/onc.2009.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karst AM, Levanon K, Drapkin R. Modeling high-grade serous ovarian carcinogenesis from the fallopian tube. Proc Natl Acad Sci U S A. 2011;108:7547–7552. doi: 10.1073/pnas.1017300108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karst AM, Drapkin R. Primary culture and immortalization of human fallopian tube secretory epithelial cells. Nat Protoc. 2012;7:1755–1764. doi: 10.1038/nprot.2012.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bahar-Shany K, Brand H, Sapoznik S, Jacob-Hirsch J, Yung Y, Korach J, Perri T, Cohen Y, Hourvitz A, Levanon K. Exposure of fallopian tube epithelium to follicular fluid mimics carcinogenic changes in precursor lesions of serous papillary carcinoma. Gynecol Oncol. 2014;132:322–327. doi: 10.1016/j.ygyno.2013.12.015. [DOI] [PubMed] [Google Scholar]
- 14.Pavri R, Nussenzweig MC. AID targeting in antibody diversity. Adv Immunol. 2011;110:1–26. doi: 10.1016/B978-0-12-387663-8.00005-3. [DOI] [PubMed] [Google Scholar]
- 15.MacCarthy T, Kalis SL, Roa S, Pham P, Goodman MF, Scharff MD, Bergman A. V-region mutation in vitro, in vivo, and in silico reveal the importance of the enzymatic properties of AID and the sequence environment. Proc Natl Acad Sci U S A. 2009;106:8629–8634. doi: 10.1073/pnas.0903803106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meng F-L, Du Z, Federation A, Hu J, Wang Q, Kieffer-Kwon K-R, Meyers RM, Amor C, Wasserman CR, Neuberg D. Convergent transcription at intragenic super-enhancers targets AID-initiated genomic instability. Cell. 2014;159:1538–1548. doi: 10.1016/j.cell.2014.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qian J, Wang Q, Dose M, Pruett N, Kieffer-Kwon K-R, Resch W, Liang G, Tang Z, Mathé E, Benner C. B cell super-enhancers and regulatory clusters recruit AID tumorigenic activity. Cell. 2014;159:1524–1537. doi: 10.1016/j.cell.2014.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Odegard VH, Schatz DG. Targeting of somatic hypermutation. Nat Rev Immunol. 2006;6:573–583. doi: 10.1038/nri1896. [DOI] [PubMed] [Google Scholar]
- 19.Ramiro AR, Jankovic M, Eisenreich T, Difilippantonio S, Chen-Kiang S, Muramatsu M, Honjo T, Nussenzweig A, Nussenzweig MC. AID is required for c-myc/IgH chromosome translocations in vivo. Cell. 2004;118:431–438. doi: 10.1016/j.cell.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 20.Robbiani DF, Bunting S, Feldhahn N, Bothmer A, Camps J, Deroubaix S, McBride KM, Klein IA, Stone G, Eisenreich TR. AID produces DNA double-strand breaks in non-Ig genes and mature B cell lymphomas with reciprocal chromosome translocations. Mol Cell. 2009;36:631–641. doi: 10.1016/j.molcel.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fritz EL, Papavasiliou FN. Cytidine deaminases: AIDing DNA demethylation? Genes Dev. 2010;24:2107–2114. doi: 10.1101/gad.1963010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bhutani N, Brady JJ, Damian M, Sacco A, Corbel SY, Blau HM. Reprogramming towards pluripotency requires AID-dependent DNA demethylation. Nature. 2010;463:1042–1047. doi: 10.1038/nature08752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ramiro AR, Barreto VM. Activation-induced cytidine deaminase and active cytidine demethylation. Trends Biochem Sci. 2015;40:172–181. doi: 10.1016/j.tibs.2015.01.006. [DOI] [PubMed] [Google Scholar]
- 24.Robbiani DF, Nussenzweig MC. Chromosome translocation, B cell lymphoma, and activation-induced cytidine deaminase. Annu Rev Pathol. 2013;8:79–103. doi: 10.1146/annurev-pathol-020712-164004. [DOI] [PubMed] [Google Scholar]
- 25.Liu M, Duke JL, Richter DJ, Vinuesa CG, Goodnow CC, Kleinstein SH, Schatz DG. Two levels of protection for the B cell genome during somatic hypermutation. Nature. 2008;451:841–845. doi: 10.1038/nature06547. [DOI] [PubMed] [Google Scholar]
- 26.Petersen-Mahrt SK, Coker HA, Pauklin S. DNA deaminases: AIDing hormones in immunity and cancer. J Mol Med. 2009;87:893–897. doi: 10.1007/s00109-009-0496-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pauklin S, Sernandez IV, Bachmann G, Ramiro AR, Petersen-Mahrt SK. Estrogen directly activates AID transcription and function. J Cell Biol. 2009;206:99–111. doi: 10.1084/jem.20080521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marusawa H, Takai A, Chiba T. Role of activation-induced cytidine deaminase in inflammation-associated cancer development. Adv Immunol. 2011;111:109–141. doi: 10.1016/B978-0-12-385991-4.00003-9. [DOI] [PubMed] [Google Scholar]
- 29.Chiba T, Marusawa H. A novel mechanism for inflammation-associated carcinogenesis; an important role of activation-induced cytidine deaminase (AID) in mutation induction. J Mol Med (Berl) 2009;87:1023–1027. doi: 10.1007/s00109-009-0527-3. [DOI] [PubMed] [Google Scholar]
- 30.Shimizu T, Marusawa H, Matsumoto Y, Inuzuka T, Ikeda A, Fujii Y, Minamiguchi S, Miyamoto S, Kou T, Sakai Y. Accumulation of somatic mutations in TP53 in gastric epithelium with Helicobacter pylori infection. Gastroenterology. 2014;147:407–417. doi: 10.1053/j.gastro.2014.04.036. [DOI] [PubMed] [Google Scholar]
- 31.Matsumoto Y, Marusawa H, Kinoshita K, Endo Y, Kou T, Morisawa T, Azuma T, Okazaki IM, Honjo T, Chiba T. Helicobacter pylori infection triggers aberrant expression of activation-induced cytidine deaminase in gastric epithelium. Nat Med. 2007;13:470–476. doi: 10.1038/nm1566. [DOI] [PubMed] [Google Scholar]
- 32.Takai A, Toyoshima T, Uemura M, Kitawaki Y, Marusawa H, Hiai H, Yamada S, Okazaki IM, Honjo T, Chiba T. A novel mouse model of hepatocarcinogenesis triggered by AID causing deleterious p53 mutations. Oncogene. 2009;28:469–478. doi: 10.1038/onc.2008.415. [DOI] [PubMed] [Google Scholar]
- 33.Kou T, Marusawa H, Kinoshita K, Endo Y, Okazaki I-M, Ueda Y, Kodama Y, Haga H, Ikai I, Chiba T. Expression of activation-induced cytidine deaminase in human hepatocytes during hepatocarcinogenesis. Int J Cancer. 2007;120:469–476. doi: 10.1002/ijc.22292. [DOI] [PubMed] [Google Scholar]
- 34.Komori J, Marusawa H, Machimoto T, Endo Y, Kinoshita K, Kou T, Haga H, Ikai I, Uemoto S, Chiba T. Activation-induced cytidine deaminase links bile duct inflammation to human cholangiocarcinoma. Hepatology. 2008;47:888–896. doi: 10.1002/hep.22125. [DOI] [PubMed] [Google Scholar]
- 35.Muñoz DP, Lee EL, Takayama S, Coppé J-P, Heo S-J, Boffelli D, Di Noia JM, Martin DI. Activation-induced cytidine deaminase (AID) is necessary for the epithelial-mesenchymal transition in mammary epithelial cells. Proc Natl Acad Sci U S A. 2013;110:E2977–E2986. doi: 10.1073/pnas.1301021110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Okura R, Yoshioka H, Yoshioka M, Hiromasa K, Nishio D, Nakamura M. Expression of AID in malignant melanoma with BRAF(V600E) mutation. Exp Dermatol. 2014;23:347–348. doi: 10.1111/exd.12402. [DOI] [PubMed] [Google Scholar]
- 37.Hayashi A, Yashima K, Takeda Y, Sasaki S, Kawaguchi K, Harada K, Murawaki Y, Ito H. Fhit, E-cadherin, p53, and activation-induced cytidine deaminase expression in endoscopically resected early stage esophageal squamous neoplasia. J Gastroenterol Hepatol. 2012;27:1752–1758. doi: 10.1111/j.1440-1746.2012.07216.x. [DOI] [PubMed] [Google Scholar]
- 38.Ebrahimi D, Alinejad-Rokny H, Davenport MP. Insights into the motif preference of APOBEC3 enzymes. PLoS One. 2014;9:e87679–e87687. doi: 10.1371/journal.pone.0087679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roberts SA, Lawrence MS, Klimczak LJ, Grimm SA, Fargo D, Stojanov P, Kiezun A, Kryukov GV, Carter SL, Saksena G. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat Genet. 2013;45:970–976. doi: 10.1038/ng.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harris RS. Molecular mechanism and clinical impact of APOBEC3B-catalyzed mutagenesis in breast cancer. Breast Cancer Res. 2015;17:8–17. doi: 10.1186/s13058-014-0498-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Burns MB, Lackey L, Carpenter MA, Rathore A, Land AM, Leonard B, Refsland EW, Kotandeniya D, Tretyakova N, Nikas JB. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature. 2013;494:366–370. doi: 10.1038/nature11881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nik-Zainal S, Alexandrov LB, Wedge DC, Van Loo P, Greenman CD, Raine K, Jones D, Hinton J, Marshall J, Stebbings LA. Mutational processes molding the genomes of 21 breast cancers. Cell. 2012;149:979–993. doi: 10.1016/j.cell.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sakofsky CJ, Roberts SA, Malc E, Mieczkowski PA, Resnick MA, Gordenin DA. Break-induced replication is a source of mutation clusters underlying kataegis. Cell Rep. 2014;7:1640–1648. doi: 10.1016/j.celrep.2014.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leonard B, Hart SN, Burns MB, Carpenter MA, Temiz NA, Rathore A, Vogel RI, Nikas JB, Law EK, Brown WL. APOBEC3B upregulation and genomic mutation patterns in serous ovarian carcinoma. Cancer Res. 2013;73:7222–7231. doi: 10.1158/0008-5472.CAN-13-1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dickerson SK, Market E, Besmer E, Papavasiliou FN. AID mediates hypermutation by deaminating single stranded DNA. J Exp Med. 2003;197:1291–1296. doi: 10.1084/jem.20030481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stavnezer J. Complex regulation and function of activation-induced cytidine deaminase. Trends Immunol. 2011;32:194–201. doi: 10.1016/j.it.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Revelli A, Delle Piane L, Casano S, Molinari E, Massobrio M, Rinaudo P. Follicular fluid content and oocyte quality: from single biochemical markers to metabolomics. Reprod Biol Endocrinol. 2009;7:40–52. doi: 10.1186/1477-7827-7-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lada AG, Dhar A, Boissy RJ, Hirano M, Rubel AA, Rogozin IB, Pavlov YI. AID/APOBEC cytosine deaminase induces genome-wide kataegis. Biol Direct. 2012;7:47–53. doi: 10.1186/1745-6150-7-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Galandiuk S, Rodriguez-Justo M, Jeffery R, Nicholson AM, Cheng Y, Oukrif D, Elia G, Leedham SJ, McDonald SA, Wright NA. Field cancerization in the intestinal epithelium of patients with Crohn’s ileocolitis. Gastroenterology. 2012;142:855–864. doi: 10.1053/j.gastro.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lohr JG, Stojanov P, Lawrence MS, Auclair D, Chapuy B, Sougnez C, Cruz-Gordillo P, Knoechel B, Asmann YW, Slager SL. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc Natl Acad Sci U S A. 2012;109:3879–3884. doi: 10.1073/pnas.1121343109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials