

Abstract
Activation of β-platelet-derived growth factor receptor (β-PDGFR) is associated with prostate cancer (PCa) progression and recurrence after prostatectomy. Analysis of the β-PDGFR ligands in PCa revealed association between PDGF-D expression and Gleason score as well as tumor stage. During the course of studying the functional consequences of PDGF ligand-specific β-PDGFR signaling in PCa, we discovered a novel function of PDGF-D for activation/shedding of the serine protease matriptase leading to cell invasion, migration, and tumorigenesis. The present study showed that PDGF-D, not PDGF-B, induces extracellular acidification, which correlates with increased matriptase activation. A cDNA microarray analysis revealed that PDGF-D/β-PDGFR signaling upregulates expression of the acidosis regulator carbonic anhydrase IX (CAIX), a classic target of the transcriptional factor hypoxia-inducible factor-1α (HIF-1α). Cellular fractionation displayed a strong HIF-1α nuclear localization in PDGF-D-expressing cells. Treatment of vector control or PDGF-B-expressing cells with the HIF-1α activator CoCl2 led to increased CAIX expression accompanied by extracellular acidosis and matriptase activation. Furthermore, the analysis of the CAFTD cell lines, variants of the BPH-1 transformation model, showed that increased PDGF-D expression is associated with enhanced HIF-1α activity, CAIX induction, cellular acidosis, and matriptase shedding. Importantly, shRNA-mediated knockdown of CAIX expression effectively reversed extracellular acidosis and matriptase activation in PDGF-D-transfected BPH-1 cells and in CAFTD variants that express endogenous PDGF-D at a high level. Taken together, these novel findings reveal a new paradigm in matriptase activation involving PDGF-D-specific signal transduction leading to extracellular acidosis.
Keywords: PDGF-D, matriptase, acidosis, HIF-1α, CAIX
prostate cancer (PCa) is the most diagnosed noncutaneous cancer and second leading cause of cancer-related death in American men (35). This carcinoma is very much amenable to treatment at the localized stage; however, once metastasized, PCa is not as responsive to therapeutic intervention and this is reflected by the relatively poor survival rate of PCa patients with metastatic disease (35). Therefore, it is imperative to understand the molecular mechanism underlying the transition of PCa from in situ to invasive disease. Platelet-derived growth factor (PDGF) signaling has been implicated in the development and progression of PCa (23, 24, 44). This family is composed to two receptors (α-PDGFR and β-PDGFR) that form homo- or heterodimeric complexes that can be activated by their cognate ligands, PDGF-A, -B, -C, or -D. α-PDGFR is activated by PDGF-A and PDGF-C while β-PDGFR is bound by PDGF-B and PDGF-D (46). PDGF-A and -B are secreted as active homo- or heterodimers while PDGF-C and -D are released into the extracellular milieu as inactive homodimers that can be proteolytically activated by serine proteases such as matriptase and urokinase plasminogen activator (uPA) (28). In PCa, β-PDGFR is upregulated in ∼50% of bone metastatic cancer cases and is part of a five-gene signature predicting PCa recurrence postradical prostatectomy (21, 36). While the classic β-PDGFR ligand PDGF-B has not been often detected in clinical PCa specimens, our group has demonstrated that PDGF-D correlates with PCa Gleason score and tumor stage and induces β-PDGFR transformative potential (43). Furthermore, our recent study demonstrated that PDGF-D drives a more invasive program in prostate epithelial cells compared with PDGF-B, supporting the functional significance of the aforementioned clinical and preclinical findings (24). Interestingly, the PDGF-D-specific invasive phenotype was dependent on the activation of the serine protease, matriptase.
Matriptase is an epithelial-specific type II transmembrane serine protease shown to be upregulated in a many cancers including PCa (19). Matriptase's repertoire of substrates includes hepatocyte growth factor (HGF), PDGF-C, PDGF-D, uPA, and extracellular matrix (ECM) components such as collagen IV (11, 42, 43). Consequently, matriptase regulates proteolytic signaling networks as well as ECM remodeling, and thus it is being considered as a classic promoter of invasive growth of tumors (7, 8, 41, 42). Matriptase activation is a dynamic process involving autolysis, conformational change, and then complex formation with HGF activator inhibitor-1 (HAI-1) (18). As a 70-kDa transmembrane zymogen, matriptase once activated quickly carries out its enzymatic activity before being inhibited by its endogenous inhibitor HAI-1 and shed into the extracellular milieu as 110- and 95-kDa complexes (18). While the signal transduction pathways leading to matriptase activation are largely unknown, sphingosine-1-phosphase and suramin are classic activators of matriptase in various cell types (16). Particularly in PCa cells, dihydrotestosterone and ErbB-2 signaling are also implicated in matriptase activation (12, 45).
In the present study, we report a novel PDGF-D/β-PDGFR autocrine signaling loop that mediates matriptase activation. The PDGF-D-specific signaling cascade induces hypoxia-inducible factor-1α (HIF-1α) nuclear localization and transcriptional activation of carbonic anhydrase IX (CAIX), which in turn results in extracellular acidosis and matriptase activation leading to enhanced cell invasion.
MATERIALS AND METHODS
Cell culture.
Vector control (Hygro) and PDGF-B- and PDGF-D-expressing BPH cells (PDGF-B and PDGF-D BPH-1 cells, respectively) were previously described (24) and cells maintained in RPMI 1640 supplemented with 10% fetal bovine serum, 1% l-glutamine, 0.5% penicillin-streptomycin, and 200 ug/ml hygromycin (Life Technologies, Carlsbad, CA). Parental BPH-1 cells and BPH-1 CAFTD-3 and CAFTD-4 were a generous gift of Dr. Simon W. Hayward at Vanderbilt University and grown in RPMI 1640 supplemented with 5% fetal bovine serum, 1% l-glutamine, and 0.5% penicillin-streptomycin (Life Technologies).
shRNA-mediated downregulation of CAIX.
PDGF-D BPH-1 or BPH-1 CAFTD-4 cells, grown to 90% confluence, were transfected with scrambled (Open Biosystem RHS4346) or two CAIX shRNA expression vectors (Open Biosystem RHS4430-100993056 and RHS4430-100996131) using Lipofectamine 2000 (Life Technologies) per manufacturer's protocol. PDGF-D BPH-1 and BPH-1 CAFTD-4 cells were selected with 4 and 0.5 μg/ml puromycin, respectively. shRNA efficacy was determined by immunoblot analysis using whole cell lysates prepared by lysing cells for 30 min in 1× RIPA lysis buffer (Millipore, Billerica, MA) supplemented with 100 mM PMSF, 200 mM NaVO3, 1 M NaF, and 8% 50× protease inhibitor cocktail (Roche, Indianapolis, IN). Protein concentration was determined using the Pierce BCA protein quantitation assay (Pierce Biotechnology, Rockford, IL).
Reagents.
The anti-active matriptase (M69) antibody recognizes an epitope specific to the activated chain of matriptase while the total matriptase (M32) antibody recognizes both latent and active forms of matriptase and the HAI-1 antibody detects free and matriptase-complexed HAI-1 as described in Ref. 17. Anti-PDGF-B and histone H1 antibodies were from Millipore, and the anti-PDGF-D antibody was custom-designed against the growth factor domain (GD) amino acids 254–272 of PDGF-D and affinity purified (Zymed Biomedical, San Francisco, CA) as described in Ref. 44. Anti-CAIX, PTEN, and phospho- and total Akt and β-PDGFR were obtained from Cell Signaling (Boston, MA). Anti-HIF-1α was from Novus Biologicals (Littleton, CO), and anti-β-actin was from Sigma (St. Louis, MO).
Extracellular pH analysis and detection of shed/activated matriptase.
Cell lines were grown to confluence, washed with warm PBS and then treated with a HEPES-buffered Ringer solution containing the following (in mmol/l): 122.5 NaCl, 5.4 KCl, 0.8 MgCl2, 1.2 CaCl2, 1.0 NaH2PO4·2H20, 5.0 glucose, and 10 HEPES (37). At the indicated time points, conditioned media were collected and the cell debris was removed by centrifugation at 2,000 rpm for 5 min. The pH of the conditioned media was measured using a Beckman Φ300 Digital pH Meter, and pH readouts were subtracted from the pH at time zero to determine changes in extracellular pH that were further normalized to the live cell numbers (106 cells). Experiments were read in triplicates and performed in at least three separate experiments. To detect shed/activated matriptase, cells were plated and treated for 24-h as described above and conditioned media were then concentrated 50-fold using Amicon Ultra-4 centrifugal filters (Millipore) for immunoblot analysis of matriptase under nonreducing conditions.
Microarray analysis and RT-PCR validation.
Vector (Hygro), PDGF-B BPH-1, and PDGF-D BPH-1 cells were grown to confluence, serum starved for 48 h, and then RNA harvested using the Qiagen RNeasy mini kit (Valencia, CA) and subjected to the Illumnia HumanHT-12v4 array analysis. Resultant expression data were background subtracted but not normalized. A strategy was needed to determine which probes were differentially expressed between experimental conditions. We assume that the mean expression level for each probe for each experiment was normally distributed, using the respective bead mean ± SE to define the normal distribution parameters. We then generated 1,000,000 deviates for each experimental condition. P values were computed as the proportion of the resultant ratios of deviates that were greater than 1.5 or less than 2/3 (2 one-sided tests). Significant probes (P < 0.05 for either of the 2 one-sided tests) and estimated effect sizes for each planned comparison were then imported into Ingenuity for pathway analysis.
Gene validation was performed by RT-PCR using primers for CA IX: forward 5′-GGGTGTCATCTGGACTGTGTT-3′, reverse 5′-CTTCTGTGCTGCCTTCTCATC-3′; CA XII: forward 5′-CTGCCAGCAACAAGTCAG-3′, reverse 5′-ATATTCAGCGGTCCTCTC-3′; and β-actin: forward 5′-CTCACCGAGCGCGGCTACA-3′, reverse 5′-CTCCTGCTTGCTGATCCACAT-3′. PCR consisted of denaturation at 94°C for 90 s, annealing at 55°C for 30 s, and extension at 72°C for 150 s. Real-time RT-PCR was performed using QPCR SYBR Green Low ROX Mix (Thermo Fisher Scientific) according to the manufacturer's protocol. Relative values of gene expression were normalized to β-actin and calculated using the 2−ΔΔCt method.
Acidic ph and CoCl2 treatments.
Vector (Hygro) and PDGF-B BPH-1 cells were grown to confluence, washed with PBS and then treated with pH 7.4 (control) or pH 6.0 phosphate buffer. Conditioned media were collected and concentrated for immunoblot analysis. For HIF-1α activation, vector (Hygro) and PDGF-B BPH-1 cells were grown to confluence, washed with PBS and then treated with 100 μM CoCl2 (Sigma) in serum free media for 24 h. Conditioned media were harvested, concentrated, and utilized for matriptase analysis. Total RNA was collected, and cell lysates were subjected to subcellular fractionation.
Subcellular fractionation.
Cells were first lysed for 30 min in cell lysis buffer containing 1 M HEPES (pH 7.9), 1 M KCl, 0.5 M EDTA, 0.1 M EGTA, 10% NP-40, 0.1 M DTT, 100 mM PMSF, and 2% 50× protease inhibitor cocktail (Roche). Cells were then spun in a Sorvall 4°C tabletop microcentrifuge for 2 min at 13,000 rpm, and the cytoplasmic fraction was collected. Cell pellets were washed with ice-cold PBS and the remaining pellets were lysed for 30 min in nuclear extraction buffer containing 1 M HEPES (pH 7.9), 5 M NaCl, 0.5 M EDTA, 0.1 M EGTA, 0.1 M DTT, 100 mM PMSF, and 2% 50× protease inhibitor cocktail (Roche), followed by centrifugation in a Sorvall 4°C tabletop microcentrifuge at 13,000 rpm for 10 min for the collection of the nuclear fraction. Protein concentration was determined using the BCA protein quantitation assay (Pierce Biotechnology).
Matrigel cell invasion.
Live cells were counted using a Trypan Blue exclusion assay and 75,000 cells were placed in a Matrigel-coated 8-μm Transwell (BD Biosciences, San Jose, CA). Complete growth media were used as a chemoattractant and invasion permitted for 16 h with BPH-1 PDGF-D cells and 8 h with BPH-1 CAFTD-4 cells. The top surface of the Transwells were then cleaned with a Q-tip and stained in 0.9% crystal violet, and then migrated cells were quantitated using a Nikon TMS-F inverted microscope at ×100. Five high-power fields were analyzed and plotted as average cell number ± SD. Experiments were conducted in triplicates and performed in at least three separate experiments.
Cell proliferation.
Cells (2,500) were plated in a 96-well plate overnight to allow for cells attachment. Cells were then switched to serum free media, and cell proliferation was measured using the colorimetric WST-1 assay (Roche) per manufacturer's recommendations. Experiments were conducted in replicates of six and performed in at least three separate experiments.
Intracellular pH analysis.
Vector (Hygro), PDGF-B BPH-1, and PDGF-D BPH-1 cells were grown to confluence, washed with warm PBS, and either treated with complete growth media (baseline) or serum free media for 16 or 24 h. At the indicated time point, cells were trypsinized and resuspended in fresh serum free media and the membrane surface charge (zeta potential) was read using Malvern Zetasizer Nano ZS (Worcestershire, UK) as described in Ref. 34. To monitor intracellular pH using the fluorescent probe 2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein (BCECF; Life Technologies), vector (Hygro), PDGF-B BPH-1, and PDGF-D BPH-1 cells were grown in 35-mm glass bottom dishes (MatTek, Ashland, MA) to confluence and washed with PBS before being loaded with 10 μM BCECF for 10 min. Cells were then washed with PBS and fluorescence imaged using an Olympus FSX100 at ×40 magnification. Experiments were conducted in triplicates and performed in at least three separate experiments.
PDGF-D transactivation.
Conditioned media from parental BPH-1 or BPH-1 CAFTD-4 cells was obtained and concentrated as described above. Media were then incubated with or without 5 ng recombinant matriptase (rMat; R&D Systems) for 2 or 6 h. Conditioned media alone were used as a digest control. To test the functional activity of processed PDGF-D, the conditioned media reaction from above was added to NIH3T3 cells and β-PDGFR activation was assessed. rPDGF-B and serum free media alone were used as positive and negative controls, respectively. Numbers under phospho-PDGFRβ and Akt are relative densitometric values obtained using National Institutes of Health ImageJ.
Statistical analysis.
Statistical significance was determined using unpaired Student's t-test, and differences were considered significant when P < 0.05.
RESULTS
PDGF-D-specific signaling supports extracellular acidosis and matriptase activation.
Matriptase is a transmembrane zymogen and once activated it is bound by its endogenous inhibitor HAI-1 and shed into the extracellular milieu as 110- and 95-kDa complexes (18). Using the nonmalignant prostate epithelial cell line BPH-1, we observed a drastic increase in matriptase shedding/activation in response to PDGF-D overexpression but not PDGF-B (Fig. 1A). Matriptase activation was detected exclusively in the conditioned media using an antibody specific to activated matriptase as well as HAI-1, which demonstrates the presence of the high molecular weight matriptase/HAI-1 complex. Accordingly, total matriptase levels were slightly reduced in cell lysate from PDGF-D BPH-1 cells (Fig. 1A). The mechanism by which matriptase is activated and shed are largely unknown at present. Interestingly, Tseng et al. (40) recently reported that acidosis plays a key role in matriptase activation. To determine if this is a possible mechanism underlying PDGF-D-mediated matriptase activation/shedding, we measured the extracellular pH of the conditioned media. PDGF-D BPH-1 cells demonstrated a significantly acidic media at both 16 and 24 h compared with vector (Hygro) and PDGF-B BPH-1 cells (Fig. 1B). Since extracellular acidification could be a result of increased cell proliferation, we normalized extracellular pH to cell number. PDGF-D BPH-1 cells exhibited a more significant pH drop than vector (Hygro) or PDGF-B BPH-1 cells when normalized (Fig. 1C). To ascertain that extracellular acidification plays an important role for matriptase activation/shedding, vector (Hygro) and PDGF-B BPH-1 cells were exposed to either neutral (pH 7.4) or acidic (pH 6.0) phosphate buffer for 10 or 30 min. We observed a time-dependent activation of matriptase/shedding exclusively in the acidic buffer in both cell lines (Fig. 2A). In tumor cells, extracellular acidification is accompanied with a slightly alkaline intracellular pH (25). To monitor intracellular pH, we measured intracellular membrane zeta potential. Cell membrane zeta potential is usually between −20 to −25 mV and becomes less negative when exposed to positively charged particles (i.e., H+) and more negative when exposed to negatively charged particle (i.e., OH−) (34, 47). PDGF-D BPH-1 cells demonstrated negative zeta potential readings overtime suggesting the intracellular charge of the cell membrane is more basic compared with the control or PDGF-B BPH-1 cells (Fig. 2B). To corroborate the zeta potential findings, we next measured the intracellular pH using the fluorescent probe BCECF, which fluoresces more intensely under basic pH due to the unprotonated phenol and carboxylic acid functional groups (14). We observed greater fluorescent intensity in PDGF-D BPH-1 cells compared with vector (Hygro) or PDGF-B-expressing cells (Fig. 2C) indicating a basic intracellular compartment.
Fig. 1.

Platelet-derived growth factor-DPDGF-D-specific signaling induces extracellular acidosis in prostate epithelial cells. A: immunoblot analysis of matriptase and with hepatocyte growth factor activator inhibitor-1 (HAI-1) in conditioned media and total cell lysates from serum-starved vector control (Hygro), PDGF-B BPH-1, or PDGF-D BPH-1 cells. B: changes in extracellular pH of vector control (Hygro), PDGF-B BPH-1, or PDGF-D BPH-1 cells are plotted. Lines represent the mean ± SD. *P < 0.05. C: change in extracellular pH of vector control (Hygro), PDGF-B BPH-1, or PDGF-D BPH-1 cells was normalized to cell number. Lines represent the mean ± SD. *P < 0.05, comparing vector control (Hygro) and PDGF-B cells; **P < 0.05, comparing vector (Hygro) and PDGF-D cells. aMat, active matriptase; tMat, total matriptase. Ponceau S was used to evaluate equal gel loading of conditioned media.
Fig. 2.
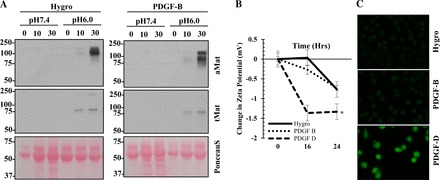
Matriptase activation/shedding is induced by extracellular acidosis in prostate epithelial cells. A: immunoblot of active and total matriptase in conditioned media from vector control (Hygro) or PDGF-B BPH-1 cells upon treatment with pH 7.4 or pH 6.0 phosphate buffer for indicated time period. Intracellular pH of vector control (Hygro), PDGF-B BPH-1, and PDGF-D BPH-1 cells was monitored through membrane zeta potential (B) and BCECF fluorescent (C) measurements. Lines represent the mean ± SD. *P < 0.05. Ponceau S was used to evaluate equal gel loading of conditioned media.
To complement our overexpression model of PDGF in BPH-1 cells, we utilized the BPH-1 transformation model described in Hayward et al. (10). Briefly, parental BPH-1 cells were grafted beneath the renal capsule with cancer-associated fibroblasts and resulting tumors were cultured to obtain the first generation of BPH-1 CAFTD variants. The first generation cell lines were then regrafted without cancer-associated fibroblasts to obtain the second generation of CAFTD variants. Within our study, we chose parental BPH-1, BPH-1 CAFTD-3, and BPH-1 CAFTD-4 as these cells demonstrated progressively transformed phenotypes including gradual increase in tumorigenesis and invasion into surrounding stroma (10). First, we assessed the protein expression profile for PDGF-B and D in the parental BPH-1 and BPH-1 CAFTD-3 and -4 variants. Immunoblot analysis showed an increase in PDGF-B expression in BPH-1 CAFTD-3 but a decrease in the more aggressive CAFTD-4 variant (Fig. 3A). It should be noted that PDGF-B was detected in cell lysates of CAFTD variants, while it was barely detected in conditioned media, likely due to its binding to ECM proteins or internalization through autocrine signaling. Unlike PDGF-B that is intracellularly processed and secreted as a biologically active dimer, PDGF-D is secreted as a latent dimer containing the NH2-terminal CUB domain and the COOH-terminal growth factor domain. The full-length PDGF-D (50 kDa) can be processed into the active 18-kDa growth factor domain and eventually the inactive 15-kDa species by extracellular serine proteases (43). Immunoblot analysis detected full-length PDGF-D in the conditioned media to gradually increase from parental BPH-1 to BPH-1 CAFTD-3 to CAFTD-4 (Fig. 3A). To confirm the 50-kDa band detected in the BHP-1 CAFTD variants is indeed the latent PDGF-D proteins, BPH-1 CAFTD-4 conditioned media were incubated with recombinant matriptase, a well-characterized activator of PDGF-D. Using an anti-PDGF-D antibody raised against the growth factor domain (44), we confirmed that the full-length PDGF-D in CAFTD-4 conditioned media can be processed into the growth factor domain (Fig. 3B). To test the biological activity of BPH-1 CAFTD-4-derived PDGF-D, recombinant matriptase-treated conditioned media from BPH-1 parental or CAFTD-4 cells were used to stimulate the β-PDGFR in NIH3T3 cells. Recombinant matriptase-mediated proteolytic processing resulted in enhanced β-PDGFR activation, especially in BPH-1 CAFTD-4 samples (Fig. 3C).
Fig. 3.

Enhanced PDGF-D expression in the BPH-1 transformation model correlates with matriptase shedding. A: immunoblot analysis of PDGF-D and PDGF-B in conditioned media or cell lysates from serum-starved parental BPH-1, BPH-1 CAFTD-3, and BPH-1 CAFTD-4 variants. B: BPH-1 CAFTD-4 conditioned media (CM) were incubated with 5 ng recombinant matriptase (rMat) for 2 or 6 h. Conditioned media alone were used as a digest control. PDGF-D processing was assessed using immunoblot analysis. Ponceau S was used as a loading control. C: NIH3T3 cells were treated with digested or undigested conditioned media from parental or BPH-1 CAFTD-4 cells and β-PDGFR activation assessed. rPDGF-B and serum free media (SFM) alone were used as positive and negative controls, respectively. Numbers under phospho-PDGFRβ and Akt are relative densitometric values obtained using National Institutes of Health ImageJ. D: matriptase expression profile in the BPH-1 transformation model. Ponceau S was used to evaluate equal gel loading of conditioned media.
Next, we asked whether increased PDGF-D expression correlates with matriptase activation/shedding and/or extracellular acidosis. Consistent with our overexpression model, BPH-1 CAFTD-4 cells, which expressed higher PDGF-D, displayed greater matriptase activation and shedding as well as enhanced extracellular acidosis (Figs. 3D and 4A), even after the normalization to the cell numbers (Fig. 4B). Similar to our overexpression model, matriptase activation/shedding and acidosis were associated with PDGF-D expression but not with PDGF-B as BPH-1 CAFTD-3 variant cells, which express higher PDGF-B, did not exhibit high levels of matriptase activation/shedding compared with the parental BPH-1 cells. Previously, we demonstrated that PDGF-D-expressing BPH-1 cells were more invasive than their PDGF-B counterparts (24). To assess if this is true in the in vivo derived BPH-1 transformation model cell lines, we performed Matrigel invasion assays and observed greater invasive potential in the BPH-1 CAFTD variants, specifically CAFTD-4, correlating the PDGF-D expression profile of these variant with cell invasion (Fig. 4C). Thus both ectopic and endogenous PDGF-D expression models support the notion that PDGF-D signaling mediates extracellular acidosis and concomitant matriptase activation supporting cell invasiveness.
Fig. 4.

Extracellular acidosis and increased cell invasion are evident in the BPH-1 transformation model cells expressing high PDGF-D. Changes in extracellular pH (A) were monitored in BPH-1 parental and BPH-1 variant cells then normalized to cell number (B). C: Matrigel invasion was assessed in BPH-1 parental and BPH-1 variant cells. Values represent the mean ± SD. HPF, high-power field. *P < 0.05, between BPH-1 parental and BPH-1 CAFTD-3; **P < 0.05, between BPH-1 parental and BPH-1 CAFTD-4.
PDGF-D-specific signaling enhances CAIX expression.
In an effort to determine the molecular mechanisms by which PDGF-D-specific signaling mediates extracellular acidosis, we performed a cDNA microarray analysis of vector (Hygro), PDGF-B BPH-1, and PDGF-D BPH-1 cells and analyzed the expression levels of different pH regulators (Fig. 5A). Among those, we found that CAIX expression increased at both the RNA and protein levels in response to PDGF-D overexpression in BPH-1 cells (Fig. 5, B and C). In agreement with previous reports (4), CAIX proteins were detected, ranging from 35 to 58 kDa. Next, we wanted to confirm these findings in the BPH-1 transformation model, specifically in BPH-1 CAFTD-4 cells that express PDGF-D at high levels. RT-PCR demonstrated an increase in CAIX expression, and not CAXII, and these findings were corroborated through immunoblot analysis (Fig. 5, D and E).
Fig. 5.
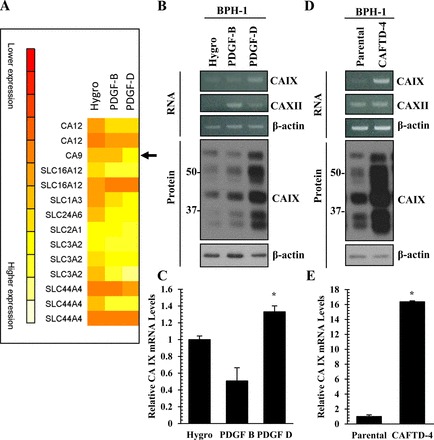
Carbonic anhydrase IX (CAIX) expression is enhanced in response to PDGF-D upregulation. A: list of pH regulating genes from a cDNA microarray analysis of vector control (Hygro), PDGF-B BPH-1, and PDGF-D BPH-1 cells. Qualitative and quantitative RT-PCR as well as immunoblot analysis of carbonic anhydrase (CA) family members in vector control (Hygro), PDGF-B and PDGF-D BPH-1 (B and C), and BPH-1 CAFTD variant (D and E) cells. *P < 0.05.
PDGF-D-mediated extracellular acidosis is dependent on CAIX expression.
CAIX hydrates metabolically released CO2 into H+ and HCO3−. While HCO3− is imported into the cell by different bicarbonate transporters such as anion exchangers, excess of H+ on the outside of tumor cells results in an acidic extracellular milieu promoting invasive and metastatic behavior (2, 9). To assess the functional role CAIX plays in PDGF-D-mediated extracellular acidosis, we stably downregulated CAIX expression using two separate shRNA constructs and then monitored extracellular acidosis and cell invasion. Attenuation of CAIX expression in PDGF-D BPH-1 (Fig. 6, A–C) and BPH-1 CAFTD-4 (Fig. 6, D–F) cells significantly abrogated extracellular acidosis, and this effect was independent of cell number. Importantly, downregulation of CAIX reduced matriptase activation/shedding in cells that express high levels of either ectopic or endogenous PDGF-D (Fig. 7, A and B). Moreover, PDGF-D-mediated Matrigel cell invasion was abrogated in response to CAIX downregulation (Fig. 7, C and D). Taken together, these results support the role of CAIX in PDGF-D-mediated extracellular acidosis, matriptase activation, and cell invasion.
Fig. 6.

CAIX downregulation abrogates PDGF-D-mediated extracellular acidosis. PDGF-D BPH-1 (A) or BPH-1 CAFTD-4 (D) cells were transfected with scrambled (shScrm) or two separate CAIX shRNA and its knockdown was monitored via immunoblotting. Changes in extracellular pH were monitored then normalized to cell number in control and shRNA-mediated CAIX knockdown PDGF-D BPH-1 (B and C) or BPH-1 CAFTD-4 (E and F). Bars represent the mean ± SD. *P < 0.05, between shScrm and shCAIX-2; **P < 0.05, between shScrm and shCAIX-3.
Fig. 7.

Matriptase shedding and cell invasion is attenuated in response to CAIX knockdown. Immunoblot analysis of matriptase and HAI-1 using conditioned media of PDGF-D BPH-1 (A) and BPH-1 CAFTD-4 (B) cells without or with CAIX knockdown (shScrm and shCAIX, respectively). Ponceau S was used to evaluate equal gel loading. Matrigel invasion was monitored in control and shRNA-mediated CAIX knockdown PDGF-D BPH-1 (C) and BPH-1 CAFTD-4 (D) cells. Bars represent the mean ± SD. *P < 0.05, between shScrm and shCAIX-2; **P < 0.05, between shScrm and shCAIX-3.
PDGF-D supports nuclear translocation of HIF-1α.
Since CAIX is a classic HIF-1α target gene (25), we examined whether PDGF-D-specific signaling involves modulation of HIF-1α expression and/or subcellular localization. While there was little change in the mRNA level of HIF-1α (Fig. 8A), subcellular fractionation showed an increase in nuclear HIF-1α in response to PDGF-D overexpression in BPH-1 cells (Fig. 8B). These findings were corroborated using the BPH-1 transformation model where the more aggressive BPH-1 CAFTD-4 variant with increased PDGF-D expression displayed increased HIF-1α nuclear localization compared with BPH-1 parental cells (Fig. 8C).
Fig. 8.

PDGF-D signaling upregulates nuclear hypoxia-inducible factor-1α (HIF-1α). A: RT-PCR analysis of HIF-1α expression in vector control (Hygro), PDGF-B BPH-1, or PDGF-D BPH-1 cells. Immunoblot analyses of HIF-1α in nuclear fraction (Nuc) and cytoplasmic fraction (Cyt) of vector control (Hygro), PDGF-B BPH-1, and PDGF-D BPH-1 cells (B) or in the parental BPH-1 and CAFTD-4 variant (C). Histone H1 and PTEN were used as loading controls for the nuclear and cytoplasmic fractions, respectively.
To test the functional significance of nuclear localization of HIF-1α for CAIX expression and cellular acidosis, we treated vector (Hygro) and PDGF-B BPH-1 cells with the HIF-1α activator CoCl2. Treatment with CoCl2 mediated nuclear localization of HIF-1α (Fig. 9A), upregulated CAIX expression (Fig. 9B), and resulted in extracellular acidosis (Fig. 9, C and D) in both cell lines. These effects remained even after normalization for cell number (Fig. 9, E and F). Notably, matriptase activation was increased in response to CoCl2 exposure in both vector (Hygro) and PDGF-B BPH-1 cells (Fig. 9G). Taken together, we propose a working model that PDGF-D-specific signaling induces HIF-1α activation, which in turn upregulates CAIX expression, resulting in extracellular acidosis leading to matriptase activation and invasive phenotype (Fig. 10).
Fig. 9.
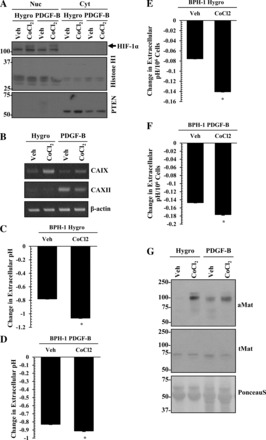
HIF-1α activation supports extracellular acidosis. A: immunoblot analysis of HIF-1α (arrow) in the nuclear and cytoplasmic fractions from vector control (Hygro) or PDGF-B BPH-1 cells with vehicle only or 100 μM CoCl2 treatment. Histone H1 and PTEN were used as loading controls for the nuclear and cytoplasmic fractions, respectively. Upon treatment of vector (Hygro) or PDGF-B BPH-1 cells with CoCl2, CA mRNA expression (RT-PCR) (B), changes in extracellular pH (C–F), and matriptase activation (G) were monitored. Values represent the mean ± SD. *P < 0.05. Ponceau S was used to evaluate equal gel loading.
Fig. 10.

A working model of PDGF-D-mediated matriptase activation. Matriptase activates full length PDGF-D (FL-PDGF-D) in a biphasic manner first yielding a PDGF-D hemidimer (HD-PDGF-D) then growth factor only PDGF-D (GD-PDGF-D). PDGF-D/β-PDGFR signaling mediates HIF-1α nuclear translocation and transcription of CAIX leading to extracellular acidosis (H+) supporting matriptase activation and enhancing further PDGF-D processing and driving a proinvasive program. L-Mat, latent matriptase.
DISCUSSION
In a growing tumor mass, dwindling nutrient supply and poor oxygen perfusion alter tumor metabolism and induce tumor-associated hypoxia (25), resulting in excessive production of lactate and carbonic acid leading to tumor-associated acidosis (3). While such harsh conditions are detrimental to the cell viability, malignant tumor cells have often adapted to the tissue microenvironment through genetic alterations such as upregulation/activation of HIF-1α (6, 27). Importantly, clinical studies have shown that hypoxia is associated with poor prognosis in many cancers and intratumoral acidosis is linked to chemotherapy resistance (25, 33).
HIF-1α is upregulated in a myriad of cancers including PCa and its expression correlates with cancer metastasis (48). This basic helix-loop-helix (bHLH) transcription factor is regulated by both oxygen-dependent and -independent mechanisms (33). While HIF-1α is an unstable protein under normoxic conditions, HIF-1α degradation is inhibited under hypoxic conditions due to inactivation of its negative regulators, prolyl hydroxylase domain-2 (PHD-2) and factor inhibiting HIF-1 (FIH) (1). HIF-1α then heterodimerizes with HIF-1β, which functions as a transcription factor and induces expression of angiogenic and cell survival factors as well as regulators of metabolism and acidosis such as GLUT1 and CAIX (3). The tumor cell's ability to regulate the cellular pH is critical for its survival, as a decrease in the intracellular pH is detrimental to the cell's biochemical and biological processes such as enzyme function and membrane integrity (6). CAIX is a transmembrane metalloenzyme whose molecular mass has been reported to range from 35 to 58 kDa (4). Our study corroborates these reports and demonstrates loss of all CAIX species in response to CAIX shRNA (Fig. 6). CAIX is responsible for hydrating metabolically released CO2 into H+ and HCO3−. Anion exchangers cooperate with CAIX activity importing HCO3− into the cell to stabilize intracellular pH leaving an excess of extracellular H+, which leads to extracellular acidification (26, 39). Consistently, clinical studies have shown increased CAIX expression levels in colorectal, ovarian, gastric, pancreatic, and breast cancers; however, little is known about its involvement in PCa (3, 22). In vitro and animal studies showed that CAIX indeed mediates extracellular acidification of colon and ovarian cancer cells and shRNA knockdown of CAIX reduced in vivo tumorigenesis (4, 38), demonstrating the functional significance of CAIX in human cancers. Importantly, the present study identified CAIX as a downstream mediator of PDGF-D-specific signaling leading to extracellular acidosis in PCa cells.
Excess H+ on the outside of the tumor cells results in an acidic extracellular milieu promoting invasive and metastatic behavior of cancer cells (9, 20). In fact, exposure of melanoma cells to acidic pH enhanced Matrix metalloproteinase (MMP)-2 and MMP-9 activity and increased lung metastatic lesions (31). Interestingly, oral administration of sodium bicarbonate inhibited the metastatic potential of PC-3M PCa cells (30). Furthermore, extracellular pH analysis of migrating cells demonstrated a decrease of extracellular pH at the leading edge of cells (37). These studies suggest that extracellular acidosis provide a biochemically favorable environment for protease activity, which in turn permits cellular invasion. This may be especially true for matriptase, which exhibits enhanced activation at pH 6.0 (40). Matriptase is often upregulated during cancer progression including PCa and has been shown to mediate PCa cell migration, invasion, and tumorigenesis (7, 8, 41). This type-II transmembrane serine protease acts on an array of substrates ranging from ECM components to growth factors (42–44). Previous work from our laboratory demonstrated that matriptase activates PDGF-D in a sequential two-step proteolytic processing, generating a hemidimer that consists of one full-length PDGF-D subunit and a growth factor domain followed by generation of growth factor domain dimer (Fig. 10). Interestingly, our previous studies showed that matriptase-activated PDGF-D signaling results in matriptase activation (24, 43), demonstrating a unique autocrine positive feedback loop between PDGF-D signaling and matriptase activation. In the current study, we report a novel pathway whereby PDGF-D signaling upregulates nuclear HIF-1α leading to CAIX gene expression, which in turn mediates extracellular acidosis responsible for matriptase activation, driving cell invasion and further PDGF-D processing (Fig. 10). These results are critical for understanding the oncogenic actions of PDGF-D as well as the molecular mechanisms underlying the regulation of HIF-1α and its downstream mediators in PCa.
In addition to its regulation by oxygen sensors, HIF-1α can also be modulated in an oxygen-independent manner (33). Growth factor-mediated signaling stimulates protein synthesis, increasing HIF-1α translation (33) while posttranslational modification stabilizes HIF-1α levels (6). Both p38 and ERK MAPK family members have been suggested to phosphorylate HIF-1α increasing HIF-1α activity (13, 29). The balance between protein degradation (O2-dependent) and synthesis and stabilization (O2-independent) controls the amount of available HIF-1α. Since our current results were performed under normoxic conditions, it is probable that PDGF-D-mediated increases in nuclear HIF-1α occur through upregulation of either protein synthesis or stabilization. In our preliminary study, vector (Hygro), PDGF-B BPH-1, and PDGF-D BPH-1 cells were treated with cycloheximide. In the absence of protein synthesis for 2 h, HIF-1α protein was undetected regardless of PDGF expression, consistent with the notion that HIF-1α is an unstable protein. Upon removal of cycloheximide in the presence or absence of the proteasome inhibitor MG132, the levels of cytoplasmic HIF-1α proteins were comparable among cells. In contrast, nuclear HIF-1α levels were significantly higher in PDGF-D-expressing cells (Fig. 11). These preliminary results suggest that PDGF-D-specific signaling may facilitate HIF-1α nuclear transport and perhaps its stability rather than increased translation of HIF-1α mRNA.
Fig. 11.
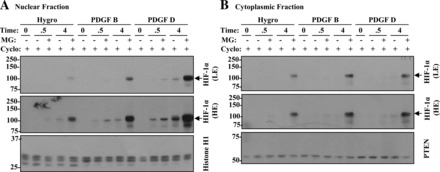
Elucidation of PDGF-D-mediated HIF-1α nuclear localization. Hygro vector control, PDGF-B BPH-1, and PDGF-D BPH-1 cells were treated with 2 μg/ml cycloheximide (Cyclo) for 2 h to inhibit protein synthesis. Cycloheximide was then removed and HIF-1α localization was monitored in the presence or the absence of 50 μM MG132 (MG) at 0.5 and 4 h. Histone H1 and PTEN were used as loading controls for the nuclear (A) and cytoplasmic (B) fractions, respectively.
The present study helps explain how a growth factor utilizes an intracellular signaling program to create a tissue microenvironment that is favorable for activation of proteolytic cascades promoting tumor cell invasion. These results also have implications for the efficacy of current therapeutic modalities. Since most cytotoxic agents are weak bases, extracellular acidosis neutralizes these agents before they can reach their targets (25). In fact, acidosis has been reported to mediate methotrexate resistance in KHT sarcoma and B16F1 melanoma cells (32). We envision that blocking PDGF-D would not only reverse PDGF-D-mediated cell signaling for the previously characterized PDGF-D functions such as tumor cell survival, motility, and angiogenesis but also reduce intratumoral acidosis and thereby abrogate matriptase activity and reduce tumorigenic potential. It should be noted that the PDGF-D downstream mediators we identified in this study, CAIX and matriptase, have been used as noninvasive biomarkers to detect tumors (5, 15). In colon cancer for instance, active matriptase is specifically detected in aggressive disease using a fluorescently labeled matriptase antibody (15) while fluorescently labeled CAIX inhibitors have shown great selectivity in preclinical trials detecting hypoxic tumors (5).
Taken together, we have identified a novel PDGF-D-mediated signaling pathway involving HIF-1α for CAIX regulation, resulting in extracellular acidosis and matriptase activation, leading to a more invasive cellular program in PCa (Fig. 10), and providing valuable information as to PDGF-D isoform-specific functions.
GRANTS
This work was supported by the National Cancer Institute Grant CA-123362, the McLaren Fund (to H. R. Kim), and the Ruth L. Kirschstein National Research Service Award F32-CA-142038-01A1 (to A. J. Najy).
DISCLOSURES
C.-Y. Lin is an inventor on US Patent No. 6077938 (Monoclonal Antibody to an 80-kDa Protease), 6677377 (Structure Based Discovery of Inhibitors of Matriptase for the Cancer Diagnosis and Therapy by Detection and Inhibition of Matriptase Activity), and 7355015 (Matriptase, a Serine Protease and Its Applications).
AUTHOR CONTRIBUTIONS
Author contributions: A.J.N., B.P.J., and H.-R.C.K. conception and design of research; A.J.N. performed experiments; A.J.N., G.D., B.P.J., and H.-R.C.K. analyzed data; A.J.N. and H.-R.C.K. interpreted results of experiments; A.J.N. prepared figures; A.J.N. drafted manuscript; A.J.N., C.-Y.L., and H.-R.C.K. edited and revised manuscript; H.-R.C.K. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Michael D. Johnson for critical reading of the current manuscript and Amanda Flack and Kenneth Lewis for technical assistance assessing intracellular pH.
REFERENCES
- 1.Berra E, Ginouves A, Pouyssegur J. The hypoxia-inducible-factor hydroxylases bring fresh air into hypoxia signalling. EMBO Rep 7: 41–45, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brahimi-Horn MC, Bellot G, Pouyssegur J. Hypoxia and energetic tumour metabolism. Curr Opin Genet Dev 21: 67–72, 2011. [DOI] [PubMed] [Google Scholar]
- 3.Chiche J, Brahimi-Horn MC, Pouyssegur J. Tumour hypoxia induces a metabolic shift causing acidosis: a common feature in cancer. J Cell Mol Med 14: 771–794, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chiche J, Ilc K, Laferriere J, Trottier E, Dayan F, Mazure NM, Brahimi-Horn MC, Pouyssegur J. Hypoxia-inducible carbonic anhydrase IX and XII promote tumor cell growth by counteracting acidosis through the regulation of the intracellular pH. Cancer Res 69: 358–368, 2009. [DOI] [PubMed] [Google Scholar]
- 5.Dubois L, Lieuwes NG, Maresca A, Thiry A, Supuran CT, Scozzafava A, Wouters BG, Lambin P. Imaging of CA IX with fluorescent labelled sulfonamides distinguishes hypoxic and (re)-oxygenated cells in a xenograft tumour model. Radiother Oncol 92: 423–428, 2009. [DOI] [PubMed] [Google Scholar]
- 6.Fang JS, Gillies RD, Gatenby RA. Adaptation to hypoxia and acidosis in carcinogenesis and tumor progression. Semin Cancer Biol 18: 330–337, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Forbs D, Thiel S, Stella MC, Sturzebecher A, Schweinitz A, Steinmetzer T, Sturzebecher J, Uhland K. In vitro inhibition of matriptase prevents invasive growth of cell lines of prostate and colon carcinoma. Int J Oncol 27: 1061–1070, 2005. [PubMed] [Google Scholar]
- 8.Galkin AV, Mullen L, Fox WD, Brown J, Duncan D, Moreno O, Madison EL, Agus DB. CVS-3983, a selective matriptase inhibitor, suppresses the growth of androgen independent prostate tumor xenografts. Prostate 61: 228–235, 2004. [DOI] [PubMed] [Google Scholar]
- 9.Gatenby RA, Gillies RJ. A microenvironmental model of carcinogenesis. Nat Rev Cancer 8: 56–61, 2008. [DOI] [PubMed] [Google Scholar]
- 10.Hayward SW, Wang Y, Cao M, Hom YK, Zhang B, Grossfeld GD, Sudilovsky D, Cunha GR. Malignant transformation in a nontumorigenic human prostatic epithelial cell line. Cancer Res 61: 8135–8142, 2001. [PubMed] [Google Scholar]
- 11.Hurst NJ Jr, Najy AJ, Ustach CV, Movilla L, and Kim HR. Platelet-derived growth factor-C (PDGF-C) activation by serine proteases: implications for breast cancer progression. Biochem J 441: 909–918, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kiyomiya K, Lee MS, Tseng IC, Zuo H, Barndt RJ, Johnson MD, Dickson RB, Lin CY. Matriptase activation and shedding with HAI-1 is induced by steroid sex hormones in human prostate cancer cells, but not in breast cancer cells. Am J Physiol Cell Physiol 291: C40–C49, 2006. [DOI] [PubMed] [Google Scholar]
- 13.Kwon SJ, Song JJ, Lee YJ. Signal pathway of hypoxia-inducible factor-1alpha phosphorylation and its interaction with von Hippel-Lindau tumor suppressor protein during ischemia in MiaPaCa-2 pancreatic cancer cells. Clin Cancer Res 11: 7607–7613, 2005. [DOI] [PubMed] [Google Scholar]
- 14.Lanz E, Slavik J, Kotyk A. 2′,7′-Bis-(2-carboxyethyl)-5(6)-carboxyfluorescein as a dual-emission fluorescent indicator of intracellular pH suitable for argon laser confocal microscopy. Folia Microbiol (Praha) 44: 429–434, 1999. [DOI] [PubMed] [Google Scholar]
- 15.LeBeau AM, Lee M, Murphy ST, Hann BC, Warren RS, Delos Santos R, Kurhanewicz J, Hanash SM, VanBrocklin HF, Craik CS. Imaging a functional tumorigenic biomarker in the transformed epithelium. Proc Natl Acad Sci USA 110: 93–98, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee MS, Kiyomiya K, Benaud C, Dickson RB, Lin CY. Simultaneous activation and hepatocyte growth factor activator inhibitor 1-mediated inhibition of matriptase induced at activation foci in human mammary epithelial cells. Am J Physiol Cell Physiol 288: C932–C941, 2005. [DOI] [PubMed] [Google Scholar]
- 17.Lee MS, Tseng IC, Wang Y, Kiyomiya K, Johnson MD, Dickson RB, Lin CY. Autoactivation of matriptase in vitro: requirement for biomembrane and LDL receptor domain. Am J Physiol Cell Physiol 293: C95–C105, 2007. [DOI] [PubMed] [Google Scholar]
- 18.Lin CY, Tseng IC, Chou FP, Su SF, Chen YW, Johnson MD, Dickson RB. Zymogen activation, inhibition, and ectodomain shedding of matriptase. Front Biosci 13: 621–635, 2008. [DOI] [PubMed] [Google Scholar]
- 19.List K. Matriptase: a culprit in cancer? Future Oncol 5: 97–104, 2009. [DOI] [PubMed] [Google Scholar]
- 20.Martinez-Zaguilan R, Seftor EA, Seftor RE, Chu YW, Gillies RJ, Hendrix MJ. Acidic pH enhances the invasive behavior of human melanoma cells. Clin Exp Metastasis 14: 176–186, 1996. [DOI] [PubMed] [Google Scholar]
- 21.Mathew P, Thall PF, Bucana CD, Oh WK, Morris MJ, Jones DM, Johnson MM, Wen S, Pagliaro LC, Tannir NM, Tu SM, Meluch AA, Smith L, Cohen L, Kim SJ, Troncoso P, Fidler IJ, Logothetis CJ. Platelet-derived growth factor receptor inhibition and chemotherapy for castration-resistant prostate cancer with bone metastases. Clin Cancer Res 13: 5816–5824, 2007. [DOI] [PubMed] [Google Scholar]
- 22.McDonald PC, Winum JY, Supuran CT, Dedhar S. Recent developments in targeting carbonic anhydrase IX for cancer therapeutics. Oncotarget 3: 84–97, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Najy AJ, Jung YS, Won JJ, Conley-LaComb MK, Saliganan A, Kim CJ, Heath E, Cher ML, Bonfil RD, Kim HR. Cediranib inhibits both the intraosseous growth of PDGF D-positive prostate cancer cells and the associated bone reaction. Prostate 72: 1328–1338, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Najy AJ, Won JJ, Movilla LS, Kim HR. Differential tumorigenic potential and matriptase activation between PDGF B versus PDGF D in prostate cancer. Mol Cancer Res 10: 1087–1097, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 10: 767–777, 2011. [DOI] [PubMed] [Google Scholar]
- 26.Parks SK, Chiche J, Pouyssegur J. pH control mechanisms of tumor survival and growth. J Cell Physiol 226: 299–308, 2011. [DOI] [PubMed] [Google Scholar]
- 27.Pouyssegur J, Dayan F, Mazure NM. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 441: 437–443, 2006. [DOI] [PubMed] [Google Scholar]
- 28.Reigstad LJ, Varhaug JE, Lillehaug JR. Structural and functional specificities of PDGF-C and PDGF-D, the novel members of the platelet-derived growth factors family. FEBS J 272: 5723–5741, 2005. [DOI] [PubMed] [Google Scholar]
- 29.Richard DE, Berra E, Gothie E, Roux D, Pouyssegur J. p42/p44 mitogen-activated protein kinases phosphorylate hypoxia-inducible factor 1alpha (HIF-1alpha) and enhance the transcriptional activity of HIF-1. J Biol Chem 274: 32631–32637, 1999. [DOI] [PubMed] [Google Scholar]
- 30.Robey IF, Baggett BK, Kirkpatrick ND, Roe DJ, Dosescu J, Sloane BF, Hashim AI, Morse DL, Raghunand N, Gatenby RA, Gillies RJ. Bicarbonate increases tumor pH and inhibits spontaneous metastases. Cancer Res 69: 2260–2268, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rofstad EK, Mathiesen B, Kindem K, Galappathi K. Acidic extracellular pH promotes experimental metastasis of human melanoma cells in athymic nude mice. Cancer Res 66: 6699–6707, 2006. [DOI] [PubMed] [Google Scholar]
- 32.Schlappack OK, Zimmermann A, Hill RP. Glucose starvation and acidosis: effect on experimental metastatic potential, DNA content and MTX resistance of murine tumour cells. Br J Cancer 64: 663–670, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer 3: 721–732, 2003. [DOI] [PubMed] [Google Scholar]
- 34.Shin L, Basi N, Jeremic A, Lee JS, Cho WJ, Chen Z, Abu-Hamdah R, Oupicky D, Jena BP. Involvement of vH(+)-ATPase in synaptic vesicle swelling. J Neurosci Res 88: 95–101, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Siegel R, Naishadham D, Jemal A. Cancer statistics. CA Cancer J Clin 63: 11–30, 2013. [DOI] [PubMed] [Google Scholar]
- 36.Singh D, Febbo PG, Ross K, Jackson DG, Manola J, Ladd C, Tamayo P, Renshaw AA, D′Amico AV, Richie JP, Lander ES, Loda M, Kantoff PW, Golub TR, Sellers WR. Gene expression correlates of clinical prostate cancer behavior. Cancer Cell 1: 203–209, 2002. [DOI] [PubMed] [Google Scholar]
- 37.Stock C, Mueller M, Kraehling H, Mally S, Noel J, Eder C, Schwab A. pH nanoenvironment at the surface of single melanoma cells. Cell Physiol Biochem 20: 679–686, 2007. [DOI] [PubMed] [Google Scholar]
- 38.Svastova E, Hulikova A, Rafajova M, Zat'ovicova M, Gibadulinova A, Casini A, Cecchi A, Scozzafava A, Supuran CT, Pastorek J, Pastorekova S. Hypoxia activates the capacity of tumor-associated carbonic anhydrase IX to acidify extracellular pH. FEBS Lett 577: 439–445, 2004. [DOI] [PubMed] [Google Scholar]
- 39.Swietach P, Wigfield S, Supuran CT, Harris AL, Vaughan-Jones RD. Cancer-associated, hypoxia-inducible carbonic anhydrase IX facilitates CO2 diffusion. BJU Int 101, Suppl 4: 22–24, 2008. [DOI] [PubMed] [Google Scholar]
- 40.Tseng IC, Xu H, Chou FP, Li G, Vazzano AP, Kao JP, Johnson MD, Lin CY. Matriptase activation, an early cellular response to acidosis. J Biol Chem 285: 3261–3270, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsui KH, Chang PL, Feng TH, Chung LC, Hsu SY, Juang HH. Down-regulation of matriptase by overexpression of bikunin attenuates cell invasion in prostate carcinoma cells. Anticancer Res 28: 1977–1983, 2008. [PubMed] [Google Scholar]
- 42.Uhland K. Matriptase and its putative role in cancer. Cell Mol Life Sci 63: 2968–2978, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ustach CV, Huang W, Conley-LaComb MK, Lin CY, Che M, Abrams J, Kim HR. A novel signaling axis of matriptase/PDGF-D/ss-PDGFR in human prostate cancer. Cancer Res 70: 9631–9640, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ustach CV, Taube ME, Hurst NJ Jr, Bhagat S, Bonfil RD, Cher ML, Schuger L, Kim HR. A potential oncogenic activity of platelet-derived growth factor d in prostate cancer progression. Cancer Res 64: 1722–1729, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu SR, Cheng TS, Chen WC, Shyu HY, Ko CJ, Huang HP, Teng CH, Lin CH, Johnson MD, Lin CY, Lee MS. Matriptase is involved in ErbB-2-induced prostate cancer cell invasion. Am J Pathol 177: 3145–3158, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yu J, Ustach C, Kim HR. Platelet-derived growth factor signaling and human cancer. J Biochem Mol Biol 36: 49–59, 2003. [DOI] [PubMed] [Google Scholar]
- 47.Zhang Y, Yang M, Portney NG, Cui D, Budak G, Ozbay E, Ozkan M, Ozkan CS. Zeta potential: a surface electrical characteristic to probe the interaction of nanoparticles with normal and cancer human breast epithelial cells. Biomed Microdevices 10: 321–328, 2008. [DOI] [PubMed] [Google Scholar]
- 48.Zhong H, De Marzo AM, Laughner E, Lim M, Hilton DA, Zagzag D, Buechler P, Isaacs WB, Semenza GL, Simons JW. Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res 59: 5830–5835, 1999. [PubMed] [Google Scholar]