

Abstract
Idiopathic pulmonary fibrosis (IPF) is a chronic lung disease characterized by progressive decline in lung function, resulting in significant morbidity and mortality. Current concepts of the pathogenesis of IPF primarily center on dysregulated epithelial cell repair and altered epithelial-mesenchymal communication and extracellular matrix deposition following chronic exposure to cigarette smoke or environmental toxins. In recent years, increasing attention has been directed toward the role of the intercellular junctional complex in determining the specific properties of epithelia in pulmonary diseases. Additionally, recent genomewide association studies suggest that specific genetic variants predictive of epithelial cell dysfunction may confer susceptibility to the development of sporadic idiopathic pulmonary fibrosis. A number of genetic disorders linked to pulmonary fibrosis and familial interstitial pneumonias are associated with loss of epithelial integrity. However, the potential links between extrapulmonary clinical syndromes associated with defects in epithelial cells and the development of pulmonary fibrosis are not well understood. Here, we report a case of hereditary mucoepithelial dysplasia that presented with pulmonary fibrosis and emphysema on high-resolution computed tomography. This case illustrates a more generalizable concept of epithelial disintegrity in the development of fibrotic lung diseases, which is explored in greater detail in this review article.
Keywords: pulmonary fibrosis, epithelial barrier dysfunction, cell-cell adhesion, epithelial junctional complex
interstitial lung diseases represent a heterogeneous group of diffuse lung diseases characterized by chronic, progressive dyspnea that occurs primarily in older adults. Idiopathic pulmonary fibrosis (IPF) is the most common among the idiopathic interstitial pneumonias (IIPs) and is characterized by usual interstitial pneumonia on high-resolution computed tomography (HRCT) and lung biopsy (45). Two drugs, pirfenidone and nintedanib, were approved by the United States Food and Drug Administration for IPF based on reduction in the rate of lung function decline (24, 46); however, these drugs do not appear to arrest (or reverse) fibrosis. The mechanisms of their antifibrotic actions remain unclear, and treatment efficacy may be influenced by effects on the epithelium and/or mesenchymal cells.
The fibrosing varieties of IIPs are thought to be associated with alveolar epithelial cell (AEC) dysfunction, characterized by progressive loss of the normal alveolar architecture. The current paradigm is that recurrent injury to AECs followed by aberrant repair/regeneration of epithelial barrier, persistence of activated fibroblasts, and alterations in extracellular matrix (ECM) result in progressive pulmonary fibrosis (52, 71). In recent years, increasing attention has been directed toward the role of the intercellular junctional complex in determining the specific properties of epithelia in pulmonary diseases (19, 23, 26, 29, 50, 54). Identification of familial cases of interstitial pneumonias with gene mutations in surfactant protein C (40) or telomerase (3), and clinical syndromes such as Hermansky-Pudlak syndrome (11, 67), further support a critical role for AEC injury in disease pathogenesis. Additionally, recent population studies in patients with IIP demonstrate multiple susceptibility loci that indicate an increasingly important role of specific genetic variants associated with defects in host defense and cell-to-cell adhesion (8, 17, 41).
A broad spectrum of inherited and acquired conditions, including infections or autoimmune diseases, in which essential components of intercellular junctional complexes are missing or structurally altered, can lead to epithelial barrier disintegrity. In this report, we describe a case characterized by epithelial cell defects: hereditary mucoepithelial dysplasia (HMD), presenting with features of pulmonary fibrosis. Translating this finding “bedside to bench,” we explore the role of dysfunction of components of the intercellular junctional complex in the alveolar epithelium in the development of fibrotic lung diseases, in general. Furthermore, we discuss emerging concepts in the involvement of epithelial cell defects and polymorphisms of genes associated with lung epithelium in the pathogenesis of IPF.
Case Report
A 39-yr-old Caucasian male was evaluated for progressively worsening dyspnea on exertion and persistent nonproductive cough for over 5 years. He has had alopecia, skin scaling, and visual disturbances since childhood. He denied any other rashes, joint pains, or mucosal lesions. He was a lifelong nonsmoker without identifiable environmental exposures. He has a family history of HMD confirmed by pathology reports in a sibling (sister) who died at age 12 of pulmonary complications. His father also died at an early age (24 yr) of an unknown lung disease. With this family history and characteristic clinical presentation during childhood, he was given the diagnosis of HMD prior to his presentation to our institution. Physical exam at presentation to our clinic was remarkable for fine crackles and scant expiratory wheezes bilaterally, digital clubbing, and skin scaling of the arms. Pulmonary function tests (PFTs) revealed severe obstruction [forced expiratory volume in 1 s (FEV1) = 1.57 liters (37%); forced vital capacity (FVC) = 3.36 liters (59%); FEV1/FVC ratio = 47%], air trapping [total lung capacity (TLC) = 6.45 liters (81%); residual volume (RV) = 3.10 (135%)] and moderately reduced diffusion [carbon monoxide diffusion capacity (DlCO) = 13 (43%)]. During the 6-min walk test, he was able to walk 351 m without oxygen desaturation. HRCT scans showed predominantly reticulations with honeycombing and traction bronchiectasis in a peripheral and basilar distribution with coexistent paraseptal emphysema (Fig. 1). Complete blood count and routine chemistry panels were normal. Tests for specific antibodies including antinuclear factor (ANA), anti-SS-A and SS-B antibodies, antineutrophilic cytoplasmic antibodies (ANCA), anti-DNA antibodies, anti-Jo-1, anti-Scl-70, and anticyclic citrullinated peptide (CCP) antibodies were all negative. Over the course of 4 years of follow-up in our clinic, his PFTs showed a decline in FVC to 2.40 liters (43%), TLC 4.82 liters (61%), and DlCO 9.5 (35%). He has been treated with inhaled corticosteroids and bronchodilators.
Fig. 1.

High-resolution computed tomography sections of a patient with hereditary mucoepithelial dysplasia demonstrating subpleural reticulation with honeycombing and traction bronchiectasis in a peripheral and basilar distribution with coexistent paraseptal emphysema.
Pathogenesis of Hereditary Mucoepithelial Dysplasia
HMD is a dyshesive, dyskeratotic epithelial syndrome caused by an abnormality in desmosomes and gap junctions (GJs) with autosomal dominant inheritance (64). It can present with phenotypic variants involving mucosae, skin, hair, lungs, and eyes. Patients have severe airflow obstruction with combined interstitial fibrosis and emphysema; lung involvement commonly presents with spontaneous pneumothorax from rupture of giant bullae (4, 32, 63, 64). There are fewer reports of an association with pulmonary fibrosis, perhaps related to availability of HRCT until more recently. A 1979 report indicated the presence of fibrosis with thickened septa and small cysts throughout the lung parenchyma on postmortem examination of lung tissue section in a patient with HMD (63); in this report, histopathology of oral and vaginal mucosa demonstrated dyshesive epithelium with lack of maturation and atrophy, dyskeratosis, and unusual cytoplasmic inclusions. Ultrastructural studies showed a paucity of desmosomes and the presence of perinuclear filamentous inclusions resembling internalized GJ and desmosome material in dyskeratotic cells (63). A more recent report of mucosal biopsies of eight patients with HMD indicated the presence of numerous cytoplasmic vacuoles and the filament bundles interspersed between these vacuoles expressing keratin; however, the expression of junctional and cytoskeletal proteins was normal (4). These findings suggest that this disease likely represents a defect in the development of cytoskeletal components and/or assembly of intercellular GJs. The desmosomal cadherin gene cluster in chromosome 18q12.1 including desmoglein and desmocollin was excluded as possible candidate genes in the haplotype analysis of one family (4). However, the specific genetic mutation(s) involved in HMD is yet to be identified.
Epithelial Barrier Composition and Dynamics in Pulmonary Fibrosis
Persistent exposure to cigarette smoke or environmental toxins results in AEC injury with associated basement membrane damage. This may lead to dysregulated repair/regeneration and altered epithelial-mesenchymal communication with subsequent progressive fibrosis (13, 25). Cell-cell adhesion is critical for maintaining the integrity of alveolar epithelium, thus maintaining its protective barrier function against toxic agents or pathogens and allowing interepithelial transfer of molecules and signals. Cell adhesion molecules (CAMs), a group of specialized proteins, are found on epithelial cell surfaces and mediate the adhesive cell-cell interactions between adjacent epithelial cells. The intercellular junction complexes at cell-cell contact sites of epithelial cells are comprised of the tight junction (TJ), adherens junction (AJ), GJ, and desmosomes (Fig. 2).
Fig. 2.
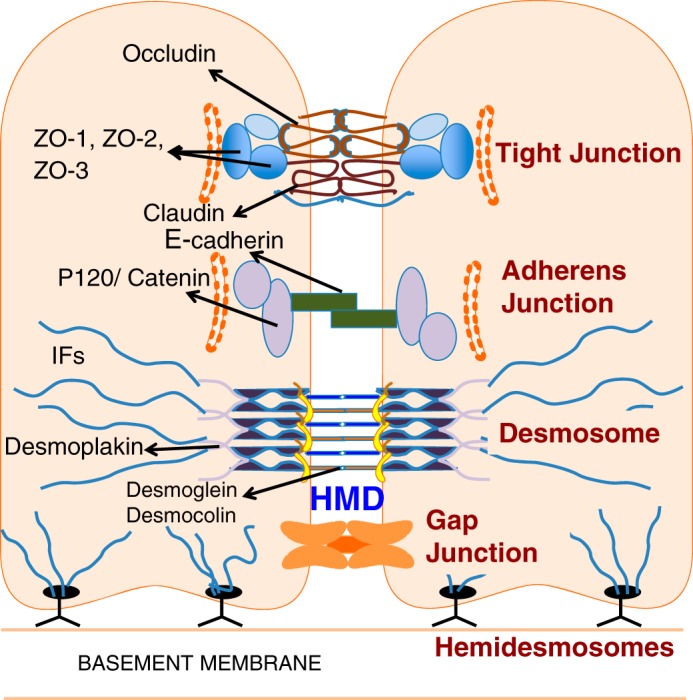
Cell-cell junctions: Tight junctions, adherens junctions, and desmosomes are the three main junctional complexes connecting adjacent epithelial cells. Tight junctions are the most apical protein complexes and regulate epithelial barrier paracellular permeability. Adherens junctions and desmosomes stabilize cell-to-cell adhesion and maintain lung homeostasis. They also regulate the actin-cytoskeletal organization and confer mechanical strength to the alveolar epithelial barrier. Gap junctions are essential for intercellular communication and secretion of surfactant. Hemidesmosomes bind basal epithelial cells to the basement membrane. Hereditary mucoepithelial dysplasia (HMD) affects desmosomal structures. ZO, zona occludens; IFs, intermediate filaments.
Epithelial barrier dysfunction due to repetitive tissue injury leads to host responses involving a myriad of interactions among various cells and soluble factors that are orchestrated to restore normal lung structure and function. Few studies have examined the potential role of intercellular junctional complex proteins in maintenance of the epithelial barrier integrity and development of pulmonary fibrosis following loss of this barrier function. In this section, we will briefly review the role of each component of the alveolar epithelial intercellular junctional complex.
Tight junctions.
TJs form the apical component of the junction complex and are essential for innate immunity, as well as cellular differentiation and proliferation. They comprise the membrane proteins (claudins and occludins) in addition to scaffold proteins known as zona occludens (ZO-1, ZO-2, and ZO-3) (48). Claudins, particularly claudin 18, are the major proteins contributing to the epithelial barrier function of TJs in the lungs and maintain alveolar fluid homeostasis (33, 47). Disruption of TJs can result in increased paracellular permeability, thus permitting entry of antigens, toxins, and protein-rich fluid into alveolar spaces. Reduced expression of claudins, particularly claudin-18, along with lower levels of mRNA encoding TJ proteins was reported in an experimental bleomycin-induced lung injury model (42). Differential claudin and cadherin expression in hyperplastic AECs compared with normal AECs during an aberrant repair process suggests focal changes in permeability of this barrier (19, 29). Although the lower expression of claudins could simply be due to epithelial cell death from bleomycin exposure, the structural disruption of TJs in fibrotic lesions suggests that bleomycin injury causes alveolar barrier dysfunction by other possible mechanisms. Transforming growth factor-β1 (TGF-β1), a well-established profibrotic cytokine, has been shown to cause disruption of TJs in human alveolar epithelial cells and induce epithelial-to-mesenchymal transition (EMT) (42). Additionally, TGF-β1-induced TJ disruption was augmented in a bleomycin injury model of phosphatase and tensin homolog (pten)-null mice (38). In this study, pten-null mice demonstrated disassembly of TJs of AECs and exacerbated lung fibrosis following injury. Furthermore, this study also demonstrated decreased PTEN expression in AECs of human IPF lungs. Taken together, these studies suggest that structural disruption of TJs with subsequent loss of alveolar epithelial integrity play an important role in the development of pulmonary fibrosis.
In addition to preservation of barrier function, adequate expression of claudins may be essential to restore alveolar epithelial barrier during normal injury-repair responses. Increased expression of both TJ and AJ proteins was demonstrated in regenerative alveolar epithelium (29). However, expression of claudin-1, claudin-3, and claudin-4 in fibrotic lung was shown to be similar to or even lower than that measured in the healthy controls (29). Interestingly, claudin knockout mice demonstrated impaired alveologenesis and alveolar barrier dysfunction (28). It is possible that the diminished capacity of epithelial cells to produce claudin could lead to incomplete repair and differentiation of epithelial cells, thus resulting in hyperplastic type II AECs seen in pulmonary fibrosis.
Adherens junctions.
Located more basal to TJs, AJs consist of cadherin and the nectin family CAMs; they are linked to the actin cytoskeleton through the binding proteins catenins and afadin, respectively. AJs function to stabilize cell-cell adhesion and regulate actin cytoskeletal organization, intracellular signaling, and gene transcription. Epithelial cadherin (E-cadherin) is a calcium-dependent CAM with pivotal roles in epithelial cell behavior (20), and it is often used as a marker of epithelial cells. Expression of E-cadherin is reduced in lung sections of patients with IPF; cytoplasmic localization of this protein during EMT is associated with disruption of epithelial barrier function and increased cell migration (20, 61). Cigarette smoke impairs the proteins, ZO-1, ZO-2, and E-cadherin in human bronchial epithelial cells, thus resulting in altered permeability of the epithelial barrier and, potentially, mesenchymal differentiation of epithelial cells (49, 69). Thoracic radiation and bleomycin-induced lung injury can also decrease E-cadherin and aquaporin-5 expression in AECs, increasing plasma/water permeability into alveolar spaces (2, 9). Immunohistochemistry of lung tissue in aquaporin-5 knockout mice demonstrated fibrosis with increased deposition of type I collagen in alveolar walls (9). Although these changes in permeability and increased alveolar fluid accumulation are typically associated with acute lung injury, persistent injury to the epithelial barrier may lead to a cascade of reactions with release of soluble factors that promote myofibroblast differentiation and ECM deposition.
α3β1 Integrin is a laminin receptor that promotes cell-cell communications through its interactions with the E-cadherin/β-catenin complex (34, 59). It was recently reported that α3β1 integrin interacts with E-cadherin and the TGF-β receptor to form a trimolecular complex that triggers phosphorylation of β-catenin at Y654 in AECs (23). Phosphorylated β-catenin subsequently interacts with phosphorylated Smad2, a TGF-β receptor-regulated effector protein, to induce fibrotic gene expression. This study suggests a role for interactions between α3β1, E-cadherin, and β-catenin signaling in the development of lung fibrosis. Additionally, increased expression of cadherin-11 (CDH11) in IPF patients and animal models of lung fibrosis have been reported (50); treatment with CDH11-blocking antibody or genetic deletion of Cdh11 protects mice against bleomycin injury-induced lung fibrosis. These data support a pivotal role of CDH11 in pulmonary fibrosis. Given the fact that multiple cell populations including AECs, fibroblasts, and macrophages express CDH11 in pulmonary fibrosis, it is likely that CDH11 regulates multiple steps in the fibrotic process.
Gap junctions.
GJs are essential for intercellular communication and secretion of surfactant necessary for barrier function. They consist of an array of transmembrane channels composed of connexins (Cx) that connect to similar structures in the adjacent cells. Differential expression of various connexins in the lung, especially Cx43 and Cx46, has an important role in the regulation of normal lung homeostasis and remodeling in response to epithelial cell injury (1, 31). Fibroblasts from patients with IPF demonstrate significant reduction in Cx43 mRNA with alteration to GJ intercellular communication compared with fibroblasts from normal subjects (53). Additionally, mice deficient in vascular endothelial cell-specific Cx43 and Cx40 develop spontaneous fibrosis with fibroblast accumulation and aberrant alveolar remodeling (26). Further studies are required to determine whether abnormalities in alveolar epithelial cell-specific connexins and epithelial cell GJs predispose to lung fibrosis.
Hemidesmosomes.
Hemidesmosomes are specialized multiprotein transmembrane complexes that facilitate the binding of keratin intermediate filament (IF) in epithelial cells to the underlying basement membrane and ECM. This binding is essential in maintenance of integrity and mechanical stability of the lung. The hemidesmosomes are formed by integrin α6β4, laminin 5, and tetraspanin CD151 (37, 56). Tetraspanins belong to a family of proteins that form multimolecular complexes with a variety of other proteins including integrins.
Tetraspanin CD151 is predominantly expressed in the basolateral surface of epithelial cells and is crucial for the maintenance of epithelial integrity via adhesion of the basal surface of AECs to basement membrane. Deletion of CD151 in AECs results in alterations to the cell structure and degradation of the epithelial integrity due to impaired adhesion to basement membrane (54). In this study, CD151 knockout mice were shown to spontaneously develop age-related pulmonary fibrosis; AECs from these mice exhibit fibroblast-like changes through upregulation of TGF-β1 signaling and augmented phosphorylated Smad2. Furthermore, decreased CD151 expression was observed in AECs from patients with IPF (54), supporting a role for loss of CD151 and epithelial integrity in at least a subset of IPF patients. Interestingly, tetraspanin CD151-integrin α3β1 association has been shown to be functionally important for α3β1-integrin-mediated cell migration and matrix remodeling (21). Further studies to understand the role of tetraspanin CD151-integrin interactions and their effects on TGF-β1 in the development of pulmonary fibrosis are warranted.
Genetic Variants Affecting Epithelial Cell Integrity
Over the past decade, there has been remarkable progress in the understanding of IPF pathogenesis. In addition to acquired defects in epithelial barrier integrity, there have been recent advances in identifying polymorphisms of genes associated with lung epithelium and their association with higher risk of IPF. Population studies initially implicated the role of specific genetic variants including MUC5B, TERT, TERC, SFTPC, and SFTPA2 in the development of IPF and other fibrosing IIPs (8, 17, 41). Additional genetic variants including desmoplakin (DSP) and dipeptidyl peptidase 9 (DPP9) genes associated with cell-to-cell adhesion were recently identified in patients with fibrotic IIP (8).
Desmosomes are intercellular junctions located in the basolateral membranes of epithelial cells. There is calcium-dependent transmembrane interaction between the extracellular domains of the desmosomal cadherins between adjacent cells. The cadherin cytoplasmic tails then associate with the linker proteins plakoglobin and plakophilins. Linkage of this desmosomal assembly to the cytoskeleton is mediated through a series of interactions between binding proteins, desmoplakin, and linker proteins (7). Thus desmosomes primarily provide mechanical support for maintenance of tissue architecture by tethering the keratin IF network to the plasma membrane (10). This is particularly important in maintaining the integrity of tissues that experience mechanical stress such as peripheral portions of the lungs, myocardium, skin, bladder, and gastrointestinal mucosa.
Mutations in expression of DSP gene that encodes for desmoplakin have been associated with several skin diseases including keratoderma, alopecia, and severe acantholytic epidermolysis bullosa (18, 60). More importantly, DSP mutations that affect exon 24 encoding the COOH-terminal domain have been associated with cardiac interstitial fibrosis and arrhythmogenic right ventricular dysplasia/cardiomyopathy (35, 66). This was shown to be due to disruption of mechanical linkage between cells and modifications to cell-cell adhesion proteins. The minor allele of variant rs2076295 in intron 5 has been associated with decreased whole lung DSP expression and higher risk of IPF (8, 36). The differential expression of DSP in association with this variant suggests that disruption of desmosomal integrity with resultant impairment of cell-cell adhesion and aberrant epithelial barrier injury-repair response may participate in the development of IPF.
DSP has been shown to inhibit Wnt/β-catenin signaling pathway through regulation of plakoglobin, β-catenin, and matrix metalloproteinase 14 in a lung cancer model (65). Aberrant activation of Wnt/β-catenin signaling pathway has been implicated in the development of pulmonary fibrosis (6) and is, thus, one potential mechanism for a profibrotic role of DSP in IPF. Interestingly, increased DSP gene expression was demonstrated in lung tissue of IPF patients without the DSP gene variant rs2076295 (36). This increase could be the consequence of epithelial cell injury-repair response to maintain epithelial barrier integrity in response to persistent epithelial injury. Desmosomes have intra- and extracellular components, and changes to any part of this structure could lead to alterations in yet-undetermined cell signaling pathways.
DPP9 is an enzyme ubiquitously expressed by epithelial cells and fibroblasts and is necessary for intracellular signaling, cell adhesion, and migration (68). DPP9 gene silencing or enzyme inhibition has been shown to suppress the adhesion-signaling pathway through decreased phosphorylation of focal adhesion kinase and paxillin (68). Thus alterations in this gene may result in impaired cell movement during repair and lead to aberrant healing. Taken together, these findings suggest that genetic variations in the expression of DSP and DPP9 contribute to biochemical and biomechanical modifications that alter cell-cell adhesion in the lung. The presence of these genetic variations may increase epithelial cell susceptibility to barrier disintegrity in response to persistent injury, thus resulting in pulmonary fibrosis.
Epithelial-Mesenchymal Cross Talk
Epithelial and mesenchymal cell interactions are critical in the process of lung development, homeostasis during adulthood, and injury-repair responses (5). Loss of epithelial barrier integrity and the altered alveolar microenvironment disrupts the tightly orchestrated temporal and spatial regulation of epithelial-mesenchymal cross talk (52). Injured alveolar epithelial cells and macrophages release/activate profibrotic mediators, in particular TGF-β1 (22, 30). TGF-β1, activated by AEC-integrin-mediated process (39) or by biomechanical signals (62), induces myofibroblast differentiation and activation. Recent studies indicate that myofibroblasts in IPF acquire a senescent and apoptosis-resistant phenotype (15). These mesenchymal cells secrete additional paracrine factors, such as hydrogen peroxide (16, 55), angiotensin-II (43, 57), Fas-ligand (12, 58), and TGF-β1 (14, 51), that may potentiate or perpetuate injury/apoptosis of AECs. In turn, the accumulation of a highly cross-linked and stiff matrix may perpetuate mesenchymal activation and progressive fibrosis (27, 70) (Fig. 3).
Fig. 3.
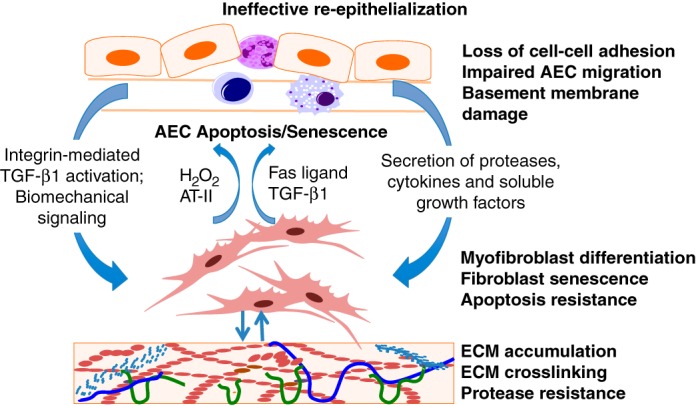
Epithelial-mesenchymal cross talk in pulmonary fibrosis: Cellular homeostasis of the alveolar structure is dependent on bidirectional signaling between alveolar epithelial cells (AECs) and mesenchymal cells. Loss of homeostasis and ineffective reepithelialization may occur with loss of cell-cell adhesion, impaired AEC migration, senescence, and/or apoptosis. The resultant epithelial disintegrity leads to integrin-mediated activation of transforming growth factor-β1 (TGF-β1), altered biomechanics, and release of proteases, cytokines, and growth factors from the epithelium that activate the underlying mesenchyme. In turn, activated fibroblasts and myofibroblasts that acquire an apoptosis-resistant phenotype secrete a number of soluble factors that can induce apoptosis/senescence of AECs; these soluble factors include TGF-β1, hydrogen peroxide (H2O2), angiotensin-II (AT-II), and Fas ligand, thus perpetuating the injury-repair cycle leading to extracellular matrix (ECM) modification and accumulation.
Future Directions
In summary, there is increasing evidence indicating a role for developmental and acquired defects in epithelial cell-cell adhesion in the pathogenesis of fibrotic lung diseases. In this review, we highlight a unique case of a young man with a known genetic defect that results in loss of cell-cell adhesion that resulted in early and severe fibrosis of the lungs. We posit that developmental or acquired dysfunction of epithelial intercellular junctional complexes may have pivotal role in the pathogenesis of pulmonary fibrosis. Whether changes in the expression of specific intercellular junctional proteins are causally involved in the development of pulmonary fibrosis is yet to be elucidated. Persistent alveolar epithelial cell injury from chronic exposure to cigarette smoke or environmental toxins could alter the expression of proteins critical in maintenance of cell-cell adhesion and alveolar epithelial integrity. Although data exploring the molecular mechanisms associated with these alterations and development of IPF are limited, emerging evidence suggests that failure of normal alveolar injury-repair responses culminate in perpetuating cycles of myofibroblast activation and ECM deposition. Recent progress in identifying genetic variants, specifically the genes associated with alterations in cell-cell adhesion, could transform our current understanding of the pathogenesis of IPF.
An integrated approach including genotyping, environmental risk factor assessment, and proteomic analyses of epithelial cell junctional proteins is needed to develop personalized approaches to diagnosis and treatment of patients with IPF. It is not known whether defects in cell-cell adhesion in distal airway/bronchiolar epithelia contribute to disease pathogenesis; for example, the previously reported association between a MUC5B promoter polymorphism and development of IPF with improved survival (44) suggests loss of epithelial homeostasis in the distal airway. Further investigation of specific mechanisms involved in epithelial disintegrity would be invaluable in providing genetic and molecular targets for the development of interventions that prevent/ameliorate disease progression in IPF.
GRANTS
This research was supported by National Heart, Lung, and Blood Institute Grants P01 HL114470, R01 AG046210, and R01 HL124076.
DISCLOSURES
T. Kulkarni has no conflicts of interest to disclose. V. J. Thannickal and Y. Zhou have received research grants from NIH. J. de Andrade has received research grants from the NIH, Genentech, Boehringer Ingelheim, and Fibrogen and consulting fees from Genentech, Boehringer Ingelheim, and Immuneworks, outside the submitted work. T. Luckhardt has received research grants from the NIH, the Pulmonary Fibrosis Foundation, Gilead, Celgene, and Boehringer Ingelheim, outside the submitted work.
AUTHOR CONTRIBUTIONS
T.K., J.A.d.A., Y.Z., T.R.L., and V.J.T. conception and design of research; T.K. prepared figures; T.K. drafted manuscript; T.K., J.A.d.A., Y.Z., T.R.L., and V.J.T. edited and revised manuscript; T.K., J.A.d.A., Y.Z., T.R.L., and V.J.T. approved final version of manuscript.
REFERENCES
- 1.Abraham V, Chou ML, DeBolt KM, Koval M. Phenotypic control of gap junctional communication by cultured alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol 276: L825–L834, 1999. [DOI] [PubMed] [Google Scholar]
- 2.Almeida C, Nagarajan D, Tian J, Leal SW, Wheeler K, Munley M, Blackstock W, Zhao W. The role of alveolar epithelium in radiation-induced lung injury. PLoS One 8: e53628, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Armanios MY, Chen JJ, Cogan JD, Alder JK, Ingersoll RG, Markin C, Lawson WE, Xie M, Vulto I, Phillips JA 3rd, Lansdorp PM, Greider CW, Loyd JE. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med 356: 1317–1326, 2007. [DOI] [PubMed] [Google Scholar]
- 4.Boralevi F, Haftek M, Vabres P, Lepreux S, Goizet C, Leaute-Labreze C, Taieb A. Hereditary mucoepithelial dysplasia: clinical, ultrastructural and genetic study of eight patients and literature review. Br J Dermatol 153: 310–318, 2005. [DOI] [PubMed] [Google Scholar]
- 5.Chapman HA. Epithelial-mesenchymal interactions in pulmonary fibrosis. Annu Rev Physiol 73: 413–435, 2011. [DOI] [PubMed] [Google Scholar]
- 6.Chilosi M, Poletti V, Zamo A, Lestani M, Montagna L, Piccoli P, Pedron S, Bertaso M, Scarpa A, Murer B, Cancellieri A, Maestro R, Semenzato G, Doglioni C. Aberrant wnt/beta-catenin pathway activation in idiopathic pulmonary fibrosis. Am J Pathol 162: 1495–1502, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Delva E, Tucker DK, Kowalczyk AP. The desmosome. Cold Spring Harb Perspect Biol 1: a002543, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fingerlin TE, Murphy E, Zhang W, Peljto AL, Brown KK, Steele MP, Loyd JE, Cosgrove GP, Lynch D, Groshong S, Collard HR, Wolters PJ, Bradford WZ, Kossen K, Seiwert SD, du Bois RM, Garcia CK, Devine MS, Gudmundsson G, Isaksson HJ, Kaminski N, Zhang Y, Gibson KF, Lancaster LH, Cogan JD, Mason WR, Maher TM, Molyneaux PL, Wells AU, Moffatt MF, Selman M, Pardo A, Kim DS, Crapo JD, Make BJ, Regan EA, Walek DS, Daniel JJ, Kamatani Y, Zelenika D, Smith K, McKean D, Pedersen BS, Talbert J, Kidd RN, Markin CR, Beckman KB, Lathrop M, Schwarz MI, Schwartz DA. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat Genet 45: 613–620, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gabazza EC, Kasper M, Ohta K, Keane M, D'Alessandro-Gabazza C, Fujimoto H, Nishii Y, Nakahara H, Takagi T, Menon AG, Adachi Y, Suzuki K, Taguchi O. Decreased expression of aquaporin-5 in bleomycin-induced lung fibrosis in the mouse. Pathol Int 54: 774–780, 2004. [DOI] [PubMed] [Google Scholar]
- 10.Getsios S, Huen AC, Green KJ. Working out the strength and flexibility of desmosomes. Nat Rev Mol Cell Biol 5: 271–281, 2004. [DOI] [PubMed] [Google Scholar]
- 11.Gochuico BR, Huizing M, Golas GA, Scher CD, Tsokos M, Denver SD, Frei-Jones MJ, Gahl WA. Interstitial lung disease and pulmonary fibrosis in Hermansky-Pudlak syndrome type 2, an adaptor protein-3 complex disease. Mol Med 18: 56–64, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Golan-Gerstl R, Wallach-Dayan SB, Amir G, Breuer R. Epithelial cell apoptosis by fas ligand-positive myofibroblasts in lung fibrosis. Am J Respir Cell Mol Biol 36: 270–275, 2007. [DOI] [PubMed] [Google Scholar]
- 13.Gross TJ, Hunninghake GW. Idiopathic pulmonary fibrosis. N Engl J Med 345: 517–525, 2001. [DOI] [PubMed] [Google Scholar]
- 14.Hagimoto N, Kuwano K, Inoshima I, Yoshimi M, Nakamura N, Fujita M, Maeyama T, Hara N. Tgf-beta 1 as an enhancer of fas-mediated apoptosis of lung epithelial cells. J Immunol 168: 6470–6478, 2002. [DOI] [PubMed] [Google Scholar]
- 15.Hecker L, Logsdon NJ, Kurundkar D, Kurundkar A, Bernard K, Hock T, Meldrum E, Sanders YY, Thannickal VJ. Reversal of persistent fibrosis in aging by targeting nox4-nrf2 redox imbalance. Sci Transl Med 6: 231ra247, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hecker L, Vittal R, Jones T, Jagirdar R, Luckhardt TR, Horowitz JC, Pennathur S, Martinez FJ, Thannickal VJ. Nadph oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat Med 15: 1077–1081, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hunninghake GM, Hatabu H, Okajima Y, Gao W, Dupuis J, Latourelle JC, Nishino M, Araki T, Zazueta OE, Kurugol S, Ross JC, San José Estépar R, Murphy E, Steele MP, Loyd JE, Schwarz MI, Fingerlin TE, Rosas IO, Washko GR, O'Connor GT, Schwartz DA. Muc5b promoter polymorphism and interstitial lung abnormalities. N Engl J Med 368: 2192–2200, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jonkman MF, Pasmooij AM, Pasmans SG, van den Berg MP, Ter Horst HJ, Timmer A, Pas HH. Loss of desmoplakin tail causes lethal acantholytic epidermolysis bullosa. Am J Hum Genet 77: 653–660, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaarteenaho-Wiik R, Soini Y. Claudin-1, -2, -3, -4, -5, and -7 in usual interstitial pneumonia and sarcoidosis. J Histochem Cytochem 57: 187–195, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kasper M, Behrens J, Schuh D, Muller M. Distribution of e-cadherin and ep-cam in the human lung during development and after injury. Histochem Cell Biol 103: 281–286, 1995. [DOI] [PubMed] [Google Scholar]
- 21.Kazarov AR, Yang X, Stipp CS, Sehgal B, Hemler ME. An extracellular site on tetraspanin cd151 determines alpha 3 and alpha 6 integrin-dependent cellular morphology. J Cell Biol 158: 1299–1309, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khalil N, O'Connor RN, Unruh HW, Warren PW, Flanders KC, Kemp A, Bereznay OH, Greenberg AH. Increased production and immunohistochemical localization of transforming growth factor-beta in idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol 5: 155–162, 1991. [DOI] [PubMed] [Google Scholar]
- 23.Kim KK, Wei Y, Szekeres C, Kugler MC, Wolters PJ, Hill ML, Frank JA, Brumwell AN, Wheeler SE, Kreidberg JA, Chapman HA. Epithelial cell alpha3beta1 integrin links beta-catenin and smad signaling to promote myofibroblast formation and pulmonary fibrosis. J Clin Invest 119: 213–224, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.King TE Jr, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, Gorina E, Hopkins PM, Kardatzke D, Lancaster L, Lederer DJ, Nathan SD, Pereira CA, Sahn SA, Sussman R, Swigris JJ, Noble PW; ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 370: 2083–2092, 2014. [DOI] [PubMed] [Google Scholar]
- 25.King TE Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet 378: 1949–1961, 2011. [DOI] [PubMed] [Google Scholar]
- 26.Koval M, Billaud M, Straub AC, Johnstone SR, Zarbock A, Duling BR, Isakson BE. Spontaneous lung dysfunction and fibrosis in mice lacking connexin 40 and endothelial cell connexin 43. Am J Pathol 178: 2536–2546, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kulkarni T, O'Reilly P, Antony VB, Gaggar A, Thannickal VJ. Matrix remodeling in pulmonary fibrosis and emphysema. Am J Respir Cell Mol Biol 54: 751–760, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.LaFemina MJ, Sutherland KM, Bentley T, Gonzales LW, Allen L, Chapin CJ, Rokkam D, Sweerus KA, Dobbs LG, Ballard PL, Frank JA. Claudin-18 deficiency results in alveolar barrier dysfunction and impaired alveologenesis in mice. Am J Respir Cell Mol Biol 51: 550–558, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lappi-Blanco E, Lehtonen ST, Sormunen R, Merikallio HM, Soini Y, Kaarteenaho RL. Divergence of tight and adherens junction factors in alveolar epithelium in pulmonary fibrosis. Hum Pathol 44: 895–907, 2013. [DOI] [PubMed] [Google Scholar]
- 30.Larson-Casey JL, Deshane JS, Ryan AJ, Thannickal VJ, Carter AB. Macrophage akt1 kinase-mediated mitophagy modulates apoptosis resistance and pulmonary fibrosis. Immunity 44: 582–596, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee YC, Yellowley CE, Li Z, Donahue HJ, Rannels DE. Expression of functional gap junctions in cultured pulmonary alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol 272: L1105–L1114, 1997. [DOI] [PubMed] [Google Scholar]
- 32.Leithauser LA, Mutasim DF. Hereditary mucoepithelial dysplasia: unique histopathological findings in skin lesions. J Cutan Pathol 39: 431–439, 2012. [DOI] [PubMed] [Google Scholar]
- 33.Li G, Flodby P, Luo J, Kage H, Sipos A, Gao D, Ji Y, Beard LL, Marconett CN, DeMaio L, Kim YH, Kim KJ, Laird-Offringa IA, Minoo P, Liebler JM, Zhou B, Crandall ED, Borok Z. Knockout mice reveal key roles for claudin 18 in alveolar barrier properties and fluid homeostasis. Am J Respir Cell Mol Biol 51: 210–222, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lubman RL, Zhang XL, Zheng J, Ocampo L, Lopez MZ, Veeraraghavan S, Zabski SM, Danto SI, Borok Z. Integrin α3-subunit expression modulates alveolar epithelial cell monolayer formation. Am J Physiol Lung Cell Mol Physiol 279: L183–L193, 2000. [DOI] [PubMed] [Google Scholar]
- 35.Mahoney MG, Sadowski S, Brennan D, Pikander P, Saukko P, Wahl J, Aho H, Heikinheimo K, Bruckner-Tuderman L, Fertala A, Peltonen J, Uitto J, Peltonen S. Compound heterozygous desmoplakin mutations result in a phenotype with a combination of myocardial, skin, hair, and enamel abnormalities. J Invest Dermatol 130: 968–978, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mathai SK, Pedersen BS, Smith K, Russell P, Schwarz MI, Brown KK, Steele MP, Loyd JE, Crapo JD, Silverman EK, Nickerson D, Fingerlin TE, Yang IV, Schwartz DA. Desmoplakin (dsp) variants are associated with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 193: 1151–1160, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Michelson PH, Tigue M, Jones JC. Human bronchial epithelial cells secrete laminin 5, express hemidesmosomal proteins, and assemble hemidesmosomes. J Histochem Cytochem 48: 535–544, 2000. [DOI] [PubMed] [Google Scholar]
- 38.Miyoshi K, Yanagi S, Kawahara K, Nishio M, Tsubouchi H, Imazu Y, Koshida R, Matsumoto N, Taguchi A, Yamashita S, Suzuki A, Nakazato M. Epithelial Pten controls acute lung injury and fibrosis by regulating alveolar epithelial cell integrity. Am J Respir Crit Care Med 187: 262–275, 2013. [DOI] [PubMed] [Google Scholar]
- 39.Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, Pittet JF, Kaminski N, Garat C, Matthay MA, Rifkin DB, Sheppard D. The integrin alpha v beta 6 binds and activates latent tgf beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell 96: 319–328, 1999. [DOI] [PubMed] [Google Scholar]
- 40.Nogee LM, Dunbar AE 3rd, Wert SE, Askin F, Hamvas A, Whitsett JA. A mutation in the surfactant protein c gene associated with familial interstitial lung disease. N Engl J Med 344: 573–579, 2001. [DOI] [PubMed] [Google Scholar]
- 41.Noth I, Zhang Y, Ma SF, Flores C, Barber M, Huang Y, Broderick SM, Wade MS, Hysi P, Scuirba J, Richards TJ, Juan-Guardela BM, Vij R, Han MK, Martinez FJ, Kossen K, Seiwert SD, Christie JD, Nicolae D, Kaminski N, Garcia JG. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. Lancet Respir Med 1: 309–317, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ohta H, Chiba S, Ebina M, Furuse M, Nukiwa T. Altered expression of tight junction molecules in alveolar septa in lung injury and fibrosis. Am J Physiol Lung Cell Mol Physiol 302: L193–L205, 2012. [DOI] [PubMed] [Google Scholar]
- 43.Papp M, Li X, Zhuang J, Wang R, Uhal BD. Angiotensin receptor subtype AT1 mediates alveolar epithelial cell apoptosis in response to ANG II. Am J Physiol Lung Cell Mol Physiol 282: L713–L718, 2002. [DOI] [PubMed] [Google Scholar]
- 44.Peljto AL, Zhang Y, Fingerlin TE, Ma SF, Garcia JG, Richards TJ, Silveira LJ, Lindell KO, Steele MP, Loyd JE, Gibson KF, Seibold MA, Brown KK, Talbert JL, Markin C, Kossen K, Seiwert SD, Murphy E, Noth I, Schwarz MI, Kaminski N, Schwartz DA. Association between the muc5b promoter polymorphism and survival in patients with idiopathic pulmonary fibrosis. JAMA 309: 2232–2239, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, Colby TV, Cordier JF, Flaherty KR, Lasky JA, Lynch DA, Ryu JH, Swigris JJ, Wells AU, Ancochea J, Bouros D, Carvalho C, Costabel U, Ebina M, Hansell DM, Johkoh T, Kim DS, King TE Jr, Kondoh Y, Myers J, Müller NL, Nicholson AG, Richeldi L, Selman M, Dudden RF, Griss BS, Protzko SL, Schünemann HJ; ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 183: 788–824, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, Cottin V, Flaherty KR, Hansell DM, Inoue Y, Kim DS, Kolb M, Nicholson AG, Noble PW, Selman M, Taniguchi H, Brun M, Le Maulf F, Girard M, Stowasser S, Schlenker-Herceg R, Disse B, Collard HR; INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 370: 2071–2082, 2014. [DOI] [PubMed] [Google Scholar]
- 47.Saitou M, Furuse M, Sasaki H, Schulzke JD, Fromm M, Takano H, Noda T, Tsukita S. Complex phenotype of mice lacking occludin, a component of tight junction strands. Mol Biol Cell 11: 4131–4142, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sawada N, Murata M, Kikuchi K, Osanai M, Tobioka H, Kojima T, Chiba H. Tight junctions and human diseases. Med Electron Microsc 36: 147–156, 2003. [DOI] [PubMed] [Google Scholar]
- 49.Schamberger AC, Mise N, Jia J, Genoyer E, Yildirim AO, Meiners S, Eickelberg O. Cigarette smoke-induced disruption of bronchial epithelial tight junctions is prevented by transforming growth factor-beta. Am J Respir Cell Mol Biol 50: 1040–1052, 2014. [DOI] [PubMed] [Google Scholar]
- 50.Schneider DJ, Wu M, Le TT, Cho SH, Brenner MB, Blackburn MR, Agarwal SK. Cadherin-11 contributes to pulmonary fibrosis: potential role in tgf-beta production and epithelial to mesenchymal transition. FASEB J 26: 503–512, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Solovyan VT, Keski-Oja J. Proteolytic activation of latent TGF-beta precedes caspase-3 activation and enhances apoptotic death of lung epithelial cells. J Cell Physiol 207: 445–453, 2006. [DOI] [PubMed] [Google Scholar]
- 52.Thannickal VJ, Toews GB, White ES, Lynch JP 3rd, Martinez FJ. Mechanisms of pulmonary fibrosis. Annu Rev Med 55: 395–417, 2004. [DOI] [PubMed] [Google Scholar]
- 53.Trovato-Salinaro A, Trovato-Salinaro E, Failla M, Mastruzzo C, Tomaselli V, Gili E, Crimi N, Condorelli DF, Vancheri C. Altered intercellular communication in lung fibroblast cultures from patients with idiopathic pulmonary fibrosis. Respir Res 7: 122, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tsujino K, Takeda Y, Arai T, Shintani Y, Inagaki R, Saiga H, Iwasaki T, Tetsumoto S, Jin Y, Ihara S, Minami T, Suzuki M, Nagatomo I, Inoue K, Kida H, Kijima T, Ito M, Kitaichi M, Inoue Y, Tachibana I, Takeda K, Okumura M, Hemler ME, Kumanogoh A. Tetraspanin cd151 protects against pulmonary fibrosis by maintaining epithelial integrity. Am J Respir Crit Care Med 186: 170–180, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Waghray M, Cui Z, Horowitz JC, Subramanian IM, Martinez FJ, Toews GB, Thannickal VJ. Hydrogen peroxide is a diffusible paracrine signal for the induction of epithelial cell death by activated myofibroblasts. FASEB J 19: 854–856, 2005. [DOI] [PubMed] [Google Scholar]
- 56.Walko G, Castanon MJ, Wiche G. Molecular architecture and function of the hemidesmosome. Cell Tissue Res 360: 529–544, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang R, Ramos C, Joshi I, Zagariya A, Pardo A, Selman M, Uhal BD. Human lung myofibroblast-derived inducers of alveolar epithelial apoptosis identified as angiotensin peptides. Am J Physiol Lung Cell Mol Physiol 277: L1158–L1164, 1999. [DOI] [PubMed] [Google Scholar]
- 58.Wang R, Zagariya A, Ang E, Ibarra-Sunga O, Uhal BD. Fas-induced apoptosis of alveolar epithelial cells requires ANG II generation and receptor interaction. Am J Physiol Lung Cell Mol Physiol 277: L1245–L1250, 1999. [DOI] [PubMed] [Google Scholar]
- 59.Wang Z, Symons JM, Goldstein SL, McDonald A, Miner JH, Kreidberg JA. (Alpha)3(beta)1 integrin regulates epithelial cytoskeletal organization. J Cell Sci 112: 2925–2935, 1999. [DOI] [PubMed] [Google Scholar]
- 60.Whittock NV, Wan H, Morley SM, Garzon MC, Kristal L, Hyde P, McLean WH, Pulkkinen L, Uitto J, Christiano AM, Eady RA, McGrath JA. Compound heterozygosity for non-sense and mis-sense mutations in desmoplakin underlies skin fragility/woolly hair syndrome. J Invest Dermatol 118: 232–238, 2002. [DOI] [PubMed] [Google Scholar]
- 61.Willis BC, duBois RM, Borok Z. Epithelial origin of myofibroblasts during fibrosis in the lung. Proc Am Thorac Soc 3: 377–382, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wipff PJ, Rifkin DB, Meister JJ, Hinz B. Myofibroblast contraction activates latent tgf-beta1 from the extracellular matrix. J Cell Biol 179: 1311–1323, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Witkop CJ Jr, White JG, King RA, Dahl MV, Young WG, Sauk JJ Jr. Hereditary mucoepithelial dysplasia: a disease apparently of desmosome and gap junction formation. Am J Hum Genet 31: 414–427, 1979. [PMC free article] [PubMed] [Google Scholar]
- 64.Witkop CJ Jr, White JG, Sauk JJ Jr, King RA. Clinical, histologic, cytologic, and ultrastructural characteristics of the oral lesions from hereditary mucoepithelial dysplasia. A disease of gap junction and desmosome formation. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 46: 645–657, 1978. [DOI] [PubMed] [Google Scholar]
- 65.Yang L, Chen Y, Cui T, Knosel T, Zhang Q, Albring KF, Huber O, Petersen I. Desmoplakin acts as a tumor suppressor by inhibition of the Wnt/β-catenin signaling pathway in human lung cancer. Carcinogenesis 33: 1863–1870, 2012. [DOI] [PubMed] [Google Scholar]
- 66.Yang Z, Bowles NE, Scherer SE, Taylor MD, Kearney DL, Ge S, Nadvoretskiy VV, DeFreitas G, Carabello B, Brandon LI, Godsel LM, Green KJ, Saffitz JE, Li H, Danieli GA, Calkins H, Marcus F, Towbin JA. Desmosomal dysfunction due to mutations in desmoplakin causes arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Res 99: 646–655, 2006. [DOI] [PubMed] [Google Scholar]
- 67.Young LR, Gulleman PM, Bridges JP, Weaver TE, Deutsch GH, Blackwell TS, McCormack FX. The alveolar epithelium determines susceptibility to lung fibrosis in Hermansky-Pudlak syndrome. Am J Respir Crit Care Med 186: 1014–1024, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang H, Chen Y, Wadham C, McCaughan GW, Keane FM, Gorrell MD. Dipeptidyl peptidase 9 subcellular localization and a role in cell adhesion involving focal adhesion kinase and paxillin. Biochim Biophys Acta 1853: 470–480, 2015. [DOI] [PubMed] [Google Scholar]
- 69.Zhang L, Gallup M, Zlock L, Chen YT, Finkbeiner WE, McNamara NA. Pivotal role of muc1 glycosylation by cigarette smoke in modulating disruption of airway adherens junctions in vitro. J Pathol 234: 60–73, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhou Y, Huang X, Hecker L, Kurundkar D, Kurundkar A, Liu H, Jin TH, Desai L, Bernard K, Thannickal VJ. Inhibition of mechanosensitive signaling in myofibroblasts ameliorates experimental pulmonary fibrosis. J Clin Invest 123: 1096–1108, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zoz DF, Lawson WE, Blackwell TS. Idiopathic pulmonary fibrosis: a disorder of epithelial cell dysfunction. Am J Med Sci 341: 435–438, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]