

Abstract
The stimulation of postprandial K+ clearance involves aldosterone-independent and -dependent mechanisms. In this context, serum- and glucocorticoid-induced kinase (SGK)1, a ubiquitously expressed kinase, is one of the primary aldosterone-induced proteins in the aldosterone-sensitive distal nephron. Germline inactivation of SGK1 suggests that this kinase is fundamental for K+ excretion under conditions of K+ load, but the specific role of renal SGK1 remains elusive. To avoid compensatory mechanisms that may occur during nephrogenesis, we used inducible, nephron-specific Sgk1Pax8/LC1 mice to assess the role of renal tubular SGK1 in K+ regulation. Under a standard diet, these animals exhibited normal K+ handling. When challenged by a high-K+ diet, they developed severe hyperkalemia accompanied by a defect in K+ excretion. Molecular analysis revealed reduced neural precursor cell expressed developmentally downregulated protein (NEDD)4-2 phosphorylation and total expression. γ-Epithelial Na+ channel (ENaC) expression and α/γENaC proteolytic processing were also decreased in mutant mice. Moreover, with no lysine kinase (WNK)1, which displayed in control mice punctuate staining in the distal convoluted tubule and diffuse distribution in the connecting tubule/cortical colleting duct, was diffused in the distal convoluted tubule and less expressed in the connecting tubule/collecting duct of SgkPax8/LC1 mice. Moreover, Ste20-related proline/alanine-rich kinase phosphorylation, and Na+-Cl− cotransporter phosphorylation/apical localization were reduced in mutant mice. Consistent with the altered WNK1 expression, increased renal outer medullary K+ channel apical localization was observed. In conclusion, our data suggest that renal tubular SGK1 is important in the regulation of K+ excretion via the control of NEDD4-2, WNK1, and ENaC.
Keywords: aldosterone, epithelial transport, phosphorylation, potassium, ubiquitylation, serum- and glucocorticoid-induced kinase 1, neural precursor cell expressed developmentally downregulated protein 4-2, with no lysine kinase 1, epithelial Na+ channel
maintenance of the plasma K+ level within normal physiological range (3.5–5.0 mM) is crucial for the proper function of excitable cells (e.g., neurons and skeletal and cardiac myocytes) and represents a significant homeostatic challenge that is mediated by the tight coordination of K+ excretion via the kidney (and, to a lesser extent, the colon) and K+ storage occurring mainly in skeletal muscle (67). It is well accepted that the control of K+ balance by the kidney involves both aldosterone-dependent and -independent mechanisms (55, 56, 64). Renal K+ secretion takes place essentially in the so-called aldosterone-sensitive distal nephron (ASDN), as defined by Loffing et al. (35). One of the most prominent proteins stimulated by aldosterone in the ASDN is the otherwise ubiquitously expressed serum- and glucocorticoid-induced kinase (SGK)1 (28, 34). Studies performed in cellular and animal models have suggested that SGK1 controls renal Na+ reabsorption through regulation of the epithelial Na+ channel (ENaC) and Na+-Cl− cotransporter (NCC) (3, 10, 17–19, 37, 65). This involves phosphorylation of Ser222, Ser246, and Ser328 residues of the ubiquitin protein ligase neural precursor cell expressed developmentally downregulated protein (NEDD)4-2 (5, 17, 52), known to ubiquitylate and negatively regulate ENaC and NCC (1, 3, 22, 26, 45, 51). On the other hand, it has also been suggested that SGK1 acts via phosphorylation of with no lysine kinase (WNK) kinases (12, 44, 48).
Several in vitro and in vivo reports have conferred to SGK1 a positive role in the regulation of K+ secretion. In heterologous expression systems, SGK1 is able to stimulate renal outer medullary K+ (ROMK) channel (KCNJ1) cell surface expression (38, 66). Huang et al. (23) reported that mice harboring constitutive gene inactivation of Sgk1 (Sgk1−/−) exhibited a defect in acute K+ excretion after 30–60 min of intravenous K+ load. At 6 days of high-K+ diet (HKD), Sgk1−/− mice developed hyperkalemia but excreted K+ similarly to control mice (23). Surprisingly, ROMK membrane expression was enhanced in Sgk1−/− mice (23). The ubiquitous expression of SGK1 together with the involvement of the colon, skeletal muscle, and liver in K+ homeostasis leave the possibility that the impaired ability of Sgk1−/− mice to handle K+ load was due to renal and extrarenal SGK1 (6, 29).
To decipher the specific role of renal tubular SGK1 in K+ homeostasis, we took advantage of the previously described inducible, nephron-specific Sgk1Pax8/LC1 model (18). These mice showed a Na+ losing phenotype when Na+ supply was restricted, which correlated with decreased NCC and ENaC protein levels and reduced NEDD4-2 phosphorylation (18). Here, we show that even though SGK1 is expressed in several organs involved in K+ homeostasis, the action of renal tubular SGK1 is crucial for proper renal K+ excretion and is mediated mainly by ENaC. Our data suggest that NEDD4-2 and WNK1 are key players in SGK1-mediated K+ homeostasis.
MATERIALS AND METHODS
Induction of renal tubule-specific SGK1 mutant mice and the verification of the renal tubular specificity.
Inducible, renal-tubule specific SGK1flox/flox/Pax8-rTA/LC1 knockout (KO; SGK1Pax8/LC1) or littermate control Sgk1Pax8 or Sgk1LC1 mice were generated as previously described (18). Mice were housed in a temperature-controlled facility (19–22°C) with a 12:12-h light-dark cycle. To induce gene deletion, 21- to 24-day-old mice were treated with doxycycline (2 mg/ml in 2% sucrose in drinking water) for 12 days. Male mice were used in the study after 1–3 wk following doxycycline treatment. Genotype was identified by PCR performed on ear biopsies as previously described (18). Experimental protocols were designed with respect to the Swiss Animal Welfare Act and approved by the veterinary administration of the Canton de Vaud (Switzerland); the authorization number was 2590.
Dietary manipulation.
Control (Sgk1Pax8 or Sgk1LC1) and Sgk1Pax8/LC1 doxycycline treated-mice were fed a standard diet (0.3% K+, Sniff) or HKD (5% K+ with K3+-citrate used as a K+ supplement, Sniff) for the periods indicated in the related results.
Plasma and organs collection.
Mice were anesthetized by a ketamine-xylazine intraperitoneal injection. Blood was collected by exsanguination from the retroorbital plexus in SARSTEDT heparin-containing microtubes, and plasma was separated according to the manufacturer's instructions. Mice were then humanely euthanized by cervical dislocation.
Metabolic cages and urine and plasma analysis.
After 2 days of adaptation in metabolic cages (catalog no. 3600M021, Indulab), data related to body weight and food and water intake were registered, and 24-h urine samples were collected as previously described (18). Urine analysis (Na+, K+, Ca2+, Mg2+, PO43−, creatinine, and urea) was performed by the Laboratory of Clinical Chemistry at Lausanne Hospital using a Modular Analytics System (Roche Diagnostics). Plasma Na+ and K+ levels were measured with a flame photometer (Cole-Palmer Instrument). Plasma aldosterone measurement was performed at the Service of Nephrology of the Laboratory of Clinical Chemistry at Lausanne Hospital using a radioimmunoassay kit (ALDOSTERONE-RIACT) according to the manufacturer's instructions.
Protein lysate preparation and immunoblot analysis.
Frozen tissues were homogenized using buffer containing 50 mM Tris·HCl (pH7.5), 1 mM EDTA, 1 mM EGTA, 0.27 M sucrose, 50 mM NaF, and 5 mM Na-pyrophosphate in addition to protease inhibitors purchased from Roche (Complete catalog no. 11836145001). Protein homogenates were then centrifuged at 10,000 g for 10 min at 4°C. The supernatant was collected, and protein concentration was measured using the Bradford method (catalog no. UPF86420, Uptima). For ROMK detection, an additional ultracentrifugation step at 100,000 g for 1 h was performed for membrane enrichment. Immunoblots were carried out, and proteins were quantified as previously described (45).
Antibodies.
The following antibodies were used: anti-NCC antibodies (43, 53), phosphorylated (p)Thr233 Ste20-related proline/alanine-rich kinase (SPAK) (43), total SPAK (no. 072271, Millipore), anti-αENaC (53); anti-βENaC and anti-γENaC (63), anti-NEDD4-2 (26), anti-pSer222 NEDD4-2 (18), anti-pSer328 NEDD4-2 (20), anti-pSer256 SGK1 (SC-16744, Santa Cruz Biotechnology), anti-GAPDH (MAB374, Millipore), anti-actin (A-5316, Sigma-Aldrich), anti-WNK1 [exon 12 (47)], and anti-p382WNK1/WNK4 [pan-pWNK (55)].
Generation and characterization of ROMK antibody.
Anti-ROMK antibodies are directed against a sequence of 49 residues (342–391) of rat ROMK protein (NFGKTVEVETPHCAMCLYNEKDARARMKRGYDNPNFVLSEVDETDDTQM). Guinea pigs were immunized by glutathione-S-transferase fusion proteins containing the ROMK peptide. Antibodies were generated at Cocalico Biologicals (Reamstown, PA). Sera of the immunized animals were collected and affinity purified using the Maltose Binding Fusion Protein system. The specificity of the antibodies was validated by Western blot analysis of lysates of human embryonic kidney (HEK)-293 cells transfected with a plasmid encoding ROMK and kidneys from wild-type (WT) and ROMK KO mice (Kcnj1 KO) (36) kept under a HKD for 5 days to enhance ROMK protein expression (see Fig. 5).
Fig. 5.
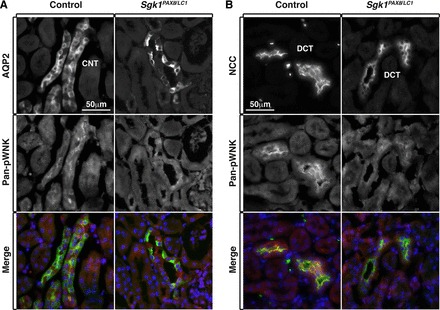
WNK phosphorylation and likely activity are decreased in both the DCT and CNT/CD of Sgk1Pax8/LC1 mice under a HKD. A: costaining of AQP2 (green) and pan-pWNK (red) showing reduced WNK phosphorylation in the CNT/CD of mutant animals compared with control animals. B: costaining of NCC (green) and pan-pWNK (red) showing decreased WNK phosphorylation in the DCT of Sgk1Pax8/LC1 animals compared with control animals.
Immunohistochemistry.
Mice were anesthetized by a ketamine-xylazine intraperitoneal injection. Cardiac perfusions with PBS followed by 4% paraformaldehyde were performed. Before being frozen, fixed kidneys were kept in 30% sucrose in PBS solution at 4°C overnight. Immunostaining was performed on 5-μm cryosections using the primary antibodies listed in the antibody section. Alexa fluor-488-conjugated goat anti-rabbit (A-11008, Life Technologies), Alexa fluor-546-conjugated goat anti-guinea pig (A11074, Thermo Fisher Scientific), or Alexa fluor-546-conjugated donkey anti-sheep (A21098, Thermo Fisher Scientific) antibodies were used. Images in the figures are representatives of data obtained from three control mice and two Sgk1Pax8/LC1 mice.
RNA extraction and TaqMan gene expression assays.
Total RNA was purified from total kidneys using TRIzol (Ambion) and precipitated with isopropanol. cDNA was synthetized from 2–5 μg total RNA using SuperScript II reverse transcriptase (Life Technologies) and random hexamers (Promega). TaqMan Gene Expression Assays (Life Technologies) were used to analyze gene expression. The primers/probes used were as follows: Nedd4l (NEDD4-2; Mm00459584_m1, ThermoFisher Scientific), Sgk1 (Mm00441380_m1), Scann1a (αENaC; Mm00803386_m1), Scann1b (βENaC; Mm00441215_m1), Scann1c (γENaC; Mm00441228_m1), renin-1 (Mm02342889_g1), and Gapdh (Mm99999915_g1).
Statistical analysis.
The significance of the metabolic parameters of the experimental groups under different diets were analyzed using two-way ANOVA followed by Bonferroni multiple-comparison tests. Values were considered significant at α < 0.05. When only one factor was involved, the data were statistically analyzed using an unpaired Student's t-test. Values were considered significant at P ≤ 0.05. Data are represented as means ± SE.
RESULTS
Sgk1Pax8/LC1 mice display suppression of SGK1 in the kidney but not in the liver.
The present model, Sgk1Pax8/LC1, has been previously described; recombination of genomic DNA was not only reported for the kidney but also the liver (18), consistent with Pax8 promotor activity, as previously reported (57). We therefore analyzed Sgk1 mRNA and SGK1 protein expression in the liver and compared the liver results with the kidney (Fig. 1). Although there was recombination, as previously described, in both organs (Fig. 1A), mRNA and protein levels of SGK1 (using an anti-pThr256 SGK1 antibody) were not affected in the liver, whereas they were strongly reduced in the kidney (Fig. 1, B–D). We therefore conclude that SGK1 is primarily deleted in the nephron; we cannot exclude, however, that there is some suppression in a minor subpopulation of liver cells.
Fig. 1.
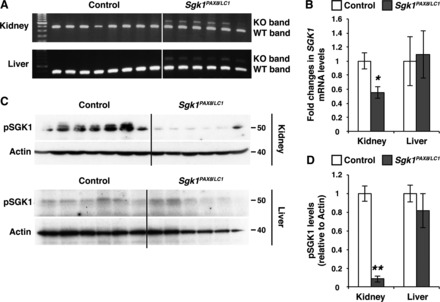
Serum- and glucocorticoid-induced kinase (Sgk)1 mRNA and SGK1 protein levels are decreased in the kidney but not in the liver of Sgk1Pax8/LC1 mutant mice. A: PCR on genomic DNA extracted from the total kidney and liver showing wild-type (WT) and mutant [knockout (KO)] Sgk1 bands after 12 days of doxycycline treatment. The intensity of the mutant band in the kidney was more pronounced than in the liver of the same animals. B: quantitative RT-PCR (TaqMan) analysis of Sgk1 mRNA in control and Sgk1Pax8/LC1 KO mice. A significant decreased of the Sgk1 mRNA level occurred in the kidney but not in the liver of the same animals (n = 7 control mice and 6 Sgk1Pax8/LC1 mice). C: Western blot (WB) analysis of phosphorylated (p)SGK1 (pThr256) in protein lysates from the total kidney (top) and liver (bottom) in control and Sgk1Pax8/LC1 KO mice. Actin was used as a loading control. D: protein quantification of the results in C showing an almost complete loss of pSGK1 in the kidney of mutant mice compared with control mice with no alteration of pSGK1 levels in the liver (n = 6 control mice and 7 Sgk1Pax8/LC1 mice). *P < 0.05; **P < 0.01.
Severe hyperkalemia correlates with diminished K+ excretion in Sgk1Pax8/LC1 mice under challenging conditions.
To understand the involvement of renal tubular SGK1 in K+ homeostasis, we assessed metabolic parameters of control and inducible Sgk1Pax8/LC1 mice under normal diet (ND; containing 0.3% K+) and HKD (5% of K+, K3+-citrate) after 48 h. We euthanized the animals at the end of the daylight period to have maximal aldosterone levels and assure the highest difference between Sgk1Pax8/LC1 and control animals. No significant differences in urine and plasma electrolytes were observed between the genotypes under the ND (Fig. 2 and Table 1). After 2 days of HKD, Sgk1Pax8/LC1 mice developed severe hyperkalemia and mild hyponatremia (Fig. 2, A and B) accompanied with elevated aldosterone levels (Fig. 2C) but no change in mRNA levels of renin 1 in the kidney (Fig. 2F). K+ excretion was enhanced upon the consumption of the HKD in both genotypes, but this increase was less important in Sgk1Pax8/LC1 mutant mice (Fig. 2D). No significant difference in Na+ excretion was observed between the two groups (Fig. 2E). After 5 days of HKD consumption, mutant mice were capable of increasing their urinary K+ secretion to levels similar to those observed in control mice, as no significant difference in K+ excretion was observed between control (1,664.2 ± 71 mmol/day) and mutant (1,319.5 ± 124 mmol/day) mice, suggesting that they came back into balance.
Fig. 2.
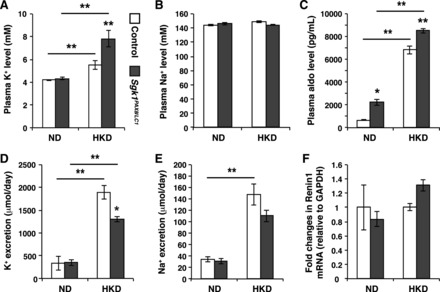
K+ excretion is defective in Sgk1Pax8/LC1 mice under a high-K+ diet (HKD) but not a normal diet (ND). A and B: plasma K+ (A) and Na+ (B) in control (open bars) and Sgk1Pax8/LC1 (solid mice) mice showing significant hyperkalemia with a slight decrease in natremia in Sgk1Pax8/LC1 mice under the HKD (n = 5 control mice and 7 Sgk1Pax8/LC1 mice) but not under a ND (n = 12 control mice and 11 Sgk1Pax8/LC1 mice). C: plasma aldosterone was increased in Sgk1Pax8/LC1 versus control mice under a ND (n = 12 control mice and 13 Sgk1Pax8/LC1 mice) and under a HKD (n = 14 control mice and 11 Sgk1Pax8/LC1 mice). D and E: 24-h urinary K+ (D) and Na+ (E) excretion indicating reduced K+ excretion in Sgk1Pax8/LC1 mutant animals with no significant difference in Na+ excretion (n = 12 control mice and 13 Sgk1Pax8/LC1 mice). *α value < 0.05; **α value < 0.01. F: quantitative RT-PCR (Taqman) on RNA isolated from kidneys of control or Sgk1Pax8/LC1 mice kept under a ND or 48 h of a HKD.
Table 1.
Metabolic parameters of control and SGK1Pax8/LC1 KO mice under a normal diet and after 2 days of a high-K+ diet
| Normal Diet (0.3% K+) |
High-K+ Diet (5% K+) |
|||
|---|---|---|---|---|
| WT | KO | WT | KO | |
| Number of animals/group | 12 | 13 | 12 | 13 |
| Body weight, g | 25.4 ± 0.5 | 24.8 ± 0.5 | 23.7 ± 0.5 | 22.4 ± 0.4 |
| 24-h Food intake/body weight, g in 24 h/g body wt | 0.14 ± 0.01 | 0.15 ± 0.004 | 0.151 ± 0.007 | 0.125 ± 0.007 |
| 24-h Water intake/body weight, ml in 24 h/g body wt | 0.18 ± 0.01 | 0.21 ± 0.01 | 0.34 ± 0.02* | 0.33 ± 0.02* |
| Urine volume, ml in 24 h | 1.62 ± 0.22 | 1.74 ± 0.26 | 2.79 ± 0.27 | 2.61 ± 0.32 |
| 24-h Pi/creatinine | 46.8 ± 3.3 | 52.7 ± 1.9 | 13.30 ± 0.5 | 13.27 ± 0.7 |
| 24-h Mg2+/creatinine | 0.75 ± 0.07 | 0.85 ± 0.07 | 0.78 ± 0.10 | 1.09 ± 0.23 |
| 24-h Ca2+/creatinine | 1.59 ± 0.5 | 2.38 ± 0.68 | 1.10 ± 0.16 | 1.01 ± 0.13 |
| 24-h Urea/creatinine | 530 ± 84 | 436 ± 77 | 307 ± 13 | 319 ± 20 |
| 24-h Creatinine/body weight, μmol in 24 h/g body wt | 162 ± 29 | 135 ± 11 | 304 ± 14 | 264 ± 18 |
Values are means ± SE. No significant differences were observed in these parameters between control and mutant mice. Note that 24-h water intake/body weight was significantly different between normal diet- and high-K+ diet-fed animals with no significant difference in urine volume between both diets, which may result from the high variability due to the evaporation in metabolic cages. WT, wild-type mice; KO, serum- and glucocorticoid-induced kinase 1 knockout mice.
P < 0.05.
Taken together, these data suggest that basal levels of K+ excretion do not require renal tubular SGK1, whereas the kinase appears to be crucial for the adjustment of K+ elimination after a high K+ load for 48 h. Moreover, the finding that Sgk1Pax8/LC1 mice showed higher circulating aldosterone but no change in kidney renin 1 mRNA levels under basal and HKD conditions suggests that the increased aldosterone levels are primarily a compensation for disturbed K+ regulation.
Aberrant ENaC cleavage, expression, and membrane localization in Sgk1Pax8/LC1 mice under HKD.
It has been previously described that Sgk1−/− mice develop hyperkalemia when subjected to a HKD (23). Electrophysiological measurements in isolated perfused cortical collecting ducts (CCDs) from Sgk1−/− animals showed lower absolute and amiloride-sensitive transepithelial potential differences, suggesting a defect in Na+-K+-ATPase and/or ENaC (23). We therefore assessed ENaC expression and localization, including full-length and proteolytically processed forms, representing ENaC maturation and activation (24, 46). Under the ND, ENaC showed similar protein expression and patterns in Sgk1Pax8/LC1 versus control mice (data now shown). However, after 2 days of the HKD, the cleavage product of αENaC and both full-length and cleaved γENaC were decreased with no change in βENaC (Fig. 3, A and B); this difference was accompanied by a decrease in αENaC apical localization and a reduction in intracellular and apical γENaC expression (Fig. 3C). mRNA levels of ENaC subunits did not change (not shown). These observations suggest that under a HKD, the deficiency of renal epithelial SGK1 causes a defect in ENaC regulation.
Fig. 3.
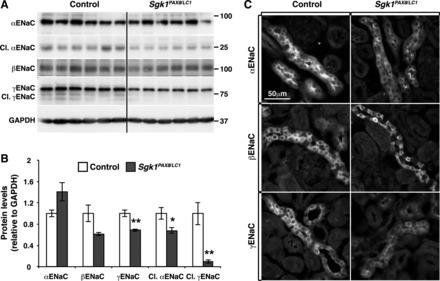
Deregulation of epithelial Na+ channels (ENaC) in Sgk1Pax8/LC1 mice after 2 days of HKD. A: WB analysis of ENaC in control and Sgk1Pax8/LC1 mice showing a decrease in cleaved (Cl.) αENaC and in full-length and cleaved γENaC without an alteration in βENaC. B: protein quantification of the results in A (n = 6 control mice and 7 Sgk1Pax8/LC1 mice). C: immunofluorescence (IF) images of ENaC in control and Sgk1Pax8/LC1mutant mice showing a decrease in the apical localization of αENaC (top) and in the total expression of γENaC (bottom). βENaC exhibited a similar pattern in both genotypes (middle). *P < 0.05; **P < 0.01.
Sgk1Pax8/LC1 mice display alterations in the NEDD4-2/WNK1pathway under HKD.
As described above, it is well documented that SGK1 phosphorylates NEDD4-2, thereby interfering with NEDD4-2-dependent inhibition of ENaC and NCC (3, 5, 8, 17, 52). Because membrane localization of αENaC and γENaC was altered in Sgk1Pax8/LC1 mice under the HKD, we evaluated the phosphorylation status of NEDD4-2 and observed a reduction in Ser222 and Ser328 phosphorylation in Sgk1Pax8/LC1 mice (Fig. 4, A and B). Interestingly, total NEDD4-2 expression was also decreased (Fig. 4, A–C) without any change in mRNA levels (not shown). No alteration in NEDD4-2 phosphorylation or protein levels was observed in the Sgk1Pax8/LC1 mice under the ND (not shown). We have recently demonstrated that WNK1 is ubiquitylated and negatively controlled by NEDD4-2. Proteins levels of the “long” kinase active isoform (L-WNK1) protein levels were increased in NeddlPax8/LC1 animals kept under a high-Na+ diet and decreased in Sgk1Pax8/LC1 mice under a low-Na+ diet (47). Under the present conditions (48 h of the HKD), Western blot analysis did not reveal a significant decrease in WNK1 in Sgk1Pax8/LC1 mice (Fig. 4, A and B), but we observed important changes by immunofluorescence analysis. The stainings in control mice showed that in the distal convoluted tubule (DCT; colocalization with NCC), WNK1 exhibited a punctate pattern (Fig. 4D). Such a localization profile had been previously described for WNK4 (55). On the other hand, in the connecting tubule (CNT)/collecting duct (CD) (colocalization with aquaporin2) of these mice, WNK1 was homogenously diffused within the cells (Fig. 4E). This segment-specific pattern of WNK1 may be related to differences in the signaling pathway associated with WNK1 in each segment. In contrast, in Sgk1Pax8/LC1 mice, WNK1 in the DCT displayed a diffuse localization (Fig. 4D). In addition, its expression was reduced in the CNT/CD compared with control littermates (Fig. 4E). These deregulations were related to a decrease in WNK activity in both the DCT and CNT/CD of mutant mice compared with control mice as evidenced by the reduced staining with p382WNK1/WNK4 (pan-pWNK) antibody, an indicator of WNK1/WNK4 activity (Fig. 5). These data suggest that SGK1 controls NEDD4-2 phosphorylation also under high-K+ conditions [as was the case with a low-Na+ diet (18)] and influences the subcellular localization/expression and activity of WNK1.
Fig. 4.
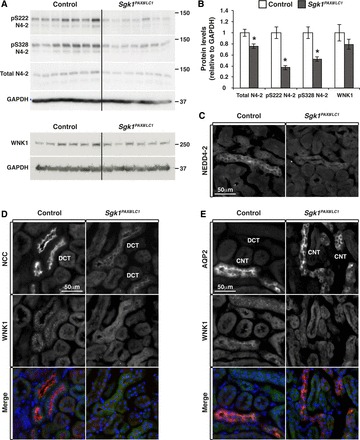
Deregulation of the neural precursor cell expressed developmentally downregulated protein (NEDD)4-2 (N4-2)/with no lysine kinase (WNK)1 pathway in Sgk1Pax8/LC1 mice after 2 days of the HKD. A: WB analysis of NEDD4-2 and WNK1 revealing a significant decrease in total NEDD4-2 and pNEDD4-2 levels in Sgk1Pax8/LC1 mice. A slight (nonsignificant) alteration was observed in WNK1 total protein levels. B: protein quantification of the results shown in A (n = 7 and 6 mice/group). *P < 0.05; **P < 0.01. C: IF images of NEDD4-2 in control and Sgk1Pax8/LC1 mutant mice showing a decrease of total NEDD4-2 expression in KO mice. D: costaining of the Na+-Cl− cotransporter (NCC; red; top) and WNK1 (green; middle). The punctate pattern of WNK1 observed in the distal convoluted tubule (DCT) of control animals disappeared in Sgk1Pax8/LC1 animals, where WNK1 exhibited a diffused pattern. E: costaining of aquaporin (AQP)2 (red) and WNK1 (green) showing the decrease in WNK1 expression in the connecting tubule (CNT)/collecting duct (CD) of mutant compared with control animals.
Sgk1Pax8/LC1 KO mice show increased ROMK expression.
SGK1 had been suggested to be a positive regulator of ROMK (32, 38, 44, 66, 68, 69). Surprisingly, however, in hyperkalemic Sgk1−/− KO mice, apical localization of ROMK was increased (23). One possible explanation for this apparent contradiction might be compensatory mechanisms taking place during nephrogenesis. We generated a novel anti-ROMK antibody in guinea pigs, as described in materials and methods, and validated the antibody by Western blot analysis (Fig. 6, A and B). The antibody recognized transfected ROMK in HEK-293 cells and in total kidney membrane extracts, whereas the corresponding bands were not visible in nontransfected cells or in kidneys from ROMK KO mice (Kcnj1 KO mice). We then analyzed ROMK in Sgk1Pax8/LC1 kidneys from mice kept under the HKD and found also in this model a slight increase in the fully glycosylated form of ROMK in Sgk1Pax8/LC1 mice (Fig. 6, C and D). Immunofluorescence analysis further revealed that ROMK protein abundance and membrane expression were enhanced both in the DCT and CNT/CD of Sgk1Pax8/LC1 animals, as indicated in the co-localization images of ROMK with either NCC or αENaC (Fig. 6, E and F). This suggests that the hyperkalemia triggered by renal SGK1 deletion is independent of ROMK.
Fig. 6.
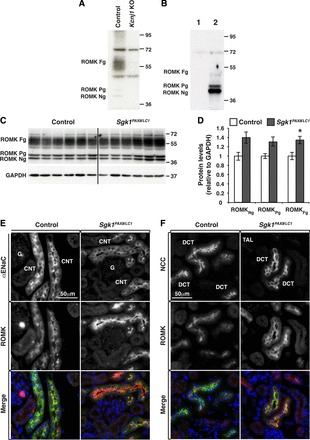
Renal outer medullary K+ (ROMK) channel localization is altered in Sgk1Pax8/LC1 mice under a HKD. A and B: validation of anti ROMK antibodies. A: WB analysis of ROMK from membrane-enriched preparations of kidney extracts of control and Kcnj1 (ROMK) KO mice after 5 days of the HKD. Fg, fully glycosylated; Pg, partially glycosylated; Ng, nonglycosylated ROMK. B: WB analysis of ROMK of untransfected human embryonic kidney (HEK)-293 cells (1) and cells transfected with ROMK (2). C: WB analysis of ROMK in control and Sgk1Pax8/LC1 mice after 2 days of the HKD. D: protein quantification of the results in A revealing a slight increase in the fully glycosylated form of ROMK. E: costaining of ROMK (red) and ENaC (green) in the CNT/CD. F: ROMK (red) and NCC (green) in the DCT. ROMK was more enhanced at the apical membrane in Sgk1Pax8/LC1 versus control mice in both ENaC- and NCC-expressing segments. Note that ROMK exhibited a more pronounced apical localization in the DCT than in the CNT/CD.
NCC and SPAK phosphorylation is altered in Sgk1Pax8/LC1 mice.
NCC dephosphorylation under K+ load has been previously described in several reports (42, 53–55, 58). To analyze the response of NCC to the observed hyperkalemia in Sgk1Pax8/LC1 mice, we analyzed NCC phosphorylation and expression both under the ND and after 2 days of the HKD. We found that under the ND, NCC phosphorylation and expression were similar in Sgk1Pax8/LC1 and control mice (not shown). Under the HKD, NCC phosphorylation levels were much weaker in mutant mice and no difference in total NCC expression was observed (Fig. 7, A–C). In addition, immunofluorescence analysis of kidney sections revealed that NCC was less localized to the apical membrane in KO mice (Fig. 7C). We then asked if this weak NCC phosphorylation correlated with changes in SPAK phosphorylation, a kinase known to phosphorylate and activate NCC (43). SPAK is phosphorylated by WNK kinases on threonine (Thr233) in the T loop of the kinase domain that is crucial for SPAK activation (2, 61). Our immunofluorescence analyses revealed that similarly to WNK1, total SPAK exhibited a punctuate pattern in the DCT of control animals, which was less pronounced in Sgk1Pax8/LC1 mice (Fig. 7D). Phosphorylation at Thr233 was reduced in the DCT compared with control animals, consistent with the reduced NCC phosphorylation (Fig. 7E). Taken together, these data show that under a HKD, NCC phosphorylation and membrane localization are both diminished in Sgk1Pax8/LC1 mice, coinciding with a reduction in SPAK activation.
Fig. 7.
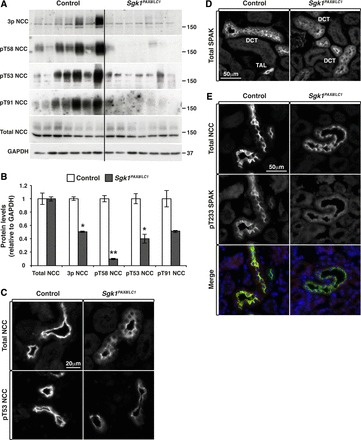
Alterations in Ste20-related proline/alanine-rich kinase (SPAK) and NCC phosphorylation and cellular localization after 2 days of the HKD. A: WB analysis of NCC in control and Sgk1Pax8/LC1 mice showing reduced NCC phosphorylation of several phosphorylation sites without alterations in NCC total expression in Sgk1Pax8/LC1 mice. B: protein quantification of the results in A (n = 7 mice for both genotypes). *P < 0.05; **P < 0.01. C: IF images of total and pThr53 NCC in control and Sgk1Pax8/LC1 mice. The apical localization (top) and phosphorylation state of NCC (bottom) were both altered in Sgk1Pax8/LC1 animals. D: IF images of total SPAK showing a punctate pattern of SPAK in control mice, which became less pronounced in Sgk1Pax8/LC1 animals. E: costaining of NCC (green; top) and pThr233 SPAK (red; middle) showing decreased SPAK phosphorylation in the DCT of Sgk1Pax8/LC1 mice.
DISCUSSION
The present study shows that mice with inducible deletion of renal tubular SGK1 exhibit reduced K+ excretion compared with control mice, leading to severe hyperkalemia when subjected to a HKD. The reduced K+ excretion is correlated with altered NEDD4-2 and WNK1 in addition to reduced apical membrane expression of ENaC. Our observations partly confirm the previous report by Huang et al. (23) demonstrating that total Sgk1−/− mice display normal K+ handling when fed a standard diet but develop hyperkalemia, although without a difference in K+ excretion, when challenged by a HKD. We also observed elevated aldosterone levels (but no difference in renin 1 mRNA expression in the kidney) in Sgk1Pax8/LC1 mice under both standard and K+-rich diets similarly to Sgk1−/− mice (23). Given the similar K+ content in the HKD used in both studies (5% K+ ), the lower K+ excretion observed under a chronic HKD in Sgk1Pax8/LC1 but not Sgk1−/− mice suggests that renal tubular SGK1 plays a critical role in the regulation of K+ excretion under a HKD and that its role may be attenuated by compensatory mechanisms triggered during nephrogenesis in Sgk1−/− mice. Extrarenal SGK1 had been previously shown to play a role in K+ homeostasis, as suggested by experiments performed in Sgk1−/− animals demonstrating that hepatic SGK1 regulates kalemia via its role in insulin-stimulated K+ uptake (6). In our mice, we found no evidence for suppression of SGK1 in the liver; although we cannot exclude suppression of SGK1 in a subpopulation of liver cells, we consider it as unlikely that hepatic SGK1 plays a major role in this model. Finally, we cannot exclude that strain background differences may contribute to the alterations as well.
Our molecular analyses suggest that the effect of SGK1 on K+ balance is mediated at least in part by the positive regulation of ENaC, implicating phosphorylation and consequent interference with NEDD4-2 action (5, 17–19, 21, 25, 31, 52). After 2 days of the HKD, NEDD4-2 phosphorylation and total expression were reduced in Sgk1Pax8/LC1 mice with no alterations of its mRNA level. Similar observations were made in Sgk1Pax8/LC1 animals under a low-salt diet, which showed reduced NEDD4-2 phosphorylation on both Ser222 and Ser328 sites (18). However, the decrease in NEDD4-2 total expression found here might be due to the hyperkalemia and/or to increased aldosterone levels. Indeed, van der Lubbe et al. (59) showed that NEDD4-2 expression was decreased in rats fed with a HKD. Moreover, Loffing-Cueni et al. (33) described that a low-Na+ diet caused a reduction in NEDD4-2 expression and provided evidence for aldosterone-dependent inhibition of NEDD4-2 levels. Taken together, this suggests that aldosterone inhibits NEDD4-2 actions via 1) SGK1-dependent phosphorylation (likely a rapid mechanism of regulation) and 2) lowering its expression by an uncharacterized posttranscriptional mechanism (slow or late mode). NEDD4-2 phosphorylation at Ser328 interferes with the regulation of ENaC (17, 52). Consistent with this, we observed under a HKD lower expression of γENaC as well as decreased cleavage of αENaC and γENaC, features that had been associated with increased ubiquitylation of ENaC (24, 27, 49, 50). Our findings are in line with previous reports showing the decrease in γENaC cleavage in Sgk1−/− mice treated with aldosterone for 7 days (19). These Sgk1−/− mice exhibited a Na+ losing phenotype under a low-salt diet but, intriguingly, an increase in amiloride-sensitive Na+ currents, as measured by whole cell patch-clamping in the CCD. In contrast, low amiloride-sensitive transepithelial potential differences (a marker for ENaC activity) in isolated CCDs from Sgk1−/− mice under a HKD have been reported by Huang et al. (23). Similarly to Sgk1−/− mice, Sgk1Pax8/LC1 mice did not exhibit Na+ loss compared with control mice when fed a HKD despite the observed decrease in αENaC and γENaC and NCC activation. One possible explanation is an increase in Na+ reabsorption in the proximal tubules, as previously reported in Sgk1−/− mice under a low-salt diet (65). Interestingly, transgenic mice with specific deletion of αENaC in renal tubules (39) or only in the CNT (13, 41) exhibit hyperkalemia and decreased K+ excretion. In view of what precedes, we consider it as very likely that the low ENaC cleavage and membrane localization in Sgk1Pax8/LC1 mice account at least in part for the observed decrease in K+ excretion, by limiting the electrogenic driving force for K+ secretion.
We also observed that Sgk1Pax8/LC1 mice exhibited decreased phosphorylation and apical localization of NCC after 2 days of the HKD. Vallon et al. (58) showed that Sgk1−/− mice fed with a HKD (5% K+) for 7 days exhibit decreased NCC phosphorylation and total protein expression. The longer exposure to the HKD may explain this more severe NCC alteration observed by these authors. Our data suggest that the decrease in NCC apical localization in Sgk1Pax8/LC1 mice is likely an effect of reduced NEDD4-2 phosphorylation leading to increased activity toward NCC (3). The deregulation of NCC in our model was accompanied by reduced phosphorylation of Ser382 in WNK1/4 and Thr233 in SPAK. Recently, Roy et al. (47) have reported that L-WNK1 kinase, known to stimulate NCC via SPAK/OSR1 kinase, is a target of NEDD4-2 and that the expression of L-WNK1 was reduced in SgkPax8/LC1 mice fed with a low-Na+ diet. Although we did not detect a statistically significant reduction in WNK1 protein expression in SgkPax8/LC1 mice under our conditions, we did observe a striking alteration in the WNK1 localization pattern in both the DCT and CNT/CD. Taken together with the observation that WNK1 phosphorylation was reduced under HKD conditions, our data suggest that SGK1 regulates NCC phosphorylation via WNK kinases and that the NEDD4-2/WNK1/SPAK pathway participates in this process. Another mechanism for NCC regulation was proposed by Rozansky and collaborators (48), who suggested that phosphorylation of WNK4 by SGK1 on Ser1196 inhibits WNK4-dependent downregulation of NCC. However, regulation of NCC by WNK4 is complex and can be both inhibitory and stimulatory, depending on the physiological context (9). To our knowledge, no antibodies against this WNK4 phosphorylation site are currently available (32, 44); hence, we were not able to test this possibility in our model. It has also been reported that intracellular Cl− is able to inhibit WNK kinases (4, 40), and evidence was presented that low plasma K+ leads to lower intracellular Cl− concentration and stimulation of WNK kinase activity [i.e., WNK1, WNK3, and WNK4 (7, 54, 55)]. Consequently, hyperkalemia may cause an increase in intracellular Cl− concentration and inhibition of the WNK kinases. Taken together, the deregulation in NCC apical localization and phosphorylation in Sgk1Pax8/LC1 mice under a HKD may be due to decreased NEDD4-2 phosphorylation and reduced L-WNK1 expression and activity, exacerbated by hyperkalemia. Moreover, it is important to note that the reduced phosphorylation levels of NCC are likely a secondary effect of the hyperkalemia and not the cause of it.
The regulation of ROMK by SGK1 is still under debate. Several in vitro reports have suggested a positive regulation of the channel by SGK1 (11, 38, 66). However, in vivo studies have not confirmed this hypothesis (23, 56). Our data show that ROMK protein levels and membrane localization are increased in Sgk1Pax8/LC1 mice under a HKD, indicating that a deregulation of ROMK cannot account for the decrease in K+ excretion in Sgk1Pax8/LC1 animals. This is in agreement with the observation made in Sgk1−/− and aldosterone synthase KO mice, which exhibit hyperkalemia together with increased ROMK membrane expression when fed with a HKD (23, 56). Importantly, in vitro and in vivo evidences suggest that L-WNK1 inhibits ROMK by stimulating its endocytosis (14, 30, 60, 62). Consequently, the increased ROMK levels are compatible with the observed decrease of WNK1 expression and activity in our mouse model. We note that the findings regarding ROMK regulation in our mice indicate that the mechanisms of K+ excretion, affected by SGK1, may also involve other channels/transporters such as the large-conductance K+ (15) and Kir4.1 (Kcnj10) (70) channels or H+-K+-ATPase (16). This will be addressed by future studies.
Taken together, renal tubular SGK1 plays a crucial role in K+ homeostasis via the regulation of NEDD4-2-mediated ENaC inhibition. Sgk1Pax8/LC1 mice exhibit an alteration of WNK1 puncta in the DCT and a decrease of WNK1 staining in the CNT/CD. This is correlated with decreased SPAK/NCC phosphorylation and increased ROMK expression/apical localization in the DCT/CNT/CD.
GRANTS
This work was supported by Swiss National Science Foundation Grant 310030_159735 (to O. Staub) and 310030_143929/1 (to J. Loffing), the National Centre of Competence in Research “Swiss Kidney.ch” (to O. Staub and J. Loffing), networking support by COST Action ADMIRE BM1301 (to O. Staub and J. Loffing), the Novartis Foundation for medical biological research (to O. Staub), and National Institute of Diabetes and Digestive and Kidney Diseases Grant R01-DK-098145 (to A. R. Subramanya). L. Al-Qusairi was supported by a fellowship of the Marie Curie cofunding International Fellowship Program on Integrative Kidney Physiology and Pathophysiology.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
L.A.-Q. and O.S. conception and design of research; L.A.-Q., D.B., A.R., M.S., R.D.R., A.D., I.N., and M.P.M. performed experiments; L.A.-Q., D.B., A.R., M.S., J.L., A.R.S., and O.S. analyzed data; L.A.-Q., D.B., J.L., A.R.S., and O.S. interpreted results of experiments; L.A.-Q., D.B., and O.S. prepared figures; L.A.-Q., D.B., and O.S. drafted manuscript; L.A.-Q., D.B., A.R.S., and O.S. edited and revised manuscript; L.A.-Q., J.L., and O.S. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Dr. Bernard Rossier, Dr. Laurent Schild, and members of the Staub laboratory for comments and critically reading the manuscript. The authors also thank Andrée Tedjani, Sebastien Desarzens, and Jérémy Vidal for technical help and Alessia Spirli for help with the statistical analysis.
REFERENCES
- 1.Abriel H, Loffing J, Rebhun JF, Pratt JH, Schild L, Horisberger JD, Rotin D, Staub O. Defective regulation of the epithelial Na+ channel by Nedd4 in Liddle's syndrome. J Clin Invest 103: 667–673, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alessi DR, Zhang J, Khanna A, Hochdorfer T, Shang Y, Kahle KT. The WNK-SPAK/OSR1 pathway: master regulator of cation-chloride cotransporters. Sci Signal 7: re3, 2014. [DOI] [PubMed] [Google Scholar]
- 3.Arroyo JP, Lagnaz D, Ronzaud C, Vazquez N, Ko BS, Moddes L, Ruffieux-Daidie D, Hausel P, Koesters R, Yang B, Stokes JB, Hoover RS, Gamba G, Staub O. Nedd4-2 modulates Renal Na+-Cl− cotransporter via the aldosterone-SGK1-Nedd4-2 pathway. J Am Soc Nephrol 22: 1707–1719, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bazua-Valenti S, Chavez-Canales M, Rojas-Vega L, Gonzalez-Rodriguez X, Vazquez N, Rodriguez-Gama A, Argaiz ER, Melo Z, Plata C, Ellison DH, Garcia-Valdes J, Hadchouel J, Gamba G. The effect of WNK4 on the Na+-Cl− cotransporter is modulated by intracellular chloride. J Am Soc Nephrol 26: 1781–1786, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bhalla V, Daidie D, Li H, Pao AC, LaGrange LP, Wang J, Vandewalle A, Stockand JD, Staub O, Pearce D. Serum- and glucocorticoid-regulated kinase 1 regulates ubiquitin ligase neural precursor cell-expressed, developmentally down-regulated protein 4-2 by inducing interaction with 14-3-3. Mol Endocrinol 19: 3073–3084, 2005. [DOI] [PubMed] [Google Scholar]
- 6.Boini KM, Graf D, Kuhl D, Haussinger D, Lang F. SGK1 dependence of insulin induced hypokalemia. Pflügers Arch 457: 955–961, 2009. [DOI] [PubMed] [Google Scholar]
- 7.Castaneda-Bueno M, Cervantes-Perez LG, Rojas-Vega L, Arroyo-Garza I, Vazquez N, Moreno E, Gamba G. Modulation of NCC activity by low and high K+ intake: insights into the signaling pathways involved. Am J Physiol Renal Physiol 306: F1507–F1519, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chandran S, Li H, Dong W, Krasinska K, Adams C, Alexandrova L, Chien A, Hallows KR, Bhalla V. Neural precursor cell-expressed developmentally down-regulated protein 4-2 (Nedd4-2) regulation by 14-3-3 protein binding at canonical serum and glucocorticoid kinase 1 (SGK1) phosphorylation sites. J Biol Chem 286: 37830–37840, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chavez-Canales M, Zhang C, Soukaseum C, Moreno E, Pacheco-Alvarez D, Vidal-Petiot E, Castaneda-Bueno M, Vazquez N, Rojas-Vega L, Meermeier NP, Rogers S, Jeunemaitre X, Yang CL, Ellison DH, Gamba G, Hadchouel J. WNK-SPAK-NCC cascade revisited: WNK1 stimulates the activity of the Na-Cl cotransporter via SPAK, an effect antagonized by WNK4. Hypertension 64: 1047–1053, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen S, Bhargava A, Mastroberardino L, Meijer OC, Wang J, Buse P, Firestone GL, Verrey F, Pearce D. Epithelial sodium channel regulated by aldosterone-induced protein sgk. Proc Natl Acad Sci USA 96: 2514–2519, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen SY, Bhargava A, Mastroberardino L, Meijer OC, Wang J, Buse P, Firestone GL, Verrey F, Pearce D. Epithelial sodium channel regulated by aldosterone-induced protein sgk. Proc Natl Acad Sci USA 96: 2514–2519, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheng CJ, Huang CL. Activation of PI3-kinase stimulates endocytosis of ROMK via Akt1/SGK1-dependent phosphorylation of WNK1. J Am Soc Nephrol 22: 460–471, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Christensen BM, Perrier R, Wang Q, Zuber AM, Maillard M, Mordasini D, Malsure S, Ronzaud C, Stehle JC, Rossier BC, Hummler E. Sodium and potassium balance depends on αENaC expression in connecting tubule. J Am Soc Nephrol 21: 1942–1951, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cope G, Murthy M, Golbang AP, Hamad A, Liu CH, Cuthbert AW, O'Shaughnessy KM. WNK1 affects surface expression of the ROMK potassium channel independent of WNK4. J Am Soc Nephrol 17: 1867–1874, 2006. [DOI] [PubMed] [Google Scholar]
- 15.Cornelius RJ, Wen D, Li H, Yuan Y, Wang-France J, Warner PC, Sansom SC. Low Na, high K diet and the role of aldosterone in BK-mediated K excretion. PLos One 10: e0115515, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crambert G. H-K-ATPase type 2: relevance for renal physiology and beyond. Am J Physiol Renal Physiol 306: F693–F700, 2014. [DOI] [PubMed] [Google Scholar]
- 17.Debonneville C, Flores SY, Kamynina E, Plant PJ, Tauxe C, Thomas MA, Munster C, Chraibi A, Pratt JH, Horisberger JD, Pearce D, Loffing J, Staub O. Phosphorylation of Nedd4-2 by Sgk1 regulates epithelial Na+ channel cell surface expression. EMBO J 20: 7052–7059, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Faresse N, Lagnaz D, Debonneville A, Ismailji A, Maillard M, Fejes-Toth G, Naray-Fejes-Toth A, Staub O. Inducible kidney-specific Sgk1 knockout mice show a salt-losing phenotype. Am J Physiol Renal Physiol 302: F977–F985, 2012. [DOI] [PubMed] [Google Scholar]
- 19.Fejes-Toth G, Frindt G, Naray-Fejes-Toth A, Palmer LG. Epithelial Na+ channel activation and processing in mice lacking SGK1. Am J Physiol Renal Physiol 294: F1298–F1305, 2008. [DOI] [PubMed] [Google Scholar]
- 20.Flores SY, Loffing-Cueni D, Kamynina E, Daidie D, Gerbex C, Chabanel S, Dudler J, Loffing J, Staub O. Aldosterone-induced serum and glucocorticoid-induced kinase 1 expression is accompanied by Nedd4-2 phosphorylation and increased Na+ transport in cortical collecting duct cells. J Am Soc Nephrol 16: 2279–2287, 2005. [DOI] [PubMed] [Google Scholar]
- 21.Gleason CE, Frindt G, Cheng CJ, Ng M, Kidwai A, Rashmi P, Lang F, Baum M, Palmer LG, Pearce D. mTORC2 regulates renal tubule sodium uptake by promoting ENaC activity. J Clin Invest 125: 117–128, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harvey KF, Dinudom A, Cook DI, Kumar S. The Nedd4-like protein KIAA0439 is a potential regulator of the epithelial sodium channel. J Biol Chem 276: 8597–8601, 2001. [DOI] [PubMed] [Google Scholar]
- 23.Huang DY, Wulff P, Volkl H, Loffing J, Richter K, Kuhl D, Lang F, Vallon V. Impaired regulation of renal K+ elimination in the sgk1-knockout mouse. J Am Soc Nephrol 15: 885–891, 2004. [DOI] [PubMed] [Google Scholar]
- 24.Hughey RP, Mueller GM, Bruns JB, Kinlough CL, Poland PA, Harkleroad KL, Carattino MD, Kleyman TR. Maturation of the epithelial Na+ channel involves proteolytic processing of the α- and γ-subunits. J Biol Chem 278: 37073–37082, 2003. [DOI] [PubMed] [Google Scholar]
- 25.Ichimura T, Yamamura H, Sasamoto K, Tominaga Y, Taoka M, Kakiuchi K, Shinkawa T, Takahashi N, Shimada S, Isobe T. 14-3-3 proteins modulate the expression of epithelial Na+ channels by phosphorylation-dependent interaction with Nedd4-2 ubiquitin ligase. J Biol Chem 280: 13187–13194, 2005. [DOI] [PubMed] [Google Scholar]
- 26.Kamynina E, Debonneville C, Bens M, Vandewalle A, Staub O. A novel mouse Nedd4 protein suppresses the activity of the epithelial Na+ channel. FASEB J 15: 204–214, 2001. [DOI] [PubMed] [Google Scholar]
- 27.Knight KK, Olson DR, Zhou R, Snyder PM. Liddle's syndrome mutations increase Na+ transport through dual effects on epithelial Na+ channel surface expression and proteolytic cleavage. Proc Natl Acad Sci USA 103: 2805–2808, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lang F, Stournaras C, Alesutan I. Regulation of transport across cell membranes by the serum- and glucocorticoid-inducible kinase SGK1. Mol Membr Biol 31: 29–36, 2014. [DOI] [PubMed] [Google Scholar]
- 29.Lang F, Vallon V. Serum- and glucocorticoid-inducible kinase 1 in the regulation of renal and extrarenal potassium transport. Clin Exp Nephrol 16: 73–80, 2012. [DOI] [PubMed] [Google Scholar]
- 30.Lazrak A, Liu Z, Huang CL. Antagonistic regulation of ROMK by long and kidney-specific WNK1 isoforms. Proc Natl Acad Sci USA 103: 1615–1620, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liang X, Peters KW, Butterworth MB, Frizzell RA. 14-3-3 isoforms are induced by aldosterone and participate in its regulation of epithelial sodium channels. J Biol Chem 281: 16323–16332, 2006. [DOI] [PubMed] [Google Scholar]
- 32.Lin DH, Yue P, Rinehart J, Sun P, Wang Z, Lifton R, Wang WH. Protein phosphatase 1 modulates the inhibitory effect of with-no-lysine kinase 4 on ROMK channels. Am J Physiol Renal Physiol 303: F110–F119, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Loffing-Cueni D, Flores SY, Sauter D, Daidie D, Siegrist N, Meneton P, Staub O, Loffing J. Dietary sodium intake regulates the ubiquitin-protein ligase nedd4-2 in the renal collecting system. J Am Soc Nephrol 17: 1264–1274, 2006. [DOI] [PubMed] [Google Scholar]
- 34.Loffing J, Flores SY, Staub O. Sgk kinases and their role in epithelial transport. Annu Rev Physiol 68: 461–490, 2006. [DOI] [PubMed] [Google Scholar]
- 35.Loffing J, Zecevic M, Feraille E, B K, Asher C, Rossier BC, Firestone GL, Pearce D, Verrey F. Aldosterone induces rapid apical translocation of ENaC in early portion of renal collecting system: possible role of SGK. Am J Physiol Renal Physiol 280: F675–F682, 2001. [DOI] [PubMed] [Google Scholar]
- 36.Lu M, Wang T, Yan Q, Yang X, Dong K, Knepper MA, Wang W, Giebisch G, Shull GE, Hebert SC. Absence of small conductance K+ channel (SK) activity in apical membranes of thick ascending limb and cortical collecting duct in ROMK (Bartter's) knockout mice. J Biol Chem 277: 37881–37887, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Naray-Fejes-Toth A, Canessa CM, Cleaveland ES, Aldrich G, Fejes-Toth G. Sgk is an aldosterone-induced kinase in the renal collecting duct. J Biol Chem 274: 16973–16978, 1999. [DOI] [PubMed] [Google Scholar]
- 38.Palmada M, Embark HM, Yun C, Bohmer C, Lang F. Molecular requirements for the regulation of the renal outer medullary K+ channel ROMK1 by the serum- and glucocorticoid-inducible kinase SGK1. Biochem Biophys Res Commun 311: 629–634, 2003. [DOI] [PubMed] [Google Scholar]
- 39.Perrier R, Boscardin E, Malsure S, Sergi C, Maillard MP, Loffing J, Loffing DC, Sorensen MV, Koesters R, Rossier BC, Frateschi S, Hummler E. Severe salt-losing syndrome and hyperkalemia induced by adult nephron-specific knockout of the epithelial sodium channel α-subunit. J Am Soc Nephrol; doi: 10.1681/ASN.2015020154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Piala AT, Moon TM, Akella R, He H, Cobb MH, Goldsmith EJ. Chloride sensing by WNK1 involves inhibition of autophosphorylation. Sci Signal 7: ra41, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Poulsen SB, Praetorius J, Damkier HH, Miller L, Nelson RD, Hummler E, Christensen BM. Reducing αENaC expression in the kidney connecting tubule induces pseudohypoaldosteronism type 1 symptoms during K+ loading. Am J Physiol Renal Physiol 310: F300–F310, 2016. [DOI] [PubMed] [Google Scholar]
- 42.Rengarajan S, Lee DH, Oh YT, Delpire E, Youn JH, McDonough AA. Increasing plasma [K+] by intravenous potassium infusion reduces NCC phosphorylation and drives kaliuresis and natriuresis. Am J Physiol Renal Physiol 306: F1059–F1068, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Richardson C, Rafiqi FH, Karlsson HK, Moleleki N, Vandewalle A, Campbell DG, Morrice NA, Alessi DR. Activation of the thiazide-sensitive Na+-Cl− cotransporter by the WNK-regulated kinases SPAK and OSR1. J Cell Sci 121: 675–684, 2008. [DOI] [PubMed] [Google Scholar]
- 44.Ring AM, Leng Q, Rinehart J, Wilson FH, Kahle KT, Hebert SC, Lifton RP. An SGK1 site in WNK4 regulates Na+ channel and K+ channel activity and has implications for aldosterone signaling and K+ homeostasis. Proc Natl Acad Sci USA 104: 4025–4029, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ronzaud C, Loffing-Cueni D, Hausel P, Debonneville A, Malsure SR, Fowler-Jaeger N, Boase NA, Perrier R, Maillard M, Yang B, Stokes JB, Koesters R, Kumar S, Hummler E, Loffing J, Staub O. Renal tubular NEDD4-2 deficiency causes NCC-mediated salt-dependent hypertension. J Clin Invest 123: 657–665, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rossier BC, Stutts MJ. Activation of the epithelial sodium channel (ENaC) by serine proteases. Annu Rev Physiol 71: 361–379, 2009. [DOI] [PubMed] [Google Scholar]
- 47.Roy A, Al-Qusairi L, Donnelly BF, Ronzaud C, Marciszyn AL, Gong F, Chang YP, Butterworth MB, Pastor-Soler NM, Hallows KR, Staub O, Subramanya AR. Alternatively spliced proline-rich cassettes link WNK1 to aldosterone action. J Clin Invest 125: 3433–3448, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rozansky DJ, Cornwall T, Subramanya AR, Rogers S, Yang YF, David LL, Zhu X, Yang CL, Ellison DH. Aldosterone mediates activation of the thiazide-sensitive Na-Cl cotransporter through an SGK1 and WNK4 signaling pathway. J Clin Invest 119: 2601–2612, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ruffieux-Daidie D, Poirot O, Boulkroun S, Verrey F, Kellenberger S, Staub O. Deubiquitylation regulates activation and proteolytic cleavage of ENaC. J Am Soc Nephrol 19: 2170–2180, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ruffieux-Daidie D, Staub O. Intracellular ubiquitylation of the epithelial Na+ channel controls extracellular proteolytic channel activation via conformational change. J Biol Chem 286: 2416–2424, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shi PP, Cao XR, Sweezer EM, Kinney TS, Williams NR, Husted RF, Nair R, Weiss RM, Williamson RA, Sigmund CD, Snyder PM, Staub O, Stokes JB, Yang B. Salt-sensitive hypertension and cardiac hypertrophy in mice deficient in the ubiquitin ligase Nedd4-2. Am J Physiol Renal Physiol 295: F462–F470, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Snyder PM, Olson DR, Thomas BC. Serum and clucocorticoid-regulated kinase modulates Nedd4-2-mediated inhibition of the epithelial Na+ channel. J Biol Chem 277: 5–8, 2002. [DOI] [PubMed] [Google Scholar]
- 53.Sorensen MV, Grossmann S, Roesinger M, Gresko N, Todkar AP, Barmettler G, Ziegler U, Odermatt A, Loffing-Cueni D, Loffing J. Rapid dephosphorylation of the renal sodium chloride cotransporter in response to oral potassium intake in mice. Kidney Int 83: 811–824, 2013. [DOI] [PubMed] [Google Scholar]
- 54.Terker AS, Zhang C, Erspamer KJ, Gamba G, Yang CL, Ellison DH. Unique chloride-sensing properties of WNK4 permit the distal nephron to modulate potassium homeostasis. Kidney Int 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Terker AS, Zhang C, McCormick JA, Lazelle RA, Meermeier NP, Siler DA, Park HJ, Fu Y, Cohen DM, Weinstein AM, Wang WH, Yang CL, Ellison DH. Potassium modulates electrolyte balance and blood pressure through effects on distal cell voltage and chloride. Cell Metab 21: 39–50, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Todkar A, Picard N, Loffing-Cueni D, Sorensen MV, Mihailova M, Nesterov V, Makhanova N, Korbmacher C, Wagner CA, Loffing J. Mechanisms of renal control of potassium homeostasis in complete aldosterone deficiency. J Am Soc Nephrol 26: 425–438, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Traykova-Brauch M, Schonig K, Greiner O, Miloud T, Jauch A, Bode M, Felsher DW, Glick AB, Kwiatkowski DJ, Bujard H, Horst J, von Knebel Doeberitz M, Niggli FK, Kriz W, Grone HJ, Koesters R. An efficient and versatile system for acute and chronic modulation of renal tubular function in transgenic mice. Nat Med 14: 979–984, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vallon V, Schroth J, Lang F, Kuhl D, Uchida S. Expression and phosphorylation of the Na+-Cl− cotransporter NCC in vivo is regulated by dietary salt, potassium, and SGK1. Am J Physiol Renal Physiol 297: F704–F712, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.van der Lubbe N, Moes AD, Rosenbaek LL, Schoep S, Meima ME, Danser AH, Fenton RA, Zietse R, Hoorn EJ. K+-induced natriuresis is preserved during Na+ depletion and accompanied by inhibition of the Na+-Cl− cotransporter. Am J Physiol Renal Physiol 305: F1177–F1188, 2013. [DOI] [PubMed] [Google Scholar]
- 60.Vidal-Petiot E, Elvira-Matelot E, Mutig K, Soukaseum C, Baudrie V, Wu S, Cheval L, Huc E, Cambillau M, Bachmann S, Doucet A, Jeunemaitre X, Hadchouel J. WNK1-related familial hyperkalemic hypertension results from an increased expression of L-WNK1 specifically in the distal nephron. Proc Natl Acad Sci USA 110: 14366–14371, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vitari AC, Deak M, Morrice NA, Alessi DR. The WNK1 and WNK4 protein kinases that are mutated in Gordon's hypertension syndrome phosphorylate and activate SPAK and OSR1 protein kinases. Biochem J 391: 17–24, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wade JB, Fang L, Liu J, Li D, Yang CL, Subramanya AR, Maouyo D, Mason A, Ellison DH, Welling PA. WNK1 kinase isoform switch regulates renal potassium excretion. Proc Natl Acad Sci USA 103: 8558–8563, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wagner CA, Loffing-Cueni D, Yan Q, Schulz N, Fakitsas P, Carrel M, Wang T, Verrey F, Geibel JP, Giebisch G, Hebert SC, Loffing J. Mouse model of type II Bartter's syndrome. II. Altered expression of renal sodium- and water-transporting proteins. Am J Physiol Renal Physiol 294: F1373–F1380, 2008. [DOI] [PubMed] [Google Scholar]
- 64.Welling PA. Regulation of renal potassium secretion: molecular mechanisms. Semin Nephrol 33: 215–228, 2013. [DOI] [PubMed] [Google Scholar]
- 65.Wulff P, Vallon V, Huang DY, Volkl H, Yu F, Richter K, Jansen M, Schlunz M, Klingel K, Loffing J, Kauselmann G, Bosl MR, Lang F, Kuhl D. Impaired renal Na+ retention in the sgk1-knockout mouse. J Clin Invest 110: 1263–1268, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yoo D, Kim BY, Campo C, Nance L, King A, Maouyo D, Welling PA. Cell surface expression of the ROMK (Kir1.1) channel is regulated by the aldosterone-induced kinase, SGK-1, and protein kinase A. J Biol Chem 278: 23066–23075, 2003. [DOI] [PubMed] [Google Scholar]
- 67.Youn JH, McDonough AA. Recent advances in understanding integrative control of potassium homeostasis. Annu Rev Physiol 71: 381–401, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yue P, Lin DH, Pan CY, Leng Q, Giebisch G, Lifton RP, Wang WH. Src family protein tyrosine kinase (PTK) modulates the effect of SGK1 and WNK4 on ROMK channels. Proc Natl Acad Sci USA 106: 15061–15066, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yun CC, Palmada M, Embark HM, Fedorenko O, Feng Y, Henke G, Setiawan I, Boehmer C, Weinman EJ, Sandrasagra S, Korbmacher C, Cohen P, Pearce D, Lang F. The serum and glucocorticoid-inducible kinase SGK1 and the Na+/H+ exchange regulating factor NHERF2 synergize to stimulate the renal outer medullary K+ channel ROMK1. J Am Soc Nephrol 13: 2823–2830, 2002. [DOI] [PubMed] [Google Scholar]
- 70.Zhang C, Wang L, Zhang J, Su XT, Lin DH, Scholl UI, Giebisch G, Lifton RP, Wang WH. KCNJ10 determines the expression of the apical Na-Cl cotransporter (NCC) in the early distal convoluted tubule (DCT1). Proc Natl Acad Sci USA 111: 11864–11869, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]