

Carvedilol upregulates microRNA (miR)-199a-3p and miR-214 in cardiomyocytes. This miR activation is a mechanism for carvedilol-mediated p-Akt activation. The action of these carvedilol-responsive miRs on cardiomyocyte survival is mediated in part by the repression of the predictive or known targets Ddit4 and Ing4, subsequently activating Akt-Sox2 prosurvival axis.
Keywords: apoptosis, β-blocker, β-arrestin-biased β-adrenergic receptor signaling, heart disease, microRNAs
Abstract
The nonselective β-adrenergic receptor antagonist (β-blocker) carvedilol has been shown to protect against myocardial injury, but the detailed underlying mechanisms are unclear. We recently reported that carvedilol stimulates the processing of microRNA (miR)-199a-3p and miR-214 in the heart via β-arrestin1-biased β1-adrenergic receptor (β1AR) cardioprotective signaling. Here, we investigate whether these β-arrestin1/β1AR-responsive miRs mediate the beneficial effects of carvedilol against simulated ischemia/reperfusion (sI/R). Using cultured cardiomyocyte cell lines and primary cardiomyocytes, we demonstrate that carvedilol upregulates miR-199a-3p and miR-214 in both ventricular and atrial cardiomyocytes subjected to sI/R. Overexpression of the two miRs in cardiomyocytes mimics the effects of carvedilol to activate p-AKT survival signaling and the expression of a downstream pluripotency marker Sox2 in response to sI/R. Moreover, carvedilol-mediated p-AKT activation is abolished by knockdown of either miR-199a-3p or miR-214. Along with previous studies to directly link the cardioprotective actions of carvedilol to upregulation of p-AKT/stem cell markers, our findings suggest that the protective roles of carvedilol during ischemic injury are in part attributed to activation of these two protective miRs. Loss of function of miR-199a-3p and miR-214 also increases cardiomyocyte apoptosis after sI/R. Mechanistically, we demonstrate that miR-199a-3p and miR-214 repress the predictive or known apoptotic target genes ddit4 and ing4, respectively, in cardiomyocytes. These findings suggest pivotal roles for miR-199a-3p and miR-214 as regulators of cardiomyocyte survival and contributors to the functional benefits of carvedilol therapy.
NEW & NOTEWORTHY
Carvedilol upregulates microRNA (miR)-199a-3p and miR-214 in cardiomyocytes. This miR activation is a mechanism for carvedilol-mediated p-Akt activation. The action of these carvedilol-responsive miRs on cardiomyocyte survival is mediated in part by the repression of the predictive or known targets Ddit4 and Ing4, subsequently activating Akt-Sox2 prosurvival axis.
the nonselective β-adrenergic receptor antagonist (β-blocker) carvedilol is highly effective at preventing and treating heart failure (HF) in patients and has been demonstrated to reverse left ventricular failure (LVF) in numerous experimental HF models via attenuating LV remodeling, hypertrophy, fibrosis, and apoptosis (11, 52, 53). These beneficial effects of carvedilol are partially mediated by stimulation of β-arrestin-mediated β1-adrenergic receptor (β1AR) cardioprotective pathways (19, 20). We demonstrated that carvedilol is a β-arrestin-biased β1AR ligand, which activates cellular pathways in the heart independent of G protein-mediated signaling, a concept known as biased signaling (19, 30). Carvedilol has also been shown to have other pleiotropic cardioprotective effects besides β-arrestin-biased agonism. Indeed, a spectrum of anti-inflammatory, antioxidant, antifibrotic, and antiapoptotic properties of carvedilol has been reported in vitro and in vivo (28, 51). More recently, we demonstrated that carvedilol upregulates a subset of mature and premature microRNAs (miRs), but not their primary miR transcripts, through β-arrestin1 and β1AR activation (20). Although our finding suggested that this novel miR-processing pathway may be linked, in part, to the mechanism of carvedilol for cell survival, the role of β1AR/β-arrestin1-dependent miR processing in mediating the beneficial effects of carvedilol remains to be determined.
MiRs are increasingly being recognized as major regulators of physiological and pathological processes (43) and play important roles in cardiac biology and disease (4, 42). Among the five miRs we found to be regulated by carvedilol (20), miR-199a-3p and -214 have been shown to be cardioprotective during myocardial infarction (MI) and ischemia/reperfusion (I/R) injury (1, 9). The miR-199/214 cluster was also reported to be downregulated in patients with end-stage dilated cardiomyopathy (DCM) or ischemic DCM (2, 8).
Here, we investigate whether carvedilol-induced cardiomyocyte (CM) protection is mediated via the β-arrestin1/β1AR-responsive miRs, miR-199a-3p and miR-214. We provide evidence that miR-199a-3p and miR-214 act as gatekeepers of CM survival in part by repressing the predictive or known proapoptotic targets ddit4 and ing4, respectively, thus contributing to CM protection induced by carvedilol. Therefore, our data suggest that the biased β-blocker carvedilol regulates the expression of unique subsets of miRs, which may be mechanistically linked to its cardioprotective effects.
MATERIALS AND METHODS
Animal study approval.
Sprague-Dawley rats (1–2 days old) and C57BL/6 mice (8–12 wk old) were used for this study. Research on animals in this study was performed according to approved protocols and animal welfare regulations of Augusta University Institutional IACUC Committees. All animal procedures were performed to conform to the NIH guidelines (Guide for the Care and Use of Laboratory Animals). The neonatal rats were killed by decapitation under anesthesia for CM isolation. Carvedilol (Sigma-Aldrich) was dissolved in DMSO and then filled into microosmotic pumps (Alzet model 2001; DURECT) for subcutaneous delivery into adult mice at the rate of 19 mg/kg per day over a period of 7 days. In control mice, vehicle (DMSO) was used. After carvedilol administration, hearts were excised and flash frozen in liquid N2 for qRT-PCR analyses, as described previously (20).
Cell culture and transfection.
Mouse adult atrial CM HL-1 cells obtained from Dr. Claycomb and rat embryonic ventricular CM H9c2 cells were maintained as previously described (35). Primary neonatal rat ventricular CMs (NRVCs) were isolated by dissociation of hearts from 1- to 2-day-old Sprague-Dawley rats (40). The purity of NRVCs was previously shown by cell type-dependent gene expression patterns (41). To inhibit the expression of miR-199a-3p or miR-214 in CMs, we transfected mirVanna miRNA inhibitors (Life Technologies) specific to miR-199a-3p (MH11779), miR-214 (MH10347), or a miR inhibitor negative control (4464076) using Lipofectamine 2000 reagent (Invitrogen) as described previously (17). For gain-of-function studies, we transfected the cells with a miR mimic negative control, miR-199a-3p mirVanna mimic, or miR-214 mirVanna mimic (Life Technologies, MC11779 or MC10347). Transfected cells were incubated overnight in serum-free medium supplemented with 0.1% BSA, 10 mM HEPES (pH 7.4), and 1% penicillin before stimulation. Under serum starvation conditions, CMs were stimulated with carvedilol (1 μM; Sigma-Aldrich) for 1–4 h as described previously (19). To block the phosphoinositide 3-kinase (PI3K)/Akt pathway, 1 μM wortmannin (Sigma-Aldrich) was pretreated for 1 h before carvedilol stimulation. All in vitro assays were performed 48–60 h after transfection when maximum knockdown efficiency was reached.
In vitro simulated I/R.
CMs plated on coverslips or six-well plates were transfected with miR inhibitors or miR mimics, washed with 1× PBS, and placed in an ischemia buffer that contained (in mM) 118.0 NaCl, 24.0 NaH2CO3, 1.0 NaHPO4, 2.5 CaCl2, 1.2 MgCl2, 20.0 sodium lactate, 16.0 KCl, and 10.0 2-deoxyglucose (pH 6.2). CMs were then incubated in an anoxic chamber (5% CO2-0% O2) for 1 h followed by replacement of the ischemic buffer with normal cell medium and were incubated under normoxic conditions for 4 h to complete the simulated IR (sI/R) protocol as described (1). Coverslips or plates were processed for qRT-PCR, immunoblotting, and TUNEL staining.
RNA isolation and quantitative real-time RT-PCR.
Total RNA from CMs was prepared using Trizol reagent (Invitrogen) and treated with RNase-free DNase I to remove genomic DNA as described (18, 21). For detection of mature miR-199a-3p or miR-214, the TaqMan MicroRNA Reverse Transcription Kit (Life Technologies) was used to synthesize cDNA for TaqMan microRNA assays. The following probes were used to amplify and measure the amount of mature miRs by real-time RT-PCR: miR-199a-3p (002304), miR-214 (002306 or 000517), and U6 snRNA (001973) as an endogenous control. The following reaction components were used for each probe: 1.33 μl cDNA, 10.00 μl 2× TaqMan Universal PCR Master Mix (Life Technologies), 1.00 μl probe, and 7.67 μl water in a 20.00-μl total volume.
For detection of genes, cDNA was synthesized using Invitrogen SuperScript II reverse transcriptase and oligo-dT primers. Expression of genes was detected using TaqMan gene expression assays for mouse or rat (Sox2, Mm00488369_s1 or Rn01286286_g1; Nanog, Mm02019550_s1 or Rn01462825_m1; Ddit4, Mm00512504_g1 or Rn01433735_g1; Ing4, Mm00460097_m1 or Rn01185205_m1; Cav2, Mm01129337_g1 or Rn00590969_m1; Rb1, Mm00485586_m1 or Rn01753308_m1; Tfpi, Mm01334601_m1 or Rn01515299_m1; Pten, Mm00477208_m1 or Rn00477208_m1; and HPRT1, Mm00446969_m1 or Rn01527840_m1 for endogenous controls). The following reaction components were used for each probe: 2 μl cDNA, 10 μl 2× TaqMan Universal PCR Master Mix (Life Technologies), 1 μl probe, and 7 μl water in a 20-μl total volume.
Real-time PCR reactions were amplified and analyzed in triplicate using an ABI Sequence Detection System as described (21). PCR reaction conditions were as follows: step 1: 50°C for 2 min; step 2: 95°C for 10 min; step 3: 40 cycles of 95°C for 15 s followed by 60°C for 1 min. Expression relative to endogenous controls was calculated using 2−ΔΔCt, and levels were normalized to control. We performed at least five independent experiments in triplicate using different batches of RNAs each time.
Immunoblotting and detection.
CMs were washed once with 1× PBS, solubilized in 1 ml of lysis buffer (5.0 mM HEPES, 250.0 mM NaCl, 10.0% glycerol, 0.5% Nonidet P-40, 2.0 mM EDTA, and protease inhibitors) as described (19). Lysate samples were resolved by SDS-PAGE and transferred to PVDF (Bio-Rad) for immunoblotting. Antibodies for ddit4 (10638, Proteintech Group), ing4 (16188, Proteintech Group), β-actin (AC-15, Sigma-Aldrich), p-AKT (4060, Cell Signaling), and t-AKT (9272, Cell Signaling) were purchased and used at dilutions of 1:1,000 each. Detection was carried out using enhanced chemiluminescence (Amersham Biosciences).
CM apoptosis by TUNEL staining.
DNA fragmentation was detected in situ using TUNEL (34). In brief, CMs were incubated with proteinase K, and DNA fragments were labeled with fluorescein-conjugated dUTP using terminal deoxynucleotidyl transferase (Roche Diagnostics). The total number of nuclei was determined by manual counting of DAPI-stained nuclei in six random fields per coverslip (original magnification, ×200). All TUNEL-positive nuclei were counted in each coverslip. Digital photographs of fluorescence were acquired with a Zeiss microscope (ApoTome.2; Carl Zeiss) and processed with Adobe Photoshop.
In silico miR target prediction analysis.
We used several prediction algorithms based on evolutionary conservation of target sites across species including miRDB (44), PicTar (24), and Targetscan (26). Each of these algorithms predicts hundreds of possible targets for miRs, and the targets, which are important for apoptotic signals and are predicted by each of these programs, were selected for further evaluation.
Statistical analysis.
Data are expressed as means ± SE from at least four independent experiments with different biological samples per group. Statistical significance was determined by using one-way ANOVA with Bonferroni correction for multiple comparisons or Student's unpaired t-tests (GraphPad Prism, version 5). A P value <0.05 was considered statistically significant.
RESULTS
Carvedilol activates p-AKT survival signaling and pluripotent markers in CMs after sI/R.
Previous studies showed that treatment with the β-arrestin-biased β-blocker carvedilol (19, 47) reverses established LVF in experimental HF models, in association with reduced hypertrophy, fibrosis, and apoptosis (11, 50, 52, 53, 60). CXCR7-, ETAR-, or S1P1/3-mediated β-arrestin signaling has been reported to induce stem cell migration, activation, and proliferation (25, 36, 37). To test whether carvedilol-induced β-arrestin signaling can regulate CM survival signaling and resident pluripotency of CMs, we used in vitro models of sI/R in adult mouse atrial CMs (HL-1 cells) and embryonic rat ventricular CMs (H9c2 cells) treated with 1 μM carvedilol for 1 or 4 h (time points showing maximal levels of p-AKT, Sox2, and Nanog in time-course experiments, data not shown). Consistent with the previous findings by us and others to show that carvedilol activates p-AKT prosurvival signaling in mouse hearts and H9c2 cells (6, 19), sI/R induces p-AKT levels (Fig. 1A), and carvedilol increases both basal (normoxia) and sI/R-induced p-AKT levels in HL-1 cells (Fig. 1, B and C). Interestingly, the expression patterns of two stem cell markers Sox2 and Nanog in HL-1 cells were different after sI/R, in which Sox2 is decreased and Nanog is increased (Fig. 1D). However, carvedilol stimulation increases the expression of both genes in sI/R conditions, not normoxia in HL-1 cells (Fig. 1, E and F). Similar to HL-1 cells, sI/R induces p-AKT levels (Fig. 2A), and carvedilol treatment increases sI/R-induced p-AKT and Sox2 expression in H9c2 cells (Fig. 2, B and D). However, the expression of Sox2 is increased after sI/R, and carvedilol does not affect basal p-Akt levels in H9c2 cells (Fig. 2C and data not shown), which is different from adult atrial CMs. Using mouse hearts and NRVCs, we also confirmed our CM cell line data and showed that carvedilol induces the expression of Sox2 in both in vivo and primary CMs as well as p-AKT levels in NRVCs and that carvedilol-mediated Sox2 activation is blocked by an inhibitor of the PI3K/Akt pathway (Fig. 2, E–G). Along with previous reports to directly link the cardioprotective actions of carvedilol to upregulation of p-AKT/stem cell markers (6, 58) as well as to establish the Akt-Sox2 general cell survival axis (12, 16, 38), our data suggest that carvedilol may confer CM survival in part by activating the Akt-Sox2 axis in injured CMs.
Fig. 1.
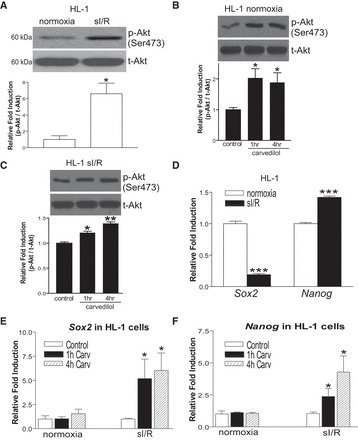
Carvedilol (Carv) activates p-AKT signaling and the expression of stem cell markers in injured HL-1 cells. HL-1 cells were treated with either vehicle (DMSO; control) or 1 μM Carv for 1 or 4 h and subjected to either normoxia (basal) or simulated ischemia/reperfusion (sI/R). Immunoblotting with antibodies for p-AKT and t-AKT (A–C) or real-time qRT-PCR for Sox2 and Nanog (D–F) was then performed. As indicated, Carv induces an increase in p-AKT levels in both normoxia (B) and sI/R (C), whereas Carv elicits upregulation of pluripotent stem cell markers (Sox2 and Nanog) only in sI/R (E and F). *P < 0.05 vs. normoxia or control (DMSO); **P < 0.01 vs. control (DMSO); ***P < 0.001 vs. normoxia; n = 4–6.
Fig. 2.
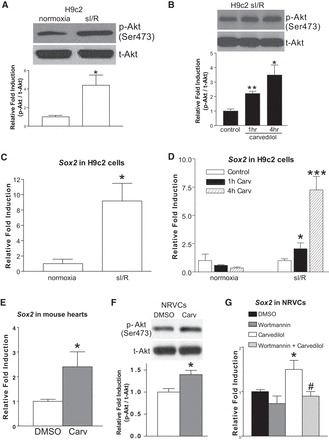
Carv activates p-AKT signaling and the expression of Sox2 in injured H9c2 cells, neonatal rat ventricular cardiomyocytes (NRVCs), and mouse hearts. A–D: H9c2 cells were treated with 1 μM Carv for 1 or 4 h and subjected to sI/R. As indicated, Carv induces an increase in p-AKT levels (B) or the expression of Sox2 (D) in sI/R, without affecting p-AKT levels (data not shown) and the expression of Sox2 (D) in normoxia. It is noted that Nanog is not expressed in H9c2 cells, and another stem cell marker (Oct4) is not expressed in both HL-1 and H9c2 cells (data not shown). E: expression of Sox2 was detected by qRT-PCR in adult mouse hearts stimulated with Carv (19 mg/kg per day) or vehicle for 7 days. Carv activates the expression of Sox2. F: NRVCs were treated with 1 μM Carv for 4 h. As indicated, Carv induces an increase in p-AKT levels. G: NRVCs were pretreated with 1 μM wortmannin for 1 h followed by 1 μM Carv for 4 h. The cells were then subjected to sI/R. Carv-mediated activation of Sox2 is blocked by wortmannin. *P < 0.05 vs. normoxia or control (DMSO); **P < 0.01 vs. control (DMSO); ***P < 0.001 vs. control (DMSO). #P <0.05 vs. Carv; n = 4–14.
Carvedilol elicits upregulation of miR-199-3p and miR-214 in CMs subjected to sI/R.
We previously showed that carvedilol upregulates miR-199a-3p and miR-214 in HEK293 cells stably expressing wild-type β1AR, and in mouse hearts, via stimulating β1AR, G protein-coupled receptor kinase 5/6, and β-arrestin1 (20). We evaluated the expression of these miRs in carvedilol-treated HL-1 and H9c2 cells. sI/R increases or decreases the expression of both miRs in HL-1 and H9c2 cells, respectively (Fig. 3, A and D). Carvedilol has no significant effect on the basal expression of these two miRs but upregulated their expression following sI/R (Fig. 3, B, C, E, and F). Interestingly, the expression of two miRs is not significantly changed after sI/R, and carvedilol does not activate their expression in NRVCs (data not shown). These data indicate that miR-199a-3p and miR-214 are sensitive to carvedilol and might mediate the effects of carvedilol on injured HL-1 and H9c2 cells.
Fig. 3.
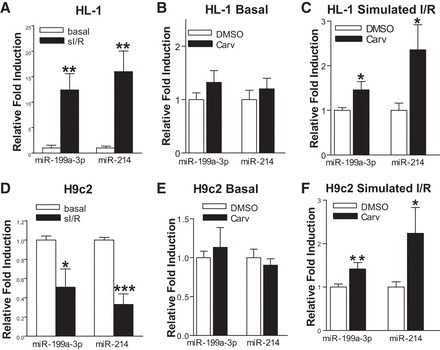
Carv induces the expression of miR-199a-3p and miR-214 under sI/R in HL-1 and H9c2 cells. A–C: HL-1 cells were treated with either vehicle (DMSO) or 1 μM Carv for 4 h and subjected to either normoxia (basal) or sI/R. The expression of mature miR-199a-3p and miR-214 was detected using TaqMan microRNA assay. Carv elicits upregulation of miR-199a-3p and miR-214 in simulated I/R, but not under normoxic (basal) conditions. D–F: H9c2 cells were treated with 1 μM Carv for 4 h and subjected to either normoxia (basal) or sI/R. The expression of mature miR-199a-3p and miR-214 was detected using TaqMan microRNA assay. Carv activates the expression of miR-199a-3p and miR-214 in sI/R, but not under basal conditions. *P < 0.05 vs. DMSO or normoxia; **P < 0.01 vs. normoxia or DMSO; ***P < 0.001 vs. normoxia; n = 5 in each group.
Carvedilol-responsive miR-199a-3p or miR-214 activates p-AKT survival signaling and the expression of a downstream pluripotency marker Sox2 in CMs after sI/R.
Carvedilol activates β1AR-mediated β-arrestin1-biased signaling to stimulate the processing of five miRs (20), and one of these miRs, miR-150, results in beneficial cardiac adaptive remodeling (41). Two other carvedilol-responsive miRs (miR-199a-3p and -214) have also been reported to induce cardioprotective effects during MI and I/R injury (1, 9). Moreover, the miR-199/214 cluster was reported to be downregulated in patients with HF (2, 8). To test the hypothesis that the beneficial effects of carvedilol might be mediated through upregulating the expression of miR-199a-3p or miR-214, we first assessed the effects of these two miRs on CM survival signaling and pluripotency. As shown in Fig. 4, A and B, overexpression of either miR (∼75-fold increase for miR-199a-3p and ∼211-fold increase for miR-214; data not shown) significantly increased p-AKT levels in HL-1 cells, which mimics the effects of carvedilol treatment (Fig. 1, B and C). Because AKT was shown to phosphorylate Sox2 and to promote its cell survival activity (12, 16, 38), we next tested whether gain of function of miR-199a-3p or miR-214 may increase Sox2 expression. Forced transient expression of miR-199a-3p indeed significantly upregulated basal and sI/R-induced Sox2 expression in HL-1 cells, whereas miR-214 upregulated basal-induced, but not sI/R, Sox2 expression (Fig. 4C). Overexpression of miR-199a-3p or miR-214 (∼63-fold increase and ∼189-fold increase, respectively; data not shown) also increased p-AKT levels in H9c2 cells subjected to sI/R, but not basal (Fig. 4, D and E), and induced Sox2 expression in H9c2 cells under normoxic conditions and following sI/R (Fig. 4F). These gain-of-function data indicate the antiapoptotic activity of the two carvedilol-responsive miRs in CMs by activating AKT-Sox2 cell survival axis.
Fig. 4.
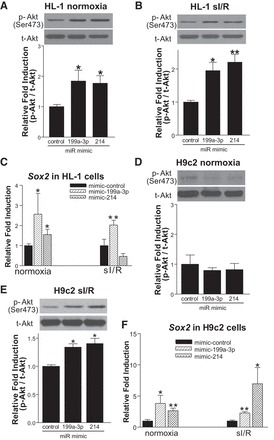
Overexpression of miR-199a-3p and miR-214 confers cardiomyocyte survival. A–C: HL-1 cells were transfected with miR-mimic control, miR-199a-3p mimic, or miR-214 mimic and subjected to either normoxia or sI/R. Gain of function of miR-199a-3p or miR-214 induces an increase in p-AKT levels in both normoxia (A) and sI/R (B). Moreover, overexpression of miR-199a-3p induces the expression of Sox2 in both normoxia and sI/R conditions, whereas overexpression of miR-214 induces the expression of Sox2 under normoxia (C). D–F: H9c2 cells were transfected with miR-mimic control, miR-199a-3p mimic, or miR-214 mimic and subjected to sI/R. Gain of function of miR-199a-3p or miR-214 increases sI/R-induced, but not basal, p-AKT levels (D and E), while inducing the expression of Sox2 under both normoxia and following sI/R (F). *P < 0.05 vs. miR mimic control; **P < 0.01 vs. miR mimic control; n = 4–6.
Expression of miR-199a-3p and miR-214 in part mediates the activation of p-AKT survival signaling by carvedilol in ischemic CMs.
A recent study reported the direct link between the cardioprotective actions of carvedilol and upregulation of p-AKT levels (6). To investigate whether loss of function of either miR-199a-3p or miR-214 modulates the beneficial effects of carvedilol after sI/R, p-AKT immunoblotting was performed. As shown in Fig. 5, carvedilol-induced activation of p-AKT in CMs is diminished after transfection with anti-miR-199a-3p or anti-miR-214 in both HL-1 (Fig. 5, A and B) and H9c2 (Fig. 5, C and D) cells subjected to sI/R, indicating that these miRs partially mediate cytoprotection induced by carvedilol in CMs.
Fig. 5.
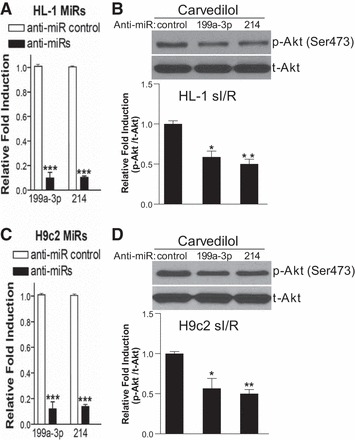
Knockdown of endogenous miR-199a-3p or miR-214 decreases Carv-mediated activation of p-AKT in injured cardiomyocytes. A: RNAs were isolated from HL-1 cells transfected with 100 nM mirvana miR-199a-3p inhibitor, miR-214 inhibitor, or a 15-mer control. The RNAs were analyzed by miR-199a-3p- or miR-214-specific qRT-PCR to access the levels of miR-199a-3p or miR-214. ***P < 0.001 vs. anti-miR control. B: HL-1 cells were transfected with anti-miR control, anti-miR-199a-3p, or anti-miR-214 and treated with 1 μM Carv for 4 h. The mouse adult atrial cardiomyocytes were then subjected to sI/R. C: RNAs were isolated from H9c2 cells transfected with 100 nM mirvana miR-199a-3p inhibitor, miR-214 inhibitor, or a 15-mer control. The RNAs were analyzed by miR-199a-3p- or miR-214-specific qRT-PCR to access the levels of miR-199a-3p or miR-214. ***P < 0.001 vs. anti-miR control. D: H9c2 cells were transfected with anti-miR control, anti-miR-199a-3p, or anti-miR-214 and treated with 1 μM Carv for 4 h. The rat embryonic ventricular cardiomyocytes were then subjected to sI/R. *P < 0.05 vs. anti-miR control; **P < 0.01 vs. anti-miR control; n = 4 in each group.
MiR-199a-3p or miR-214 protects CMs against apoptosis.
To determine the importance of miR-199a-3p and miR-214 for CM survival, we performed TUNEL staining of CMs in vitro in conjunction with sI/R. Loss of function of miR-199a-3p in HL-1 cells increased adult CM apoptosis under both basal conditions and following sI/R (Fig. 6, A–D), whereas loss of function of miR-214 increased HL-1 cell apoptosis only following sI/R (Fig. 6, A–D). Moreover, treatment with anti-miR-199a-3p or anti-miR-214 in NRVCs increased apoptosis under both basal and sI/R conditions (Fig. 7, A–C). These data suggest that the two miRs function as regulators of survival in CMs.
Fig. 6.
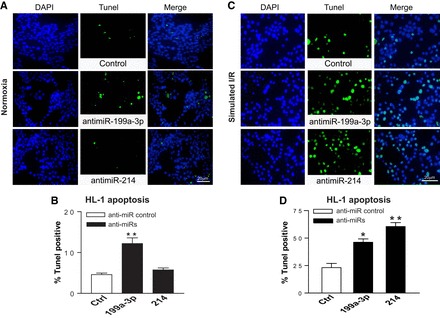
MiR-199a-3p or miR-214 functions as a gatekeeper of HL-1 cell survival. Adult mouse cardiomyocyte HL-1 cells were transfected with anti-miRs and subjected to sI/R. TUNEL assays were then performed under both normoxic (A and B) and sI/R conditions (C and D). The percentage of TUNEL-positive cells was calculated by normalizing DAPI-positive cells. All images were taken in the same magnification and were shown in the same size. *P < 0.05 vs. anti-miR control (Ctrl); **P < 0.01 vs. anti-miR control; n = 5 in each group.
Fig. 7.
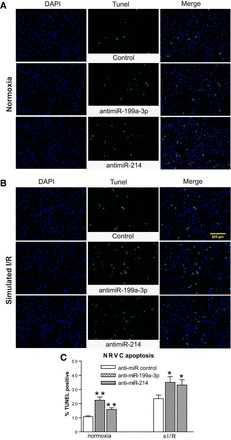
MiR-199a-3p or miR-214 functions as a gatekeeper of NRVC survival. NRVCs were transfected with anti-miRs and subjected to sI/R. TUNEL assays were then performed under both normoxic (A and C) and sI/R conditions (B and C). The percentage of TUNEL-positive cells was calculated by normalizing DAPI-positive cells. All images were taken in the same magnification and were shown in the same size. *P < 0.05 vs. anti-miR control; **P < 0.01 vs. anti-miR control; n = 5 in each group.
MiR-199a-3p or miR-214 regulates proapoptotic ddit4 or ing4.
To identify potential gene targets of miR-199a-3p and miR-214, we used several prediction algorithms including miRDB (44), PicTar (24), and Targetscan (26). In silico ingenuity pathway analysis (46) showed that one of the top associated network functions of the predicted targets of the two miRs is antiproliferation, cell cycle arrest, or apoptosis. Accordingly, we focused on apoptosis-related genes and found that caveolin-2, rb1, ddit4, and tfpi, which were predicted by all three target algorithms, were potential targets of miR-199a-3p. Pten and ing4, which were predicted by all three target algorithms, were potential targets of miR-214. The binding sites of these two miRs were well conserved among target mRNAs from mouse, rat, and human, which elevates the potential importance of these miRs in regulating the target genes.
We next postulated that identifying the functional targets of miR-199a-3p and miR-214 in primary CMs may elucidate the mechanisms whereby they elicit cardioprotective signaling. Accordingly, we performed loss- and gain-of-function studies in NRVCs (Fig. 8, A and B). One predicted target of miR-199a-3p, ddit4, was upregulated with miR-199a-3p inhibition and downregulated with miR-199a-3p overexpression (Fig. 8, A–C). In contrast, expression of cav2, rb1, and tfpi was unaffected by modulation of miR-199a-3p (Fig. 8, A, B, and D). Similarly, one predicted target of miR-214, ing4, was upregulated with miR-214 inhibition and downregulated with miR-214 overexpression (Fig. 8, A–C), whereas pten expression was unaffected (Fig. 8, A, B, and D). The mRNA results were confirmed by immunoblotting analysis that demonstrated concordant alterations in protein levels of ddit4 or ing4 after transfection of either miR mimics or anti-miRs for miR-199a-3p or miR-214, respectively (Fig. 8, E–H).
Fig. 8.
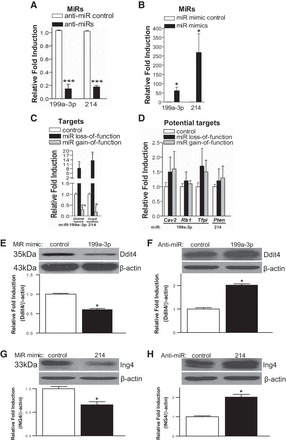
MiR-199a-3p or miR-214 represses proapoptotic ddit4 or ing4 in primary cardiomyocytes. A–D: RNAs were isolated from NRVCs transfected with 100 nM mirvana miR-199a-3p inhibitor, miR-214 inhibitor, or 15-mer control (A) or miR-199a-3p mimic, miR-214 mimic, or 15-mer control (B). The RNAs were then analyzed by miR-199a-3p- or miR-214-specific qRT-PCR to access the levels of miR-199a-3p or miR-214. *P < 0.05 vs. miR mimic control; ***P < 0.001 vs. anti-miR control. Levels of miR-199a-3p and miR-214 predictive target mRNAs are indicated in C and D. Data were normalized to HPRT1 and expressed relative to control (anti-miR control or miR mimic control). Results are representative of 4 independent experiments with different biological samples. *P < 0.05; #P < 0.05; ##P < 0.01 vs. control. E–H: modulation of miR-199a-3p and miR-214 in primary NRVCs concomitantly regulated Ddit4 (E and F) and Ing4 protein levels (G and H), respectively. Results are representative of 4 independent experiments with different biological samples. *P < 0.05 vs. control.
Our in vivo and NRVC data also showed that carvedilol decreased the expression of ddit4 and ing4 (Fig. 9, A–C), suggesting that carvedilol may inhibit these apoptotic genes by upregulating cardioprotective miR-199a-3p or miR-214. Interestingly, we also found that ddit4 and ing4 were upregulated after sI/R in NRVCs (Fig. 9, B and C). Our results indicate that ddit4 and ing4 may be functional CM targets of miR-199a-3p and miR-214, respectively. This idea is further supported by previous reports that 1) miR-199/214 cluster was downregulated in patients with end-stage dilated cardiomyopathy (DCM), ischemic DCM, or acute HF (2, 8, 31); 2) ddit4 was upregulated in both an in vivo cardiac ischemia model and an in vitro CM hypoxia model (22); 3) ddit4 was shown to induce CM apoptosis both in vitro and in vivo (5, 59) and to inhibit cardioprotective p38 MAPK- and PI3K/Akt-dependent mammalian target of rapamycin (mTOR) pathways (15); and 4) ing4 was recently suggested to negatively regulate CM function (54). Taken together, our data along with the previous studies indicate that downregulation of proapoptotic ddit4 or ing4, which is an evolutionarily conserved predictive and experimentally validated target, in part contributes to the beneficial actions of miR-199a-3p or miR-214 in CMs.
Fig. 9.
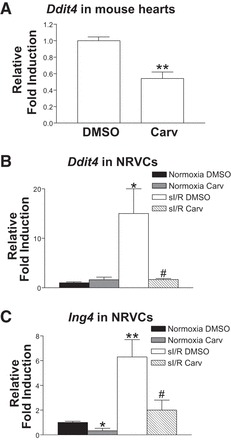
Carv inhibits the expression of ddit4 or ing4 in mouse hearts or NRVCs. A: expression of ddit4 was detected by real-time qRT-PCR in adult mouse hearts stimulated with Carv (19 mg/kg per day) or vehicle for 3 days. Carv inhibits the expression of ddit4 without significantly affecting ing4 expression in mouse hearts (data not shown). B and C: NRVCs were treated with 1 μM Carv for 4 h and subjected to either normoxia (basal) or sI/R. qRT-PCR analysis for ddit4 or ing4 was then performed. Carv inhibits the expression of ddit4 only in sI/R (B), while reducing the expression of ing4 in both basal and sI/R (C). *P < 0.05 vs. basal DMSO; **P < 0.01 vs. DMSO; #P < 0.05 vs. sI/R DMSO; n = 6.
DISCUSSION
In this study, we report a role for miR-199a-3p or miR-214 in mediating p-AKT prosurvival signaling in CMs following carvedilol stimulation. We also demonstrate that these miRs are ischemic stress-responsive protectors against CM apoptosis. CMs lacking miR-199a-3p or miR-214 have an increased sensitivity to sI/R-induced apoptosis, whereas CMs overexpressing miR-199a-3p or miR-214 have increased p-AKT prosurvival signaling and elevated expression of a downstream pluripotent gene Sox2. Moreover, we identify that a molecular mechanism of CM protection by these miRs is the inhibition of proapoptotic ddit4 (for miR-199a-3p) and ing4 (for miR-214), which are target genes supported by our bioinformatics and experimental evidences. Our findings suggest that the identified miR-target pairs may be involved in carvedilol-mediated cardioprotective signaling (Fig. 10).
Fig. 10.
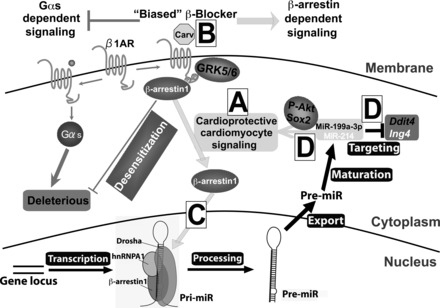
Carv regulates the levels of miR-199a-3p and miR-214 in cardiac myocytes undergoing I/R: a mechanism whereby Carv mediates cardioprotective signaling. β-Arrestin-mediated β1-adrenergic receptor (β1AR) signaling confers cardioprotection [(30) and see A], and Carv is a β-arrestin-biased ligand for β1AR [(19) and see B]. Using this clinically employed biased β-blocker, we previously showed that Carv induces the processing of miR-199a-3p and miR-214 in a β1AR-, G protein-coupled receptor kinase 5/6-, or β-arrestin1-dependent manner [(20) and see C]. Here, our data suggest that β-arrestin1-biased agonism of β1AR induces processing of miR-199a-3p and miR-214 to mediate the cardioprotective effects of Carv and that miR-199a-3p and miR-214 act as protective miRs in part by repressing apoptotic genes ddit4 and ing4 as well as by activating p-AKT and Sox2 in cardiomyocytes (see D).
We previously showed that miR-199a-3p and miR-214 are β1AR/β-arrestin1-regulatable miRs, which are posttranscriptionally activated by the biased β-blocker carvedilol (Fig. 10, A–C). Together with the results presented here (Fig. 10D), we postulate that the β-arrestin1/β1AR-mediated regulation of miR processing in CMs (the only cardiac cell type in which β1ARs are expressed) may result in beneficial adaptive remodeling in failing hearts. This concept is further supported by the observation that four β1AR/β-arrestin1-regulatable miRs (miR-125b-5p, miR-150, miR-199a-3p, and miR-214) activated by carvedilol (20) are cardioprotective in vivo during MI and I/R injury (1, 9, 41, 45). Interestingly, two previous studies directly linked the cardioprotective effects of carvedilol to miR upregulation using rat models of MI (50, 60). The upregulation of miR-29b, a cardioprotective miR (56), was shown to mediate the effect of carvedilol to attenuate MI-induced fibrosis (60). Xu et al. (50) also reported that the expression of a cardioprotective miR-133 (3, 29) in rat hearts was significantly upregulated by carvedilol pretreatment and that upregulation of miR-133 mediates the antiapoptotic action of carvedilol in NRVCs. Similar additional studies are warranted to provide evidence that the cardioprotective actions of carvedilol are associated with increased levels of cardioprotective miR-199a-3p and miR-214. To fully establish carvedilol-miR axis in cardioprotection, future studies are also required to determine whether miR-29b, miR-133, miR-199a-3p, and miR-214 confer possible overlapping/compensatory effects on carvedilol-mediated cardioprotection.
Although ing4 was shown to be regulated by miR-214 in pancreatic cells and was proven to be a direct target using heterologous 293 cell and luciferase systems (55), our present study shows a novel finding that ing4 is regulated by miR-214 in CMs. Moreover, we report for the first time that ddit4 is a target of miR-199a-3p. Ddit4 (also known as Redd1) has been reported to mediate apoptotic signaling in multiple cell types (23, 39, 48). In the context of cancer, ing4 was also shown to induce apoptotic pathways (13, 27, 49). Our findings suggest that inhibition of these genes could be therapeutically beneficial for cardiac disease. Given that these two apoptotic genes are targets of miR-199a-3p and miR-214 and are upregulated in ischemic conditions, patients with HF with reduced levels of these two circulating miRs could be particularly suitable for future targeted treatments based on ddit4 and ing4. However, additional studies to directly link the cardioprotective actions of miR-199a-3p and miR-214 to repression of ddit4 and ing4 are needed before considering the identified carvedilol-responsive miR-target pairs as therapeutic options.
Interestingly, we also demonstrate in the present study that two carvedilol-responsive miRs, miR-199a-3p or miR-214, activate prosurvival AKT-Sox2 axis, and the expression of miR-199a-3p and miR-214 in part mediates the activation of p-AKT survival signaling by carvedilol in ischemic CMs (Figs. 4 and 5). Although miR-214 was shown to activate AKT pathway by targeting pten in monocytes (57), there is no previous report to show the cross talk between miR-199a-3p and AKT/Sox2 axis or between miR-214 and Sox2. Notably, ddit4 was known to inhibit AKT phosphorylation by enhancing PP2A, and this ddit4/Akt/mTOR axis was shown to be a general cell survival mechanism in various human and mouse cells (7, 14, 15). AKT was also shown to phosphorylate Sox2, thus activating its cell survival property in cancer cells (12, 16, 38) and normal cells (10, 32, 33). Given our data along with previous studies above, our working hypothesis for the detailed mechanism by which miR-199a-3p or miR-214 activates CM survival is that 1) miR-199a-3p inhibits ddit4, which subsequently activates p-AKT-Sox2 cell survival axis; and 2) miR-214 inhibits pten or ing4, which subsequently activates p-AKT-Sox2 cell survival axis. Although our data, except for no change of pten mRNA levels by miR-214 in CMs, support this hypothesis, additional studies (e.g., how ing4 regulates AKT signaling) will be needed to further clarify the mechanisms of two miRs in CM survival and carvedilol-mediated cardioprotection.
In conclusion, our results suggest that miR-199a-3p and miR-214 protect CMs against simulated ischemic injury in part via repression of ddit4 and ing4. Interestingly, previous studies in mouse models of MI and I/R reported that miR-199a-3p and miR-214 protect the heart from ischemic injury by promoting CM proliferation and cardiac regeneration (9) or regulating calcium overload and CM death (1). These studies suggest prominent roles for these two miRs in postischemic CM remodeling, which is consistent with our present study. Therefore, boosting levels of miR-199a-3p and miR-214 to attenuate CM death may provide therapeutic benefits. Moreover, induction of miR-199a-3p and miR-214 by carvedilol may in part account for the cardioprotective effects of this nonselective β-blocker.
GRANTS
This work was supported by American Physiological Society Shih-Chun Wang Young Investigator Award, American Heart Association Grant-in-Aid 12GRNT12100048, Scientist Development Grant 14SDG18970040, and National Institutes of Health R01 HL124251 to Il-man Kim as well as American Heart Association Postdoctoral Fellowship 16POST26990020 to Zuzana Broskova.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
K.-m.P., J.-p.T., Y.W., and I.-m.K. conception and design of research; K.-m.P., J.-p.T., Y.W., Z.B., and A.B. performed experiments; K.-m.P., J.-p.T., Y.W., Z.B., A.B., Y.T., H.S., N.L.W., and I.-m.K. analyzed data; K.-m.P., J.-p.T., Y.W., Y.T., H.S., N.L.W., and I.-m.K. interpreted results of experiments; K.-m.P., J.-p.T., Y.W., and I.-m.K. prepared figures; K.-m.P. and I.-m.K. drafted manuscript; K.-m.P., J.-p.T., Y.W., Z.B., A.B., Y.T., H.S., N.L.W., and I.-m.K. edited and revised manuscript; I.-m.K. approved final version of manuscript.
ACKNOWLEDGMENTS
Present affiliation for K.-m. Park: Washington University, Saint Louis, MO.
Present affiliation for Y. Wang: University of Kentucky, Lexington, KY.
REFERENCES
- 1.Aurora AB, Mahmoud AI, Luo X, Johnson BA, van Rooij E, Matsuzaki S, Humphries KM, Hill JA, Bassel-Duby R, Sadek HA, Olson EN. MicroRNA-214 protects the mouse heart from ischemic injury by controlling Ca(2)(+) overload and cell death. J Clin Invest 122: 1222–1232, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baumgarten A, Bang C, Tschirner A, Engelmann A, Adams V, von Haehling S, Doehner W, Pregla R, Anker MS, Blecharz K, Meyer R, Hetzer R, Anker SD, Thum T, Springer J. TWIST1 regulates the activity of ubiquitin proteasome system via the miR-199/214 cluster in human end-stage dilated cardiomyopathy. Int J Cardiol 168: 1447–1452, 2013. [DOI] [PubMed] [Google Scholar]
- 3.Castaldi A, Zaglia T, Di Mauro V, Carullo P, Viggiani G, Borile G, Di Stefano B, Schiattarella GG, Gualazzi MG, Elia L, Stirparo GG, Colorito ML, Pironti G, Kunderfranco P, Esposito G, Bang ML, Mongillo M, Condorelli G, Catalucci D. MicroRNA-133 modulates the beta1-adrenergic receptor transduction cascade. Circ Res 115: 273–283, 2014. [DOI] [PubMed] [Google Scholar]
- 4.Catalucci D, Gallo P, Condorelli G. MicroRNAs in cardiovascular biology and heart disease. Circ Cardiovasc Genet 2: 402–408, 2009. [DOI] [PubMed] [Google Scholar]
- 5.Chen R, Wang B, Chen L, Cai D, Li B, Chen C, Huang E, Liu C, Lin Z, Xie WB, Wang H. DNA damage-inducible transcript 4 (DDIT4) mediates methamphetamine-induced autophagy and apoptosis through mTOR signaling pathway in cardiomyocytes. Toxicol Appl Pharmacol 295: 1–11, 2016. [DOI] [PubMed] [Google Scholar]
- 6.Chen YL, Chung SY, Chai HT, Chen CH, Liu CF, Huang TH, Zhen YY, Sung PH, Sun CK, Chua S, Lu HI, Lee FY, Sheu JJ, Yip HK. Early administration of carvedilol protected against doxorubicin-induced cardiomyopathy. J Pharmacol Exp Ther 355: 516–527, 2015. [DOI] [PubMed] [Google Scholar]
- 7.Dennis MD, Coleman CS, Berg A, Jefferson LS, Kimball SR. REDD1 enhances protein phosphatase 2A-mediated dephosphorylation of Akt to repress mTORC1 signaling. Sci Signal 7: ra68, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ellis KL, Cameron VA, Troughton RW, Frampton CM, Ellmers LJ, Richards AM. Circulating microRNAs as candidate markers to distinguish heart failure in breathless patients. Eur J Heart Fail 15: 1138–1147, 2013. [DOI] [PubMed] [Google Scholar]
- 9.Eulalio A, Mano M, Dal Ferro M, Zentilin L, Sinagra G, Zacchigna S, Giacca M. Functional screening identifies miRNAs inducing cardiac regeneration. Nature 492: 376–381, 2012. [DOI] [PubMed] [Google Scholar]
- 10.Fang L, Zhang L, Wei W, Jin X, Wang P, Tong Y, Li J, Du JX, Wong J. A methylation-phosphorylation switch determines Sox2 stability and function in ESC maintenance or differentiation. Mol Cell 55: 537–551, 2014. [DOI] [PubMed] [Google Scholar]
- 11.Feuerstein GZ, Hamburger SA, Smith EF 3rd, Bril A, Ruffolo RR Jr.. Myocardial protection with carvedilol. J Cardiovasc Pharmacol 19, Suppl 1: S138–S141, 1992. [DOI] [PubMed] [Google Scholar]
- 12.Gen Y, Yasui K, Nishikawa T, Yoshikawa T. SOX2 promotes tumor growth of esophageal squamous cell carcinoma through the AKT/mammalian target of rapamycin complex 1 signaling pathway. Cancer Sci 104: 810–816, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gong A, Ye S, Xiong E, Guo W, Zhang Y, Peng W, Shao G, Jin J, Zhang Z, Yang J, Gao J. Autophagy contributes to ING4-induced glioma cell death. Exp Cell Res 319: 1714–1723, 2013. [DOI] [PubMed] [Google Scholar]
- 14.Gu Y, Kaufman JL, Bernal L, Torre C, Matulis SM, Harvey RD, Chen J, Sun SY, Boise LH, Lonial S. MLN4924, an NAE inhibitor, suppresses AKT and mTOR signaling via upregulation of REDD1 in human myeloma cells. Blood 123: 3269–3276, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hernandez G, Lal H, Fidalgo M, Guerrero A, Zalvide J, Force T, Pombo CM. A novel cardioprotective p38-MAPK/mTOR pathway. Exp Cell Res 317: 2938–2949, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jeong CH, Cho YY, Kim MO, Kim SH, Cho EJ, Lee SY, Jeon YJ, Lee KY, Yao K, Keum YS, Bode AM, Dong Z. Phosphorylation of Sox2 cooperates in reprogramming to pluripotent stem cells. Stem Cells 28: 2141–2150, 2010. [DOI] [PubMed] [Google Scholar]
- 17.Kim IM, Ackerson T, Ramakrishna S, Tretiakova M, Wang IC, Kalin TV, Major ML, Gusarova GA, Yoder HM, Costa RH, Kalinichenko VV. The Forkhead Box m1 transcription factor stimulates the proliferation of tumor cells during development of lung cancer. Cancer Res 66: 2153–2161, 2006. [DOI] [PubMed] [Google Scholar]
- 18.Kim IM, Ramakrishna S, Gusarova GA, Yoder HM, Costa RH, Kalinichenko VV. The forkhead box m1 transcription factor is essential for embryonic development of pulmonary vasculature. J Biol Chem 280: 22278–22286, 2005. [DOI] [PubMed] [Google Scholar]
- 19.Kim IM, Tilley DG, Chen J, Salazar NC, Whalen EJ, Violin JD, Rockman HA. Beta-blockers alprenolol and carvedilol stimulate beta-arrestin-mediated EGFR transactivation. Proc Natl Acad Sci USA 105: 14555–14560, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim IM, Wang Y, Park KM, Tang Y, Teoh JP, Vinson J, Traynham CJ, Pironti G, Mao L, Su H, Johnson JA, Koch WJ, Rockman HA. beta-Arrestin1-biased beta1-adrenergic receptor signaling regulates microRNA processing. Circ Res 114: 833–844, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim IM, Wolf MJ, Rockman HA. Gene deletion screen for cardiomyopathy in adult Drosophila identifies a new notch ligand. Circ Res 106: 1233–1243, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim MY, Seo EJ, Lee DH, Kim EJ, Kim HS, Cho HY, Chung EY, Lee SH, Baik EJ, Moon CH, Jung YS. Gadd45beta is a novel mediator of cardiomyocyte apoptosis induced by ischaemia/hypoxia. Cardiovasc Res 87: 119–126, 2010. [DOI] [PubMed] [Google Scholar]
- 23.Knowles LM, Yang C, Osterman A, Smith JW. Inhibition of fatty-acid synthase induces caspase-8-mediated tumor cell apoptosis by up-regulating DDIT4. J Biol Chem 283: 31378–31384, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M, Rajewsky N. Combinatorial microRNA target predictions. Nat Genet 37: 495–500, 2005. [DOI] [PubMed] [Google Scholar]
- 25.Lee E, Han J, Kim K, Choi H, Cho EG, Lee TR. CXCR7 mediates SDF1-induced melanocyte migration. Pigment Cell Melanoma Res 26: 58–66, 2013. [DOI] [PubMed] [Google Scholar]
- 26.Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell 115: 787–798, 2003. [DOI] [PubMed] [Google Scholar]
- 27.Li M, Zhu Y, Zhang H, Li L, He P, Xia H, Zhang Y, Mao C. Delivery of inhibitor of growth 4 (ING4) gene significantly inhibits proliferation and invasion and promotes apoptosis of human osteosarcoma cells. Sci Rep 4: 7380, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma XL, Yue TL, Lopez BL, Barone FC, Christopher TA, Ruffolo RR Jr, Feuerstein GZ. Carvedilol, a new beta adrenoreceptor blocker and free radical scavenger, attenuates myocardial ischemia-reperfusion injury in hypercholesterolemic rabbits. J Pharmacol Exp Ther 277: 128–136, 1996. [PubMed] [Google Scholar]
- 29.Matkovich SJ, Wang W, Tu Y, Eschenbacher WH, Dorn LE, Condorelli G, Diwan A, Nerbonne JM, Dorn GW 2nd. MicroRNA-133a protects against myocardial fibrosis and modulates electrical repolarization without affecting hypertrophy in pressure-overloaded adult hearts. Circ Res 106: 166–175, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Noma T, Lemaire A, Naga Prasad SV, Barki-Harrington L, Tilley DG, Chen J, Le Corvoisier P, Violin JD, Wei H, Lefkowitz RJ, Rockman HA. Beta-arrestin-mediated beta1-adrenergic receptor transactivation of the EGFR confers cardioprotection. J Clin Invest 117: 2445–2458, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ovchinnikova ES, Schmitter D, Vegter EL, Ter Maaten JM, Valente MA, Liu LC, van der Harst P, Pinto YM, de Boer RA, Meyer S, Teerlink JR, O'Connor CM, Metra M, Davison BA, Bloomfield DM, Cotter G, Cleland JG, Mebazaa A, Laribi S, Givertz MM, Ponikowski P, van der Meer P, van Veldhuisen DJ, Voors AA, Berezikov E. Signature of circulating microRNAs in patients with acute heart failure. Eur J Heart Fail 18: 414–423, 2015. [DOI] [PubMed] [Google Scholar]
- 32.Palumbo S, Tsai TL, Li WJ. Macrophage migration inhibitory factor regulates AKT signaling in hypoxic culture to modulate senescence of human mesenchymal stem cells. Stem Cells Dev 23: 852–865, 2014. [DOI] [PubMed] [Google Scholar]
- 33.Peltier J, Conway A, Keung AJ, Schaffer DV. Akt increases Sox2 expression in adult hippocampal neural progenitor cells, but increased sox2 does not promote proliferation. Stem Cells Dev 20: 1153–1161, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rakesh K, Yoo B, Kim IM, Salazar N, Kim KS, Rockman HA. beta-Arrestin-biased agonism of the angiotensin receptor induced by mechanical stress. Sci Signal 3: ra46, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ramakrishna S, Kim IM, Petrovic V, Malin D, Wang IC, Kalin TV, Meliton L, Zhao YY, Ackerson T, Qin Y, Malik AB, Costa RH, Kalinichenko VV. Myocardium defects and ventricular hypoplasia in mice homozygous null for the Forkhead Box M1 transcription factor. Dev Dyn 236: 1000–1013, 2007. [DOI] [PubMed] [Google Scholar]
- 36.Rosano L, Cianfrocca R, Tocci P, Spinella F, Di Castro V, Spadaro F, Salvati E, Biroccio AM, Natali PG, Bagnato A. beta-Arrestin-1 is a nuclear transcriptional regulator of endothelin-1-induced beta-catenin signaling. Oncogene 32: 5066–5077, 2012. [DOI] [PubMed] [Google Scholar]
- 37.Ryu JM, Baek YB, Shin MS, Park JH, Park SH, Lee JH, Han HJ. Sphingosine-1-phosphate-induced Flk-1 transactivation stimulates mouse embryonic stem cell proliferation through S1P/S1P-dependent beta-arrestin/c-Src pathways. Stem Cell Res 12: 69–85, 2013. [DOI] [PubMed] [Google Scholar]
- 38.Singh S, Trevino J, Bora-Singhal N, Coppola D, Haura E, Altiok S, Chellappan SP. EGFR/Src/Akt signaling modulates Sox2 expression and self-renewal of stem-like side-population cells in non-small cell lung cancer. Mol Cancer 11: 73, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sinha I, Allen JE, Pinto JT, Sinha R. Methylseleninic acid elevates REDD1 and inhibits prostate cancer cell growth despite AKT activation and mTOR dysregulation in hypoxia. Cancer Med 3: 252–264, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Su H, Li F, Ranek MJ, Wei N, Wang X. COP9 signalosome regulates autophagosome maturation. Circulation 124: 2117–2128, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tang Y, Wang Y, Park KM, Hu Q, Teoh JP, Broskova Z, Ranganathan P, Jayakumar C, Li J, Su H, Ramesh G, Kim IM. MicroRNA-150 protects the mouse heart from ischemic injury by regulating cell death. Cardiovasc Res 106: 387–397, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van Rooij E. The art of microRNA research. Circ Res 108: 219–234, 2011. [DOI] [PubMed] [Google Scholar]
- 43.van Rooij E, Marshall WS, Olson EN. Toward microRNA-based therapeutics for heart disease: The sense in antisense. Circ Res 103: 919–928, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang X. miRDB: A microRNA target prediction and functional annotation database with a wiki interface. RNA 14: 1012–1017, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang X, Ha T, Zou J, Ren D, Liu L, Zhang X, Kalbfleisch J, Gao X, Williams D, Li C. MicroRNA-125b protects against myocardial ischaemia/reperfusion injury via targeting p53-mediated apoptotic signalling and TRAF6. Cardiovasc Res 102: 385–395, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Werner T. Bioinformatics applications for pathway analysis of microarray data. Curr Opin Biotechnol 19: 50–54, 2008. [DOI] [PubMed] [Google Scholar]
- 47.Wisler JW, DeWire SM, Whalen EJ, Violin JD, Drake MT, Ahn S, Shenoy SK, Lefkowitz RJ. A unique mechanism of beta-blocker action: carvedilol stimulates beta-arrestin signaling. Proc Natl Acad Sci USA 104: 16657–16662, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wolff NC, McKay RM, Brugarolas J. REDD1/DDIT4-independent mTORC1 inhibition and apoptosis by glucocorticoids in thymocytes. Mol Cancer Res 12: 867–877, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xie YF, Sheng W, Xiang J, Zhang H, Ye Z, Yang J. Adenovirus-mediated ING4 expression suppresses pancreatic carcinoma cell growth via induction of cell-cycle alteration, apoptosis, and inhibition of tumor angiogenesis. Cancer Biother Radiopharm 24: 261–269, 2009. [DOI] [PubMed] [Google Scholar]
- 50.Xu C, Hu Y, Hou L, Ju J, Li X, Du N, Guan X, Liu Z, Zhang T, Qin W, Shen N, Bilal MU, Lu Y, Zhang Y, Shan H. beta-Blocker carvedilol protects cardiomyocytes against oxidative stress-induced apoptosis by up-regulating miR-133 expression. J Mol Cell Cardiol 75: 111–121, 2014. [DOI] [PubMed] [Google Scholar]
- 51.Yue TL, Cheng HY, Lysko PG, McKenna PJ, Feuerstein R, Gu JL, Lysko KA, Davis LL, Feuerstein G. Carvedilol, a new vasodilator and beta adrenoceptor antagonist, is an antioxidant and free radical scavenger. J Pharmacol Exp Ther 263: 92–98, 1992. [PubMed] [Google Scholar]
- 52.Yue TL, Ma XL, Gu JL, Ruffolo RR Jr, Feuerstein GZ. Carvedilol inhibits activation of stress-activated protein kinase and reduces reperfusion injury in perfused rabbit heart. Eur J Pharmacol 345: 61–65, 1998. [DOI] [PubMed] [Google Scholar]
- 53.Yue TL, Ma XL, Wang X, Romanic AM, Liu GL, Louden C, Gu JL, Kumar S, Poste G, Ruffolo RR Jr, Feuerstein GZ. Possible involvement of stress-activated protein kinase signaling pathway and Fas receptor expression in prevention of ischemia/reperfusion-induced cardiomyocyte apoptosis by carvedilol. Circ Res 82: 166–174, 1998. [DOI] [PubMed] [Google Scholar]
- 54.Zhang J, Xue X, Xu Y, Zhang Y, Li Z, Wang H. The transcriptome responses of cardiomyocyte exposed to hypothermia. Cryobiology 72: 244–250, 2016. [DOI] [PubMed] [Google Scholar]
- 55.Zhang XJ, Ye H, Zeng CW, He B, Zhang H, Chen YQ. Dysregulation of miR-15a and miR-214 in human pancreatic cancer. J Hematol Oncol 3: 46, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang Y, Huang XR, Wei LH, Chung AC, Yu CM, Lan HY. miR-29b as a therapeutic agent for angiotensin II-induced cardiac fibrosis by targeting TGF-beta/Smad3 signaling. Mol Ther 22: 974–985, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhao C, Sun W, Zhang P, Ling S, Li Y, Zhao D, Peng J, Wang A, Li Q, Song J, Wang C, Xu X, Xu Z, Zhong G, Han B, Chang YZ. miR-214 promotes osteoclastogenesis by targeting Pten/PI3K/Akt pathway. RNA Biol 12: 343–353, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhou H, Dickson ME, Kim MS, Bassel-Duby R, Olson EN. Akt1/protein kinase B enhances transcriptional reprogramming of fibroblasts to functional cardiomyocytes. Proc Natl Acad Sci USA 112: 11864–11869, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhou Y, Chen Q, Lew KS, Richards AM, Wang P. Discovery of potential therapeutic miRNA targets in cardiac ischemia-reperfusion injury. J Cardiovasc Pharmacol Ther 21: 296–309, 2015. [DOI] [PubMed] [Google Scholar]
- 60.Zhu JN, Chen R, Fu YH, Lin QX, Huang S, Guo LL, Zhang MZ, Deng CY, Zou X, Zhong SL, Yang M, Zhuang J, Yu XY, Shan ZX. Smad3 inactivation and MiR-29b upregulation mediate the effect of carvedilol on attenuating the acute myocardium infarction-induced myocardial fibrosis in rat. PLoS One 8: e75557, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]