

Abstract
Preeclampsia (PE) is a devastating disorder of pregnancy that affects up to 8% of pregnant women in the United States. The diagnosis of PE is made by the presentation of new-onset hypertension, ≥140 mmHg systolic blood pressure (BP) or ≥90 mmHg diastolic BP, and either proteinuria or another accompanying sign/symptom, such as renal insufficiency, thrombocytopenia, hepatic dysfunction, pulmonary edema, or cerebral/visual. These signs can occur suddenly and without warning. PE that presents before 34 wk of gestation is considered early onset and carries a greater risk for perinatal morbidity/mortality than late-onset PE that occurs at or after 34 wk of gestation. At this time there is no cure for PE, and the only effective treatment is delivery of the baby and placenta. If allowed to progress to eclampsia (PE with neurologic involvement), seizures will occur and possibly death through stroke. PE also carries the risk of significant fetal and neonatal morbidity/mortality in addition to long-term health risks for mother and child. Despite significant research efforts to accurately predict, diagnose, and treat PE, a cure eludes us. Elucidating the pathophysiological mechanisms that can cause PE will aid in our ability to accurately prevent, manage, and treat PE to avoid maternal and fetal losses. Intense research efforts are focused on PE, and the mouse has proven to be a useful animal model for investigating molecular mechanisms that may hold the key to unraveling the mysteries of PE in women.
Keywords: preeclampsia, placenta, trophoblast
EPIDEMIOLOGY OF PE
while preeclampsia (PE) characterizes ∼300,000 pregnancies per year in the United States (42), it affects up to 10% of pregnancies worldwide (19). Shockingly, PE is also responsible for 42% of maternal deaths worldwide, killing an estimated 76,000 women every year (49, 57). There appears to be a disparity in the incidence of maternal death associated with PE in developing countries with 99% occurring in low- to middle-income nations (42). Although genetic susceptibility cannot be ruled out, this discrepancy may simply be due to differences in perinatal care available in those countries. Even though perinatal care in developed countries is constantly improving, the incidence of PE is rising. From 1987 to 2004, the prevalence of PE diagnosis from admission to labor increased by 25% according to a United States National Hospital Discharge Survey (24).
In an effort to positively predict which women may develop PE during pregnancy, a number of conditions have been labeled as risk factors (Fig. 1). Pre-existing maternal conditions include advanced age, African-American race, higher body mass index (BMI), pregestational diabetes, chronic hypertension, renal disease, autoantibody disease (i.e., antiphospholipid antibody syndrome and systemic lupus erythematosus), family or personal history of PE, and lack of smoking (24). The presence of prepregnancy chronic hypertension can greatly complicate the diagnosis of PE, and it is important to distinguish between the two for perinatal management (4). Superimposed PE, or worsening hypertension along with an accompanying clinical feature of PE, can occur in up to 26% of pregnant women with chronic hypertension (4). Pregnancy-specific factors include nulliparity, partner-related factors (i.e., new paternity, limited sperm exposure, barrier contraception), multifetal gestation, and hydatidiform mole (24). When all of these pre-existing conditions and risk factors are considered, the increasing prevalence of PE could be attributed to a number of shifting trends in the United States and other countries. Women are looking to become pregnant later in life, and this often necessitates fertility treatments that result in multifetal gestations. Also, with the increased incidence of obesity worldwide, we see more women of childbearing age that have higher BMIs, diabetes, and hypertension as well as subfertility (24). However, the fact that an estimated two-thirds of all PE cases occur in first pregnancies that develop beyond the first trimester is staggering (15). These data strongly point toward defects in maternal adaptions to pregnancy that may be associated with first exposure of paternal antigens. The most surprising risk factor is the lack of smoking. This has been attributed to its effect on angiogenic factors (25); however, smoking before or during pregnancy would never be advised.
Fig. 1.
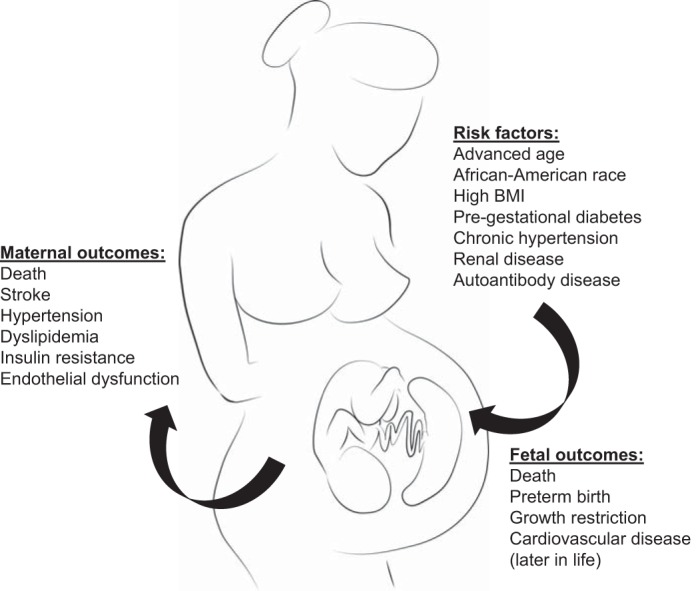
Predisposing risk factors and maternal/fetal outcomes of preeclampsia (PE). The pregnancy-specific disorder PE occurs spontaneously and without warning. Maternal conditions pre-existing before pregnancy are thought to significantly increase the risk of developing PE; however, the incidence is still unpredictable. Signs of PE resolve upon delivery of the placenta; therefore, it is considered the causative organ. This dysregulated placenta masterminds its potent effects on the fetus and mother during pregnancy but can also leave lasting detrimental effects on the health of both mother and offspring.
PE tends to have a familial disposition (7). For example, the frequency of PE in daughters is reported to be two to five times greater than in daughters-in-law (39). Studies to identify a simple acting dominant gene have been unrewarding due in part to the heterogeneity of disease presentation with PE having early or late onset and symptoms ranging from mild to moderate to severe (19). Thus, PE is considered a complex trait with inputs from environmental factors as well as fetal (paternal) genes (7, 39). Genome-wide searches are underway, and again variations are seen among and between populations (39). Determining the genetic mechanisms of PE have been complicated and frustrating.
Importantly, there is evidence that PE predisposes women to further health risks later in life. In a Scandinavian population of healthy nulliparous women with severe PE that necessitated preterm delivery, the risk of developing cardiovascular disease increased eightfold (23). A 24 to 36 yr follow-up of maternal health status in PE-affected women in Jerusalem reported a twofold increase in mortality rate from cardiovascular-related causes (1, 24). Hypertension, dyslipidemia, insulin resistance, endothelial dysfunction, and vascular impairment have all been diagnosed in women with a history of PE as early as months, and even years, after pregnancy (33). It is unclear whether PE is the primary insult in these otherwise healthy women or if PE exacerbated an underlying pathology. Regardless of the trigger for subsequent cardiovascular disease, these women have to be monitored closely for the rest of their lives.
CONSEQUENCES OF PE ON OFFSPRING
While PE presents with the signs of maternal hypertension and proteinuria, there are significant consequences on the offspring from PE pregnancies including perinatal, neonatal, and infant morbidity/mortality (2). Often the only means to halt the progression of PE is delivery of the placenta and baby, which is often preterm. In fact PE is responsible for 15–20% of preterm births per year (24). Prematurity often leads to neonatal or infant morbidity and mortality as the baby is not yet developmentally competent to live outside the uterus. One of the most serious outcomes of preterm birth is respiratory complications (2). PE is also one of the leading causes of fetal growth restriction (FGR) with ∼12–25% of growth restricted and small-for-gestational age (SGA) babies attributed to PE (24). Fetal growth is a useful marker for in utero fetal well-being, and when intrauterine FGR is present the risk of perinatal mortality rises (2). Birth weight that is below the 10th percentile, as is the case with SGA babies, has been linked to increased neonatal and infant mortality (2). Sadly, PE is also a risk factor for fetal demise and stillbirth at an estimated rate of 21 per 1,000 (2). In cases of PE where the offspring are born and survive infancy, children still have an increased risk of stroke, coronary heart disease, and metabolic syndrome in adult life (54). One phenotypic modification that has been associated with FGR is reduced nephron number, which has been linked to development of hypertension later in life (21).
Thus, PE itself represents a significant risk factor for the development of cardiovascular disease in mothers affected as well as the babies born to them. There is a strong genetic component as females born to PE-affected mothers have a greater chance of developing PE during their own pregnancies (2). The epidemic-like nature of PE and its associated diseases warrant intense research efforts into the pathophysiology of this disorder and how it exerts its long-term effects.
HISTORICAL PERSPECTIVES ON PE AND ECLAMPSIA
Interestingly, the maternal signs of PE/eclampsia have been observed and described for centuries. In ancient times, Hippocrates recorded the concept of eclampsia as “headache accompanied by heaviness and convulsions during pregnancy” (2). Unfortunately any advances in understanding PE/eclampsia were halted with the rise of Christianity and opposition to scientific and medical discovery during the Middle Ages (3). Therefore, PE/eclampsia was not classified as a disorder of pregnancy until the Renaissance period (2). During this time anatomists and artists accurately described and depicted the female reproductive tract. It is Fallopius who is credited with naming the ovaries, uterine tubes (Fallopian tubes), and the placenta (3). In the 17th century, men such as Francois Mauriceau entered the field of obstetrics and established it as a specialty (3). It is Francois Mauriceau who is credited with the first systemic description of eclampsia and the idea that primigravidas are at a greater risk for convulsions than multigravidas. By the end of the Renaissance period, convulsions of pregnancy were labeled more uniquely different than epilepsy from other causes (i.e., the head, stomach, and chilled extremities), and the word “eclampsia” first appeared in Varandaeus' treatise on gynecology (3). Nineteenth-century physicians such as Demanet, John Lever, Robert Johns, Vaquez, and Nobecourt made observations of the premonitory symptoms of the eclamptic convulsions: edema, proteinuria, headaches, and hypertension, respectively (3). Finally, the concept of PE was widely recognized. The “preeclamptic” state began appearing in textbooks in 1903 (3). Over the last 50 yr, PE has been placed in several different categories of pregnancy disorders. First, it was considered a toxemia of pregnancy and was often referred to as “pregnancy toxemia” (3). After that it was categorized as a hypertensive disorder of pregnancy, and its placement in this category was later refined as pregnancy-induced hypertension and specific criteria were defined to distinguish mild-moderate from severe PE (3). Although there are some discrepancies in the field regarding the criteria, any evidence of hypertension during pregnancy is treated as emergent. In this regard, there is a need for uniformity of diagnostic criteria and early biomarkers of PE are essential to manage these women with the goal of halting the maternal hypertension as well as preventing the need for preterm delivery.
PE, A DISEASE OF THEORIES
Given the longevity of PE in the medical literature and the mortality associated with it, PE has received significant attention as understanding its pathogenesis is crucial. The signs of PE resolve after delivery, and thus pathologies with the placenta may hold the key to unraveling this complex syndrome. Placentae from cases of severe PE where the baby is delivered preterm are small, whereas placental weights from late-gestation PE are variable (5). However, certain placental microscopic pathologies are hallmarks of PE (5). Several theories exist to describe the possible link between placental disease and development of the maternal syndrome.
A two-stage disease model has been proposed to describe the pathogenesis of PE (5). The first stage involves abnormal placentation characterized by poor trophoblast invasion, incomplete vascular remodeling of spiral arteries, and placental hypoxia. The second stage is manifested as the maternal syndrome of hypertension and proteinuria with systemic endothelial dysfunction. The transition between the two stages is thought to be due to the release of factors from the abnormally developed placenta into the maternal circulation (48). Recent evidence points toward an imbalance of pro- and antiangiogenic factors (52), as well as an immune/inflammatory imbalance in the pathophysiology of PE (20). However, the cause and onset of these mechanisms are unknown.
ANGIOGENIC FACTORS IN PE
In early pregnancy, trophoblast cells plug arterioles to protect the conceptus from excessively high oxygen levels, and thus placenta formation begins in a hypoxic state (30). The redox-sensitive transcription factor, hypoxia-inducible factor (HIF)-1α, plays a significant role in this process (31). Once the intra-arterial plugs are removed, oxygenated blood perfuses the newly formed placenta. Prolonged hypoxia can have damaging effects, including induction of apoptosis, oxidative stress, and expression of antiangiogenic factors, and this is hypothesized to occur in PE (51). In fact, placental explants subjected to hypoxic conditions secrete the antiangiogenic factor, soluble fms-like tyrosine kinase 1 (sFlt) (31, 36, 38). Some PE patients show increased levels of circulating sFlt early in pregnancy, and this is thought to induce a state of angiogenic imbalance by binding to the proangiogenic factors, vascular endothelial growth factor (VEGF) and placental growth factor (PGF) (32). Increased sFlt is not observed in all PE patients; therefore, studies are ongoing to determine the origins of increased antiangiogenic factors in PE and the apparent lack of proangiogenic factors. Inadequate uterine angiogenesis/vascularity at the time of implantation has been suggested (53). However, early pregnancy events, such as implantation, cannot be thoroughly investigated in human pregnancies.
IMMUNOREGULATORY MECHANISMS DURING PE
One of the most remarkable features of pregnancy is maternal tolerance of the semiallogeneic fetus (12, 34). The uterine environment undergoes significant physiological adaptations to ensure survival of the developing conceptus. Embryo-uterine interactions trigger the recruitment and activation of key immune cells, such as decidual natural killer (dNK) cells, at the maternal-fetal interface that will facilitate spiral artery transformation and ensure proper placental blood flow (12, 20). Pregnancies characterized by PE have inadequate trophoblast invasion and remodeling of spiral arteries within the decidua, resulting in poor placental perfusion and hypoxia (5). Placental ischemia during PE results in an increase in proinflammatory immune cells and cytokines as well as a decrease in regulatory immune cells and anti-inflammatory cytokines leading to unrestrained inflammation (20).
The most abundant immune cells at the maternal-fetal interface, dNK cells, secrete angiogenic factors, VEGF and PGF, as well as the cytokine interferon-γ (IFN-γ) (12, 35). VEGF and PGF promote angiogenesis beginning in early pregnancy, while dNK cell-derived IFN-γ is thought to directly modify and promote vasodilation of decidual spiral arteries (35). Women with PE are reported to have fewer mature dNK cells (58). Furthermore, an association of impaired chemoattraction of trophoblasts by dNK cells from pregnancies that had high uterine artery Doppler resistance index (RI), an indicator of impaired spiral artery remodeling, has been demonstrated (55). These data hold promise as women with high uterine artery RI have increased risk of developing PE (55). Therefore, dNK cells likely play a prominent role in the pathogenesis of PE.
In addition to playing a crucial role in placental angiogenesis and vascular remodeling, dNK cells also work at the maternal-fetal interface to dampen local inflammation (14). One proposed mechanism by which dNK cells mediate this is the antagonism of T helper 17 (Th17) cells and the expansion of T regulatory cells (14). Preeclamptic women have been shown to have a decrease in circulatory T regulatory cells and an increase in Th17 cells as well as proinflammatory cytokines, including interleukin (IL)-17, IL-6, and tumor necrosis factor-α (56). This robustly proinflammatory status observed during PE is thought to contribute to the activation of the maternal syndrome via increased trophoblast cell death, increased endothelial activation and dysfunction, increased production of reactive oxygen species (ROS), and amplified responsiveness to components of the renin-angiotensin system (RAS) (20). Intense research effort is focused on investigating perturbations of these immunoregulatory pathways, including the role of inflammatory cells, cytokines, and angiogenic factors involved at the maternal-fetal interface during PE. The rodent models described below are valuable in understanding these critical pregnancy mechanisms.
RODENT MODELS TO INVESTIGATE PE
The initiation of fetoplacental pathologies has been extraordinarily difficult to study in humans because of logistical and ethical challenges associated with examining the first trimester. Therefore, animal models of PE are critically important for longitudinal investigation of early events that are essential for healthy pregnancy outcomes (Fig. 2). Despite some differences in pregnancy physiology, rodents have been widely used for studying human pregnancy disorders, such as PE. Some rodent models of PE involve surgical or pharmacological induction of the maternal syndrome at mid- to late gestation (18, 32), thus making it impossible to study key early pregnancy events that drive placenta formation. Recently, several transgenic mouse strains have been developed that phenocopy the maternal syndrome along with other key features of PE (10, 37). These mice allow the opportunity to investigate early pregnancy events in the context of PE; however, due to the heterogeneity of the disease presentation it is unlikely that a single gene is responsible for its development.
Fig. 2.
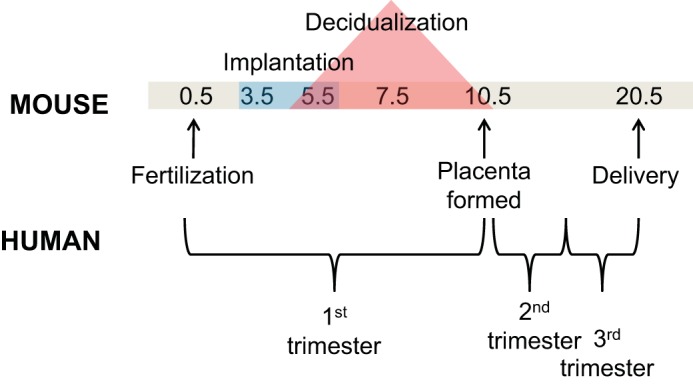
Timeline of mouse gestation and analogous trimester demarcations in human pregnancy. Mice have a relatively short length of gestation (∼20 days) compared with humans at 9 mo. Important 1st-trimester early pregnancy milestones leading up to placenta formation make up half of the gestation length in mice. A thorough understanding of the significance of each day in mouse gestation leading up to embryonic day 10.5 is crucial to translating these findings to the human PE disease process.
Rat Model of Placental Ischemia, Reduced Uterine Perfusion Pressure
Remodeling of spiral arteries during early pregnancy is essential to deliver adequate blood flow to the developing fetoplacental unit. In the case of PE, evidence points toward incomplete remodeling of spiral arteries by invasive trophoblast cells and thus inadequate uteroplacental blood flow to the placenta and fetus (5). To mimic reduced uteroplacental perfusion in vivo, Crews and colleagues (6) developed the reduced uterine perfusion pressure (RUPP) rat model of PE. At embryonic day (e)14.5, silver clips are placed around the aorta proximal to the iliac bifurcation and also around the right and left uterine arcade to prevent compensatory blood flow to the placenta. Blood flow to the gravid uterus is reduced by 40% (28). This surgical intervention results in late gestational (∼e18.5) hypertension and proteinuria as well as decreased litter sizes and evidence of FGR. In addition to these maternal and fetal consequences of PE, the RUPP rat model also recapitulates the angiogenic imbalance seen in women with PE, including increased antiangiogenic factors sFlt1 and soluble endoglin (sENG) as well as decreased VEGF and PGF (16, 17). Importantly though, these rodents do not have abnormal placentation (28). Therefore, the RUPP rat model provides a useful tool for studying placental ischemia and late gestational outcomes, including potential therapeutic approaches for managing the maternal syndrome, but not necessarily the origins of placental defects.
Adoptive Transfer Model
PE is often described as an inflammatory process with increased expression of proinflammatory cytokines locally in the placenta as well as systemically in the maternal circulation (20). Therefore, animal models that can mimic this scenario specifically during pregnancy are of great value. Zenclussen et al. (60) described the adoptive transfer model of PE, which involved intravenous injection of activated Th1-switched splenocytes on e10.5 and e12.5 of interstrain pregnancy (BALB/c females mated with C57/BL6 males). This induced an abrupt increase in maternal blood pressure and urinary protein excretion during pregnancy that was not seen when adoptive transfer was performed on nonpregnant female mice (60). However, pregnant female mice were already prehypertensive on day 1 of pregnancy (mean systolic BP of 125 mmHg); thus, this model may fall into the category of superimposed PE. Injection of Th1-switched splenocytes provoked unspecific activation of the immune system in female pregnant mice leading to the rapid migration of immune cells to the kidney and maternal-fetal interface (60). Importantly, placental pathologies were observed after adoptive transfer, such as thickening of decidual vessels (60). Further studies showed increased expression of RAS receptors, similar to what is seen in women with PE, in the maternal kidney (44). Although this model supports the critical role of immunoregulatory mechanisms in the development of the maternal PE presentation, it is limited to late gestation investigations as injection of activated Th1-switched splenocytes occurs at time points in mouse gestation after the placenta has been formed (see Fig. 2). Therefore, it could not be utilized to examine the origins of PE but rather possible novel interventions after the diagnosis of PE has been made.
Chronic Arginine Vasopressin Infusion Model
The arginine vasopressin (AVP) pathway has recently been brought forward as a focus for PE investigation. AVP-dependent hypertension is characterized by low circulating renin-angiotensin system activity, which is also seen in preeclamptic women relative to nonpreeclamptic pregnant women (42). Chronic infusion with AVP beginning at the onset of pregnancy (e0.5) until e16.5 was sufficient to induce the PE phenotype in normal healthy C57 female mice. Systolic BP and urinary protein excretion were increased at e15.0/16.0 and e17.0, respectively. This was associated with robust renal glomerular endotheliosis, a pathognomonic finding in PE (42). There were also data to support FGR in AVP-infused pregnancies. Fetal masses measured at late gestation (e18.5) were significantly smaller after chronic AVP infusion (42). This model supports the novel concept that central mediators of hypertension are activated in early pregnancy, and thus it has the potential to be an exciting new tool for elucidating the origins of PE. Chronic AVP infusion during pregnancy warrants further exploration to determine if inhibition of AVP secretion or activation can prevent and/or treat PE.
STOX1-overexpressing Mice
While animal models that mimic the maternal syndrome provide a tool for investigating late gestation pathologies and end-stage treatment strategies, they provide little insight into the origins of PE. Transgenic mouse models that phenocopy the maternal syndrome are emerging; one such is the STOX1-overexpressing model (10). STOX1 has been found to be overexpressed in first-trimester placenta from pregnancies that went on to develop PE (13). The human STOX1 cDNA was used to generate transgenic mice, and hSTOX1 was preferentially overexpressed in the placenta (10). Wild-type (WT) female mice crossed with transgenic males were used to restrict transgene expression to the fetoplacental unit. A steady rise in systolic BP was observed in STOX1-overexpressing pregnancies beginning at e0.5, peaking at delivery, and rapidly returning to baseline within days after parturition. Pregnant females also exhibit increases in urinary albumin/creatinine ratios at mid- and late gestation. In addition to displaying the cardinal features of the PE maternal syndrome, WT female mice carrying fetuses expressing the transgene had elevations in circulating levels of sFlt1 and sEng and significant renal pathologies at e16.5 compared with WT × WT females. As is often seen in human cases of PE, these transgenic mice appeared to suffer from fetal demise, as evident by smaller litter sizes, and structural abnormalities of the placentae. Interestingly, chronic aspirin therapy ameliorated the maternal syndrome in WT female mice carrying fetuses expressing the transgene (10). Because this model does not require intervention to provoke the maternal syndrome, it provides a tool to assess treatment strategies beginning early in pregnancy. However, PE is a multifactorial disease that is unlikely to be monogenic, and this must be considered when making any conclusions from these results.
Matrix Metalloproteinase 9-null Mice
Animal models that mimic the maternal PE syndrome without intervention during pregnancy are critical to understanding the early origins of this disorder. Single gene mutations can be engineered in mice that then develop distinct pregnancy-associated pathologies, such as the matrix metalloproteinase 9 (MMP9)-null mouse that has been shown to phenocopy features of PE and intrauterine growth restriction (IUGR) (37). MMP9-null mice were generated by a functional knockout of the active and zinc-binding domain of MMP9 (11). Homozygous matings between MMP9-null mice resulted in pregnancies with resorptions and poorly developed fetoplacental units at midgestation (e10.5), which was attributed to impaired differentiation of trophoblast cells (37). MMP9 production by trophoblast giant cells is needed for proper trophoblast invasion into the decidua; therefore, it was an important finding that MMP-null implantation sites had inadequate remodeling of the decidua and morphologically abnormal maternal-embryonic connections at e7.5 (37). These seminal results are important in shifting the focus of PE research on the origins of this disease process rather than the late-gestational development of the maternal syndrome, which is believed to occur long after the initiating pathologies. However, MMP9-null mice exhibit increased systolic BP compared with heterozygotes at pre-, early, mid- and late gestation without a significant rise from prepregnancy to any stage of pregnancy (37). Furthermore, they have pre-existing renal pathologies including the pathognomic-PE sign of glomerular endotheliosis (37). These two signs may complicate studies where interventions are made early in pregnancy prior to the onset of the maternal syndrome. Interestingly, increased maternal BP early in pregnancy has been linked to lower birth-weight and SGA babies in women with PE (29), so this model may be promising for investigating not just PE but also FGR.
Mice Deficient for IDO
Transgenic mouse models are widely used to study human disease states by systemically altering one gene at a time. Recently, experts in the field have engineered indoleamine 2, 3-dioxygenase (IDO-KO) mice to study PE (41). Placentae from women with PE have demonstrated reduced IDO mRNA and activity (26). IDO has been shown to be expressed at the maternal-fetal in early pregnancy in mice and may be essential for mediating essential T cell functions (50) at the level of the placenta. Together, this led the authors to hypothesize that IDO deficiency may play a role in the pathogenesis of PE. To test their hypothesis, they utilized Cre-Lox recombination technology to generate IDO-KO mice by deleting the majority of the IDO coding region in C57/Bl6 mice (41). Importantly, while IDO deletion was confirmed in a number of tissues, it was not reported to be deleted in the placenta. Thus, pregnant IDO-KO mice were shown to “partially phenocopy PE” with proteinuria and pathognomonic glomerular endotheliosis at gestational day (GD) 17 and 18, respectively, but without maternal hypertension as measured by systolic BP recordings (41). However, pregnant IDO-KO mice do exhibit pregnancy-specific endothelial dysfunction and IUGR. Histological analyses of the placenta were comparable between IDO-KO and control C57Bl/6 mice. This mouse model highlights the heterogeneity of PE disease presentation and the observation that this pregnancy-specific disorder is unlikely to be monogenic. Targeted gene deletion prior to pregnancy was hypothesized by the authors to trigger chronic compensatory mechanisms that would prevent the fulminant PE presentation. Introducing relevant environmental stressors, such as high-fat diet, during pregnancy in a single gene transgenic model, e.g., the IDO-KO mouse, could be very useful in elucidating the mechanisms underlying PE if the partial PE model would become complete.
BPH/5 Mouse Model of PE
Our laboratory discovered the first strain of mice to spontaneously recapitulate the maternal syndrome of PE, BPH/5 (8). These mice were one of several mouse strains developed at the University of Kansas by the geneticist Dr. Gunther Schlager (43). Hypotensive and hypertensive mice were generated by inbreeding eight different strains of normotensive mice and selecting for low and high BP over 23 generations. This resulted in two new and distinct genetically inbred strains of mice, BPL (Blood Pressure Low)/1 and BPH (Blood Pressure High)/2. Several sublines of BPH/2 were further developed with varying intermediate blood pressures. BPH/5 mice exhibited a mildly elevated baseline BP profile, and because this is a known risk factor in the development of PE in women, Davisson et al. (8) hypothesized that BPH/5 mice would develop the maternal syndrome of PE during gestation.
Discovery of a novel spontaneous mouse model of PE, BPH/5.
BPH/5 mice are prehypertensive, without signs of renal disease, prior to pregnancy (8). Beginning at e14.0, pregnant BPH/5 mice display a significant rise in mean arterial pressure that returns to baseline after delivery of the pups and placentae. These mice also exhibit late-gestational proteinuria with renal histopathological findings consistent with glomerulosclerosis (8). Similar to PE in people that experience perinatal morbidity/mortality, BPH/5 mice also have small litter sizes due to in utero fetal demise at midgestation. Pups that are born live have low birth weight compared with C57 control mice, indicative of FGR (8). Another feature of the maternal PE syndrome is widespread endothelial dysfunction (40). Importantly, endothelium-intact mesenteric arteries taken from BPH/5 pregnant females at e19.5 showed diminished relaxation in response to acetylcholine, suggestive of systemic endothelial dysfunction (8). Because BPH/5 females are chronically prehypertensive before pregnancy, they may more accurately be described as a model of superimposed PE. However, superimposed PE, with worsening of pre-existing hypertension, is associated with the same adverse maternal and fetal outcomes as classical PE (4). Thus the BPH/5 mouse emerged as a novel spontaneous genetic model with a PE-like phenotype that recapitulates-key pathophysiological findings seen in women with PE and their offspring.
Fetoplacental abnormalities in the BPH/5 mouse model.
It is widely accepted that the placenta plays a causal role in the pathogenesis of PE (7). In addition to the cardinal features of PE, BPH/5 mice also develop placental pathologies that precede the maternal syndrome (9). Between e9.5 and e14.5, BPH/5 pregnant mice have a 40–50% reduction in placental mass compared with C57 placenta (9). Also, fetal weight is significantly decreased in BPH/5 pregnant females at e12.5, e14.5, and e18.5 vs. C57 (9). Therefore, the lower-birth-weight pups first documented by Davisson et al. (8) are a result of in utero FGR. Histological analyses of the BPH/5 placentae at e10.5 revealed inadequately remodeled decidual vessels with thickened walls that retained actin-positive cells and thus narrowed lumens compared with C57 decidual vessels (9). Placentae at e12.5 show decreased depth of trophoblast invasion into the maternal decidua and a decrease in the fractional area occupied by the junctional zone (9). Both these findings are indicative of impaired endovascular invasion and interstitial invasion, respectively. These two pathologies of the BPH/5 placenta would suggest reduced placental perfusion, and indeed this was reflected with Doppler ultrasound (9). Uterine arteries from BPH/5 mothers at e16.5 had elevated pulsatility index, which indicates placental vascular insufficiency and impairment of the maternal-fetal circulation compared with C57 (9). Furthermore, the labyrinth region in BPH/5 placentae at e12.5 had attenuated and irregular branching morphogenesis and reduced expansion (9). Therefore, defects in both the fetal and maternal compartments characterize the BPH/5 placenta, and this occurs in pregnancy before the onset of the maternal syndrome.
BPH/5 mice have oxidative stress and an angiogenic imbalance during pregnancy.
BPH/5 mice demonstrate the cardinal signs of the maternal PE syndrome (8), as well as placental abnormalities identified in women with PE (8, 9). After this PE-like phenotype was characterized, studies were undertaken to demonstrate a causal role for pathways implicated in the pathogenesis of PE. First, oxidative stress and ROS were investigated during BPH/5 gestation. BPH/5 midgestation placentae have increased levels of superoxide and reduced levels and activity of the antioxidant enzyme superoxide dismutase (SOD) (22). Placental oxidative stress was attenuated by chronic Tempol (SOD mimetic) treatment, and this was associated with a decrease in BPH/5 resorptions, an increase in BPH/5 litter size, and normalization of BPH/5 placental and fetal weights (22). Furthermore, Tempol treatment ameliorated the maternal syndrome of late-gestational hypertension and proteinuria (22). These findings provide evidence for a causal role of oxidative stress in the pathogenesis of the maternal PE syndrome as well poor pregnancy outcomes, such as fetal demise and poor fetoplacental development.
Next, angiogenic factors were considered as a potential mechanism behind the maternal syndrome of PE and poor fetoplacental outcomes in this model. BPH/5 females were found to have decreased circulating and placental levels of VEGF protein at early and midgestation (59). Another proangiogenic factor, PGF, was measured and found to be decreased in the circulation and in placental mRNA abundance at early gestation (59). However, this was not associated with an increase in the antiangiogenic factor sFlt but, rather, a decrease in plasma and placental mRNA (59). Delivery of an adenovirus encoding VEGF121 (Ad-VEGF) early in gestation at e7.5 blunted the maternal syndrome in BPH/5 females while also decreasing midgestation resorptions and improving litter size at term vs. C57 (59). Interestingly, serum collected at e12.5 from BPH/5 Ad-VEGF-treated mothers showed a marked improvement in angiogenic potential as measured by the endothelial tube formation assay (59). These data highlight the importance of angiogenic factors in the BPH/5 mouse model and support the hypothesis that VEGF plays a key role in the maternal PE syndrome as well as fetal outcomes. Moreover, delivery of Ad-VEGF early in pregnancy provides strong rationale for investigating early pregnancy events and their relationship to downstream pregnancy outcomes in BPH/5.
Abnormalities during the peri-implantation period in the BPH/5 mouse model.
The spontaneous BPH/5 mouse model provides us with the opportunity to interrogate early pregnancy events prior to the onset of the maternal syndrome. Thus we are able to assess any stage of pregnancy and probe for dysregulated events that may have ripple effects that promote poor pregnancy outcomes. In this model, we have recently identified key defective embryo-uterine interactions during the peri-implantation period that were preceded by aberrant ovarian hormone signaling (47). Abnormal peri-implantation immunoregulatory events, including natural killer cell activation at the maternal-fetal interface, have also been described in BPH/5 mice (46). We have recently published data supporting the central role of Cox2 and IL-15 overexpression at the maternal-fetal interface and adverse pregnancy outcomes in BPH/5 mice, including maternal hypertension and poor fetoplacental development (45). Ongoing studies in the BPH/5 model include restoration of critical peri-implantation signaling events in early pregnancy and amelioration of adverse pregnancy outcomes.
SUMMARY
PE is a widely recognized multifactorial disorder of pregnancy. While the diagnosis of PE is made by the clinical presentation of the maternal syndrome, the origins of PE occur much earlier in pregnancy. Even though this devastating disorder of pregnancy has been described for thousands of years, the mechanisms involved in PE still baffle clinical and basic scientists alike. Placenta formation occurs in the first trimester of pregnancy, so this very critical time point in gestation may hold the key to the dysregulated events that lead to PE. The use of certain animal models affords researchers the opportunity to investigate key early pregnancy events, such as uterine angiogenesis during implantation and decidualization that may contribute to the development of PE. No one single animal model is perfect for studying PE, but through the use of unique rodent models that utilize surgical, pharmacological, and genetic manipulations, significant strides are being made in the field to understand the mechanisms that regulate this tragic disease of pregnancy and beyond.
GRANTS
National Institutes of Health Grants T32OD-011000 (J. L. Sones), R01 HL-063887 (R. L. Davisson), and HL-084207 (R. L. Davisson); American Heart Association Grant 12POST11250010 (J. L. Sones); and March of Dimes Grant 6-FY14-411 (R. L. Davisson).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
J.L.S. and R.L.D. conception and design of research; J.L.S. and R.L.D. prepared figures; J.L.S. drafted manuscript; J.L.S. and R.L.D. edited and revised manuscript; J.L.S. and R.L.D. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Vera Rinaldi for illustration expertise.
REFERENCES
- 1.Al-Jameil N, Aziz Khan F, Fareed Khan M, Tabassum H. A brief overview of preeclampsia. J Clin Med Res 6: 1–7, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Backes CH, Markham K, Moorehead P, Cordero L, Nankervis CA, Giannone PJ. Maternal preeclampsia and neonatal outcomes. J Pregnancy 2011: 214365, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bell MJ. A historical overview of preeclampsia-eclampsia. J Obstet Gynecol Neonatal Nurs 39: 510–518, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bramham K, Seed PT, Lightstone L, Nelson-Piercy C, Gill C, Webster P, Poston L, Chappell LC. Diagnostic and predictive biomarkers for pre-eclampsia in patients with established hypertension and chronic kidney disease. Kidney Int 89: 874–885, 2016. [DOI] [PubMed] [Google Scholar]
- 5.Cheng MH, Wang PH. Placentation abnormalities in the pathophysiology of preeclampsia. Expert Rev Mol Diagn 9: 37–49, 2009. [DOI] [PubMed] [Google Scholar]
- 6.Crews JK, Herrington JN, Granger JP, Khalil RA. Decreased endothelium-dependent vascular relaxation during reduction of uterine perfusion pressure in pregnant rat. Hypertension 35: 367–372, 2000. [DOI] [PubMed] [Google Scholar]
- 7.Cross JC. The genetics of pre-eclampsia: a feto-placental or maternal problem? Clin Genet 64: 96–103, 2003. [DOI] [PubMed] [Google Scholar]
- 8.Davisson RL, Hoffmann DS, Butz GM, Aldape G, Schlager G, Merrill DC, Sethi S, Weiss RM, Bates JN. Discovery of a spontaneous genetic mouse model of preeclampsia. Hypertension 39: 337–342, 2002. [DOI] [PubMed] [Google Scholar]
- 9.Dokras A, Hoffmann DS, Eastvold JS, Kienzle MF, Gruman LM, Kirby PA, Weiss RM, Davisson RL. Severe feto-placental abnormalities precede the onset of hypertension and proteinuria in a mouse model of preeclampsia. Biol Reprod 75: 899–907, 2006. [DOI] [PubMed] [Google Scholar]
- 10.Doridot L, Passet B, Mehats C, Rigourd V, Barbaux S, Ducat A, Mondon F, Vilotte M, Castille J, Breuiller-Fouche M, Daniel N, le Provost F, Bauchet AL, Baudrie V, Hertig A, Buffat C, Simeoni U, Germain G, Vilotte JL, Vaiman D. Preeclampsia-like symptoms induced in mice by fetoplacental expression of STOX1 are reversed by aspirin treatment. Hypertension 61: 662–668, 2013. [DOI] [PubMed] [Google Scholar]
- 11.Dubois B, Arnold B, Opdenakker G. Gelatinase B deficiency impairs reproduction. J Clin Invest 106: 627–628, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Erlebacher A. Immunology of the maternal-fetal interface. Annu Rev Immunol 31: 387–411, 2013. [DOI] [PubMed] [Google Scholar]
- 13.Founds SA, Conley YP, Lyons-Weiler JF, Jeyabalan A, Hogge WA, Conrad KP. Altered global gene expression in first trimester placentas of women destined to develop preeclampsia. Placenta 30: 15–24, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fu B, Li X, Sun R, Tong X, Ling B, Tian Z, Wei H. Natural killer cells promote immune tolerance by regulating inflammatory TH17 cells at the human maternal-fetal interface. Proc Natl Acad Sci USA 110: E231–E240, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Funai EF, Paltiel OB, Malaspina D, Friedlander Y, Deutsch L, Harlap S. Risk factors for pre-eclampsia in nulliparous and parous women: the Jerusalem perinatal study. Paediatr Perinat Epidemiol 19: 59–68, 2005. [DOI] [PubMed] [Google Scholar]
- 16.Gilbert JS, Babcock SA, Granger JP. Hypertension produced by reduced uterine perfusion in pregnant rats is associated with increased soluble fms-like tyrosine kinase-1 expression. Hypertension 50: 1142–1147, 2007. [DOI] [PubMed] [Google Scholar]
- 17.Gilbert JS, Gilbert SA, Arany M, Granger JP. Hypertension produced by placental ischemia in pregnant rats is associated with increased soluble endoglin expression. Hypertension 53: 399–403, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Granger JP, LaMarca BB, Cockrell K, Sedeek M, Balzi C, Chandler D, Bennett W. Reduced uterine perfusion pressure (RUPP) model for studying cardiovascular-renal dysfunction in response to placental ischemia. Methods Mol Med 122: 383–392, 2006. [DOI] [PubMed] [Google Scholar]
- 19.Grill S, Rusterholz C, Zanetti-Dallenbach R, Tercanli S, Holzgreve W, Hahn S, Lapaire O. Potential markers of preeclampsia–a review. Reprod Biol Endocrinol 7: 70, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harmon AC, Cornelius DC, Amaral LM, Faulkner JL, Cunningham MW Jr, Wallace K, LaMarca B. The role of inflammation in the pathology of preeclampsia. Clin Sci (Lond) 130: 409–419, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herrera-Garcia G, Contag S. Maternal preeclampsia and risk for cardiovascular disease in offspring. Curr Hypertens Rep 16: 475, 2014. [DOI] [PubMed] [Google Scholar]
- 22.Hoffmann DS, Weydert CJ, Lazartigues E, Kutschke WJ, Kienzle MF, Leach JE, Sharma JA, Sharma RV, Davisson RL. Chronic tempol prevents hypertension, proteinuria, and poor feto-placental outcomes in BPH/5 mouse model of preeclampsia. Hypertension 51: 1058–1065, 2008. [DOI] [PubMed] [Google Scholar]
- 23.Irgens HU, Reisaeter L, Irgens LM, Lie RT. Long term mortality of mothers and fathers after pre-eclampsia: population based cohort study. BMJ 323: 1213–1217, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jeyabalan A. Epidemiology of preeclampsia: impact of obesity. Nutr Rev 71, Suppl 1: S18–S25, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jeyabalan A, Powers RW, Durica AR, Harger GF, Roberts JM, Ness RB. Cigarette smoke exposure and angiogenic factors in pregnancy and preeclampsia. Am J Hypertens 21: 943–947, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kudo Y, Boyd CA, Sargent IL, Redman CW. Decreased tryptophan catabolism by placental indoleamine 2,3-dioxygenase in preeclampsia. Am J Obstet Gynecol 188: 719–726, 2003. [DOI] [PubMed] [Google Scholar]
- 27.Leeman L, Dresang LT, Fontaine P. Hypertensive disorders of pregnancy. Am Fam Physician 93: 121–127, 2016. [PubMed] [Google Scholar]
- 28.Li J, LaMarca B, Reckelhoff JF. A model of preeclampsia in rats: the reduced uterine perfusion pressure (RUPP) model. Am J Physiol Heart Circ Physiol 303: H1–H8, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Macdonald-Wallis C, Tilling K, Fraser A, Nelson SM, Lawlor DA. Associations of blood pressure change in pregnancy with fetal growth and gestational age at delivery: findings from a prospective cohort. Hypertension 64: 36–44, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Malassine A, Frendo JL, Evain-Brion D. A comparison of placental development and endocrine functions between the human and mouse model. Hum Reprod Update 9: 531–539, 2003. [DOI] [PubMed] [Google Scholar]
- 31.Maltepe E, Krampitz GW, Okazaki KM, Red-Horse K, Mak W, Simon MC, Fisher SJ. Hypoxia-inducible factor-dependent histone deacetylase activity determines stem cell fate in the placenta. Development 132: 3393–3403, 2005. [DOI] [PubMed] [Google Scholar]
- 32.Maynard SE, Min JY, Merchan J, Lim KH, Li J, Mondal S, Libermann TA, Morgan JP, Sellke FW, Stillman IE, Epstein FH, Sukhatme VP, Karumanchi SA. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest 111: 649–658, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McDonald SD, Malinowski A, Zhou Q, Yusuf S, Devereaux PJ. Cardiovascular sequelae of preeclampsia/eclampsia: a systematic review and meta-analyses. Am Heart J 156: 918–930, 2008. [DOI] [PubMed] [Google Scholar]
- 34.Moffett A, Loke C. Immunology of placentation in eutherian mammals. Nat Rev Immunol 6: 584–594, 2006. [DOI] [PubMed] [Google Scholar]
- 35.Murphy SP, Tayade C, Ashkar AA, Hatta K, Zhang J, Croy BA. Interferon gamma in successful pregnancies. Biol Reprod 80: 848–859, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nevo O, Soleymanlou N, Wu Y, Xu J, Kingdom J, Many A, Zamudio S, Caniggia I. Increased expression of sFlt-1 in in vivo and in vitro models of human placental hypoxia is mediated by HIF-1. Am J Physiol Regul Integr Comp Physiol 291: R1085–R1093, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Plaks V, Rinkenberger J, Dai J, Flannery M, Sund M, Kanasaki K, Ni W, Kalluri R, Werb Z. Matrix metalloproteinase-9 deficiency phenocopies features of preeclampsia and intrauterine growth restriction. Proc Natl Acad Sci USA 110: 11109–11114, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Redman CW, Sargent IL. Placental stress and pre-eclampsia: a revised view. Placenta 30, Suppl A: S38–S42, 2009. [DOI] [PubMed] [Google Scholar]
- 39.Roberts JM, Gammill HS. Preeclampsia: recent insights. Hypertension 46: 1243–1249, 2005. [DOI] [PubMed] [Google Scholar]
- 40.Roberts JM, Pearson GD, Cutler JA, Lindheimer MD, National Heart Lung, and Blood Institute. Summary of the NHLBI Working Group on Research on Hypertension During Pregnancy. Hypertens Pregnancy 22: 109–127, 2003. [DOI] [PubMed] [Google Scholar]
- 41.Santillan MK, Pelham CJ, Ketsawatsomkron P, Santillan DA, Davis DR, Devor EJ, Gibson-Corley KN, Scroggins SM, Grobe JL, Yang B, Hunter SK, Sigmund CD. Pregnant mice lacking indoleamine 2,3-dioxygenase exhibit preeclampsia phenotypes. Physiol Rep 3: e12257, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Santillan MK, Santillan DA, Scroggins SM, Min JY, Sandgren JA, Pearson NA, Leslie KK, Hunter SK, Zamba GK, Gibson-Corley KN, Grobe JL. Vasopressin in preeclampsia: a novel very early human pregnancy biomarker and clinically relevant mouse model. Hypertension 64: 852–859, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schlager G, Sides J. Characterization of hypertensive and hypotensive inbred strains of mice. Lab Anim Sci 47: 288–292, 1997. [PubMed] [Google Scholar]
- 44.Schmid M, Sollwedel A, Thuere C, Wafula PO, Zenclussen ML, Muller DN, Gratze P, Woiciechowsky C, Volk HD, Zenclussen AC. Murine pre-eclampsia induced by unspecific activation of the immune system correlates with alterations in the eNOS and AT1 receptor expression in the kidneys and placenta. Placenta 28: 688–700, 2007. [DOI] [PubMed] [Google Scholar]
- 45.Sones JL, Cha J, Woods AK, Bartos A, Heyward CY, Lob HE, Isroff CE, Butler SD, Shapiro SE, Dey SK, Davisson RL. Decidual Cox2 inhibition improves fetal and maternal outcomes in a preeclampsia-like mouse model. J Clin Invest Insight 1: e75351, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sones JL, Lob HE, Isroff CE, Davisson RL. Role of decidual natural killer cells, interleukin-15, and interferon-gamma in placental development and preeclampsia. Am J Physiol Regul Integr Comp Physiol 307: R490–R492, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sones JL, Zhou Y, Woods AK, Isroff CE, Knudsen MG, Davisson RL. Ovarian hormone signaling defects are associated with asynchronous peri-implantation events in a mouse model of preeclampsia, BPH/5. Reprod Sci Suppl 20: 2013. [Google Scholar]
- 48.Soto E, Romero R, Kusanovic JP, Ogge G, Hussein Y, Yeo L, Hassan SS, Kim CJ, Chaiworapongsa T. Late-onset preeclampsia is associated with an imbalance of angiogenic and anti-angiogenic factors in patients with and without placental lesions consistent with maternal underperfusion. J Matern Fetal Neonatal Med 25: 498–507, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Steegers EA, von Dadelszen P, Duvekot JJ, Pijnenborg R. Pre-eclampsia. Lancet 376: 631–644, 2010. [DOI] [PubMed] [Google Scholar]
- 50.Suzuki S, Tone S, Takikawa O, Kubo T, Kohno I, Minatogawa Y. Expression of indoleamine 2,3-dioxygenase and tryptophan 2,3-dioxygenase in early concepti. Biochem J 355: 425–429, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tal R. The role of hypoxia and hypoxia-inducible factor-1alpha in preeclampsia pathogenesis. Biol Reprod 87: 134, 2012. [DOI] [PubMed] [Google Scholar]
- 52.Thadhani R, Mutter WP, Wolf M, Levine RJ, Taylor RN, Sukhatme VP, Ecker J, Karumanchi SA. First trimester placental growth factor and soluble fms-like tyrosine kinase 1 and risk for preeclampsia. J Clin Endocrinol Metab 89: 770–775, 2004. [DOI] [PubMed] [Google Scholar]
- 53.Torry DS, Hinrichs M, Torry RJ. Determinants of placental vascularity. Am J Reprod Immunol 51: 257–268, 2004. [DOI] [PubMed] [Google Scholar]
- 54.Uzan J, Carbonnel M, Piconne O, Asmar R, Ayoubi JM. Pre-eclampsia: pathophysiology, diagnosis, management. Vasc Health Risk Manag 7: 467–474, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wallace AE, Host AJ, Whitley GS, Cartwright JE. Decidual natural killer cell interactions with trophoblasts are impaired in pregnancies at increased risk of preeclampsia. Am J Pathol 183: 1853–1861, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wallace K, Richards S, Dhillon P, Weimer A, Edholm ES, Bengten E, Wilson M, Martin JN Jr, LaMarca B. CD4+ T-helper cells stimulated in response to placental ischemia mediate hypertension during pregnancy. Hypertension 57: 949–955, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Whitehead N. Biomedicine. Proteins and a pregnancy woe. Science 345: 249, 2014. [DOI] [PubMed] [Google Scholar]
- 58.Williams PJ, Bulmer JN, Searle RF, Innes BA, Robson SC. Altered decidual leucocyte populations in the placental bed in pre-eclampsia and foetal growth restriction: a comparison with late normal pregnancy. Reproduction 138: 177–184, 2009. [DOI] [PubMed] [Google Scholar]
- 59.Woods AK, Hoffmann DS, Weydert CJ, Butler SD, Zhou Y, Sharma RV, Davisson RL. Adenoviral delivery of VEGF121 early in pregnancy prevents spontaneous development of preeclampsia in BPH/5 mice. Hypertension 57: 94–102, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zenclussen AC, Fest S, Joachim R, Klapp BF, Arck PC. Introducing a mouse model for pre-eclampsia: adoptive transfer of activated Th1 cells leads to pre-eclampsia-like symptoms exclusively in pregnant mice. Eur J Immunol 34: 377–387, 2004. [DOI] [PubMed] [Google Scholar]