

Abstract
Nonalcoholic fatty liver disease (NAFLD) and its more advanced form nonalcoholic steatohepatitis (NASH) are the most common chronic liver diseases in developed countries. Moreover, NAFLD and NASH are considerable risk factors for atherosclerosis, the most frequent vascular pathology in these and other metabolic diseases. Despite this strong connection, current knowledge of the relationship between NAFLD/NASH and atherosclerosis is scarce. Recently, we studied hyperlipidemic Apoe knockout mice with endothelial-specific gain of transient receptor potential canonical 3 channel function (TgESTRPC3/ApoeKO) and found that these animals had increased burden of advanced aortic atherosclerosis (16 wk on high-fat diet) compared with nontransgenic ApoeKO littermate controls (non-Tg/ApoeKO), whereas early lesions (10 wk on high-fat diet) were not different. Here, we report that at the early stage when differences in aortic atherosclerosis are not yet manifest, the livers of TgESTRPC3/ApoeKO mice show steatosis, fibrosis, and altered hepatic enzymes compared with non-Tg/ApoeKO animals. Because differences in liver pathology were noticeable long before differences in atherosclerosis were evident, our studies suggest that TRPC3-related endothelial mechanisms that promote steatohepatitis may also contribute to atherosclerosis progression. In vitro, downregulation of TRPC3 in liver sinusoid endothelial cells reduces their susceptibility to endoplasmic reticulum stress-induced apoptosis, suggesting that a proapoptotic effect of TRPC3 may add to other fibrogenic factors in vivo. These novel findings show a positive association between augmented expression of an endothelial TRPC channel, development of early steatohepatitis, and atherosclerotic burden in a hyperlipidemic mouse model of NAFLD fed conventional Western-type diet.
Keywords: TRPC channels, NAFLD, NASH, atherosclerosis, endothelium
nonalcoholic fatty liver disease (NAFLD) has an extensive histological spectrum ranging from simple fat accumulation in liver (steatosis) to nonalcoholic steatohepatitis (NASH), the latter being characterized by liver inflammation, presence of apoptotic cells, and fibrosis (1). NAFLD is the most common chronic liver disease in adults and children of developed countries (2). About 30% of individuals in Western societies have NAFLD, and it is estimated that 20% of them will progress to NASH (12). NAFLD and NASH also contribute to development and/or progression of atherosclerosis, the main cause of coronary artery disease and most frequent vascular complication in these and other metabolic disorders. Likewise, atherosclerosis is a major determinant of mortality in NAFLD/NASH patients (12). In individuals with NAFLD, progression of liver inflammation and fibrosis correlates positively with plaque burden, independently of cardiometabolic risk factors (6). Despite the strong association between NAFLD/NASH and atherosclerosis, how these two diseases interplay remains poorly understood, to a great extent due to lack of animal models that simultaneously develop the liver and the vascular pathologies.
Transient receptor potential canonical 3 (TRPC3) is a nonselective Ca2+-permeable channel that belongs to the TRPC family (TRPC1-7) of cation channels (15). TRPC3 has important functions in endothelium and participates in numerous events associated to cardiovascular physiology and disease (15). TRPC3 is activated downstream of receptor-stimulated phospholipases and also exhibits significant constitutive function (15). Understanding the roles of TRPC3 in metabolic and cardiovascular diseases requires the identification of cellular and/or molecular events that are affected by TRPC3 function. Existing evidence on potential roles of TRPC3 in regulating metabolic processes is only of an associative nature, and direct causative roles have not been demonstrated (22). There is, however, compelling in vitro and in vivo work demonstrating functions of TRPC3 in cardiovascular pathologies, many of which are often complications of metabolic diseases (15). In a recent study using hyperlipidemic Apoe knockout (ApoeKO) mice with endothelial-specific gain of TRPC3 function (TgESTRPC3/ApoeKO) we found that advanced aortic atherosclerotic plaques (after 16 wk on high-fat diet, “HFD”) in these mice were of bigger size and cellularity compared with nontransgenic ApoeKO littermates (non-Tg/ApoeKO; Ref. 17). Early atherosclerotic lesions (10 wk on HFD) were not different between the two groups of animals, but notably, at this early stage the endothelium of TgESTRPC3/ApoeKO mice already showed augmented inflammation and signs of increased endoplasmic reticulum (ER) stress compared with non-Tg/ApoeKO animals (17). In the present work we report that at the early stage of atherosclerosis, when differences in aortic plaques are not yet noticeable, the livers of TgESTRPC3/ApoeKO mice show, compared with non-Tg/ApoeKO, significant steatosis, fibrosis, augmented cell apoptosis, and altered liver enzymes, i.e., long before differences in aortic atherosclerosis become evident. This suggests that endothelial TRPC3-dependent effects exist that, directly or indirectly, promote steatohepatitis and may also contribute to atherosclerosis progression. This is also the first evidence of a positive association between augmented expression of an endothelial TRPC channel and development of steatohepatitis in a hyperlipidemic mouse model of NAFLD fed conventional Western-type diet.
MATERIALS AND METHODS
Animals.
All animal studies described in this work conform to the National Institutes of Health Guide for Care and Use of Laboratory Animals and were approved by University of Toledo Institutional Animal Care and Use Committee. Generation and characterization of ApoeKO mice with endothelial-specific overexpression of human TRPC3 (TgESTRPC3/ApoeKO) are described in Ref. 17. Aortic root sectioning and histomorphometric analysis of atherosclerotic lesions are described in detail in Ref. 17.
Measurement of metabolic parameters.
Blood was collected by submandibular vein puncture after a 12 h fasting period. Total plasma cholesterol and triglycerides were determined as in Ref. 17 using commercial kits (Wako Chemicals) following manufacturer's instructions. Nonesterified fatty acids (NEFAs) were determined as described in Ref. 11.
Liver histology and function.
Hepatic triglycerides and cholesterol were determined as in Ref. 11. Plasma alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were evaluated with commercial kits (Abcam) following manufacturer's instructions. Wet liver weight was determined immediately after death. Livers were embedded in optimal cutting temperature compound and frozen in the Peltier stage of the cryostat (Thermo-Scientific R.Allan-HM550), and sections (10 μm) were collected onto Fisher Superfrost-Plus-coated slides. Histological features were examined by hematoxylin-eosin (H&E) staining as in Ref. 17. Steatosis and inflammation were graded by two operators blinded to the study, using the 0–3 scale (3 = highest; NASH scoring system, NIDDK-NASH Clinical-Research-Network; Ref. 5). Alternatively, images of oil-red O (ORO)-stained sections were captured with a digital camera (Micropublisher 3.3-Megapixel Cooled-CCD Color-Digital-Camera coupled to Zeiss-Axiovert40CL inverted microscope), and ORO-stained areas were measured with image analysis software (NIS-Elements-D). The inflammatory genes TNF-α, IL-1β, and IL-6 were evaluated by quantitative (q)RT-PCR using protocols and primers described in Ref. 18. Presence of apoptotic cells was determined by in situ terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) as in Ref. 17.
Liver fibrosis.
Liver sections were stained for collagen deposition (Masson's trichrome), and the extent of fibrosis estimated from relative area of collagen-staining respect to liver area, as we described in Ref. 17. Hydroxyproline was determined by photometry in liver hydrolysates as in Ref. 20. mRNA levels of collagen type I (Col1A) and transforming growth factor-β (TGF-β) were determined by qRT-PCR using primers described in Ref. 10.
Isolation and transfection of liver sinusoid endothelial cells.
Liver sinusoid endothelial cells (LSECs) were isolated by collagenase perfusion, iodixanol-density-gradient centrifugation, and centrifugal elutriation as in Ref. 21. Viability was >95%, purity >98% based on uptake of formaldehyde-treated albumin, a function specific to LSECs (4). siRNA oligonucleotides for TRPC3, 6, or 7 (siGENOME-SMART-pool, Dharmacon) were delivered to LSECs with Lipofectamine2000 (Invitrogen), 100 nM final concentration (as in Ref. 16), and cells were used 48 h later. Downregulation of TPRC3, 6, and 7 was confirmed by qRT-PCR and Western blot as in Ref. 16.
Statistical analysis.
Values were compared by a two-tailed t-test for two means (Graph Pad Software, San Diego, CA). P values < 0.05 were considered significant.
RESULTS
In a recent study using ApoeKO mice with endothelial-specific gain of TRPC3 function (TgESTRPC3/ApoeKO) we observed that whereas early atherosclerotic lesions (10 wk on HFD) were not different between transgenic and nontransgenic mice (17), at this early stage the endothelium of TgESTRPC3/ApoeKO mice showed more inflammation and increased ER stress compared with non-Tg/ApoeKO animals (17). Importantly, these changes occurred long before differences in aortic atherosclerotic burden were evident between TgESTRPC3/ApoeKO and non-Tg/ApoeKO mice, which occurred after 16 wk on HFD. Based on this and considering that when maintained on HFD ApoeKO mice develop NAFLD (14), we wished to examine whether endothelial gain of TRPC3 had an impact on the liver of TgESTRPC3/ApoeKO animals, and if so, how this related to progression of atherosclerosis burden. Six-week-old female TgESTRPC3/ApoeKO mice or their non-Tg/ApoeKO sex-matched littermates were placed on an HFD (21% fat, 0.2% cholesterol; TD.88137-Harlan Laboratories) for 10 or 16 wk to allow for development, respectively, of early and advanced atherosclerotic lesions. Figure 1 (top panels) shows that after 10 wk on HFD the livers of TgESTRPC3/ApoeKO mice had significant steatosis, as assessed by a NASH scoring system (5) (score: 1.71 ± 0.3 vs. 0.35 ± 0.15, for TgESTRPC3/ApoeKO vs. non-Tg/ApoeKO, respectively; P < 0.01, n = 8) or by quantification of ORO-stained areas (20 ± 3 vs. 8 ± 2% ORO-stained area, for TgESTRPC3/ApoeKO vs. non-Tg/ApoeKO, respectively; P < 0.01, n = 8). As reported by others (13, 14) ApoeKO fed regular chow diet did not develop steatosis, regardless of the TRPC3 expression status (not shown). In agreement with our previous findings (17), after 10 wk on HFD there were no differences in the already hyperlipidemic profile of the two groups of animals (Table 1) or in plaque burden (Fig. 2). In line with the histological observations, hepatic triglyceride content after 10 wk on HFD was higher in transgenic vs. non-Tg/ApoeKO mice (Fig. 1). Hepatic cholesterol (Fig. 1), gain in body weight and circulating NEFAs were similar between the two groups (Table 1). After 16 wk on HFD liver steatosis was slightly augmented (Fig. 1, bottom panels), which coincided with significant progression of atherosclerosis. Similar to our previous observations (17), advanced aortic plaques in TgESTRPC3/ApoeKO mice were larger compared with non-Tg/ApoeKO animals (Fig. 2). After 16 wk on HFD hepatic triglycerides remained higher in transgenic vs. nontransgenic mice (Fig. 1); hepatic cholesterol, although slightly increased compared with levels at 10 wk, was not significantly different between groups (Fig. 1). Gain in body weight and lipid profile were also similar between groups (Table 1). H&E staining revealed scattered inflammatory infiltration (score: 1.38 ± 0.32 vs. 0.51 ± 0.21, for TgESTRPC3/ApoeKO vs. non-Tg/ApoeKO, respectively; P < 0.01, n = 8). Levels of the inflammatory genes TNF-α, IL-1β, and IL-6 were augmented by 2.3-, 3.1-, and 2.8-fold, respectively, in livers from TgESTRPC3/ApoeKO vs. non-Tg/ApoeKO mice (n = 5, P < 0.01).
Fig. 1.

Apoe knockout mice with endothelial-specific gain of transient receptor potential canonical 3 function (TgESTRPC3/ApoeKO) or their nontransgenic littermates (non-Tg/ApoeKO) were maintained on a high-fat diet (HFD) for 10 (top panels) or 16 (bottom panels) wk. At the end of the diet periods mice were killed and liver sections (10 μm) prepared and stained with oil-red O (hematoxylin and eosin counterstaining) to evaluate lipid content. The bar graphs show values for hepatic triglycerides and cholesterol content (means ± SE, n = 8) for these groups of mice after the indicated diet periods. *P < 0.001 vs. controls.
Table 1.
| Non-Tg/ApoeKO | TgESTRPC3/ApoeKO | |
|---|---|---|
| Cholesterol, mg/dl | 1,121 ± 93 | 1,105 ± 90 |
| 1,096 ± 84 | 1,117 ± 89 | |
| Triglycerides, mg/dl | 335 ± 37 | 319 ± 42 |
| 341 ± 40 | 309 ± 36 | |
| Nonesterified fatty acids, mmol/l | 1.3 ± 0.6 | 1.5 ± 0.7 |
| 1.1 ± 0.8 | 1.6 ± 0.5 | |
| Body weight before diet, g | 19.5 ± 1.0 | 18.2 ± 1.1 |
| 10 wk on high-fat diet | 28.7 ± 2.2 | 27.7 ± 1.8 |
| 16 wk on high-fat diet | 29.4 ± 1.2 | 29.6 ± 2.0 |
Values are means ± SE, n = 10 mice. Mice were fed a high-fat diet for 10 (1st row of values) and 16 wk (2nd row of values), and the indicated parameters were measured as described in materials and methods. TgESTRPC3/ApoeKO, Apoe knockout mice with endothelial-specific gain of transient receptor potential canonical 3 channel function; non-Tg/ApoeKO, nontransgenic ApoeKO littermate controls.
Fig. 2.

Aortic root sections from TgESTRPC3/ApoeKO mice (n = 10) or their transgenic littermates (non-Tg/ApoeKO, n = 10) that were maintained on a high-fat diet for 10 or 16 wk, were subjected to histomorphometric analysis to determine total plaque area as in Ref. 17. Shown are mean values and corresponding SE.
Next we examined whether the livers of TgESTRPC3/ApoeKO mice showed evidence of fibrosis. Figure 3 shows that already after 10 wk on HFD there is collagen deposition in livers from TgESTRPC3/ApoeKO mice, but not in non-Tg/ApoeKO mice (Fig. 3, top panels). The fibrosis augmented and was more disseminated by 16 wk on HFD (Fig. 3, bottom panels), i.e., as aortic atherosclerosis progressed. Hepatic hydroxyproline content was higher in TgESTRPC3/ApoeKO mice vs. non-Tg/ApoeKO (respectively, 0.91 ± 0.02 vs. 0.58 ± 0.03 mg/100 mg liver; n = 6, P < 0.01). These histological and biochemical findings were positively correlated with increased levels of fibrogenic genes Col1A and TGF-β in livers from transgenic mice (5.22 ± 1.8- and 3.24 ± 0.9-fold increase in normalized mRNA levels in livers from TgESTRPC3/ApoeKO vs. non-Tg/ApoeKO mice, for Col1A and TGF-β, respectively; n = 5, P < 0.01). These results indicate that, compared with non-Tg/ApoeKO, TgESTRPC3/ApoeKO mice develop early steatohepatitis with fibrosis. Wet liver weights were similar between TgESTRPC3/ApoeKO and non-Tg/ApoeKO (not shown). Plasma AST and ALT levels were higher in TgESTRPC3/ApoeKO mice compared with non-Tg/ApoeKO (AST: 162 ± 12 vs. 55 ± 9 U/l; ALT: 115 ± 9 vs. 45 ± 7 U/l, for TgESTRPC3/ApoeKO vs. non-Tg/ApoeKO, respectively; P < 0.01, n = 7), again long before differences in atherosclerosis burden were evident. Counting of TUNEL+ cells in liver sections showed higher numbers of apoptotic cells in livers of TgESTRPC3/ApoeKO mice compared with non-Tg/ApoeKO (Fig. 4A). The identity of these cells (i.e., endothelial, parenchymal) remains to be determined. Recently, we showed that TRPC3 contributes to mechanisms of ER stress-induced apoptosis in coronary endothelial cells (3). To examine if a similar effect exists in LSECs, we isolated LSECs from livers of ApoeKO mice and transfected them with siRNA specific for TRPC3, its close relatives TRPC6 and 7, or scrambled oligonucleotides (controls) under conditions previously shown by us to downregulate these proteins efficiently (16). After transfection LSECs were exposed (24 h) to oxidized LDL or tunicamycin to trigger ER stress-induced apoptosis. Figure 4B shows that downregulation of TRPC3, but not TRPC6 or 7, markedly reduced the susceptibility of LSECs to apoptosis.
Fig. 3.

TgESTRPC3/ApoeKO or their nontransgenic littermates (non-Tg/ApoeKO) were maintained on a high-fat diet for 10 (top panels) or 16 (bottom panels) wk. At the end of the diet periods mice were killed and liver sections (10 μm) prepared and stained with Mason's trichrome to evaluate collagen content. Fibrotic areas (blue staining) are indicated by arrows. The bar graph shows relative fibrosis area (% of total liver area) as means ± SE, n = 8. *P < 0.001 vs. controls.
Fig. 4.
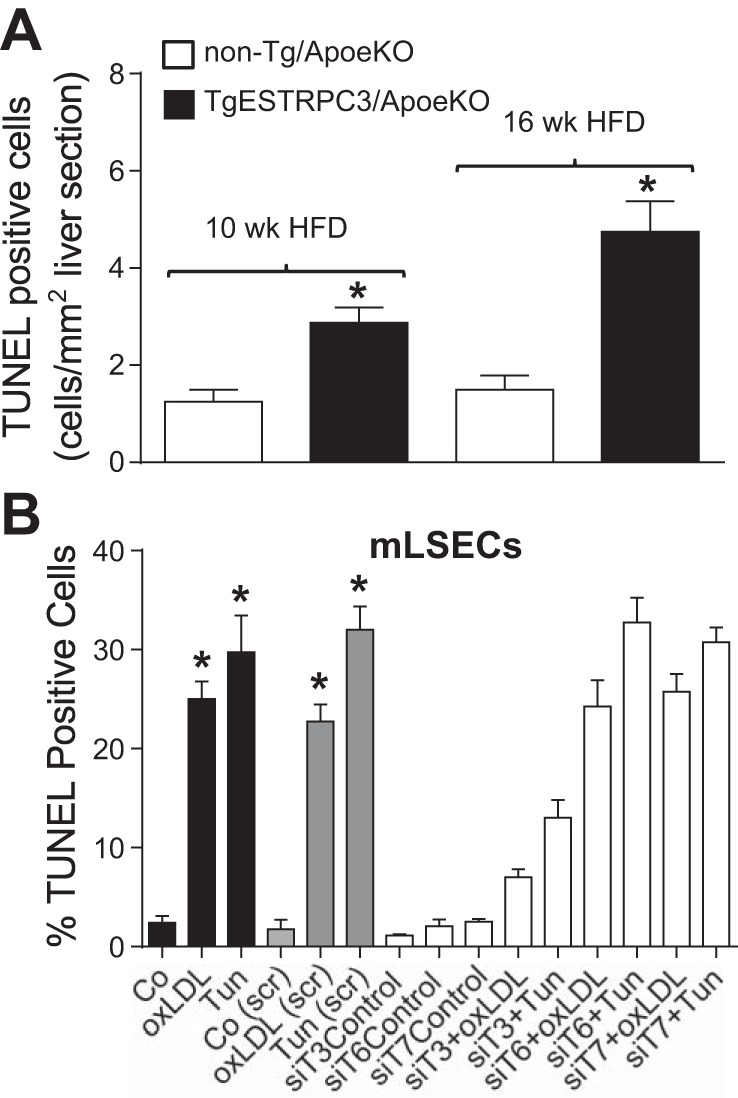
In A, liver sections (10 μm) from TgESTRPC3/ApoeKO mice or nontransgenic littermates (non-Tg/ApoeKO) that were maintained on a high-fat diet for 10 or 16 wk were stained for in situ terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL), and the number of apoptotic (TUNEL+) cells were counted and normalized per mm2 of section area. Shown are means ± SE (n = 5), *P < 0.05 vs. corresponding control. In B, liver sinusoid endothelial cells isolated from ApoeKO mice (mLSECs) were incubated in EBM-2 (Control) or EBM-2 containing oxidized LDL (oxLDL, 50 μg/ml) or tunicamycin (Tun, 10 μg/ml) for 24 h and processed for TUNEL assay (black bars). Alternatively, mLSECs were transfected with scrambled oligonucleotides (gray bars) or with siRNA specific for TRPC3, 6, or 7 (siT3, siT6, or siT7; empty bars) and 48 h posttransfection subjected to the above described treatments and processed for TUNEL assay. Shown are means ± SE (n = 3). *P < 0.001 vs. corresponding control. Differences for “siT3+oxLDL” and “siT3+Tun” vs. corresponding scrambled-transfected controls have P < 0.0001. In the presence of siT6 or siT7, the effect of oxLDL and Tun was not different from corresponding scrambled-transfected controls.
DISCUSSION
The present studies show that ApoeKO mice with endothelial-specific gain of TRPC3 (TgESTRPC3/ApoeKO) progress from NAFLD to steatohepatitis with fibrosis after only 10 wk on a conventional HFD. The pathological liver phenotype of the TgESTRPC3/ApoeKO mice was manifest long before differences in aortic atherosclerotic burden were noticeable. This suggests that in a hyperlipidemic setting, TRPC3-dependent effects occur in the hepatic endothelium contributing to mechanisms mediating the progression of NAFLD to NASH. The early aggravation of the liver pathology might then add to the existing atherogenic factors to accelerate atherosclerosis progression. Similar to our previous findings (17), the early atherosclerotic lesions, 10 wk on HFD, in aortic roots of TgESTRPC3/ApoeKO mice were not different from non-Tg/ApoeKO animals. However, at this time point, the steatohepatitis, fibrosis, and altered liver function were already present in TgESTRPC3/ApoeKO mice compared with non-Tg/ApoeKO animals. Importantly, this occurred without significant differences between the two groups of mice in regards to body weight or lipid profiles. Although parameters of energy intake/expenditure were not evaluated, these results suggest that the overall energy metabolism is not different between TgESTRPC3/ApoeKO and non-Tg/ApoeKO mice. In ApoeKO mice, often used as a model of NAFLD, transition from simple steatosis to steatohepatitis, and in particular, the appearance of fibrosis, requires either extended periods on HFD or drastic dietary manipulations (9, 13, 14). In this context, the outcome of our studies is also especially interesting, as we observe progression from NAFLD to NASH with fibrosis after feeding the mice with a conventional HFD for only 10 wk.
The livers in TgESTRPC3/ApoeKO mice were clearly more steatotic than in non-Tg/ApoeKO mice. Because steatosis can promote liver fibrosis (19), it is possible that the increased steatosis in TgESTRPC3/ApoeKO mice serves as a profibrogenic factor in these animals. Increased circulating NEFAs are a consistent steatogenic factor in NAFLD; however, plasma NEFAs and lipid profiles were similar between transgenic and nontransgenic mice. Of note, TNF-α, which is prosteatogenic, was elevated in livers from TgESTRPC3/ApoeKO mice. Additional studies are needed to determine what causes increased steatosis in the TgESTRPC3/ApoeKO mice. Apoptosis of LSECs can also lead to fibrosis (7). In NAFLD/NASH the hepatic endothelium is chronically exposed to ER stressors, and endothelial cell apoptosis occurs throughout most stages of the disease (8). Our in vitro studies in isolated LSECs, although limited in that they do not recapitulate the environment to which these cells are exposed in vivo, suggest that TRPC3 has a permissive role in ER stress-induced apoptosis. We speculate that by virtue of this role, augmented TRPC3 expression in LSECs of TgESTRPC3/ApoeKO mice may add to the plethora of mechanisms mediating fibrosis in vivo.
The pathophysiological/translational relevance of these findings in the context of excessive caloric intake, NAFLD/NASH, and atherosclerosis can be better appreciated in the light of our previous studies showing that the proinflammatory conditions found in hyperlipidemic ApoeKO mice result in upregulation of endothelial TRPC3 (16). Thus, the findings here add to those in our previous report (17) supporting the notion that augmented TRPC3 expression may have pathological relevance.
In sum, the present studies in a hyperlipidemic mouse model of NAFLD show that endothelial-specific gain of TRPC3 function is positively associated with early transition of the liver pathology to steatohepatitis with fibrosis. This suggests that endothelial TRPC3 participates in mechanisms mediating progression of NAFLD to NASH. Since this transition precedes the aggravation of atherosclerosis, our data support the notion that NAFLD/NASH are important atherogenic factors. Despite the strong connection between NAFLD/NASH and atherosclerosis, current understanding of the relationships between these diseases is poor. Our results indicate that the TgESTRPC3/ApoeKO mouse is an attractive model for further investigation of these interactions and the functions of TRPC3, in a hyperlipidemic setting and without drastic dietary manipulations.
GRANTS
Work supported by National Heart, Lung, and Blood Institute Grant R01HL-111877 (G. Vazquez) and University of Toledo College of Medicine and Life Sciences.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
K.S., P.D., and G.V. performed experiments; K.S., P.D., and G.V. approved final version of manuscript; G.V. conception and design of research; G.V. analyzed data; G.V. interpreted results of experiments; G.V. prepared figures; G.V. drafted manuscript; G.V. edited and revised manuscript.
REFERENCES
- 1.Ahmed M. Non-alcoholic fatty liver disease in 2015. World J Hepatol 7: 1450–1459, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alkhouri N, McCullough AJ. Noninvasive diagnosis of NASH and liver fibrosis within the spectrum of NAFLD. Gastroenterol Hepatol (NY) 8: 661–668, 2012. [PMC free article] [PubMed] [Google Scholar]
- 3.Ampem PT, Smedlund KB, Vazquez G. Pharmacological evidence for a role of the transient receptor potential canonical 3 (TRPC3) channel in endoplasmic reticulum stress-induced apoptosis of human coronary artery endothelial cells. Vascul Pharmacol 76: 42–52, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blomhoff R, Eskild W, Berg T. Endocytosis of formaldehyde-treated serum albumin via scavenger pathway in liver endothelial cells. Biochem J 218: 81–86, 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bondini S, Kleiner DE, Goodman ZD, Gramlich T, Younossi ZM. Pathologic assessment of non-alcoholic fatty liver disease. Clin Liver Dis 11: 17–23, 2007. [DOI] [PubMed] [Google Scholar]
- 6.Chen Y, Xu M, Wang T, Sun J, Sun W, Xu B, Huang X, Xu Y, Lu J, Li X, Wang W, Bi Y, Ning G. Advanced fibrosis associates with atherosclerosis in subjects with nonalcoholic fatty liver disease. Atherosclerosis 241: 145–150, 2015. [DOI] [PubMed] [Google Scholar]
- 7.DeLeve LD. Liver sinusoidal endothelial cells in hepatic fibrosis. Hepatology 61: 1740–1746, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gilbert RE. Augmenting endothelial repair in diabetes: role of bone marrow-derived cells. Can J Diabetes 37: 315–318, 2013. [DOI] [PubMed] [Google Scholar]
- 9.Imajo K, Yoneda M, Kessoku T, Ogawa Y, Maeda S, Sumida Y, Hyogo H, Eguchi Y, Wada K, Nakajima A. Rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Int J Mol Sci 14: 21833–21857, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kohli R, Kirby M, Xanthakos SA, Softic S, Feldstein AE, Saxena V, Tang PH, Miles L, Miles MV, Balistreri WF, Woods SC, Seeley RJ. High-fructose, medium chain trans fat diet induces liver fibrosis and elevates plasma coenzyme Q9 in a novel murine model of obesity and nonalcoholic steatohepatitis. Hepatology 52: 934–944, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Najjar SM, Ledford KJ, Abdallah SL, Paus A, Russo L, Kaw MK, Ramakrishnan SK, Muturi HT, Raphael CK, Lester SG, Heinrich G, Pierre SV, Benndorf R, Kleff V, Jaffa AA, Lévy E, Vazquez G, Goldberg IJ, Beauchemin N, Scalia R, Ergün S. Ceacam1 deletion causes vascular alterations in large vessels. Am J Physiol Endocrinol Metab 305: E519–E529, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paschos P, Paletas K. Non alcoholic fatty liver disease and metabolic syndrome. Hippokratia 13: 9–19, 2009. [PMC free article] [PubMed] [Google Scholar]
- 13.Schierwagen R, Maybuchen L, Zimmer S, Hittatiya K, Back C, Klein S, Uschner FE, Reul W, Boor P, Nickenig G, Strassburg CP, Trautwein C, Plat J, Lutjohann D, Sauerbruch T, Tacke F, Trebicka J. Seven weeks of Western diet in apolipoprotein-E-deficient mice induce metabolic syndrome and non-alcoholic steatohepatitis with liver fibrosis. Sci Rep 5: 12931, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shibata M, Shibata E, Fujioka S, Harada-Shiba M. Apolipoprotein E-knockout mice as a lifestyle-related disease model of atherosclerosis and non-alcoholic fatty liver disease. Int J Lab Med Res 1: 107–109, 2015. [Google Scholar]
- 15.Smedlund K, Bah M, Vazquez G. On the role of endothelial TRPC3 channels in endothelial dysfunction and cardiovascular disease. Cardiovasc Hematol Agents Med Chem 10: 265–274, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smedlund K, Tano JY, Vazquez G. The constitutive function of native TRPC3 channels modulates vascular cell adhesion molecule-1 expression in coronary endothelial cells through nuclear factor kappaB signaling. Circ Res 106: 1479–1488, 2010. [DOI] [PubMed] [Google Scholar]
- 17.Smedlund KB, Birnbaumer L, Vazquez G. Increased size and cellularity of advanced atherosclerotic lesions in mice with endothelial overexpression of the human TRPC3 channel. Proc Natl Acad Sci USA 112: E2201–E2206, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Solanki S, Dube PR, Tano JY, Birnbaumer L, Vazquez G. Reduced endoplasmic reticulum stress-induced apoptosis and impaired unfolded protein response in TRPC3-deficient M1 macrophages. Am J Physiol Cell Physiol 307: C521–C531, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tolman KG, Dalpiaz AS. Treatment of non-alcoholic fatty liver disease. Ther Clin Risk Manag 3: 1153–1163, 2007. [PMC free article] [PubMed] [Google Scholar]
- 20.Trebicka J, Hennenberg M, Laleman W, Shelest N, Biecker E, Schepke M, Nevens F, Sauerbruch T, Heller J. Atorvastatin lowers portal pressure in cirrhotic rats by inhibition of RhoA/Rho-kinase and activation of endothelial nitric oxide synthase. Hepatology 46: 242–253, 2007. [DOI] [PubMed] [Google Scholar]
- 21.Wang L, Wang X, Xie G, Wang L, Hill CK, DeLeve LD. Liver sinusoidal endothelial cell progenitor cells promote liver regeneration in rats. J Clin Invest 122: 1567–1573, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu Z, Luo Z, Ma S, Liu D. TRP channels and their implications in metabolic diseases. Pflügers Arch 461: 211–223, 2011. [DOI] [PubMed] [Google Scholar]