

Abstract
Expression of a cohort of disease-associated genes, some of which are active in fetal myocardium, is considered a hallmark of transcriptional change in cardiac hypertrophy models. How this transcriptome remodeling is affected by the common genetic variation present in populations is unknown. We examined the role of genetics, as well as contributions of chromatin proteins, to regulate cardiac gene expression and heart failure susceptibility. We examined gene expression in 84 genetically distinct inbred strains of control and isoproterenol-treated mice, which exhibited varying degrees of disease. Unexpectedly, fetal gene expression was not correlated with hypertrophic phenotypes. Unbiased modeling identified 74 predictors of heart mass after isoproterenol-induced stress, but these predictors did not enrich for any cardiac pathways. However, expanded analysis of fetal genes and chromatin remodelers as groups correlated significantly with individual systemic phenotypes. Yet, cardiac transcription factors and genes shown by gain-/loss-of-function studies to contribute to hypertrophic signaling did not correlate with cardiac mass or function in disease. Because the relationship between gene expression and phenotype was strain specific, we examined genetic contribution to expression. Strikingly, strains with similar transcriptomes in the basal heart did not cluster together in the isoproterenol state, providing comprehensive evidence that there are different genetic contributors to physiological and pathological gene expression. Furthermore, the divergence in transcriptome similarity versus genetic similarity between strains is organ specific and genome-wide, suggesting chromatin is a critical buffer between genetics and gene expression.
Keywords: cardiac hypertrophy, transcriptome, genetic diversity, chromatin
investigations of the mechanisms underpinning complex physiological phenotypes generally take one of two approaches: either a given pathway (new or previously observed in another cell) is interrogated using gain-/loss-of-function approaches or omics-based discovery experiments are used to determine groups of molecules involved in a phenomenon. Particularly when one approach is marshaled as validation for the other, these dichotomous methods have revealed the molecular basis for various disease processes. The advent of systems genetics in mouse models (an example in the latter category), in which multiple inbred mouse strains with characterized genetic diversity are systematically phenotyped in response to a stress, concomitant with gene expression analysis, allows for genome-wide association analyses (12) and data-driven discovery of genetic networks (27). In the present study, we utilize systems genetics to test hypotheses regarding the differential onset of cardiac disease and to discover novel principles of gene expression. The disease in question is cardiac hypertrophy and the biological process is chromatin-dependent regulation of transcription.
In response to chronic stresses and/or acute injuries, the mammalian heart hypertrophies, increasing the size of individual myocytes. This compensatory response plays a beneficial role in vivo but is also a common precursor to the deleterious condition of heart failure. The following three pieces of rationale related to this disease condition serve as the basis for the present investigation: 1) multiple models of cardiac hypertrophy have been shown to be accompanied by “fetal gene reprogramming,” in which adult diseased muscle expresses genes/isoforms associated with an earlier developmental state (25); 2) an extensive cadre of genes has been implicated in cardiac hypertrophy on the basis of knockout and overexpression studies in cells and mice (33); and 3) human and mouse studies indicate that cardiac hypertrophy and failure have large genetic components of unexplained mechanism (i.e., they are heritable, but we do not know how the heritability manifests at the molecular level) (8, 22). In this study, we test hypotheses that incorporate these three distinct observations. Specifically, we examine whether the panel of genes altered in a single genetic background behaves similarly when examined in the real-world scenario of common genetic variation. Second, we ask how genes known to be capable of modulating cardiac phenotype in gain-/loss-of-function studies correlate with cardiac phenotype in the setting of naturally occurring genetic and phenotypic diversity. Lastly, we investigate how chromatin proteins and transcription factors are affected by common genetic variation, and the relationship in turn between the transcriptomes of these molecules and cardiac phenotype.
MATERIALS AND METHODS
All experiments involving animals conform to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the UCLA Animal Research Committee.
Analysis of data from the hybrid mouse diversity panel.
Microarray [RNA isolated from left ventricle (LV)] and phenotypic data from 84 classical inbred and recombinant strains of mice in the basal state or after treatment with isoproterenol (ISO) were analyzed. Isoproterenol was administered continuously at 30 mg/kg/day for 3 wk via osmotic mini-pump to 8–10 wk old female mice. N = 2 for microarray (RNA from 2 mice/strain/condition were combined and analyzed on a single microarray). N = 2 for control phenotype and n = 4 for ISO phenotype (26). Additional microarray data were obtained from macrophages before and after LPS stimulation (16 wk, male) (23), bone marrow (16 wk, male) (9), striatum, and hippocampus (8 wk, male) (24). Microarrays were performed as follows: cardiac and brain tissue, Illumina ref 8v2; macrophages, Affymetrix mouse 430a; bone marrow, Illumina ref 6v1. Change in expression with isoproterenol was calculated as the difference between the normalized, log2 transformed values for isoproterenol and basal. Cardiac mass and echocardiography parameters were used to classify mice into four phenotype categories based on their response to isoproterenol. Resistant mice were defined as exhibiting minimal change in response to isoproterenol (as compared with the entire panel). Hypertrophic and failing mice were classified based on measurements of their state after isoproterenol and in the change (ISO-Basal) in parameters in response to isoproterenol. Strains whose traits were congruent with different conditions or that resembled different phenotypes when we considered their isoproterenol state versus their ISO-Basal state were left unclassified.
We used a stepwise process, incorporating multiple morphological and functional phenotypes, to classify the response of mice to cardiac stress. Strains were ranked from smallest to largest percent change in normalized total heart weight, a range that spanned 85% to 164% [ISO as a percentage of basal heart weight/body weight (HW/BW)]. Strains with ISO heart sizes close to basal heart sizes (102% to 118%; representing 14% of strains) were subset for later consideration as either resistant or failing based on other parameters (only one strain, CXB-13/HiAJ, had a heart smaller after ISO). Strains with yet larger hearts after ISO (135–164%; representing 38% of strains) were subset for consideration as either hypertrophic or failing. These thresholds of 118 and 135% were determined based on the values of AXB-4/PgnJ and C3H/HeJ, respectively, which we had previously classified as resistant and failing according to a less systematic approach based solely on mass. Next, strains were ranked based on changes in normalized LV weight after ISO (giving an LV/BW range of 75 to 171% of basal size), and strains with minimal change in LV size (100–110%; representing 8% of strains) were subset to be potentially classified as resistant; those with LV >130% of basal after ISO were subset as failing or hypertrophic (130–138% as potentially failing; >140% as potentially hypertrophic or failing, representing 52% of strains). For left ventricular internal diameter in diastole (LVIDd; which gave a range of 85 to 136% of basal after ISO), strains with small diameters (85–103%, 17% of strains) were subset as potentially hypertrophic, whereas strains with LVIDd ≥104% (7% of strains) were subset as either hypertrophic or resistant, those unchanging (105–107% of basal after ISO, 17% of strains) subset as potentially resistant, and those with enlarged diameters (117–136% of basal after ISO, 18% of strains) were subset as potentially failing. For posterior wall thickness (PWTH; ranging from 24% to 293% of basal after ISO), strains with thinner walls (24% to 79%; 24% of strains) were subset as potentially failing, those with minimal change in PWTH (97–107%, 20% of strains) were subset as potentially resistant and those with increased PWTH (120–293%, 32% of strains) were subset as potentially hypertrophic (although PWTH ≤135% was allowable in a strain to be classified as resistant, provided the other metrics did not warrant classification as hypertrophic). Lastly, for ejection fraction (EF; ranging from 56 to 138% of basal after ISO), strains with depressed EF (56–86% of basal after ISO, 15% of strains) were subset as potentially failing, those with minimal change in EF (97–109%, 32% of strains) were subset as potentially resistant and those with improved EF (109–138%, 35% of strains) were subset as potentially hypertrophic.
Based on the above parameters, we assigned strains to a single category. For each strain, we counted the number of phenotypes that matched the resistant, hypertrophic, or failing category and assigned said strain to the most appropriate group, provided there were not multiple conflicting classifications. For example, a strain would not be assigned resistant, even if the majority of parameters were labeled resistant, if there were multiple examples of parameters being classified otherwise. As a result, a strain's classification would be based not on a single parameter. Finally, we repeated this analysis using the ISO only values (as opposed to change between ISO and basal, as described above) and adjusted strain classifications if the results of the ISO phenotype data strongly conflicted with the change in expression data. Our objective was a core set of strains that were confidently identifiable as hypertrophic, failing, or resistant. The cost was that we left almost 50% of strains unclassified, but what we gained was the ability to reliably make conclusions about the expression patterns of the other three categories, since we had stringently defined phenotype groups. The purpose of this entire classification exercise was to study the syndrome of cardiac pathology more closely to how it presents in the clinic, which is as a spectrum of phenotypes across a population of humans, rather than as a single morphological or functional endpoint.
Identification of genes with consistent response to isoproterenol within phenotype group.
All 25,697 probes on the microarray were classified as upregulated (ISO-Basal > 1), downregulated (ISO-Basal < −1) or unchanged (−1 < ISO-Basal < 1) in each hypertrophic, failing, or resistant strain after isoproterenol. We next searched for probes that were either up- or downregulated in the majority of strains within at least one phenotype group. Majority required 77% of failing (10 of 13 strains), 77% of hypertrophic (17 of 22 strains), or 75% of resistant strains (6 of 8 strains) to have the same response. In total, 21 probes met this condition. By contrast, 24,887 were unchanged in the majority of all three phenotype groups, with 12,257 probes unchanged in all strains. We confirmed that all 21 of these genes are expressed [fragments per kilobase of transcript per million mapped reads (FPKM) ranges from 2 to 13, median of 6] in the adult mouse heart by comparing our data to RNA-seq data from ENCODE (ENCFF742HJE).
Correlation of gene expression change with disease phenotype.
We generated comprehensive lists of different functional subsets of genes that have been implicated based off of manual curation of the literature to be involved in cardiac remodeling during hypertrophy and failure: fetal genes (representing not just genes expressed in development, but specifically demonstrated to change in hypertrophy and be used as biomarkers of hypertrophy in the literature); cardiac transcription factors; hypertrophic regulators (based on previous gain- and loss-of-function studies in the heart); and chromatin regulators. Genes from other subsets that were functionally validated in mice were also included as hypertrophic regulators. For the purpose of the hypertrophic regulator gene list, we focused on the disease outcome, not etiology, identifying genes associated with cardiac hypertrophy or failure based on published mouse models. We examined the Pearson correlation of expressions (basal, isoproterenol, or change with isoproterenol) of these subsets of cardiac genes with 49 phenotypes (basal, isoproterenol, or change with isoproterenol) across hybrid mouse diversity panel (HMDP) mouse strains. We also computed correlations between each gene's expression and the total number of genes up- or downregulated after isoproterenol treatment (ISO-Basal > 1.5 or < −1.5) for each strain.
To determine whether any gene subsets (fetal genes, cardiac transcription factors, hypertrophic regulators, chromatin regulators) were enriched with genes that were correlated to phenotype, we discretized the correlation of individual genes into “significant” and “nonsignificant” based on a significance threshold of alpha = 0.05 and performed a hypergeometric test on the group followed by a false discovery rate (FDR) correction using fdrtool (30, 31).
To examine whether changes in expression of individual genes correlate with overall disease state, we used our strain categorization to plot change in expression across strains that are resistant, undergoing hypertrophy, or in failure. The Kruskal-Wallis test was used to quantify statistical significance of the differences between changes in individual gene expression across disease states, and we corrected for multiple testing using the FDR (fdrtool).
Histone clusters and expression in the different phenotype groups.
Change in expression of histones was used to cluster the hypertrophic, failing, and resistant strains using hierarchical ordered partitioning and collapsing hybrid (HOPACH), generating five clusters of strains. HOPACH was developed in part to optimize clustering samples based on gene subsets by combining hierarchical and partitioning clustering (34). We tested for contribution of population structure to our clustering by comparing the kinship coefficients of strains within a group to the coefficients between groups and found no significant difference for any of our clusters. Enrichment of the three disease states (hypertrophic, failing, or resistant) in each histone cluster was determined by the binomial test. Separately, change in expression with isoproterenol was calculated for each histone variant across all strains in a disease state (hypertrophic, failing, or resistant). Presence of significant differences between the three disease states was determined for each variant using the Kruskal-Wallis test. For those variants with significant differences between the groups, the Mann-Whitney test was used to determine which disease states exhibited the difference.
Linear regression modeling of expression versus phenotype.
We used the R package glmnet (11) to identify microarray probes that may be better predictors of cardiac phenotypes when taken together. The difference between log2 transformed isoproterenol and basal expression of all microarray probes in 82 strains of mice was scaled and used as the pool of potential predictors. Response was change in normalized total heart weight (isoproterenol HW/BW minus basal HW/BW). The cvfit function was set to nfold = 10 such that a model is built on 90% of the strains and validated on 10% and this process repeated 10 times. We used default parameters of cvfit with s = “lambda.min” and increasing values of alpha to empirically determine the appropriate alpha to generate ∼100 predictors for analysis. For each alpha 0.1–1 (increments of 0.1), we ran cvfit 1,000 times and found alpha = 0.2 generated 98 predictors (microarray probes) that were present in 800 of the 1,000 models (with each model representing 10 fold cross-validation). The 98 predictors were then subsetted and used to rerun cvfit, resulting in 74 probes in the final model. The accuracy of this model was validated by its ability to accurately predict the difference in total heart weight of the 82 strains. The 74 returned predictors were searched using Princeton University GO Term Finder (4) and DAVID Bioinformatics (13, 14). Neither generated a Gene Ontology (GO) term with enrichment after Benjamini correction. Motif enrichment for the promoter regions of these genes [2 kb upstream of transcription start site (TSS)] was determined using CentriMo in the MEME suite, using the vertebrate motif database. We then attempted a similar analysis using EF instead of HW/BW as the outcome to predict. However, the glmnet algorithm did not generate an optimized model with our input parameters when using cross-validation. Instead, the model was highly strain dependent, and we therefore did not feel confident drawing conclusions from the model without further optimization.
Multiple organ clustering.
To explore features across organs, we analyzed the 37 strains with microarray data from all tissues (heart, macrophage, bone marrow, striatum and hippocampus). Cluster, a package for R, was used to cluster strains into a predetermined number of groups (k = 5) with partitioning around medoids using the Euclidean metric. Clustering was performed on microarray data for all core histone variant probes on the array in the tissue being analyzed. Separately, clustering was performed using the entire transcriptome or using the panel of chromatin regulators. The Euclidean distance between each strain-by-strain comparison was converted to a rank from the most-closely related strain pair to the least-similar pair. Rankings were used to cluster organs based on similar “expression relatedness” between strains and genetic relatedness [based on kinship matrix derived from single nucleotide polymorphisms (SNPs)] using heatmap.2, a function of gplots. The kinship matrix was derived using EMMA (16), and serves to generate a kinship coefficient for each pairwise comparison between strains that estimates the proportion of the genome that is identical between two strains due to common ancestry (19).
RESULTS
Expression of individual genes does not correlate with disease severity.
A recent genome-wide association study (GWAS) on mice treated with the beta-adrenergic agonist isoproterenol revealed significant genetic loci, and new genes, associated with cardiac hypertrophy across a panel of inbred mouse strains (26). This study provided a unique opportunity to subclassify cardiac pathology based on the various phenotypes measured in these mice; for the present analysis, we have done this classification based on in vivo measurements of cardiac function, to mimic the clinical situation in which patients with comparable environmental risk present distinct extent of disease and in some cases, no disease. We chose phenotypes related to heart size and function recorded after 3 wk of isoproterenol infusion and measured by echocardiography (34a), combined with postmortem measurements of heart mass (26), and divided strains into four disease states: hypertrophic (mass increased but function was preserved, n = 22); failing (mass increased and function deteriorated, n = 13); resistant (minimal change in mass or function, n = 9); or unclassified (mass, function and other disease measurements were not in agreement with each other and the spectrum of phenotypes in the animal thus included some that appeared diseased and others that appeared healthy, n = 40). The distribution of strains between these groups for six measurements of cardiac size and function is shown in Fig. 1A. Population structure did not contribute significantly to disease state classifications, as determined by genetic similarities within groups versus between groups (measured by kinship coefficients).
Fig. 1.

The fetal gene program does not coincide with cardiac hypertrophy in diverse genetic backgrounds. A: 84 inbred strains of mice were phenotyped after treatment with the beta-adrenergic agonist isoproterenol. Mice were categorized as hypertrophic or failing if the majority of their traits matched human clinical measurements of these conditions. Mice with minimal change after isoproterenol were classified as resistant. Strains whose traits were congruent with different conditions were left unclassified. Each dot represents a strain. Bar represents group mean. LVIDd, left ventricular internal diameter in diastole; EF, ejection fraction; PWTH, posterior wall thickness; normalized TH, total heart weight/body weight; normalized LV, left ventricular weight/body weight; IVSd, interventricular septum thickness at diastole. B: cardiac microarray data from the same 84 strains of mice were used to analyze change in expression of the fetal genes. With the exception of alpha myosin heavy chain (α-MHC), there was an equal number of strains upregulating or downregulating the fetal genes, even when controlling for disease state, suggesting they are not good markers of isoproterenol-induced hypertrophy across genetic backgrounds. Each bar represents a strain. Expected direction of change in expression is represented by the black arrows to the left. The commonly studied mouse strains BALB/cJ and C57BL/6J do not display expected trends in expression, while DBA/2J and FVB/NJ do (right panels). ANF, atrial natriuretic factor; SERCA, sarcoplasmic reticulum calcium ATPase.
Cardiac disease in animal models is often associated with characteristic alterations in gene expression, called a “fetal gene program,” because the change in gene expression mimics aspects of earlier developmental stages of the heart. (As an aside, it is noteworthy that there are many differences in the transcriptomes of fetal, adult, and diseased hearts, and the term “fetal genes,” used to refer to a small cohort that may behave in the diseased adult heart more similarly to the healthy fetal heart, is thus somewhat of a misnomer. We use the term fetal genes to distinguish the canonical marker genes measured as indicators of hypertrophy from what we refer to herein as “hypertrophic regulators,” which in this study include genes whose genetic manipulation in mouse studies implicate them as components of hypertrophic signaling.) Some human studies have shown similar gene expression changes in heart failure patients (28, 32). However, the role of common genetic variation to influence this gene expression change is unknown. We tested the hypothesis that this gene expression program would be conserved across genetic backgrounds as a common mechanism of disease pathology. To test this, we examined a representative subset of genes often used experimentally to evaluate this phenomenon: alpha myosin heavy chain (α-MHC, predicted to decrease with disease), beta myosin heavy chain (β-MHC, predicted to increase), atrial natriuretic factor (ANF, predicted to increase) and the sarcoplasmic reticulum calcium ATPase (SERCA, predicted to decrease). Because of the differential disease susceptibility across the panel of mice, we were able to examine fetal gene expression in different disease states, despite having microarray data from only two time points (basal and after 3 wk of ISO). To our surprise, these fetal genes were poor predictors of cardiac pathology across genetically distinct mice (Fig. 1B), with only α-MHC displaying the expected trend, in which most of the animals with disease exhibited a decrease in expression. Notably, commonly used strains including BALB/cJ and C57BL/6J were inconsistent, whereas FVB/NJ and DBA/2J were consistent, with the expected changes in expression of these fetal genes (Fig. 1B, right panels).
Complementary to the aforementioned manual classification, we also used two computational approaches to cluster strains. The first was unsupervised hierarchical clustering of Euclidean distances based on the percent change in phenotype after ISO for the same six phenotypes used to manually group strains. This scenario generated five clusters of strains, which we examined for their expression of ANF, SERCA, α-MHC, and β-MHC, asking if >50% of strains followed the expected trend (up- or downregulated) for individual genes. Only one cluster had at least two genes meeting this minimal criteria (n = 23/41 strains upregulating ANF and n = 34/41 strains downregulating α-MHC). This cluster also contained 18 of the 22 strains we had manually classified as hypertrophic. The second method we used was also an unsupervised clustering approach (called partitioning around medoids), using as input a different set of phenotypes (normalized total HW, normalized LV weight, normalized lung weight, LVIDd, and EF; expressed as the ISO value minus that in basal state) to produce five clusters of mice. In this scenario as well, ANF, SERCA, and β-MHC all failed to show consistent up- or downregulation in any of these clusters. One of the groups had over 50% of strains upregulating ANF (57% of strains upregulated), while two groups had SERCA downregulated in >50% of strains (60% of strains in one group, 58% of strains in the second group). No group had β-MHC upregulated in over 37% of strains, and all groups had α-MHC downregulated in ∼71% of strains. Thus, two unsupervised clustering methods and one manual clustering approach all failed to produce groups of mice whose ostensible susceptibility to cardiac pathology was accompanied by the canonical changes in fetal gene expression. Switching back to the phenotype classifications shown in Fig. 1, then, we examined all microarray probes to determine if other genes serve as better markers of response to isoproterenol treatment across genetic backgrounds (Fig. 2). The limited size of this list (21 probes) suggests that single genes serve as poor predictors of cardiac disease state.
Fig. 2.

Genes with shared expression response within phenotype groups. We identified genes that showed the same expression response to isoproterenol in at least 75% of strains within any 1 phenotype group and cross-referenced the genes with patient heart failure expression data. Colored boxes represent genes were at least 75% of strains within a phenotype group showed a consistent gene expression response (green is upregulated, ISO-basal >1; red is downregulated, ISO-basal <1; yellow is no change, ISO-basal between −1 and 1). White boxes represent genes where, for that phenotype, there was not a consistent response. The value represents the mean ISO-basal value. Note that the mean is not always representative of the majority of strains due to outliers; the trend of the majority is indicated by the color. ICM, ischemic cardiomyopathy; DCM, dilated cardiomyopathy.
We wanted to further explore this observation by probing genes as a group as well as considering individual phenotypes as opposed to overall disease states. We expanded the list of fetal genes examined from four to 37 and also tested additional cohorts of genes: regulators of hypertrophic signaling (as determined from knockout and/or transgenesis experiments in mice, n = 142, see Supplemental Table S1 for citations and phenotype information from previous mouse studies), cardiac transcription factors (with known effects on cardiac phenotype, n = 31), and chromatin regulators (see Table 1 for a list of genes in all these groups).1 Because many of the changes in gene expression (and phenotype) across genetic backgrounds cannot be explained through the actions of SNPs acting in cis (i.e., the variant base is situated in or near the modified gene), we reasoned that alterations in the expression of chromatin modifiers could be a mechanistic explanation for some of these differences in gene expression. To test this, we also included a list of genes for epigenetic and chromatin structural modifiers (referred to as chromatin regulators, n = 124; Table 1) in our analyses. We examined the expression of these genes in the basal setting, after isoproterenol and the difference between these points as three independent measurements, rather than looking only at the difference as in Fig. 1B.
Table 1.
List of genes analyzed for each gene subset
| Cardiac TF | Chromatin Regulators | Fetal Genes | Hypertrophy Regulators | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gata4 | Aicda | Ezh1 | Jmjd2b | Rnf40 | Acadm | Slc2a4 | Adrb1 | Frap1 | Map3k1 | Pdpk1 | Slc9a1 |
| Gata5 | Arid1a | Ezh2 | Jmjd2c | Ruvbl1 | Acta1 | Slc8a1 | Adrbk1 | Gata4 | Map3k7 | Pik3ca | Smad1 |
| Gata6 | Arid2 | Hat1 | Jmjd2d | Ruvbl2 | Acta2 | Smad3 | Agtr1a | Gata6 | Mapk1 | Plcb1 | Smad2 |
| Hand1 | Arid3a | Hdac10 | Jmjd3 | Set | Actc1 | Ucp2 | Agtr1b | Gdf15 | Mapk10 | Pln | Smad3 |
| Hand2 | Arid3b | Hdac11 | Jmjd4 | Setd1a | Adss | Ucp3 | Agtr2 | Gna11 | Mapk14 | Ppara | Smad6 |
| Irx4 | Arid3c | Hdac2 | Jmjd5 | Setd1b | Ankrd1 | Vdac1 | Akt1 | Gnaq | Mapk3 | Pparg | Smad7 |
| Isl1 | Arid4a | Hdac3 | Jmjd6 | Setd2 | Atp2a2 | Atp2a2 | Gsk3a | Mapk7 | Ppargc1a | Smarca4 | |
| Mef2b | Arid4b | Hdac4 | Kat5 | Setd3 | Ckm | Bnip3 | Gsk3b | Mapk8 | Ppargc1b | Sp1 | |
| Mef2c | Arid5a | Hdac5 | Lmna | Setd4 | Col1a2 | Brd4 | Hand1 | Mapk9 | Ppp2ca | Tbx20 | |
| Mef2d | Arid5b | Hdac6 | Lmnb1 | Setd5 | Col3a1 | Cacna1c | Hand2 | Mb | Ppp3ca | Tead1 | |
| Mesp1 | Bmi1 | Hdac7 | Lmnb2 | Setd6 | Cpt1a | Cacna1g | Hdac2 | Mef2c | Ppp3cb | Tgfb1 | |
| Myocd | Brd4 | Hdac8 | Mecp2 | Setd7 | Cpt1b | Calm1 | Hdac3 | Mef2d | Ppp3cc | Tlr4 | |
| Nfat5 | Carm1 | Hdac9 | Mst1 | Setd8 | Ctgf | Camk2d | Hdac4 | Mmp2 | Prkca | Tmod1 | |
| Nfatc1 | Cbx2 | Hmga1 | Ncl | Setdb1 | Fhl1 | Camk2g | Hdac5 | Mmp9 | Prkcb | Tpm2 | |
| Nfatc2 | Cbx3 | Hmga2 | Pbrm1 | Setdb2 | Fos | Camta2 | Hdac6 | Mov10l1 | Prkcd | Trpc6 | |
| Nfatc3 | Cbx4 | Hmgb1 | Prdm1 | Sirt1 | Gnas | Cdc42 | Hdac9 | Mtpn | Prkce | Vegfa | |
| Nfatc4 | Cbx5 | Hmgb2 | Prdm10 | Sirt2 | Gys1 | Cib1 | Hey2 | Mybpc3 | Prkcm | Yy1 | |
| Nfkb1 | Cbx6 | Hmgb2l1 | Prdm14 | Sirt6 | Hspa8 | Ckm | Hif1a | Myl2 | Pten | Zfpm2 | |
| Nfkb2 | Cbx7 | Hmgb3 | Prdm16 | Sirt7 | Jun | Crebbp | Hmga1 | Myl4 | Ptk2 | ||
| Nkx2-3 | Cbx8 | Hmgb4 | Prdm4 | Smarca4 | Mlycd | Csrp3 | Hopx | Myocd | Ptpn11 | ||
| Nkx2-5 | Chd4 | Hmgn1 | Prdm5 | Smarcd3 | Myc | Ctf1 | Il6 | Myoz2 | Rac1 | ||
| Nkx2-6 | Crebbp | Hmgn2 | Prdm6 | Smc1a | Myh6 | Ctgf | Il6ra | Nfatc2 | Raf1 | ||
| Smad1 | Ctcf | Hmgn3 | Prdm9 | Smc3 | Myh7 | Ctnnb1 | Il6st | Nfatc3 | Rasa1 | ||
| Smad6 | Dnmt1 | Jarid1b | Prmt2 | Smyd1 | Myl7 | Ctsk | Irx4 | Nfatc4 | Rhoa | ||
| Sp1 | Dnmt3a | Jarid1c | Prmt3 | Suv39h1 | Ndufb10 | Dgkz | Jmjd2a | Nfkb1 | Rock1 | ||
| Srf | Dnmt3b | Jarid1d | Prmt5 | Suv39h2 | Nppa | Dscr1 | Lif | Nfkb2 | Rock2 | ||
| Tbx20 | Dnmt3l | Jarid2 | Prmt6 | Suv420h1 | Nppb | Ep300 | Map2k1 | Nkx2-5 | Rps6kb1 | ||
| Tbx5 | Dpf3 | Jmjd1a | Prmt7 | Suv420h2 | Pdk2 | Fgf16 | Map2k3 | Nppa | Ryr2 | ||
| Tead1 | Ehmt1 | Jmjd1b | Prmt8 | Suz12 | Pdk4 | Fgf2 | Map2k4 | Parp1 | S100a1 | ||
| Xbp1 | Ehmt2 | Jmjd1c | Rad1 | Utx | Ppara | Foxo1 | Map2k5 | Paxip1 | Sirt1 | ||
| Yy1 | Ep300 | Jmjd2a | Rnf20 | Wiz | Slc2a1 | Foxo3 | Map2k6 | Pde5a | Sirt3 | ||
TF, transcription factors.
Unlike the previous analysis looking for genes consistently up- or downregulated across strains, here we looked for linear correlations between the level of gene expression and the severity of the cardiac phenotypes. Using expression data for probes representing each gene cohort and individual phenotypes for all strains, we derived Pearson correlations for the relationships between gene expression and phenotypes. We took the fraction of probes for each gene cohort that were deemed significantly correlated with the given phenotype (percentages are indicated for cardiac mass and function parameters in Fig. 3A) and measured enrichment by comparing to the fraction of significantly correlated probes from the entire microarray (Table 2 lists P values for enrichment for the complete set of phenotypes examined in this study). While individual genes remained poor predictors of cardiac disease (few genes show significant correlation, indicated by P < 0.05, for any given phenotype), when examined as groups, the fetal gene subset and chromatin regulator subset were significantly enriched in genes correlated with multiple cardiac phenotypes compared with the correlation exhibited by all genes detected on the microarray (Fig. 3A and Table 2). Microarray probes for the fetal genes showed significant enrichment for correlations with left atrial mass (enriched 2.97-fold compared with entire transcriptome), change in total heart mass (3.14-fold), and change in LV mass (3.81-fold) after isoproterenol. Individually, considering the seven heart mass and function phenotypes in three conditions (ISO, basal, change with ISO), 65% of the fetal genes correlate with at least three of the 21 comparisons, an enrichment over the correlation of all genes in the genome. Expression of chromatin remodelers correlated with right ventricular mass under both basal and isoproterenol conditions (1.36-fold and 1.42-fold, respectively) and fractional shortening (1.50-fold) with isoproterenol.
Fig. 3.

Gene groups, but not individual fetal genes, correlate with cardiac hypertrophy across diverse genetic backgrounds. A: mRNA abundance for individual probes on the microarray were compared with cardiac parameters across all 84 strains and plotted to identify genes whose expression, or change in expression, was significantly correlated with a trait in either basal or isoproterenol-treated hearts (Pearson correlation, P < 0.05). Plotted is the distribution of P values for individual probes. Tables show the percentage of probes within a subset of genes that demonstrate significance. Gene subsets that were enriched for probes that were correlated with phenotype were determined by normalizing to the level of correlation across all probes on the microarray. * or red indicates P < 0.05 for enrichment of gene subset with respect to the entire array. B: the changes in expression after isoproterenol for individual cardiac transcription factors (TF) (n = 31), fetal genes (n = 37), hypertrophic regulators (n = 142), and chromatin regulators (n = 124) reveal no significant difference between resistant, hypertrophic, or failing mice. Shown are examples of genes with nonsignificant positive and negative trends across these groups. Red indicates failing, green resistant, and blue hypertrophic strains. Horizontal lines represent change of 0.
Table 2.
P values for enrichment of correlation of gene groups with cardiac parameters for basal, isoproterenol, and change with isoproterenol state
| BASAL |
ISO |
ISO - BASAL |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Phenotype | Cardiac TF | Chr | Fetal | Hyp | Cardiac TF | Chr | Fetal | Hyp | Cardiac TF | Chr | Fetal | Hyp |
| Total heart | 0.51 | 0.41 | 0.44 | 0.31 | 0.47 | 0.32 | 0.0058 | 0.40 | 0.36 | 0.58 | 0.039 | 0.36 |
| Left ventricle | 0.41 | 0.33 | 0.43 | 0.27 | 0.54 | 0.27 | 0.028 | 0.31 | 0.32 | 0.52 | 3.53E-04 | 0.28 |
| Right ventricle | 0.53 | 0.09 | 0.28 | 0.020 | 0.60 | 0.06 | 0.017 | 0.16 | 0.48 | 0.43 | 0.32 | 0.06 |
| Left atrium | 0.50 | 0.12 | 0.51 | 0.62 | 0.43 | 0.64 | 6.93E-04 | 0.32 | 0.19 | 0.65 | 0.042 | 0.07 |
| Right atrium | 0.63 | 0.57 | 0.036 | 0.64 | 0.36 | 0.60 | 0.20 | 0.17 | 0.29 | 0.28 | 0.14 | 0.21 |
| Lung | 0.0069 | 0.46 | 0.63 | 0.28 | 0.66 | 0.45 | 0.0061 | 0.52 | 0.05 | 0.64 | 0.27 | 0.14 |
| Liver | 0.43 | 0.64 | 0.44 | 0.32 | 0.54 | 0.57 | 0.09 | 0.07 | 0.19 | 0.49 | 0.31 | 0.31 |
| Adrel | 0.38 | 0.60 | 0.63 | 0.35 | 0.43 | 0.26 | 0.57 | 0.51 | 0.59 | 0.51 | 0.59 | 0.64 |
| Normalized TH | 0.36 | 0.22 | 0.38 | 0.19 | 0.46 | 0.27 | 0.09 | 0.66 | 0.43 | 0.32 | 0.0048 | 0.50 |
| Normalized LV | 0.14 | 0.26 | 0.37 | 0.33 | 0.55 | 0.19 | 0.26 | 0.59 | 0.34 | 0.31 | 4.59E-04 | 0.44 |
| Normalized RV | 0.63 | 0.021 | 0.15 | 0.031 | 0.54 | 0.042 | 0.24 | 0.12 | 0.52 | 0.52 | 0.53 | 0.32 |
| Normalized LA | 0.53 | 0.59 | 0.63 | 0.61 | 0.50 | 0.55 | 0.0018 | 0.56 | 0.24 | 0.65 | 0.07 | 0.16 |
| Normalized RA | 0.11 | 0.57 | 0.41 | 0.20 | 0.36 | 0.60 | 0.21 | 0.45 | 0.35 | 0.19 | 0.19 | 0.22 |
| Normalized Lung | 0.65 | 0.61 | 0.61 | 0.63 | 0.13 | 0.61 | 0.050 | 0.37 | 0.0044 | 0.65 | 0.27 | 0.13 |
| Normalized liver | 0.24 | 0.46 | 0.54 | 0.21 | 0.18 | 0.49 | 0.27 | 0.11 | 0.14 | 0.54 | 0.07 | 0.35 |
| Normalized Adrel | 0.48 | 0.63 | 0.48 | 0.63 | 0.32 | 0.13 | 0.66 | 0.57 | 0.47 | 0.65 | 0.61 | 0.65 |
| Fibrosis (score) | 0.55 | 0.61 | 0.55 | 0.39 | 0.61 | 0.61 | 0.0093 | 0.52 | 0.46 | 0.64 | 0.023 | 0.60 |
| TG | 0.37 | 0.41 | 0.61 | 0.53 | 0.61 | 0.63 | 0.28 | 0.66 | 0.66 | 0.60 | 0.26 | 0.62 |
| Cholesterol | 0.61 | 0.63 | 0.19 | 0.19 | 0.63 | 0.43 | 0.043 | 0.43 | 0.63 | 0.36 | 0.59 | 0.60 |
| HDL | 0.61 | 0.50 | 0.10 | 0.14 | 0.63 | 0.27 | 0.21 | 0.39 | 0.58 | 0.20 | 0.51 | 0.52 |
| UC | 0.55 | 0.52 | 2.04E-04 | 0.41 | 0.59 | 0.62 | 0.13 | 0.46 | 0.60 | 0.50 | 0.53 | 0.61 |
| FFA | 0.27 | 0.12 | 0.59 | 0.22 | 0.64 | 0.36 | 0.06 | 0.63 | 0.22 | 0.046 | 0.30 | 0.0042 |
| Glucose | 0.64 | 0.49 | 0.11 | 0.66 | 0.38 | 0.56 | 0.51 | 0.020 | 0.44 | 0.36 | 0.45 | 0.57 |
| Fibrosis (area) | 0.21 | 0.61 | 0.06 | 0.35 | 0.59 | 0.52 | 0.06 | 0.36 | 0.52 | 0.40 | 0.050 | 0.31 |
| Heart rate | 0.27 | 0.63 | 0.18 | 0.49 | 0.54 | 0.64 | 0.31 | 0.43 | 0.37 | 0.22 | 0.24 | 0.20 |
| IVSd | 0.40 | 0.33 | 0.06 | 0.47 | 0.31 | 0.07 | 0.52 | 0.53 | 0.10 | 0.48 | 0.57 | 0.36 |
| LVIDd | 0.39 | 0.56 | 0.10 | 0.24 | 0.62 | 0.60 | 0.0022 | 0.36 | 0.55 | 0.66 | 0.09 | 0.55 |
| PWd | 0.66 | 0.13 | 0.06 | 0.29 | 0.54 | 0.21 | 0.44 | 0.0045 | 0.54 | 0.58 | 0.30 | 0.024 |
| IVSs | 0.61 | 0.0033 | 0.050 | 0.13 | 0.31 | 0.0045 | 0.56 | 0.030 | 0.52 | 0.62 | 0.52 | 0.39 |
| LVIDs | 0.36 | 0.61 | 0.16 | 0.31 | 0.56 | 0.34 | 0.016 | 0.26 | 0.62 | 0.36 | 0.20 | 0.32 |
| PWs | 0.65 | 0.60 | 0.40 | 0.62 | 0.58 | 0.0068 | 0.59 | 0.0026 | 0.18 | 0.20 | 0.41 | 0.0018 |
| ET | 0.65 | 0.29 | 0.52 | 0.61 | 0.55 | 0.67 | 0.17 | 0.32 | 0.35 | 0.30 | 0.49 | 0.35 |
| E | 0.63 | 0.29 | 0.59 | 0.52 | 0.39 | 0.65 | 0.20 | 0.31 | 0.27 | 0.12 | 0.27 | 0.27 |
| A | 0.59 | 0.040 | 0.05 | 0.13 | 0.45 | 0.022 | 0.53 | 0.11 | 0.15 | 0.46 | 0.45 | 0.62 |
| E/A | 0.39 | 0.0040 | 0.28 | 0.20 | 0.41 | 0.05 | 0.56 | 0.28 | 0.21 | 0.21 | 0.57 | 0.64 |
| A/E | 0.46 | 0.0014 | 0.28 | 0.10 | 0.61 | 0.27 | 0.56 | 0.56 | 0.13 | 0.64 | 0.22 | 0.44 |
| FS | 0.43 | 0.50 | 0.21 | 0.36 | 0.41 | 0.032 | 0.26 | 0.30 | 0.48 | 0.43 | 0.19 | 0.38 |
| IVS/PWd | 0.60 | 0.44 | 0.60 | 0.56 | 0.31 | 0.66 | 0.58 | 0.62 | 0.34 | 0.61 | 0.51 | 0.35 |
| IVS/PWs | 0.26 | 0.0017 | 0.15 | 0.0053 | 0.31 | 0.61 | 0.31 | 0.54 | 0.36 | 0.53 | 0.61 | 0.20 |
| RWTd | 0.24 | 0.32 | 0.19 | 0.16 | 0.42 | 0.19 | 0.43 | 0.12 | 0.64 | 0.65 | 0.44 | 0.23 |
| PWTH | 0.59 | 0.15 | 0.13 | 0.15 | 0.64 | 0.63 | 0.31 | 0.63 | 0.06 | 0.31 | 0.47 | 0.36 |
| Vold | 0.32 | 0.61 | 0.12 | 0.43 | 0.61 | 0.62 | 0.010 | 0.56 | 0.56 | 0.67 | 0.09 | 0.35 |
| Vols | 0.27 | 0.57 | 0.17 | 0.32 | 0.36 | 0.31 | 0.012 | 0.48 | 0.61 | 0.63 | 0.48 | 0.30 |
| EF | 0.52 | 0.60 | 0.11 | 0.36 | 0.39 | 0.07 | 0.11 | 0.35 | 0.50 | 0.37 | 0.21 | 0.33 |
| LVM | 0.57 | 0.39 | 0.19 | 0.26 | 0.65 | 0.32 | 0.0015 | 0.16 | 0.43 | 0.046 | 0.06 | 0.54 |
| LVMc | 0.57 | 0.39 | 0.19 | 0.26 | 0.65 | 0.32 | 0.0015 | 0.16 | 0.43 | 0.047 | 0.06 | 0.54 |
| Vcf | 0.23 | 0.60 | 0.35 | 0.22 | 0.32 | 0.0069 | 0.58 | 0.11 | 0.61 | 0.53 | 0.32 | 0.31 |
| MNSER | 0.31 | 0.51 | 0.46 | 0.21 | 0.36 | 0.027 | 0.60 | 0.24 | 0.62 | 0.60 | 0.34 | 0.43 |
| Upreg genes | 0.47 | 0.30 | 0.31 | 0.16 | 0.19 | 0.31 | 0.66 | 0.41 | 0.19 | 0.16 | 0.27 | 0.010 |
| Downreg genes | 0.61 | 0.21 | 0.29 | 0.12 | 0.20 | 0.012 | 0.46 | 5.30E-06 | 0.033 | 0.028 | 0.43 | 0.15 |
Subset of probes representing cardiac transcription factors (TF), chromatin regulators, fetal gene program, and hypertrophic regulators were analyzed by Pearson correlation to determine significance with association to phenotype. Here, enrichment of these gene groups was compared with respect to all probes that had detectable expression on the microarray. Measured phenotypes include: mass of heart and indicated chambers and other tissues (these were, in addition, normalized to body weight); fibrosis, measured independently by visual scoring and area quantification; plasma triglycerides (TG), cholesterol, HDL cholesterol, unesterified cholesterol (UC), glucose, free fatty acids (FFA); echocardiogram parameters including heart rate, diastolic and systolic interventricular septal thickness (IVSd, IVSs), left ventricular internal diameter (LVIDd, LVIDs), posterior wall thickness (PWd, PWs), ejection time (ET), early and late ventricular filling velocities (E, A) and their ratios, fractional shortening (FS), diastolic relative wall thickness (RWTd), posterior wall thickening (PWTH), end diastolic and systolic end volume (Vold, Vols), ejection fraction (EF), left ventricular mass (LVM) and LVW corrected for growth (LVMc), mean velocity of circumferential fiber shortening (Vcf), and mean normalized systolic ejection rate (MNSER). Additionally, the numbers of up- and downregulated genes after isoproterenol treatment for each strain (Upreg and Downreg) were measured to determine whether overall changes in transcription are correlated to gene groups. Significant enrichment is indicated in boldface.
When we investigated relationships in the expression of individual genes among the different disease states, trends did emerge for some of the cardiac transcription factors, the expanded fetal gene list, hypertrophic regulators, and chromatin modifiers, but these were not significant (Fig. 3B), highlighting again that individual genes do not correlate well with overall state. From these observations we made two hypotheses. First, genetic variation buffers the expression of individual genes, such that the effect of a gene's absolute mRNA abundance on phenotype is dependent on its stoichiometry with other interacting genes and therefore is a poor indicator of phenotype when analyzed individually. Second, part of the buffering effect of genetic variation to influence cardiac phenotype may be mediated by chromatin. To test this second hypothesis further, we examined genes encoding chromatin proteins in more detail, focusing on histone variants, which we previously found to exhibit altered stoichiometry in a mouse model of cardiac hypertrophy and failure (10).
Specific histone variants are consistently regulated by disease state.
Of the chromatin remodeling genes, 58 are histone-modifying enzymes, of which, 43% are correlated with at least three of the seven cardiac size a nd function phenotypes in any of the conditions (ISO, basal, change with ISO). To investigate whether histone variants in addition to histone modification may be regulating susceptibility to cardiac pathology, we performed the antithetic clustering analysis: rather than cluster strains by disease state and examine histone expression, we organized strains according to histone expression (basal value subtracted from isoproterenol value) and observed whether phenotype patterns emerged. Figure 4A shows the five clusters of strains that emerge based on histone variant expression; the rows are histone variants and the shading of the cells corresponds to the change in expression. We checked for contribution of population structure and found no genetic bias between strains within clusters. When each of these clusters was examined for the distribution of disease states, cluster 4 (marked by minimal change in histone expression; Fig. 4A) was significantly enriched in hypertrophic mice and depleted of resistant mice (Fig. 4B); however, the other histone clusters did not discriminate between phenotypic outcome to isoproterenol, suggesting only weak correlation between histone stoichiometry and susceptibility to isoproterenol. To test this, we examined histone variants as groups between the disease states. No differences were observed in the contribution of variant families to the overall stoichiometry of histones between the different phenotypes (Fig. 4C); however, when individual histone variants were examined, three showed significant expression differences between disease states (Fig. 4D). This is in contrast to the fetal genes, cardiac transcription factors, hypertrophic regulators, and chromatin regulators, for which there was no single gene with significant difference in expression between disease states. (Note, the analyses in Figs. 3B and 4D use lower thresholds than that of the consistently changing genes in Fig. 2, in that Fig. 2 requires 75% of strains in a disease state to exhibit the same trend, while these figures require only a significant difference in trends between disease states.) Together, this suggests that like the other gene groups analyzed, a general relationship between histone stoichiometry and disease outcome does not exist across different genetic backgrounds despite histones having been implicated in hypertrophy in individual strains (7).
Fig. 4.

Clustering mice by histone expression reveals patterns of change associated with phenotype. A: hierarchical ordered partitioning and collapsing hybrid (HOPACH) clustered strains into 5 groups based on histone expression change with isoproterenol. Rows indicate individual histone variants, while shading represents changes in expression; n = 12, cluster 1; n = 9, cluster 2; n = 3, cluster 3; n = 7, cluster 4; n = 13, cluster 5. B: for each histone cluster, the composition of phenotype group is shown. Cluster 4 is significantly enriched (enriched 2.57-fold, P = 0.0068) for hypertrophic mice and depleted for resistant mice (P = 0.0585, not significant). C: abundance of all variants for each of the core histones was pooled across strains in the basal (light) or isoproterenol-treated (dark) hearts, revealing no gross differences between phenotypic groups. D: 3 histone variants were differentially regulated by isoproterenol between phenotype groups (*P < 0.05).
Because we had previously characterized, using mass spectrometry, histone variant and chromatin protein expression in another model of cardiac hypertrophy and failure induced by pressure overload hypertrophy (10), we sought to determine whether the modules of proteins identified with altered chromatin association in this study were recapitulated in the transcriptome data. In short, none of the chromatin protein modules identified by quantitative proteomics exhibited conserved transcriptome regulation in the present study (data not shown), a not completely unexpected observation, given the tiers of cellular regulation between gene expression and protein occupancy on chromatin.
Unbiased identification of disease predictors do not share common ontology.
We next asked if we could identify genes better correlated with cardiac phenotype. We used linear regression modeling to identify genes whose expressions are predictive of change in normalized total heart weight with isoproterenol. This exercise identified 74 probes that were present in 80% of our models and successfully predicted phenotype but found that they shared no common pathways or functions (based on no significant GO results), suggesting that the relevance of important cardiac signaling pathways may be masked by genetic diversity in the transcriptional response to isoproterenol such that the changes in stoichiometry of key genes within functional modules are genotype dependent (Table 3). We compared our data to RNA-seq data from the adult mouse heart (ENCODE, ENCFF742HJE): 34 of the genes were expressed with an FPKM > 1. GO analysis of these 34 also returned no significant terms. Furthermore, motif analyses of the promoters (2 kb upstream of TSS) of these 34 genes revealed no enriched vertebrate motifs.
Table 3.
Gene predictors of heart mass are not members of a single biological process
| Illumina | Gene | Coefficient | Illumina | Gene | Coefficient |
|---|---|---|---|---|---|
| ILMN_2934549 | BC055324 | −6.79E-05 | ILMN_2444594 | Cdh3 | −2.17E-05 |
| ILMN_2816271 | Dcpp2 | 6.22E-05 | ILMN_2890915 | EG331493 | −2.15E-05 |
| ILMN_2687716 | 4930433I11Rik | −5.99E-05 | ILMN_2751822 | Dsg1b | 2.11E-05 |
| ILMN_3072147 | Foxo6 | −5.77E-05 | ILMN_2509644 | Tpk1 | 2.10E-05 |
| ILMN_2482494 | Trim16 | 5.75E-05 | ILMN_2795644 | Tlr11 | −2.06E-05 |
| ILMN_2977849 | Fank1 | −4.57E-05 | ILMN_1253791 | Kcnv2 | −2.05E-05 |
| ILMN_1234692 | 1300007F04Rik | 4.29E-05 | ILMN_2719803 | Stap1 | 1.82E-05 |
| ILMN_2780323 | Tkt | 4.27E-05 | ILMN_2859908 | Olfr335 | −1.79E-05 |
| ILMN_2792089 | Srebf1 | −4.21E-05 | ILMN_2686924 | Epha1 | 1.78E-05 |
| ILMN_1213351 | Gsdma1 | 4.10E-05 | ILMN_3162671 | EG432555 | −1.75E-05 |
| ILMN_1238397 | Olfr126 | −4.01E-05 | ILMN_2824954 | OTTMUSG00000000421 | −1.65E-05 |
| ILMN_2742887 | Scrib | 3.89E-05 | ILMN_2540726 | Olfr670 | −1.60E-05 |
| ILMN_3072427 | Il1rn | 3.70E-05 | ILMN_2703720 | Bclaf1 | 1.55E-05 |
| ILMN_2965612 | Abca6 | −3.55E-05 | ILMN_2701712 | Plcxd3 | −1.48E-05 |
| ILMN_1223335 | Ano10 | −3.52E-05 | ILMN_1254646 | Sox6 | −1.48E-05 |
| ILMN_2734661 | Hagh | 3.49E-05 | ILMN_2806235 | Gm813 | 1.44E-05 |
| ILMN_1240675 | Rbm12b | 3.43E-05 | ILMN_2911123 | Itga2b | −1.40E-05 |
| ILMN_1214602 | Sfrp2 | 3.12E-05 | ILMN_3062163 | Rab11fip5 | 1.38E-05 |
| ILMN_2642418 | Mest | −3.10E-05 | ILMN_2675623 | Mrfap1 | −1.32E-05 |
| ILMN_1245040 | 9030617O03Rik | −3.02E-05 | ILMN_1218034 | Tmco4 | 1.31E-05 |
| ILMN_2898062 | Olfr108 | 2.96E-05 | ILMN_1239583 | Wins2 | −1.29E-05 |
| ILMN_1213265 | 2610208M17Rik | 2.96E-05 | ILMN_2863437 | 1110038F14Rik | −1.19E-05 |
| ILMN_2833993 | 4921501E09Rik | −2.91E-05 | ILMN_3048689 | Rffl | −1.19E-05 |
| ILMN_2779272 | Olfr313 | −2.86E-05 | ILMN_2805051 | Upk3a | 1.18E-05 |
| ILMN_2708142 | Xkr6 | 2.82E-05 | ILMN_2693946 | Olfr1347 | −9.91E-06 |
| ILMN_2753867 | Scgb3a2 | 2.76E-05 | ILMN_2742311 | Cyp39a1 | −9.27E-06 |
| ILMN_2670398 | Eif4ebp1 | 2.70E-05 | ILMN_1214065 | Slco1a6 | −8.32E-06 |
| ILMN_2900653 | Gadd45b | 2.69E-05 | ILMN_1223591 | Zfp202 | −7.79E-06 |
| ILMN_2938373 | Tas2r116 | −2.68E-05 | ILMN_2601758 | Gsto2 | −6.97E-06 |
| ILMN_2682493 | Bmp5 | 2.57E-05 | ILMN_2678714 | Id4 | 6.61E-06 |
| ILMN_1259759 | Olfr672 | 2.57E-05 | ILMN_2657207 | Hey2 | −5.61E-06 |
| ILMN_2527490 | LOC381375 | 2.51E-05 | ILMN_2661495 | Tmem44 | 3.59E-06 |
| ILMN_1242281 | Txndc2 | 2.43E-05 | ILMN_2726837 | Nppb | 3.50E-06 |
| ILMN_1221960 | Gtf2ird1 | −2.38E-05 | ILMN_2700468 | Pcdhgc4 | −3.37E-06 |
| ILMN_2790241 | Herpud1 | −2.28E-05 | ILMN_2705242 | Wee2 | 2.22E-06 |
| ILMN_2630521 | Hist1h1a | −2.24E-05 | ILMN_2863849 | C1qtnf3 | 2.19E-06 |
| ILMN_1223384 | Nadsyn1 | 2.20E-05 | ILMN_2596998 | Lypd6b | 1.67E-06 |
Linear regression analysis, using the glmnet package, was performed on change in expression with isoproterenol for all probes on the microarray to predict change in normalized total heart mass in 82 strains. We used 10-fold cross-validation to build each model, and a total of 1,000 models were performed to identify 98 probes with predictive capacity in 800 of the 1,000 models. The model incorporates 74 of these probes and fits the actual data for the 82 strains with an r-squared of 0.99. Coefficient indicates weight of the contribution in final model. GO analysis on these genes returned no significant enrichment for biological processes or cellular components.
Cardiac transcriptome patterns are not reflective of genetics.
In addition to dissecting the genetically conserved expression changes that predict cardiac phenotype, we next tested if expression, both conserved and strain specific, was in fact correlated with genetics by expanding our analysis to expression data for the HMDP from bone marrow (9), macrophages (23), striatum, and hippocampus (24). We asked: if two strains have similar gene expression in one organ (due to genetics), do they also share similar transcriptomes in other organs? When two organs cluster in the dendrogram (such as control macrophages and hippocampus; Fig. 5A, left), it indicates that strains with similar expression in one organ also have similar expression in the other organ. Dendrograms were made with expression data for chromatin regulators, histones, or all genes (Fig. 5A).
Fig. 5.

The role of genetics in chromatin gene expression is organ specific. A: we asked if 2 strains showing similar expression in 1 organ are also closely associated in another organ. We clustered 37 strains with microarray data in 5 tissues based on histone expression (middle). The relatedness in expression between each strain-by-strain comparison was compared across organs. Strains with similar histone expression in basal macrophages also show similar expression after LPS treatment (control and LPS-treated are neighbors); this is not the case for the heart before and after isoproterenol stimulation. This analysis was repeated using expression of chromatin regulators (left) or of all genes (right) to cluster strains. B: heatmap x-axis represents each strain-by-strain comparison. Scale is the ranking of Euclidean distance in the organ and gene subset from 1 (green, most-closely related strain pair) to 667 (red, most-distant strain pair). “Genetics” indicates relatedness based on kinship matrix derived from single nucleotide polymorphisms. Diagram below represents observations from macrophage and heart expression datasets (in boldface) with respect to genetic similarities between strains. As in the dendrograms, we see that LPS stimulus of macrophages does not disrupt coexpression relatedness between strains, but isoproterenol stimulation of the heart does. Similarly, macrophages exhibit the greatest overlap between expression relatedness and genetic relatedness.
For each organ we ranked strain-by-strain transcriptome comparisons from the most similar strain pair (1, green) to the most different (667, red), and generated a heatmap to display how these rankings vary by organ (Fig. 5B). Surprisingly, similar transcriptomes between strains in the basal heart did not predict similar expression after isoproterenol (control and ISO hearts do not cluster, Fig. 5B), suggesting the genetic determinants of physiological and pathological gene expression are different. By contrast, basal and LPS-stimulated macrophages do cluster. We also included a ranking of genetic similarity using kinship matrices based on SNPs. The similarity between the transcriptomes of two strains was only weakly dependent on the genes analyzed, as all gene subsets clustered together for a given organ. Secondly, the relatedness between strains as calculated by gene expression was markedly different from the genetic relatedness determined by SNPs (with the exception of macrophages), though gene expression of all genes was consistent with chromatin gene expression. Together these two patterns suggest that the relationship between genetics and gene expression is buffered by a mechanism that is both organ dependent and globally acting across multiple genes. We postulate this is due to the effects of cell/organ-specific epigenetic programming.
DISCUSSION
Previous studies have shown a diversity of cardiac phenotype across distinct mouse strains (3, 35) and linked them to gene expression differences (1). Here we have expanded these analyses to look across a larger panel of strains to determine whether these differences can still be attributed to the changes in a single set of genes. We found that, unexpectedly, the majority of cardiac-associated genes implicated with cardiac disease do not share consistent expression changes across strains with similar phenotypes. Instead, our results highlighted the roles of gene cohorts, including those involved with chromatin regulation.
Genetics and chromatin combine to influence gene expression patterns and phenotype, although the mechanisms are incompletely understood (Fig. 6), in no small part because animal studies often examine only a single genetic background. Here, we show that phenotype cannot be predicted solely by expression of individual genes when a genetically diverse population is examined. Neither genes that have been implicated in regulating hypertrophic signaling nor the fetal genes associate with overall disease state after isoproterenol. In a comparable study using C57BL/6J and DBA/2J administered ISO for 2 wk, both strains displayed significant increases in HW, fractional shortening, and EF (17). These phenotypes are consistent with what we observe, as are the changes in α-MHC expression (no change in the C57BL/6J strain and decrease in DBA/2J). In contrast to our observations, this study reported an increase in ANF in C57BL/6, a difference that may be attributable to variation in the time course of the study.
Fig. 6.
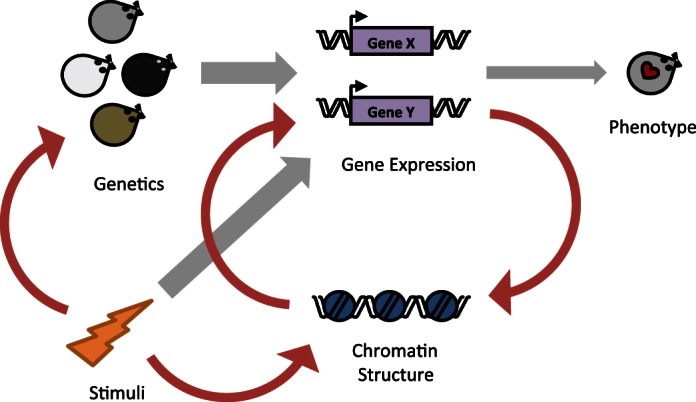
Chromatin proteins modulate the relationship between genetics and gene expression. The existing framework (gray arrows) for explaining cardiac phenotype is that genetics and environmental stimulation are the driving regulators of gene expression, which in turn determines phenotype. Our data support this model, showing that cardiac gene expression does not correlate with phenotype across all genetic backgrounds due to contributions by chromatin regulatory inputs. Additionally, we build on this model (red arrows) showing that chromatin protein levels feedback to influence gene expression, based on our observation that gene expression “relatedness” is organ dependent. We further posit that there is a direct interaction between environmental stimuli and genetics, based on our observation that different genetic components are responsible for basal or isoproterenol-induced gene expression in the heart.
We further find that the genetic drivers of gene expression are different after isoproterenol. Strains with similar expression patterns in the basal heart no longer share expression similarities after stimulus. A similar observation is seen when comparing gene expression similarities across organs. Our analyses suggest that chromatin, in addition to environmental stimulus, is an important independent modifier of the genetic contribution to gene expression.
The HMDP demonstrates that strains that are most closely related genetically do not always exhibit the most similar transcriptional responses to pathogenic stimuli. To identify the source of this discrepancy, we report three separate findings that point to chromatin as the mechanism. Firstly, we show that the discrepancy between shared genetics and shared transcriptomes is due to a mechanism that has a strong organ-dependent component. Secondly, we show that the genetic relationships act similarly to control the expression of different gene subsets, suggesting a genome-wide mechanism. Finally, our analyses of gene subsets demonstrate that genetic buffering diminishes the correlation between a single gene and cardiac phenotype, and yet, we still find examples of histone variants with conserved expression changes across disease states and show that chromatin regulators as a group correlate with several disease phenotypes. Although not highlighted in our analyses, DNA methylation serves as another regulator for gene expression and was included in our analyses of chromatin regulators. We find that modulators of DNA methylation, including DNA methyltransferases and methylation binding proteins, correlate with aspects of cardiac hypertrophy. This matches our previous observation that there is differential cardiac DNA methylation in the HMDP (6).
We see two explanations for the lack of correlation between mRNA expression and phenotype, disease state, and genetics. The first is that there are posttranscriptional events that result in a disconnect between mRNA abundance and functional protein levels. Future studies are needed to test these relationships at the protein level. Coexpression modules from microarray have been shown to be inconsistent with protein interaction networks from the heart (5), whereas recent detailed bioinformatics analyses indicate that transcriptome and proteome levels are often quite similar (18). This question can only be resolved experimentally for each protein. Previous studies have indicated that for select genes, the protein levels are very good indicators of heart failure in humans. BNP and NTproBNP, for example, have been shown to be effective biomarkers for ruling out heart failure in patients referred for suspected heart failure by their general practitioner (38). In these cases, differences between tissue mRNA levels (not measured in humans) and circulating plasma protein levels could arise due to the multiple regulatory steps between transcription and subsequent secretion. However, we also propose that for some genes, genetics is playing a major role in disrupting the correlation between the expression of disease-causing genes and phenotype.
Previous work on the HMDP has been successful using GWAS analysis to identify 24 significant or suggestive loci regulating cardiac hypertrophy and fibrosis (26). None of the 24 candidate genes in these loci showed a significant linear correlation with either total HW or LV weight, with only one (Srpx, P = 0.049) showing modest correlation with fibrosis due to isoproterenol treatment. Independently, we found 579 genes (3% of genes detectable on the microarray) showed significant correlation with fibrosis in the isoproterenol state, including Col3a1, Ctgf, and Postn (genes implicated in cardiac fibrosis), which also showed significant correlations with total heart and LV masses. One major implication from our work is that analysis of individual genes should be complemented with future studies that include a middle-ground approach that takes into account many interacting SNPs (as we do here with kinship matrices) while still examining individual SNPs with functional roles (as in GWAS). Such approaches are being developed, wherein large cohorts of SNPs (in this case all SNPs on a single chromosome or in the entire genome) are related to phenotype and have proven more successful at explaining the majority of the heritability of common traits (37).
The low level of causative SNPs identified by GWAS for heart failure (21) has been attributed to the theory that an interaction between multiple SNPs, each with small effect size, is necessary to explain certain complex traits (20). This theory is in line with our observation that gene subsets serve as better predictors of cardiac traits than do individual genes. Importantly, GWAS analysis by the CHARGE consortium also demonstrates that the genetic determinants of heart failure incidence are different between ethnicities (21). Here we attempted to ask a similar question in mice, that is: are hypertrophic or failing mice sick due to the same pathological transcriptome, independent of genetics? In other words, can the same genes serve as predictors of cardiac phenotype across the HMDP? We were surprised to find poor correlation between abundance of individual genes and specific cardiac phenotypes. In light of this, we attempted to further subdivide strains based on overall disease state, re-examining the data for potential relationships between gene expression and phenotype. This step was taken to address the fact that the different strains may be at different stages of disease progression (i.e., a variation in temporal onset of disease), in addition to being overall more or less resistant to pathology (i.e., variation in severity of disease). This classification inherently induces bias, and thus it is possible that the grouping of strains we performed does not reflect the type of distribution in a human population. It may be that more precise classification strategies exist that would reveal cohorts of animals that have better correlation for the classes of genes tested herein. For the subdivision we present here, however, we find poor correlation between specific genes and overall disease state. This refutes the hypothesis that all mouse strains show altered cardiac size upon isoproterenol by similar transcriptional changes and supports a model where different gene stoichiometry can result in similar phenotypes (36).
It is known that both the incidence of cardiovascular risk factors, such as hypertension, and the incidence of advanced-stage disease outcomes, including heart failure, differ by race (2, 15). Understanding genetic differences in cardiovascular disease is necessary to better assess risk and tailor treatment to individual patients. We show that in addition to differences in heart failure susceptibility, mice of diverse genetic backgrounds also undergo diverse transcriptional responses to achieve a similar phenotypic outcome. One ramification of this is that fetal genes, as well as the entire transcriptome, are poor predictors of cardiac phenotype when analyzed individually across different genetic backgrounds. In our study, chromatin emerges as a mediator of the response to isoproterenol, integrating genetic variation with environmental stress.
GRANTS
This study was supported by National Heart, Lung, and Blood Institute (NHLBI) Grants HL-105699 (T. M. Vondriska), HL-115238 (T. M. Vondriska), HL-129639 (T. M. Vondriska, Y. Wang), HL-28481 (A. J. Lusis), HL-123295 (A. J. Lusis, Y. Wang), and HL-114437 (J. N. Weiss). E. Karbassi, E. Monte, M. Rosa Garrido, R. Lopez, and C. D. Rau were supported by American Heart Association Fellowships. D. J. Chapski was supported by NHLBI Training Grant T32 HL-69766.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
E.K., E.M., J.N.W., Y.W., A.J.L., and T.M.V. conception and design of research; E.K., E.M., R.L., M.R.G., J.K., C.D.R., and J.J.W. performed experiments; E.K., E.M., D.J.C., M.R.G., N.W., C.D.R., J.J.W., and T.M.V. analyzed data; E.K., E.M., D.J.C., N.W., and T.M.V. interpreted results of experiments; E.K., E.M., D.J.C., and T.M.V. prepared figures; E.K., E.M., and T.M.V. drafted manuscript; E.K., E.M., and T.M.V. edited and revised manuscript; E.K., E.M., D.J.C., R.L., M.R.G., J.K., N.W., C.D.R., J.J.W., J.N.W., Y.W., A.J.L., and T.M.V. approved final version of manuscript.
Supplementary Material
Footnotes
The online version of this article contains supplemental material.
REFERENCES
- 1.Auerbach SS, Thomas R, Shah R, Xu H, Vallant MK, Nyska A, Dunnick JK. Comparative phenotypic assessment of cardiac pathology, physiology, and gene expression in C3H/HeJ, C57BL/6J, and B6C3F1/J mice. Toxicol Pathol 38: 923–942, 2010. [DOI] [PubMed] [Google Scholar]
- 2.Bahrami H, Kronmal R, Bluemke DA, Olson J, Shea S, Liu K, Burke GL, Lima JA. Differences in the incidence of congestive heart failure by ethnicity: the multi-ethnic study of atherosclerosis. Arch Intern Med 168: 2138–2145, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barnabei MS, Palpant NJ, Metzger JM. Influence of genetic background on ex vivo and in vivo cardiac function in several commonly used inbred mouse strains. Physiol Genomics 42A: 103–113, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boyle EI, Weng S, Gollub J, Jin H, Botstein D, Cherry JM, Sherlock G. GO::TermFinder–open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics 20: 3710–3715, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Camargo A, Azuaje F. Linking gene expression and functional network data in human heart failure. PLoS One 2: e1347, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen H, Orozco LD, Wang J, Rau CD, Rubbi L, Ren S, Wang Y, Pellegrini M, Lusis AJ, Vondriska TM. DNA methylation indicates susceptibility to isoproterenol-induced cardiac pathology and is associated with chromatin states. Circ Res 118: 786–797, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen IY, Lypowy J, Pain J, Sayed D, Grinberg S, Alcendor RR, Sadoshima J, Abdellatif M. Histone H2A.z is essential for cardiac myocyte hypertrophy but opposed by silent information regulator 2alpha. J Biol Chem 281: 19369–19377, 2006. [DOI] [PubMed] [Google Scholar]
- 8.Dorn GW., 2nd Genetics of common forms of heart failure. Curr Opin Cardiol 26: 204–208, 2011. [DOI] [PubMed] [Google Scholar]
- 9.Farber CR, Bennett BJ, Orozco L, Zou W, Lira A, Kostem E, Kang HM, Furlotte N, Berberyan A, Ghazalpour A, Suwanwela J, Drake TA, Eskin E, Wang QT, Teitelbaum SL, Lusis AJ. Mouse genome-wide association and systems genetics identify Asxl2 as a regulator of bone mineral density and osteoclastogenesis. PLoS Genet 7: e1002038, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Franklin S, Chen H, Mitchell-Jordan S, Ren S, Wang Y, Vondriska TM. Quantitative analysis of the chromatin proteome in disease reveals remodeling principles and identifies high mobility group protein B2 as a regulator of hypertrophic growth. Mol Cell Proteom 11: M111 014258, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Friedman J, Hastie T, Tibshirani R. Regularization paths for generalized linear models via coordinate descent. J Stat Softw 33: 1–22, 2010. [PMC free article] [PubMed] [Google Scholar]
- 12.Ghazalpour A, Rau CD, Farber CR, Bennett BJ, Orozco LD, van Nas A, Pan C, Allayee H, Beaven SW, Civelek M, Davis RC, Drake TA, Friedman RA, Furlotte N, Hui ST, Jentsch JD, Kostem E, Kang HM, Kang EY, Joo JW, Korshunov VA, Laughlin RE, Martin LJ, Ohmen JD, Parks BW, Pellegrini M, Reue K, Smith DJ, Tetradis S, Wang J, Wang Y, Weiss JN, Kirchgessner T, Gargalovic PS, Eskin E, Lusis AJ, LeBoeuf RC. Hybrid mouse diversity panel: a panel of inbred mouse strains suitable for analysis of complex genetic traits. Mamm Genome 23: 680–692, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37: 1–13, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57, 2009. [DOI] [PubMed] [Google Scholar]
- 15.Jones DW, Hall JE. Racial and ethnic differences in blood pressure: biology and sociology. Circulation 114: 2757–2759, 2006. [DOI] [PubMed] [Google Scholar]
- 16.Kang HM, Zaitlen NA, Wade CM, Kirby A, Heckerman D, Daly MJ, Eskin E. Efficient control of population structure in model organism association mapping. Genetics 178: 1709–1723, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kiper C, Grimes B, Van Zant G, Satin J. Mouse strain determines cardiac growth potential. PLoS One 8: e70512, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li JJ, Biggin MD. Gene expression. Statistics requantitates the central dogma. Science 347: 1066–1067, 2015. [DOI] [PubMed] [Google Scholar]
- 19.Lynch M, Ritland K. Estimation of pairwise relatedness with molecular markers. Genetics 152: 1753–1766, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, Cho JH, Guttmacher AE, Kong A, Kruglyak L, Mardis E, Rotimi CN, Slatkin M, Valle D, Whittemore AS, Boehnke M, Clark AG, Eichler EE, Gibson G, Haines JL, Mackay TF, McCarroll SA, Visscher PM. Finding the missing heritability of complex diseases. Nature 461: 747–753, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McNamara DM, London B. GWAS applied to heart failure: bigger will be better… eventually. Circ Cardiovasc Genet 3: 226–228, 2010. [DOI] [PubMed] [Google Scholar]
- 22.Monte E, Vondriska TM. Epigenomes: the missing heritability in human cardiovascular disease? Proteom Clin Appl 8: 480–487, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Orozco LD, Bennett BJ, Farber CR, Ghazalpour A, Pan C, Che N, Wen P, Qi HX, Mutukulu A, Siemers N, Neuhaus I, Yordanova R, Gargalovic P, Pellegrini M, Kirchgessner T, Lusis AJ. Unraveling inflammatory responses using systems genetics and gene-environment interactions in macrophages. Cell 151: 658–670, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park CC, Gale GD, de Jong S, Ghazalpour A, Bennett BJ, Farber CR, Langfelder P, Lin A, Khan AH, Eskin E, Horvath S, Lusis AJ, Ophoff RA, Smith DJ. Gene networks associated with conditional fear in mice identified using a systems genetics approach. BMC Syst Biol 5: 43, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rajabi M, Kassiotis C, Razeghi P, Taegtmeyer H. Return to the fetal gene program protects the stressed heart: a strong hypothesis. Heart Fail Rev 12: 331–343, 2007. [DOI] [PubMed] [Google Scholar]
- 26.Rau CD, Wang J, Avetisyan R, Romay MC, Martin L, Ren S, Wang Y, Lusis AJ. Mapping genetic contributions to cardiac pathology induced by Beta-adrenergic stimulation in mice. Circ Cardiovasc Genet 8: 40–49, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rau CD, Wisniewski N, Orozco LD, Bennett B, Weiss J, Lusis AJ. Maximal information component analysis: a novel non-linear network analysis method. Front Genet 4: 28, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Razeghi P, Young ME, Alcorn JL, Moravec CS, Frazier OH, Taegtmeyer H. Metabolic gene expression in fetal and failing human heart. Circulation 104: 2923–2931, 2001. [DOI] [PubMed] [Google Scholar]
- 29.Rosa-Garrido M, Karbassi E, Monte E, Vondriska TM. Regulation of chromatin structure in the cardiovascular system. Circ J 77: 1389–1398, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Strimmer K. fdrtool: a versatile R package for estimating local and tail area-based false discovery rates. Bioinformatics 24: 1461–1462, 2008. [DOI] [PubMed] [Google Scholar]
- 31.Strimmer K. A unified approach to false discovery rate estimation. BMC Bioinformatics 9: 303, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taegtmeyer H, Sen S, Vela D. Return to the fetal gene program: a suggested metabolic link to gene expression in the heart. Ann NY Acad Sci 1188: 191–198, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Berlo JH, Maillet M, Molkentin JD. Signaling effectors underlying pathologic growth and remodeling of the heart. J Clin Invest 123: 37–45, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van der Laan MJ, Pollard KS. A new algorithm for hybrid hierarchical clustering with visualization and the bootstrap. J Stat Plan Infer 117: 275–303, 2003. [Google Scholar]
- 34a.Wang JJ, Rau C, Avetisyan R, Ren S, Romay MC, Stolin G, Gong KW, Wang Y, Lusis AJ. Genetic dissection of cardiac remodeling in an isoproterenol-induced heart failure mouse model. PLoS Genet 12: e1006038, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Waters SB, Diak DM, Zuckermann M, Goldspink PH, Leoni L, Roman BB. Genetic background influences adaptation to cardiac hypertrophy and Ca(2+) handling gene expression. Front Physiol 4: 11, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weiss JN, Karma A, MacLellan WR, Deng M, Rau CD, Rees CM, Wang J, Wisniewski N, Eskin E, Horvath S, Qu Z, Wang Y, Lusis AJ. “Good enough solutions” and the genetics of complex diseases. Circ Res 111: 493–504, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang J, Manolio TA, Pasquale LR, Boerwinkle E, Caporaso N, Cunningham JM, de Andrade M, Feenstra B, Feingold E, Hayes MG, Hill WG, Landi MT, Alonso A, Lettre G, Lin P, Ling H, Lowe W, Mathias RA, Melbye M, Pugh E, Cornelis MC, Weir BS, Goddard ME, Visscher PM. Genome partitioning of genetic variation for complex traits using common SNPs. Nat Genet 43: 519–525, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zaphiriou A, Robb S, Murray-Thomas T, Mendez G, Fox K, McDonagh T, Hardman SM, Dargie HJ, Cowie MR. The diagnostic accuracy of plasma BNP and NTproBNP in patients referred from primary care with suspected heart failure: results of the UK natriuretic peptide study. Eur J Heart Fail 7: 537–541, 2005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.