

Abstract
Descending controls on spinal nociceptive processing play a pivotal role in shaping the pain experience after tissue injury. Secondary hypersensitivity develops within undamaged tissue adjacent and distant to damaged sites. Spinal neuronal pools innervating regions of secondary hypersensitivity are dominated by descending facilitation that amplifies spinal inputs from unsensitized peripheral nociceptors. Cyclooxygenase–prostaglandin (PG) E2 signaling within the ventrolateral periaqueductal gray (vlPAG) is pronociceptive in naive and acutely inflamed animals, but its contributions in more prolonged inflammation and, importantly, secondary hypersensitivity remain unknown. In naive rats, PG EP3 receptor (EP3R) antagonism in vlPAG modulated noxious withdrawal reflex (EMG) thresholds to preferential C-nociceptor, but not A-nociceptor, activation and raised thermal withdrawal thresholds in awake animals. In rats with inflammatory arthritis, secondary mechanical and thermal hypersensitivity of the hindpaw developed and was associated with spinal sensitization to A-nociceptor inputs alone. In arthritic rats, blockade of vlPAG EP3R raised EMG thresholds to C-nociceptor activation in the area of secondary hypersensitivity to a degree equivalent to that evoked by the same manipulation in naive rats. Importantly, vlPAG EP3R blockade also affected responses to A-nociceptor activation, but only in arthritic animals. We conclude that vlPAG EP3R activity exerts an equivalent facilitation on the spinal processing of C-nociceptor inputs in naive and arthritic animals, but gains in effects on spinal A-nociceptor processing from a region of secondary hypersensitivity. Therefore, the spinal sensitization to A-nociceptor inputs associated with secondary hypersensitivity is likely to be at least partly dependent on descending prostanergic facilitation from the vlPAG.
SIGNIFICANCE STATEMENT After tissue damage, sensitivity to painful stimulation develops in undamaged areas (secondary hypersensitivity). This is found in many painful conditions, particularly arthritis. The periaqueductal gray (PAG) is an important center that controls spinal nociceptive processing, on which secondary hypersensitivity depends. Prostaglandins (PGs) are mediators of inflammation with pronociceptive actions within the PAG under normal conditions. We find that secondary hindpaw hypersensitivity in arthritic rats results from spinal sensitization to peripheral A-nociceptor inputs. In the PAG of arthritic, but not naive, rats, there is enhanced control of spinal A-nociceptor processing through PG EP3 receptors. The descending facilitatory actions of intra-PAG PGs play a direct and central role in the maintenance of inflammatory secondary hypersensitivity, particularly relating to the processing of A-fiber nociceptive information.
Keywords: arthritis, descending facilitation, inflammation, periaqueductal grey, prostaglandins, secondary hyperalgesia
Introduction
The midbrain periaqueductal gray (PAG) and downstream nuclei within the medulla and brainstem, such as the rostroventral medulla, form a descending pain modulatory system that can augment or inhibit spinal processing of nociceptive information. This system affects pain experience by regulating the spinal processing of nociceptive inputs and the subsequent transmission of nociceptive information to the brain (Millan, 2002).
The balance of descending inhibitory and facilitatory control of spinal nociceptive processing contributes to the enhanced pain that develops after tissue injury. For example, sensitization to noxious stimulation can develop within undamaged tissue adjacent/distant to the damaged site, termed secondary hypersensitivity (also discussed as secondary hyperalgesia and/or allodynia; Treede and Magerl, 2000; Tracey and Mantyh, 2007). Spinal dorsal horn neurons that have receptive fields within the region of secondary hypersensitivity (referred to as “secondary sites” herein) are subject to both descending facilitatory and inhibitory controls from the midbrain. However, facilitation often exerts more influence than inhibition on inputs from secondary sites (Urban et al., 1996; Vanegas and Schaible, 2004). Maladaptation of these descending controls, resulting in net facilitation, is a major contributor to the development and maintenance of chronic pain (De Felice et al., 2011; Staud, 2013).
Prostaglandin E2 (PGE2) is a lipid signaling molecule with well described peripheral and spinal functions in nociception, particularly in inflammatory arthritis (Vanegas and Schaible, 2001; Korotkova and Jakobsson, 2014). One identified, but not yet fully characterized, tonic descending facilitatory system from the PAG uses endogenous PGs (Leith et al., 2007). Cyclooxygenase (COX) enzymes, key enzymes in the production of PGs, and all four PGE2 receptors, known as EP receptors 1–4 (Kennedy et al., 1982), are reportedly expressed in the PAG (Breder et al., 1992; Breder et al., 1995; Vaughan, 1998; Oliva et al., 2006; Palazzo et al., 2011; Myren et al., 2012). Unfortunately, complete information on cellular localization is not available due to a lack of available high-quality, specific antibodies (Myren et al., 2012). The endogenous tonic COX-1-PGE2 system within the ventrolateral PAG (vlPAG) primarily facilitates spinal processing of C-nociceptor inputs in naive rats, in that PGE2 injection in the vlPAG produces hyperalgesia (Heinricher et al., 2004; Leith et al., 2007) and COX-1 inhibition in the PAG results in analgesia (Tortorici and Vanegas, 1995; Leith et al., 2007). EP receptor signaling in the PAG contributes to both acute primary inflammatory hyperalgesia and neuropathic pain in the rat (Oliva et al., 2006; Palazzo et al., 2008).
PGE2 signaling within the vlPAG modulates ON cell activity within the rostral ventral medulla (RVM) (Heinricher et al., 2004), an important area controlling both descending facilitation and inhibition of the spinal cord (Heinricher et al., 2009). The RVM is critical in the development of secondary hypersensitivity (Urban et al., 1999; Vanegas, 2004; Ambriz-Tututi et al., 2011). However, the contribution of the PG/COX descending facilitatory system to secondary hypersensitivity is not known. Systemic nonsteroidal anti-inflammatory drugs (NSAIDs), which inhibit COX activity, can block sensitization in secondary sites in both humans and animal models (Petersen et al., 1997; Koppert et al., 2004; Stubhaug et al., 2007).
A-nociceptors and C-nociceptors play distinct roles in the initiation and maintenance of secondary hypersensitivity, respectively (Ziegler et al., 1999), and in the perceived quality of pain (Torebjörk, 1985). Peripheral C-nociceptors innervating joints in inflammatory arthritis are sensitized and display dramatically altered properties (Schaible et al., 2009). The resulting increase in C-nociceptor drive is hypothesized to sensitize central neurons to A-nociceptor inputs from secondary sites of undamaged tissue, resulting in secondary, usually mechanical, hypersensitivity (Ziegler et al., 1999; Magerl et al., 2001; You et al., 2010). However, our understanding of the additional contribution of descending systems to enhanced spinal A-nociceptor processing and the impact of this on secondary hypersensitivity remains limited.
In this study, we hypothesized that facilitatory vlPAG PG signaling through intra-PAG EP3 receptors (EP3Rs) contributes to secondary hypersensitivity of the hindpaw by augmenting the spinal processing of A-nociceptor, but not C-nociceptor, inputs from undamaged tissue.
Materials and Methods
Animal preparation
A total of 125 male Wistar rats (250–350 g) were used for all experiments. Any experimental animals subsequently excluded from analysis, and the reasons for doing so, are noted in the experimental procedures below. All procedures were performed in laboratories at the University of Bristol and Glaxo-Smith-Kline (GSK) in accordance with the UK Animals (Scientific Procedures) Act of 1986 plus associated guidelines and with the approval of the University of Bristol and GSK Ethical Review Groups. Animals were housed at 21°C and 55% relative humidity with a 12 h light/dark cycle. Food and water were provided ad libitum.
Implantation of chronic intracerebral guide cannulae
Intracerebral cannulae were implanted in 20 naive male rats. Anesthesia was induced with 3–5% isofluorane in O2 and then maintained at 1–2% via a nose cone placed around the snout. Once anesthetized, all animals received antibiotic treatment (1.0 ml/kg subcutaneous Synulox (clavulanic acid 35 mg/ml and amoxicillin 140 mg/ml; Pfizer) and preemptive analgesia with 0.4 ml of Rimadyl (carprofen50 mg/ml; Pfizer). Body temperature was maintained within physiological limits by means of a feedback-controlled heating blanket and rectal probe. Animals were then positioned in a stereotaxic frame and a guide cannula implanted into the PAG. A craniotomy was performed over the periaqueductal gray using a small hand-held drill (RS Components) fitted with a fine dental burr. Three further holes were drilled through the skull and screws inserted to provide stability for the guide cannula once in place. A stainless steel guide cannula (26 gauge, with the cannula cut 8 mm below the pedestal; tubing inner diameter 0.24 mm, outer diameter 0.46 mm; part number C315G, Plastics One) was slowly lowered into the brain aimed at the left vlPAG (coordinates relative to bregma were 7.4–7.5 mm caudal, 0.9 mm left of the midline, 6.5 mm below the skull surface). The guide cannula was then secured to the skull and screws with cyanoacrylate gel (RS Components). The area was thoroughly cleaned and the incision closed using absorbable suture (Ethicon Vicryl Rapide 4-0; Johnson & Johnson). A stylet cut to the same length as the implanted cannula (part number C315DC; Plastics One) was inserted into the implanted cannula and screwed tightly onto the cannula to maintain its patency until behavioral testing. After surgical procedures, animals received saline rehydration (5 ml subcutaneously) and were placed in a warmed environment until recovery of the righting reflex. Animals were then housed singly to minimize damage to the implanted cannula assembly and overlying wound and received 5 d of postoperative care consisting of soft paper bedding, a soft wet diet, and monitoring of body weight. No further experimental procedures were undertaken for at least 5 d and until preoperative body weight had been achieved.
Experimental protocol for microinjection of drugs into the PAG and nociceptive behavioral testing in naive animals
Baseline paw withdrawal thresholds to a linear thermal ramp stimulus (22–50°C ramp at 1°C/s delivered to the plantar paw surface using a Peltier device (contact area 3 × 4 cm) to the left hindpaw; equipment built in-house at GSK) were measured three times (Leith et al., 2014). The temperature at which the paw was withdrawn from the thermal stimulus was taken as the end point. A minimum of 30 min was left between repeated thermal stimuli on the same animal.
Animals were assigned randomly into two experimental groups (each n = 10) for drug administration. Compounds were administered via the implanted guide cannula, vehicle (30% DMSO in physiological saline), or GW671021B (EP3R antagonist; 250 nm), as used previously (Leith et al., 2007; Leith et al., 2014), in a total volume of 300 nl. The experimenter was blinded to the identity of the drug administered during the testing phase. Compounds were injected into the PAG using an internal “injector” guide cannula cut to project 0.5 mm beyond the end of the implanted guide cannula (Plastics One) connected to a 1 μl syringe (Scientific Glass Engineering). Animals were held securely and the stylet removed from the implanted guide cannula. Compounds were injected over 1 min and the injector was left in place for an additional minute after the completion of the injection to prevent backflow of the compound up the cannula. The stylet was then replaced into the implanted cannula. Paw withdrawal thresholds to the thermal ramp device were tested again 30 min after drug administration. At the end of the behavioral experiments, animals were killed by placement in an enclosure containing normal room air and were subject to a rising concentration of carbon dioxide gas, followed by confirmation of death by cessation of the circulation. Brains were removed and fixed in 4% paraformaldehyde in 0.1 m phosphate buffer for at least 24 h, then cryoprotected in 30% sucrose solution for at least 24 h, before sectioning at 60 μm. PAG injection sites were localized with reference to a rat brain atlas (Paxinos and Watson, 2006). Animals in which the cannula was found to have been outwith the vlPAG were used as a control for the regional effect of drug injection (n = 3). Animals receiving vehicle injection outside of the PAG were excluded (n = 4). Data from the vehicle-injected animals have been described previously for comparison with intra-PAG ketoprofen injection (Leith et al., 2014).
Experimental protocol for induction of secondary inflammatory hypersensitivity, nociceptive behavioral testing, and acute electrophysiological study
Inflammation was induced in a total of 50 animals. To induce secondary hyperalgesia of the hindpaw, animals received a single 100 μl intra-articular injection of complete Freund's adjuvant (CFA; 1 mg/ml; catalog #F5881, Sigma-Aldrich) into the left knee intra-articular space using a U100 needle (29G, U100, Terumo) under isoflurane anesthesia (2–3% in O2).
In a subset of the arthritic animals (n = 11) at 7 d after CFA, the knee width (n = 7) and the hindpaw thickness (n = 5) of the inflamed limb was measured using micrometer calipers (Camlab) and compared with measurements taken from age-matched naive animals (n = 5) to assess the extent of tissue edema. Before induction and 1, 3, and 7 d after intra-articular injection, 7 CFA animals also underwent nociceptive testing to assess the development of hindpaw secondary hyperalgesia/allodynia. Animals were habituated to the holding apparatus and experimenter beginning 3 d before the start of testing. For thermal hyperalgesia testing, the Hargreaves apparatus (Ugo Basile) was used to assess the development of thermal hyperalgesia after inflammation (Hargreaves et al., 1988). The latency to withdrawal from the radiant heat source directed onto the plantar surface of the left hindpaw was recorded and compared before and after inflammation. On the baseline testing day, animals were allowed to habituate to the chamber for 5–10 min before testing began. The radiant heat source was placed directly below the plantar surface and hindfoot behind the footpad. The intensity of the radiant heat source was adjusted so that animals gave withdrawal latencies at 12 ± 2 s. Three consecutive measurements were recorded and then averaged to give the withdrawal latency for that animal. An interstimulus interval (ISI) of at least 8 min was allowed to prevent sensitization. On experimental testing days, the same intensity of radiant heat was used and three consecutive recordings were made and averaged. We saw no evidence of acute or chronic sensitization of the hindpaw to thermal stimulation at this ISI (see Fig. 3, which shows stable withdrawal latencies between 1 and 7 d and very little variation in latency at each tested time). For mechanical hyperalgesia/allodynia testing, von Frey hairs (Ugo Basile) were used to deliver a known gram force to the middle of the plantar surface of the left hindpaw (middle of foot pad). Each hair was applied five times in an ascending order of force and the occurrence of a withdrawal (or display of nocifensive behavior) recorded. The delivery of a stimulus was stopped immediately after withdrawal/occurrence of nocifensive behavior. The percentage of withdrawals evoked by each hair was then calculated (between 0% and 100%). A stimulus–response curve was generated by application of a range of forces (typically 0.6–60 g) to give a full stimulus response curve from 0% to 100%. Stimulus–response curves were plotted for each animal, at each time point, and the 50% withdrawal threshold calculated as grams. These values were then compared across the inflammatory period. The distribution of weight across the hind limbs was measured using an incapacitance meter (incapacitance tester, Linton Instrumentation; Kobayashi et al., 2003) in 11 animals. Animals were habituated to a Perspex box that positioned animals so that the hindpaws rested on two force transducers. The forepaws were supported on an inclined Perspex slope. The weight distributed on the left leg was calculated as a percentage of the total weight distributed across both hind limbs. Two consecutive readings were taken at each experimental time point and the mean calculated. The proportion of total body weight borne on the hindpaws did not vary significantly over the course of the experiment.
Figure 3.

CFA-induced knee joint inflammation leads to behavioral secondary hyperalgesia and allodynia of the hindpaw. A, B, A single intra-articular knee joint injection of CFA (100 μl) leads to an increase in the width of the knee joint (Wilcoxon's signed-rank test, W = −28.0, p = 0.016, Shapiro–Wilk normality test for naive data, W = 0.58, p = 0.002; n = 7; A), but not in the hindpaw (unpaired t test, p = 0.11, t = 1.8, df = 8, n = 5; B). C, D, Knee joint inflammation produced a reduction in the withdrawal threshold to both thermal (C) and mechanical (D) stimuli delivered to the plantar surface of the hindpaw of the inflamed limb [repeated-measures ANOVA, p < 0.0001 (for both), F(3,18) = 39.69 (for C) and F(3,18) = 14.80 (for D) with Bonferroni's post hoc tests, ****p < 0.0001, ***p < 0.001, *p < 0.05, n = 7/group). E, Knee joint inflammation leads to a persistent reduction in the weight borne on the inflamed limb (Friedman test, Q = 21.99, p < 0.0001 followed by Dunn's multiple-comparison test, **p < 0.01, ***p < 0.001, Shapiro–Wilk normality test for day 0 data, W = 0.78, p = 0.005, n = 11). F, Withdrawal threshold to preferential A-nociceptor, but not C-nociceptor, activation was significantly lower in secondary sites in arthritic animals versus naive animals (one-way ANOVA, p < 0.0001, F(3,60) = 39.26 with Bonferroni's post hoc test ****p < 0.0001, n = 16).
Acute electrophysiological experiments in naive and arthritic animals
A total of 42 naive and 60 arthritic animals were used in these experiments. In 15 arthritic animals, EMG experiments could not be performed due unattainable or unstable baseline EMG thresholds and these were excluded from further analyses. All surgical preparation was performed under initial isoflurane anesthesia (2–3% in O2) and consisted of: (1) external jugular branch cannulation for anesthetic maintenance, (2) external carotid artery branch cannulation for blood pressure measurement, and (3) tracheal cannulation to maintain clear airways. Once an intravenous anesthetic line had been established, anesthesia was switched to a continuous infusion of alphaxalone diluted in physiological saline (1:2; ∼40 mg/kg/h; Alfaxan, Jurox) for the remainder of the experiments. Body temperature was maintained within physiological limits (37–38°C) by means of a feedback-controlled heating mat and rectal probe. Animals were then placed in a stereotaxic frame and a craniotomy was performed over the caudal PAG (−7.56 to −7.92 mm from bregma) using a dental drill (RS Components) and burr (catalog #014; Emile Lange) to allow for delivery of drugs to the PAG. Some animals (n = 21) also received a laminectomy of lumbar vertebrae 1 and 2 and thoracic vertebra 13 to allow for electrophysiological extracellular recordings of spinal dorsal horn neurons.
Recording of EMG activity in terminally anesthetized animals
Evoked thresholds for EMG activity recorded in the hindleg bicep femoris was assessed and used to quantitate noxious thermal withdrawal thresholds in 39 naive and 24 arthritic anesthetized animals (subsets of the total number of animals given above). For EMG recordings, a custom-made bipolar electrode was made from Teflon-coated steel wire (0.075 mm diameter; Advent Research Materials) and inserted into the bicep femoris of the left hindleg. The signal across the electrodes was amplified (x1K, A-B configuration; Neurolog NL104 amplifier, Digitimer), filtered (50 Hz–5 kHz, NL125, Neurolog), and raw data digitized via a CED 1401 and stored for off-line analysis using Spike2 (CED).
Recording of dorsal horn neuronal activity in terminally anesthetized animals
Extracellular recordings were made of spinal wide dynamic range (WDR) neurons in 21 arthritic animals (a subset of the total number of animals given above) under continuous alphaxalone anesthesia. The vertebral column was clamped on the vertebrae dorsal processes at each end of the laminectomy and at the lateral edges of the vertebral column around the recording site to stabilize the preparation. To locate the spinal site to perform electrophysiological recordings, spinal neuronal activity was electrically evoked (∼5 V, 0.1 ms, 0.1 Hz) from the contralateral hindpaw dorsum using subcutaneous bipolar stimulating electrodes. A low impedance ball electrode was tracked along the rostrocaudal extent of the exposed spinal cord and used to determine the location at which maximum dorsum potentials could be recorded. Once the site was determined, the dura was removed, a pool made with skin flaps, and the whole area filled with agar to further stabilize the preparation and to prevent excessive loss of tissue fluid. Once set, a recording window was cut out of the agar over the desired recording site and filled with warm paraffin oil. Using an electronic microstepper, a tungsten microelectrode (∼5 MΩ; Microprobes) (Merrill and Ainsworth, 1972) was lowered into the cord at the rostrocaudal location at which maximum dorsum potentials had been observed. Single-unit neuronal activity was amplified (x5K, A-B configuration, Neurolog NL104, Digitimer), filtered (300–5000 Hz, NL125, Digitimer), passed through a 50 Hz noise eliminator (Hum-Bug, Quest Scientific), and the raw data digitized via a CED 1401 and captured by a computer running Spike5 software.
Preferential A- and C-nociceptor activation
A- and C-heat nociceptors were activated preferentially using a custom-made heat lamp placed in contact with dorsal surface of the hindpaw (McMullan et al., 2004; Leith et al., 2007; Leith et al., 2014). This is a noninvasive method for preferential activation of either C-nociceptors (with slow rates of contact skin heating) or A-nociceptors (with fast rates of heating). The rates of skin heating used here reliably reproduce the same subcutaneous heating rates as those described previously that preferentially activate A- versus C-nociceptors at the single-fiber level (Yeomans and Proudfit, 1996; McMullan et al., 2004; Leith et al., 2007). One advantage of this noninvasive preferential activation method is the removal of the potential confound of different stimulus modalities used to activate different sensory neurons (thermal/mechanical) from the interpretation of the findings. This method also activates populations of neurons rather than individual fibers using a more physiological stimulus than, for example, electrical stimulation. It should be borne in mind, however that this method is a preferential activation and not a selective activation of different afferent groups. Briefly, a custom-built stainless steel casing positioned a sputter-coated 12 V bulb (halogen lamp, 15 v, 150 w; Phillips) over a copper disk (contact area 45 mm2) that was placed in contact with the surface of the paw. A constant voltage was applied to the lamp to provide fast (7.5 ± 1°C/s) or slow rates of heating (2.5 ± 1°C/s), which activate A-fiber (myelinated, capsaicin-insensitive) and C-fiber (unmyelinated, capsaicin-sensitive) heat nociceptors preferentially, respectively (McMullan et al., 2004; Leith et al., 2007). A T-type thermocouple attached to the copper disk was used to monitor temperature at the heat lamp–skin border during heating. The heat ramp apparatus was placed in contact with the paw for at least 60 s before the start of heating to allow for adaptation of low-threshold mechanoreceptors. Stimuli were delivered with an ISI of at least 8 min to prevent tissue damage and sensitization. An ISI of at least 8 min has been determined to be sufficient to prevent paw sensitization during the use of this stimulation method (McMullan et al., 2004; McMullan and Lumb, 2006a, 2006b; Leith et al., 2007; Géranton et al., 2009; Hughes et al., 2013; Leith et al., 2014). In addition, a feedback-controlled cutoff was set at 58°C for fast thermal ramps and 55°C for slow thermal ramps. In recordings in which thermal ramps reached cutoff temperature with no observable EMG activity, threshold was recorded as cutoff +2°C (Leith et al., 2007).
Experimental protocols and data analysis
EMG experiments.
Naive and arthritic (7 d) animals were anesthetized and underwent preparatory surgery for acute electrophysiological experiments as described above. After 1 h of postsurgery rest (under continued anesthesia), animals received alternating preferential A- and C-nociceptor stimulation of the hindpaw dorsum of the left leg. Alternating fast and slow thermal ramps were delivered with an interstimulus of at least 8 min until 3 consecutive thresholds within ±2°C were achieved. The mean withdrawal thresholds for A- and C-nociceptor activation were calculated and then compared between arthritic and naive animals. To assess effects of manipulating COX and EP3R function on A- and C-nociceptor evoked EMG thresholds, some animals underwent additional surgery and a craniotomy was performed as described above to deliver drugs to the midbrain PAG. In some animals (naive: 2 for vlPAG sulprostone, 5 for vlPAG GW671021B; arthritic: 4 for dorsolateral PAG (dlPAG) sulprostone, 6 for vlPAG ketoprofen, 7 for vlPAG vehicle, 6 for vlPAG GW671021B) drug effects on EMG (withdrawal) threshold to both A- and C-nociceptor activation were assessed in the same animal. After rest, mean baseline withdrawal thresholds to alternating A- and C-nociceptor activation were determined as above. Once a stable baseline had been established, a glass micropipette containing drug or control solution was lowered vertically into the vlPAG ipsilateral to the stimulated hindpaw. Solutions were injected into the PAG slowly (300 nl, >1 min) using a custom-made paraffin-filled pressure injection system connected to a 1 μl syringe (Hamilton). Alternating A- and C-nociceptor stimulation of the ipsilateral hindpaw was resumed at 1 min after drug delivery and withdrawal thresholds measured up to 120 min after delivery. The order of A- and C-nociceptor stimulation was counterbalanced across experiments. In some experiments, the effect of PAG-delivered drugs on EMG thresholds to A- or C-nociceptor stimulation was assessed separately in different rather than the same animals. The numbers of animals in which only A- or C-nociceptor stimulation was used were as follows: naive: 4 for vlPAG vehicle + A-nociceptor stimulation, 5 for vlPAG vehicle + C-nociceptor stimulation, 1 for vlPAG sulprostone + A-nociceptor stimulation, 3 for vlPAG sulprostone + C-nociceptor stimulation, 1 for vlPAG GW671021B + A-nociceptor stimulation, 2 for vlPAG GW671021B + C-nociceptor stimulation; and arthritic: 1 for vlPAG GW671021B + A-nociceptor stimulation. (Both A- and C-stimulation was used in other animals in each experimental group.) At the end of the experiments, animals were killed by overdose with sodium pentobarbital (30 mg, i.v. bolus; Sigma-Aldrich) and brains were dissected and processed as described above for injection site localization.
The threshold temperature (in degrees Celsius) at which EMG activity occurred was taken as the withdrawal threshold and was plotted against time to display the time course of any drug or vehicle effects. In arthritic EMG experiments, the timing of postdrug A- or C-nociceptor stimulations varied between animals; that is, ISIs were often >8 min due to counterbalancing. To more accurately display the data, mean postdrug stimulation and SEM values were calculated and plotted as x-axis error. The same analysis was not performed for EMG experiments in naive animals because postdrug A-/C-nociceptor stimulations were consistently delivered with an ISI of 8 min. Finally, because our experimental hypothesis specifically related to drug versus vehicle effects on EMG thresholds rather than the time course of such action, individual net area under the curve (AUC) for drug and vehicle effects were calculated from baseline values for individual animals and mean net AUC compared with determine specificity of drug actions.
Spinal neuronal electrophysiological experiments.
Effects of intra-PAG delivery of GW671021B/vehicle on action potential firing threshold of spinal dorsal horn WDR neurons to peripheral A- or C-nociceptor stimulation was assessed in 21 arthritic animals. Drug or vehicle effects on WDR neuron firing thresholds to peripheral A- or C-nociceptor stimulation were assessed in separate animals with an ISI of at least 10 min.
Functional classification of spinal WDR (class 2) neurons
WDR neurons were located in arthritic animals using a low-threshold search stimulus (brush, tap, prod) applied to the ipsilateral hindpaw to activate spinal neurons while an electrode was tracked vertically down through the spinal dorsal horn (250–1000 μm from the dorsal surface of the spinal cord) using an electronic microstepper. Once a cell was located and isolated, its receptive field was mapped using low-threshold stimuli to ensure that its excitatory receptive field could encompass the heat ramp contact area (>45 mm2). The spinal neuron was further characterized using limb movement, noxious, and non-noxious mechanical (26 g von Frey and brush) and noxious cold (ethyl chloride) to determine the neuronal type (proprioceptive, low-threshold, WDR, or nociceptive specific). Only cells that responded to both low- and high-threshold stimuli (including heat) were characterized further, corresponding to the class 2 neurons described previously (Menétrey et al., 1977; Menétrey et al., 1979). Nociceptive-specific (class 3) were not identified because a low-threshold search stimulus was used and therefore no class 3 neurons were isolated.
Neuronal activity of class 2 neurons was recorded continuously at 20 K samples/s using Spike2. A ‘wavemark’ template was created so that all matching spikes from the same cell were written to a separate channel at 20 K samples/s. This channel was duplicated and displayed both as the waveform of each event and as firing frequency from which the rate of spontaneous activity and the responses of the cell to peripheral stimuli could then be measured.
Spinal neuronal baseline responses to preferential A- or C-nociceptor activation were determined by delivering three consecutive thermal ramps by determination of the contact temperature at which single neuronal firing was stimulated. Either A- or C-nociceptor activation alone was used in each animal. During the ramping thermal stimulus, the activation threshold was taken as the temperature at which the evoked firing rate exceeded and remained elevated for >10% of the total stimulus time (see Fig. 7). Spontaneous neuronal activity was calculated over the period −10 to −5 s before the initiation of the thermal ramp stimulus.
Figure 7.

In the arthritic rat, EP3R antagonism in the vlPAG increases the firing threshold of spinal dorsal horn WDR neurons to C-nociceptor, but not A-nociceptor, activation in the hindpaw secondary hypersensitive site. A, B, Example traces of spinal WDR neuronal recordings and their responses to peripheral A-nociceptor (A) and C-nociceptor (C) activation before (baseline) and after injection of the EP3R antagonist into the vlPAG. Digitized data traces show: firing frequency of sorted spikes of individual WDR neurons, examples of action potential spike trains of spinal WDR neurons from which the firing frequency traces are derived, and changes in hindpaw dorsum contact skin temperature in response to the ramping thermal stimuli used to activate A- and C-nociceptors preferentially. Horizontal cursors in on firing frequency traces mark the mean firing rate in the 10 s preceding the start of thermal stimulus (e.g., 0.1 and 0.9 Hz in A). The thermal firing threshold is taken at the point in which thermally evoked activity exceeded the mean baseline firing rate and remained elevated for at least 10% of the total duration of thermal stimuli (shown by the vertical cursors (1) in A and B). WDR neuron thermal firing threshold temperatures (in degrees Celsius) are thus indicated using the horizontal cursors in skin contact temperature trace (e.g., 44.4°C and 43.1°C in A) as the skin temperature at which these conditions are met. Digitized data examples for both baseline responses and after EP3R antagonist injection into vlPAG are shown for A-nociceptor activation (A) and C-nociceptor activation (C). C, D, Injection of the EP3R antagonist GW6789101B had no significant effect on the firing threshold of spinal WDR neuronal firing thresholds to A-nociceptor stimulation (n = 6 for drug and 4 for vehicle; C) but increased WDR neuronal firing threshold to C-nociceptor stimulation (two-way ANOVA, main effect of treatment p < 0.0001, F(1,63) = 20.72 with Sidak's post hoc test **p < 0.01, n = 7 for drug and 4 for vehicle; D). E, Overall effect of EP3R antagonism on WDR neuron firing thresholds to C-nociceptor activation was significantly greater than that of a vehicle injection and greater when evoked by C- versus A-nociceptor activation (Kruskal–Wallis test, H = 11.31, p = 0.01 with Dunn's post test, *p < 0.05 Shapiro–Wilk normality test for C-nociceptor + EP3 antagonist data, W = 0.79, p = 0.03).
Once a stable baseline to heat stimulation was established (thresholds within 2°C of each other) a glass micropipette containing drug (EP3R antagonist GW671021B) or vehicle was lowered into the ipsilateral caudal vlPAG and injected as described above. Preferential A- or C-nociceptor activation was resumed 1 min after drug delivery and continued for 60 min with an ISI of at least 8 min. Mean (two to three stimulations) baseline and postdrug/vehicle firing thresholds were plotted against time to display the time course of any effects on spinal WDR neurons. For consistency with analysis of EMG data, the overall drug or vehicle effects on WDR firing threshold over the 60 min of recording (net AUC) were calculated and compared statistically to determine the specificity of drug actions.
Drugs
Stock solutions of all drugs (including vehicles) for microinjection were made up in dimethyl sulfoxide (DMSO; Sigma-Aldrich) at 10 mm and stored at −20°C until required. On the day of the experiment, an aliquot was thawed and diluted to working concentrations in vehicle solution (>90% PBS, <10% DMSO and containing 3–5% w/v pontamine sky blue dye to mark injection sites in electrophysiological experiments; 70% physiological saline, 30% DMSO vehicle was used in behavioral experiments). Despite some differences in DMSO concentration and the addition of pontamine sky blue, the vehicle control injections used in anesthetized and behavioral experiments produced no observable effects on EMG, spinal neuronal firing threshold (see Figs. 2, 4, 6, 7), or paw withdrawal threshold (see Fig. 1), respectively. All drugs were delivered in a final volume of 300 nl with the final drug doses based on effective doses as described by others and/or selectivity data for rat EP receptors. Drugs were as follows: ketoprofen (nonspecific COX inhibitor) 10 mm (10% DMSO); GW671021B 250 nm (<1% DMSO); rat EP3R antagonist selective over other EP, TP, FP, and DP receptors (Juteau et al., 2001; Belley et al., 2006; Su et al., 2008) made in-house at GSK; sulprostone, 1 nm (<1% DMSO); potent full EP3 agonist displaying activity at <1 nm in the rat, with only partial agonist activity at the EP1 receptor at doses considerably higher than used here (Coleman et al., 1994; Boie et al., 1997).
Figure 2.

In the anesthetized naive rat, modulation of vlPAG EP3R signaling affects the spinal processing of only C-nociceptor inputs. A, B, Injection of the EP3 agonist sulprostone into the vlPAG decreased only C-nociceptor-evoked withdrawal thresholds (EMG responses) (two-way ANOVA main effect of treatment, p = 0.0098, F(1,53) = 7.19, Sidak's post hoc test ns at all time points, sulprostone n = 3, vehicle n = 4) (N.B. significance attributable entirely to the last three data points). B Two-way ANOVA main effect of treatment p < 0.0001, F(1,92) = 69.67, Sidak's post hoc test **p < 0.01, sulprostone n = 5, vehicle n = 5; B). C, D, Similarly, the EP3R antagonist GW671021B increased only C-nociceptor-evoked withdrawal thresholds (two-way ANOVA main effect of treatment, p < 0.0001, F(1,100) = 132.0, effect of time p = 0.0016, F(12,100) = 2.90, interaction p = 0.005, F(12,100) = 3.29 with Sidak's post hoc test ****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05, GW n = 6, vehicle n = 4; GW, n = 6; vehicle, n = 5). E, The overall effect of agonist and antagonist on A-nociceptor-evoked withdrawals was not significantly larger than that of vehicle (one-way ANOVA of AUC in A and C, p = 0.27, F(2,10) = 1.49). F, Conversely, the overall effect of agonist and antagonist on C-nociceptor-evoked withdrawals was significantly different from that of vehicle (one-way ANOVA of AUC in B and D, p < 0.0001, F(2,14) = 47.27 with Bonferroni's post hoc test, ****p < 0.0001, *p < 0.05).
Figure 4.

In the arthritic rat, the ventrolateral and not the dorsolateral columns of the PAG mediate prostanergic effects on spinal nociception. A, B, Ketoprofen injected into the dlPAG had no notable effect on EMG-derived withdrawal thresholds to preferential A- and C-nociceptor activation in the hindpaw secondary site (n = 4 for both groups). C, D, Ketoprofen injected into the vlPAG increased withdrawal threshold to A- and C-nociceptor activation in the hindpaw secondary site (C: two-way ANOVA main effect of treatment p < 0.0001, F(1,89) = 26.23, effect of time p = 0.0051, F(8,89) = 2.99, interaction p = 0.0061, F(8,89) = 2.92 with Sidak's post hoc test **p < 0.01, *p < 0.05, n = 7 for vehicle and n = 6 for ketoprofen; D: two-way ANOVA main effect of treatment p < 0.0001, F(1,88) = 40.57, effect of time p = 0.048, F(7,88) = 2.13 with Sidak's post hoc test ***p < 0.001, *p < 0.05, n = 5 for ketoprofen, n = 7 for vehicle). E, Ketoprofen injected into the vlPAG had a significantly greater effect on EMG thresholds evoked by A-nociceptors than that of a vehicle injection. In addition, when delivered into the vlPAG, ketoprofen had a significantly greater effect on EMG thresholds evoked by A-nociceptors compared with delivery into the dlPAG (one-way ANOVA p = 0.0001, F(2,14) = 18.98 with Bonferroni's post hoc test ***p < 0.001, **p < 0.01; n = 4 for dlPAG, n = 6 for vlPAG ket, n = 7 for vlPAG vehicle). F, Ketoprofen injected into the vlPAG had a greater effect on EMG thresholds evoked by C-nociceptor activation than that of vehicle. In addition, when delivered into the vlPAG, ketoprofen had a greater effect on EMG thresholds evoked by C-nociceptor activation compared with delivery into the dlPAG (one-way ANOVA p = 0.0012, F(2,13) = 11.83 with Bonferroni's post hoc test **p < 0.01; n = 4 for dlPAG, n = 5 for vlPAG ket, n = 7 for vlPAG vehicle).
Figure 6.

In the arthritic rat, EP3R antagonism in the vlPAG increases EMG withdrawal thresholds to A- and C-nociceptor activation in the hindpaw secondary hypersensitive site. Injection of the EP3 antagonist GW671021B into the vlPAG increased EMG thresholds evoked by preferential A-nociceptor and C-nociceptor activation in the hindpaw secondary site (A: two-way ANOVA main effect of treatment p < 0.0001, F(1,86) = 38.53 with Sidak's post hoc test **p < 0.01, *p < 0.05, n = 6 for GW671021B and 7 for vehicle; B: two-way ANOVA main effect of treatment p < 0.0001, F(1,78) = 56.51, interaction p = 0.41 F(7,78) = 2.22 with Sidak's post hoc test ****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05, n = 5 for GW671021B and n = 7 for vehicle). C, EP3R antagonist had a greater effect on EMG thresholds evoked by A- and C-nociceptor activation than vehicle (one-way ANOVA p < 0.0001, F(3,21) = 20.84 with Bonferroni's post test ****p < 0.0001, **p < 0.01, n = 6 and 5 for EP3 A- and C-nociceptor groups, respectively, and n = 7 for both vehicle groups). D, Effect of the EP3R antagonist on EMG thresholds evoked by C-nociceptor stimulation was equivalent in arthritic and naive animals. However, the effect of the EP3R antagonist on EMG thresholds evoked by A-nociceptor stimulation was greater in arthritic versus naive animals (one-way ANOVA p = 0.0037, F(3,20) = 6.2 with Bonferroni's post test, **p < 0.01; n = 6 for A-nociceptor naive and arthritic, n = 7 for C-nociceptor naïve, and n = 5 for C-nociceptor arthritic).
Figure 1.

Blockade of EP3 signaling within the vlPAG increases thermal withdrawal threshold in the awake naive rat. Delivery of the PG E2 EP3R antagonist (GW671021B) to the vlPAG increased thermal withdrawal thresholds evoked from the hindpaw (Kruskal–Wallis test p = 0.002, H = 18.64 with Dunn's post hoc test, **p < 0.01, *p < 0.05, Shapiro–Wilk normality test for EP3 antagonist postdrug data, p = 0.006, W = 0.72; vehicle, n = 6; EP3R antagonist, n = 7; outside PAG, n = 3).
Tissue isolation and PAG mRNA analysis
In five arthritic and five naive animals, tissue samples of the functional PAG columns (ventrolateral and dorsolateral) were isolated from whole PAG using a custom-built tissue puncher. Briefly, the sharp tip of a 21 G needle (internal diameter 0.5 mm; Terumo) was removed using a Dremel and cutting disk, the newly cut edge sharpened to create a cutting edge, and the needle center bored out to create a tissue punch with an internal diameter of 0.5 mm. Clean working habits were followed to prevent contamination of the apparatus. The work area and apparatus was cleaned with 70% alcohol before and after work was conducted on each sample, a new apparatus (including microtome blade, cutting disk, and slides) were used for each sample, and sterile disposables and filter pipette tips were used whenever possible (e.g., gloves, needles, syringes, etc.). Animals were killed with an overdose of intraperitoneal barbiturate (60 mg/kg). Brains were dissected and the PAG removed with a clean razor blade aided by a Perspex guide block (1 mm brain slicer; Zivic Instruments). Using a freezing microtome, and with reference to images from the rat brain atlas (Paxinos and Watson, 2006), 50 μm sections were cut from the caudal end of PAG to the level at which the ventrolateral columns ended. At ∼8.3 mm from bregma, a 1-mm-thick section containing vlPAG columns was carefully cut using the microtome, placed onto a glass slide, and the sample flash frozen on dry ice. A second 1-mm-thick section (caudal end of the section ∼7.30 mm caudal to bregma) was then made immediately at the point where the dlPAG is clearly defined. Aided by a dissecting microscope, ventrolateral and dorsolateral holes were punched out from respective sections using the premade tissue puncher. Coordinates used were made with reference to the rat brain atlas (Paxinos and Watson, 2006) to ensure tissue section contained relevant PAG columns throughout its thickness. Each final punched sample was 1 mm thick with a 0.5 mm diameter. Clean syringes and tissue punches were used for each sample. Samples were stored at −80°C until further processing.
Punched tissue samples from vlPAG and dlPAG were homogenized in 1 ml of TRIzol/50 mg of tissue and RNA was extracted following the manufacturer's instructions. Extracted RNA was treated with DNase I to eliminate any genomic DNA and then reverse-transcribed using MMLV reverse transcriptase. To detect COX-1, COX-2, and EP3R mRNA in the vlPAG, PCR was performed (PTC-200 Peltier Thermal Cycler; MJ Research) using previously published primers (Donaldson et al., 2001; Guo et al., 2006; Myren et al., 2010) and cycling conditions shown in Table 1. COX-2 primers were designed using the online primer design tool PRIMER-BLAST. GAPDH PCR was used as an internal control and “no RT” controls were included in all reactions to ensure that there was no genomic DNA contamination of the samples. PCR products were run and visualized (Gel Doc Ez Imager; Bio-Rad) on 2% agarose gels (w/v) in Tris-acetate-EDTA buffer containing ethidium bromide. PCR amplicon intensities were quantitated using ImageJ. Data are shown as intensity relative to GAPDH intensity in the same sample.
Table 1.
PCR primers, conditions, and amplicon sizes
| Target | Primer sequences | Amplification conditions (dehaturation, annealing, extension, cycle number) | Amplicon size (bp) |
|---|---|---|---|
| Cox-1 | 94°C 30 s | 116 | |
| Upstream | GGCGTTGCACATCCATCTACTC | 50°C 30 s | |
| Downstream | AGCATCTGTGAGCAGTACCGG | 72°C 60 s | |
| 35 cycles | |||
| Cox-2 | 94°C 30 s | 98 | |
| Upstream | GATGACGAGCGACTGTTCCA | 45°C 30 s | |
| Downstream | TGGTAACCGCTCAGGTGTTG | 72°C 60 s | |
| 35 cycles | |||
| EP3 | 94°C 30 s | 144 | |
| Upstream | GAGACGGCTATCCAGCTTAT | 60°C 30 s | |
| Downstream | ATTGCACTCCTTCTCCTTTC | 72°C 60 s | |
| 35 cycles | |||
| GAPDH | 94°C 30 s | 180 | |
| Upstream | CAACTACATGGTTTACATGTTC | 50°C 30 s | |
| Downstream | GCCAGTGGACTCCACGAC | 72°C 60 s | |
| 35 cycles |
Statistics
All data are displayed as mean and SEM unless otherwise stated. Data were tested for normality and results are reported in figure legends for significant findings only. Shapiro–Wilk tests were used with groups of sufficient size, but in some instances where there were smaller groups, the Kolmogorov–Smirnov test was used, as stated in the figure legends. Normally distributed data with equal variance were compared using GraphPad Prism (version 4/5) and paired/unpaired Student's t test or ordinary or repeated-measures one-way ANOVA followed by adjusted post hoc tests for multiple comparisons, respectively, as detailed in the text. Two-way ANOVA was used to compare the effect of drugs on EMG withdrawal thresholds over time (see Figs. 2, 4, 6, 7), followed by post hoc tests for multiple comparisons as detailed in the text and figure legends. For two-way ANOVA, normality of data was assumed. If the data did not meet the assumptions of parametric tests (non-Gaussian, unequal variances), then nonparametric test equivalents (Mann–Whitney U test, Wilcoxon signed-rank test, Kruskal–Wallis test, or Friedman's test) were used, as stated in the figure legends. For mRNA expression data, normality was not tested or assumed and Mann–Whitney U tests were used to determine the exact p-values. The probability at which significance was accepted (α) was adjusted to account for the multiple tests used (two) and therefore set at p = 0.025 (p = 0.05/2).
Results
Blockade of the PG EP3R within the vlPAG modulates withdrawal reflex thresholds in conscious naive rats
We have previously shown that inhibition of COX in the vlPAG increases paw withdrawal thresholds in both awake rats (Leith et al., 2014) and anesthetized rats (Leith et al., 2007), suggesting a tonic PAG COX-dependent pronociceptive system. Delivery of the EP3R antagonist GW671021B into the vlPAG increased thermal paw withdrawal thresholds compared with both baseline and vehicle (EP3R antagonist baseline vs postdrug = 44.9 ± 0.1 vs 45.9 ± 0.2°C, vehicle baseline vs postvehicle = 44.4 ± 0.3 vs 44.6 ± 0.2°C; Fig. 1). Delivery of GW671021B into the area outside of the PAG did not significantly alter paw withdrawal thresholds (baseline vs postdrug = 44.9 ± 0.3 vs 45.0 ± 0.1°C; Fig. 1).
EP3Rs within the vlPAG modulate withdrawal reflexes evoked by peripheral C-nociceptor, but not A-nociceptor, activation in naive rats
The EP3 agonist sulprostone injected into the vlPAG had no effect on withdrawal thresholds evoked by A-nociceptor activation (Fig. 2A,E), but decreased the withdrawal threshold to C-nociceptor activation compared with baseline (52.2 ± 0.4°C to a minimum of 49.5 ± 0.5°C at t = 33 min; Fig. 2B,F). Sulprostone injection into the dlPAG had no effect on either A- or C-nociceptor withdrawal thresholds [A-nociceptor: baseline (55 ± 0.8°C) vs 15 min (54 ± 3.2°C) vs 45 min (56 ± 0.9 °C) vs 90 min (54 ± 1.4°C) post drug; C-nociceptor: baseline (55 ± 1.1°C) vs 15 min (57 ± 0.3°C) vs 45 min (54 ± 1.5°C) vs 90 min (54 ± 1.9°C) postdrug, n = 3 for both]. The EP3R antagonist GW671021B had no effect on withdrawal thresholds evoked by A-nociceptor activation (Fig. 2C,E), but increased the withdrawal threshold to C-nociceptor activation compared with baseline (52 ± 0.4°C to 56 ± 0.4°C at t = 33 min; Fig. 2D). Comparison of the overall effect of the two compounds, the EP3R agonist sulprostone and the EP3R antagonist GW671021B, confirmed that they had no significant effect on A-nociceptor evoked withdrawal thresholds compared with vehicle (AUC EP3 agonist vs AUC vehicle = −10.0 ± 43.7 vs 41.4 ± 24.0°C · min, AUC EP3 antagonist vs AUC vehicle = 89.9 ± 41.1 vs 41.4 ± 24.0°C · min; Fig. 2E). The effects of EP3 agonist and EP3 antagonist on C-nociceptor evoked withdrawal thresholds were significantly greater than vehicle (AUC EP3 agonist vs AUC vehicle = −164.6 ± 38.50°C · min vs −6.2 ± 18.50°C · min, AUC EP3 antagonist vs AUC vehicle = 302.9 ± 39.27 vs 6.2 ± 18.50°C · min; Fig. 2F).
CFA-induced knee joint arthritis leads to a secondary hyperalgesia and allodynia of the hindpaw associated with an enhanced spinal processing of A-nociceptor, but not C-nociceptor, inputs
As shown previously on many occasions (Rees et al., 1996; Martindale et al., 2007), knee joint inflammatory arthritis resulted in an increase in joint swelling (mesio-lateral distance measured across the joint, baseline vs 7 d = 7.6 ± 0.2 vs 9.1 ± 0.2 mm; Fig. 3A). The thickness of the hindpaw of the inflamed limb did not change in size as a result of knee joint inflammation (baseline vs 7 d = 4.1 ± 0.1 vs 4.2 ± 0.1 mm, p = 0.118; Fig. 3B). Inflammatory arthritis led to a significant fall in hindpaw thermal withdrawal latency (Fig. 3C), as described previously (Davis et al., 1994; Vermeirsch et al., 2007) apparent at 24 h and persisting for at least 7 d (baseline vs 24 h vs 7 d withdrawal latencies = 12.4 ± 0.4 vs 8.1 ± 0.5 vs 8.3 ± 0.4 s, respectively; Fig. 3C). Arthritis also resulted in a significant hindpaw mechanical allodynia over the same time frame (baseline vs 24 h vs 7 d withdrawal thresholds = 22 ± 2 vs 14 ± 4 vs 4 ± 1 g, respectively; Fig. 3D) and a decrease in the weight borne on the inflamed limb (baseline vs 7 d = 49 ± 0.5 vs 23 ± 1.5%; Fig. 3E).
As we have also shown in a different cohort of animals using the same model (Hsieh et al., 2015), anesthetized arthritic animals had significantly lower withdrawal thresholds only to A-nociceptor stimulation in the hindpaw, the secondary hyperalgesic site (A-nociceptor-evoked withdrawal threshold naive vs arthritic, secondary hyperalgesia = 57 ± 0.2 vs 54 ± 0.3°C, respectively; Fig. 3F). C-nociceptor-evoked withdrawal thresholds were unchanged in arthritic rats (naive vs arthritic = 54 ± 0.2 vs 54 ± 0.1°C, p = 0.266; Fig. 3F).
vlPAG is the site of prostanergic descending facilitation in the anesthetized inflamed rat
We have found previously that the vlPAG, and not the dlPAG, forms the site of prostanergic descending facilitation in the naive anesthetized rat (Leith et al., 2007). To determine whether a similar anatomical delineation extended to arthritic animals, the nonspecific COX inhibitor ketoprofen was injected into the dlPAG and vlPAG to determine its effects on EMG withdrawal thresholds to A- or C-nociceptor activation in the area of secondary hypersensitivity. Ketoprofen injected into the dlPAG had no effect on withdrawal thresholds to either A- or C-nociceptor activation [A-nociceptor baseline (55 ± 0.7°C) vs 9.5 min (55 ± 0.7°C) vs 42.5 min (54 ± 0.7°C) vs 75 min (52.7 ± 0.8°C); Fig. 4A; C-nociceptor baseline (52.7 ± 0.5°C) vs 5.3 min (53.0 ± 0.9°C) vs 38.8 min (52.7 ± 0.5°C) vs 86.3 min (53 ± 0.6°C); Fig. 4B]. These datasets had low variability, but also low numbers of animals, which would not allow for variability to be seen if present. We performed post hoc power calculations and determined that the actual power of these experiments was 0.99 (both A- and C-stimulation). Sample size calculations using the data generated in these experiments also suggested that 22 animals (A-nociceptor stimulation) and 48 animals (C-nociceptor stimulation) would be necessary to see statistical significance on these measures. We therefore concluded that any fluctuations in measures in these experiments were not likely to be physiologically significant and that the lack of effect of ketoprofen in the dlPAG is a true negative rather a type II statistical error.
When delivered into the vlPAG, ketoprofen increased withdrawal thresholds to both A- and C-nociceptor stimulation (A-nociceptor baseline vs 74 min = 52.8 ± 0.6 vs 57.8 ± 1°C, Fig. 4D; C-nociceptor baseline vs 72.5 min = 52.6 ± 0.6 vs 55.8 ± 0.7, Fig. 4C). The effect of vlPAG ketoprofen on withdrawal thresholds to A- and C-nociceptor stimulation was significantly greater than vehicle injection (A-nociceptor AUC vlPAG ket vs AUC vlPAG vehicle = 375.6 ± 66.1 vs −61.1 ± 33.0°C · min, Fig. 4E; C-nociceptor AUC vlPAG ket vs AUC vlPAG vehicle = 239.0 ± 45.6 vs −3.9 ± 34.5°C · min; Fig. 4F). The vlPAG expression of COX-1 was increased (Fig. 5A), but COX-2 mRNA was unchanged, in arthritic animals (Fig. 5B). Surprisingly despite the lack of functional effects on nociceptive reflexes of COX inhibition in the dlPAG, COX-1 mRNA was upregulated, and COX-2 mRNA downregulated in the dlPAG of arthritic animals.
Figure 5.
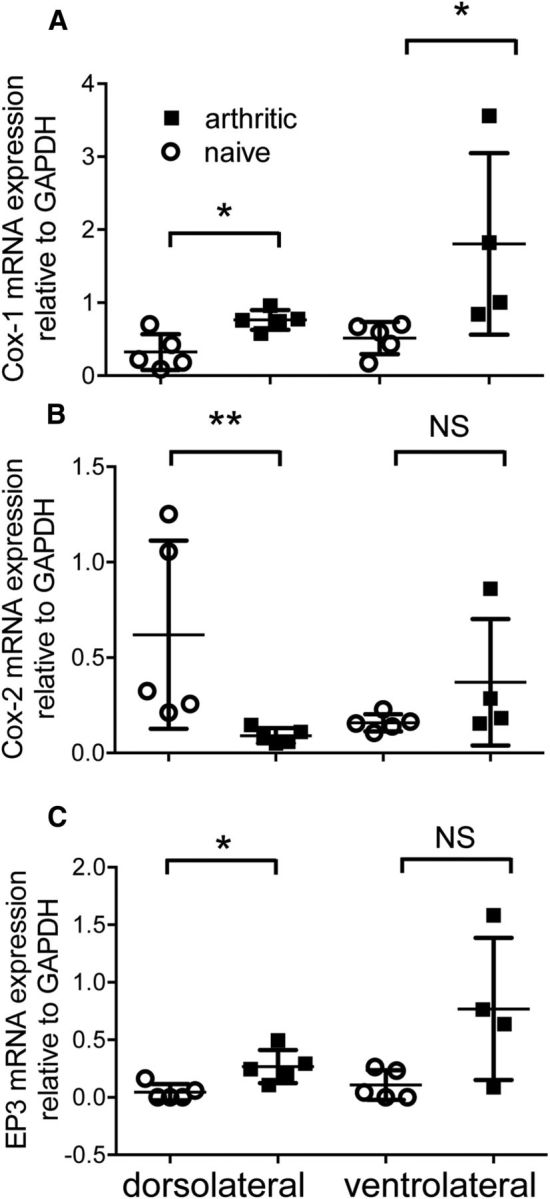
COX enzyme and EP3R mRNA expression are regulated within the PAG after knee joint arthritis. A, COX-1 mRNA expression was significantly increased in both vlPAG and dlPAG 7 d after induction of knee joint arthritis [Mann–Whitney U = 0 (sum of ranks, 15, 30; vlPAG) and U = 1 (16,39; dlPAG), p = 0.016 for both (α = 0.025, corrected for multiple comparisons)]. B, COX-2 mRNA expression was significantly decreased in dlPAG [U = 0 (40, 15), p = 0.008], but unchanged in vlPAG [U = 3 (18, 27), p = 0.11]. C, EP3 mRNAs had a tendency to increase in vlPAG [U = 2 (17, 28), p = 0.06] and was significantly increased in dlPAG [U = 1 (16, 39), p = 0.02].
In the anesthetized arthritic rat, vlPAG EP3R antagonism affects the spinal processing of both A- and C-nociceptor inputs from an area of secondary hypersensitivity
EP3R antagonist reversed the lowered withdrawal thresholds to A-nociceptor activation in the hindpaw area of secondary hypersensitivity in arthritic rats (baseline vs postdrug = 53.8 ± 0.5°C vs 58.7 ± 0.7°C at 4.5 min postdrug; Fig. 6A). Thresholds were raised to higher values than those seen in naive animals (cf. Figs. 2C, 6A) and the overall effect of EP3R antagonist was significantly greater in inflammatory secondary hypersensitivity compared with naive animals (A-nociceptor AUC naive vs AUC arthritic = 89.9 ± 41.1 vs 375.6 ± 66.1°C · min; Fig. 6D). In contrast, the prostanergic facilitatory tone on C-nociceptor reflex withdrawal thresholds seen in naive animals was maintained at the same level in secondary hypersensitivity as seen in naive animals, as intra-vlPAG EP3R antagonist increased withdrawal thresholds to C-nociceptor activation (baseline vs postdrug = 53.0 ± 0.8°C vs 56 ± 1.0°C at 60 min postdrug; Fig. 6B) to a degree equivalent to that seen in the naive animals (C-nociceptor AUC naive vs AUC arthritic = 302.9 ± 39.27 vs 239.0 ± 45.6°C · min; Fig. 6D). The overall effect of EP3R antagonism on A- and C-nociceptor-evoked thresholds in inflammatory secondary hypersensitivity was equivalent in inflammatory arthritis and significantly greater than vehicle (EP3R A-nociceptor vs vehicle A-nociceptors = 375.6 ± 66.1 vs −61.1 ± 33.0°C · min, EP3R C-nociceptor vs vehicle C-nociceptors = 239.0 ± 45.6 vs −3 ± 34.5°C · min; Fig. 6C). EP3R mRNA expression was unchanged in vlPAG in arthritis. Like COX-1, EP3R mRNA was upregulated in the dlPAG in arthritic rats (Fig. 5C).
In the anesthetized arthritic rat, vlPAG EP3R antagonism modulates spinal dorsal horn neuronal responses to C- but not A-nociceptor inputs from an area of secondary hypersensitivity
Intra-vlPAG EP3R antagonist had no effect on the firing threshold of WDR neurons in knee arthritic rats to preferential A-nociceptor activation on the hindpaw [baseline (46.0 ± 1.2°C) vs 10 (45.83 ± 1.49°C) vs 30 (45.17 ± 1.89°C) vs 60 (43.67 ± 0.67°C) mins postdrug, evoking responses at thresholds similar to those previously reported (McMullan and Lumb, 2006a; Leith et al., 2014); Fig. 7A,C,E]. In contrast, EP3R antagonist increased neuronal firing threshold in response to preferential activation of hindpaw C-nociceptors (baseline vs 20 min postdrug = 45.7 ± 1.4°C and 51.0 ± 2°C; Fig. 7B,D,E). The effect of EP3R antagonism was significantly greater on C-nociceptor compared with A-nociceptor responses and significantly greater than vehicle (EP3 C-nociceptor vs EP3 A-nociceptor = 175.7 ± 39.75 vs −39.17 ± 48.76°C · min; EP3 C-nociceptor vs vehicle C-nociceptor = 175.7 ± 39.7 vs −38.73 ± 55.7°C · min; Fig. 7E).
The thresholds of evoked EMG withdrawals are higher than, but encompass the range of, thresholds of WDR neurons in this study, (e.g., A-nociceptor evoked WDR firing threshold range = 42–55°C; A-nociceptor evoked EMG thresholds = 52–56°C). Values for EMG and WDR thresholds are consistent with our previous studies (Leith et al., 2007; Leith et al., 2014). Hindlimb withdrawal requires coordinated, integrated activity in a multineuronal pathway spanning dorsal and ventral horns, which includes contributions from low-threshold, WDR, and nociceptive-specific neurons, not WDR neurons alone. Therefore, the threshold for limb withdrawal (EMG activity) is likely to be higher than any single neuron within that circuitry and should be closest to those spinal neurons with the highest thresholds (e.g., nociceptive specific neurons). It should also be noted that the anesthetic alphaxalone augments GABAergic inhibition, which will affect network function and exert a cumulative effect on the polysynaptic withdrawal reflex pathway. Both of these actions might be expected to raise EMG thresholds relative to WDR action potential firing thresholds.
Discussion
COX–PG signaling has well described pro-nociceptive and anti-nociceptive effects at peripheral and spinal sites, but its actions at supraspinal sites are less well defined. The PAG expresses both COX isoforms (Fig. 5; Breder et al., 1992; Breder et al., 1995) and the PG EP receptors EP1–4, although precise cellular localizations are still unclear (Palazzo et al., 2011; Myren et al., 2012). The vlPAG, rather than other PAG subregions, exerts a specific facilitatory effect over spinal nociceptive processing in naive animals (Heinricher et al., 2004; Leith et al., 2007; Palazzo et al., 2011) and in acute inflammatory and neuropathic models (Oliva et al., 2006; Palazzo et al., 2011).
Central inhibition of COX in lateral PAG (Tortorici and Vanegas, 1995) and rostroventral medulla contributes to descending inhibition through potentiation of opioid analgesia (Vaughan et al., 1997; Vaughan, 1998). Cannabinoid-stress-induced analgesia derives from multiple PAG columns (Lichtman et al., 1996; Olango et al., 2012) and can be engaged by intra-PAG COX inhibition (Vanegas et al., 2010). Our data are, however, consistent with a discrete endogenous vlPAG prostanergic-EP3R receptor mechanism directly facilitating spinal C-nociceptive processing in conscious and anesthetized naive animals and A-nociceptive processing in hindpaw secondary hypersensitivity in arthritic animals. Hindpaw secondary hypersensitivity occurs in knee-joint arthritis (Herrero and Cervero, 1996; Urban et al., 1999; Zhang et al., 2002; Martindale et al., 2007), with sensitization of the spinal processing of A-nociceptor inputs (Fig. 3F; Hsieh et al., 2015). De novo central A-nociceptor sensitization is dependent on intra-vlPAG PGs, most likely PGE2, because it is reversed with the EP3R antagonist.
The absence of effect of vlPAG EP3R antagonism on A-nociceptor-evoked WDR firing thresholds in arthritic animals was surprising considering the clear sensitization of A-nociceptor-evoked noxious withdrawals. Deep dorsal horn WDR neurons are integral to spinal reflex circuitry and activity in WDR neurons is well correlated with motor unit activity during withdrawal (You et al., 2003), hence the targeting of WDR neurons in this study. One interpretation of our findings is that the effects on A-nociceptor processing are mediated in the ventral rather than dorsal horn. The PAG has direct projections to the lumbar spinal ventral horn despite the majority of PAG projections targeting the cervical cord (Mouton and Holstege, 1994). We consider an action in the ventral horn to be unlikely as if the additional facilitation of A-nociceptor processing observed in arthritic animals were mediated at the level of the ventral horn then similar effects on C-nociceptor processing would be expected (Fig. 6D). Because A- and C-nociceptor information is integrated at this spinal level, it is therefore more likely that effects on A-nociceptive processing in arthritic animals are mediated through alternative dorsal horn neuronal populations, such as nociceptive-specific and/or WDR neurons located in superficial laminae. Descending inhibitory pathways originating from the vlPAG modulate both A- and C-nociceptor evoked c-fos expression in superficial laminae (indicative of spinal neuronal activation), but affect only C-nociceptor-evoked c-fos expression in deeper laminae (Koutsikou et al., 2007).
Peripheral nociceptors innervating secondary sites do not display significant sensitization (Baumann et al., 1991; LaMotte et al., 1992; Torebjörk et al., 1992; also see Serra et al., 2004). The development of secondary hypersensitivity is critically dependent on central sensitization, including potentiation of synaptic efficacy, and engagement of descending controls (Urban et al., 1999; Sandkühler, 2009; Naka et al., 2013). Our findings are consistent with the hypothesis that activity in capsaicin-sensitive C-nociceptors (McMullan et al., 2004; Leith et al., 2007) innervating the area of tissue damage drives central sensitization (Woolf, 1983; Leith et al., 2007; You et al., 2010) and facilitates the spinal processing of A-nociceptor inputs from secondary sites, in part through descending facilitation (LaMotte et al., 1992; Treede et al., 1992; Magerl et al., 2001; Hsieh et al., 2015). Interpretation of these findings is, however, dependent on the selectivity of the heat-ramp stimulation. If a significant number of A-nociceptors were also activated by slow ramps, then we would also expect to see facilitation of slow-ramp-evoked reflexes in secondary hypersensitivity, which we do not. In addition, capsaicin, a sensitizer of TRPV1-expressing C-nociceptors, affects only slow-ramp responses; DMSO, a sensitizer of A-nociceptors, affects only fast-ramp responses. In WDR neurons, the magnitude of C-fiber response to electrical stimulation correlates with the response to slow-ramp activation (Leith et al., 2007). This evidence suggests that this method activates cutaneous C- and A-nociceptor populations preferentially (Yeomans and Proudfit, 1996).
Changes in the balance of descending inhibitory and facilitatory controls can reduce the threshold for long-term potentiation (LTP) induction, allowing noxious stimuli that are usually unable to drive central sensitization to do so (Sandkühler, 2009). For instance, electrical stimulation of peripheral nerves at A-fiber strength produces LTP at C-fiber synapses only when descending controls are disrupted (Liu et al., 1998). C-nociceptive drive from the primary injury site may affect the spinal processing of A-nociceptor inputs from the secondary sites through both spinal and supraspinal mechanisms because C-nociceptors terminate exclusively in the superficial dorsal horn and drive spinal–supraspinal–spinal loops via spinal projection neurons (Light and Perl, 1979; Suzuki et al., 2002; Mantyh and Hunt, 2004). Importantly, heterosynaptic LTP of synaptic efficacy may unmask and/or increase A-fiber drive to spinal nociceptive circuits. This could lead to the central sensitization to A-fiber inputs. Descending facilitation from the rostral ventral medulla (RVM) is critical for the maintenance of secondary hyperalgesia in rodents (Urban and Gebhart, 1999) and, notably, PG signaling within the vlPAG modulates RVM ON and OFF cell activity in naive and neuropathic rats (Heinricher et al., 2004; Palazzo et al., 2011). This suggests that a spinal/PAG/RVM/spinal loop underpins the vlPAG-prostanergic facilitatory pathway.
Secondary hypersensitivity is often suggested to be limited to mechanical stimulation (Treede et al., 1992) because inflammatory secondary thermal hyperalgesia is not present in the capsaicin model (Raja et al., 1984; Ali et al., 1996). However, secondary thermal hypersensitivity is sometimes, but not always (Pertovaara, 1998), reported in clinical studies (Bajaj et al., 2001; Schaible et al., 2009) and experimental studies (Fig. 3C; Herrero and Cervero, 1996; Urban et al., 1999; Zhang et al., 2002; Martindale et al., 2007; Hsieh et al., 2015), so it is more likely that secondary hypersensitivity is a central A-nociceptor-sensitization, not a modality-specific (thermal/mechanical) process (Hsieh et al., 2015).
Although peripheral C-nociceptors are not sensitized in secondary sites (Baumann et al., 1991; LaMotte et al., 1992; Torebjörk et al., 1992), C-nociceptor-evoked reflexes and neuronal responses are reported to be facilitated at secondary sites (Herrero and Cervero, 1996; Martindale et al., 2007). However, this may represent a potential confound of electrical C-fiber stimulation parameters in inflamed animals. The simplest interpretation of our finding of unchanged C-nociceptor-evoked reflexes in arthritic animals is that there is no additional facilitation of C-nociceptive inputs from secondary sites, in contrast to the effect on A-nociceptive processing.
In vlPAG, COX-1 mRNA levels are altered, suggesting that extension of the prostanergic facilitation to A-nociceptor inputs may result from increased intra-vlPAG COX-1-dependent PG production acting on EP3R. Altered CNS COX expression has been attributed to increased central interleukin-1beta (IL-1beta) levels found in inflammatory arthritis in humans and animals (Samad et al., 2001; Kosek et al., 2015). IL-1beta exerts different effects in different regions of the brain (An et al., 2011), for example, increased expression of COX-1 in perivascular, endothelial, and glial cells (García-Bueno et al., 2009; Matousek et al., 2010); COX-2 expression in multiple cell types including neurons (Crofford et al., 1994; Serou et al., 1999; Samad et al., 2001); and EP3Rs on CNS glial cells (Waschbisch et al., 2006). Intra-vlPAG EP1 receptors are implicated in the facilitation of neuropathic pain; surprisingly, these receptors are reportedly downregulated but remain functional in this condition (Palazzo et al., 2011).
Although COX-1, COX-2, and EP3R expression is regulated in the dlPAG in inflamed rats, inhibition of this system in dlPAG had no effect on nociceptive processing. The dlPAG is implicated in active rather than passive behavioral responses to stress/fear, including autonomic control of breathing and cardiovascular function (Dampney et al., 2013). There is no published evidence linking COXs and/or PGs with any dlPAG functional responses, although PGE2 modulates dlPAG neuronal function by EP3R-dependent inhibition of glutamatergic inputs (Lu et al., 2007).
Here, we have demonstrated that descending facilitation from the PAG affects nociceptive processing in awake animals through intra-vlPAG EP3R signaling. In the naive state, this descending facilitation is restricted to the spinal processing of C-nociceptive inputs, but extends to affect A-nociceptive inputs from secondary sites in arthritic animals. Prostanergic descending facilitation from the PAG may provide a major contribution to mechanisms of central sensitization that is critical to the inflammatory pain phenotype. These data suggest that, for centrally penetrating NSAIDs at least, their principle analgesic effect may be to reduce descending facilitation, rather than alter spinal processing, where EP receptors may exert anti-nociceptive effects (Bär et al., 2004; Natura et al., 2013). This may account for the ineffectiveness of spinally delivered NSAIDs in humans (Eisenach et al., 2010a; Eisenach et al., 2010b).
Footnotes
This work was supported by the Medical Research Council (MRC Doctoral Training Partnership Studentship to R.A.R.D. and MRC Project Grant G0801381 to B.M.L.); the Biotechnology & Biological Sciences Research Council (BBSRC CASE Studentship with GSK to J.L.L.); Yarmouk University, Yarmouk University, Jordan (scholarship to F.A.); The Wellcome Trust (Grant 071335/Z/03/A to B.M.L.); Arthritis Research UK (Project Grant 20400 to L.F.D.); and by a Biotechnology and Biological Sciences Research Council/MRC/British Pharmacological Society Strategic Skills Award (L.F.D. and B.M.L.). Data included in this manuscript have been presented in abstract form at the British Neuroscience Association Meeting 2015, at the 37th International Union of Physiological Societies 2013, and are also included in R.A.R.D.'s and J.L.L.'s doctoral theses (University of Bristol, UK, 2014 and 2008, respectively). We thank Drs. Stella Koutsikou and Nicholas Beazley-Long for technical advice on experiments in this study.
The authors declare no competing financial interests.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License Creative Commons Attribution 4.0 International, which permits unrestricted use, distribution and reproduction in any medium provided that the original work is properly attributed.
References
- Ali Z, Meyer RA, Campbell JN. Secondary hyperalgesia to mechanical but not heat stimuli following a capsaicin injection in hairy skin. Pain. 1996;68:401–411. doi: 10.1016/S0304-3959(96)03199-5. [DOI] [PubMed] [Google Scholar]
- Ambriz-Tututi M, Cruz SL, Urquiza-Marín H, Granados-Soto V. Formalin-induced long-term secondary allodynia and hyperalgesia are maintained by descending facilitation. Pharmacol Biochem Behav. 2011;98:417–424. doi: 10.1016/j.pbb.2011.02.012. [DOI] [PubMed] [Google Scholar]
- An Y, Chen Q, Quan N. Interleukin-1 exerts distinct actions on different cell types of the brain in vitro. J Inflamm Res. 2011;2011:11–20. doi: 10.2147/JIR.S15357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajaj P, Bajaj P, Graven-Nielsen T, Arendt-Nielsen L. Osteoarthritis and its association with muscle hyperalgesia: an experimental controlled study. Pain. 2001;93:107–114. doi: 10.1016/S0304-3959(01)00300-1. [DOI] [PubMed] [Google Scholar]
- Bär KJ, Natura G, Telleria-Diaz A, Teschner P, Vogel R, Vasquez E, Schaible HG, Ebersberger A. Changes in the effect of spinal prostaglandin E2 during inflammation: prostaglandin E (EP1-EP4) receptors in spinal nociceptive processing of input from the normal or inflamed knee joint. J Neurosci. 2004;24:642–651. doi: 10.1523/JNEUROSCI.0882-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann TK, Simone DA, Shain CN, LaMotte RH. Neurogenic hyperalgesia: the search for the primary cutaneous afferent fibers that contribute to capsaicin-induced pain and hyperalgesia. J Neurophysiol. 1991;66:212–227. doi: 10.1152/jn.1991.66.1.212. [DOI] [PubMed] [Google Scholar]
- Belley M, Chan CC, Gareau Y, Gallant M, Juteau H, Houde K, Lachance N, Labelle M, Sawyer N, Tremblay N, Lamontagne S, Carrière MC, Denis D, Greig GM, Slipetz D, Gordon R, Chauret N, Li C, Zamboni RJ, Metters KM. Comparison between two classes of selective EP(3) antagonists and their biological activities. Bioorg Med Chem Lett. 2006;16:5639–5642. doi: 10.1016/j.bmcl.2006.08.025. [DOI] [PubMed] [Google Scholar]
- Boie Y, Stocco R, Sawyer N, Slipetz DM, Ungrin MD, Neuschäfer-Rube F, Püschel GP, Metters KM, Abramovitz M. Molecular cloning and characterization of the four rat prostaglandin E2 prostanoid receptor subtypes. Eur J Pharmacol. 1997;340:227–241. doi: 10.1016/S0014-2999(97)01383-6. [DOI] [PubMed] [Google Scholar]
- Breder CD, Smith WL, Raz A, Masferrer J, Seibert K, Needleman P, Saper CB. Distribution and characterization of cyclooxygenase immunoreactivity in the ovine brain. J Comp Neurol. 1992;322:409–438. doi: 10.1002/cne.903220309. [DOI] [PubMed] [Google Scholar]
- Breder CD, Dewitt D, Kraig RP. Characterization of inducible cyclooxygenase in rat brain. J Comp Neurol. 1995;355:296–315. doi: 10.1002/cne.903550208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman RA, Smith WL, Narumiya S. International Union of Pharmacology classification of prostanoid receptors: properties, distribution, and structure of the receptors and their subtypes. Pharmacol Rev. 1994;46:205–229. [PubMed] [Google Scholar]
- Crofford LJ, Wilder RL, Ristimäki AP, Sano H, Remmers EF, Epps HR, Hla T. Cyclooxygenase-1 and -2 expression in rheumatoid synovial tissues: effects of interleukin-1 beta, phorbol ester, and corticosteroids. J Clin Invest. 1994;93:1095–1101. doi: 10.1172/JCI117060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dampney RA, Furlong TM, Horiuchi J, Iigaya K. Role of dorsolateral periaqueductal grey in the coordinated regulation of cardiovascular and respiratory function. Auton Neurosci. 2013;175:17–25. doi: 10.1016/j.autneu.2012.12.008. [DOI] [PubMed] [Google Scholar]
- Davis AJ, Kelly D, Perkins MN. The induction of des-Arg9-bradykinin-mediated hyperalgesia in the rat by inflammatory stimuli. Braz J Med Biol Res. 1994;27:1793–1802. [PubMed] [Google Scholar]
- De Felice M, Sanoja R, Wang R, Vera-Portocarrero L, Oyarzo J, King T, Ossipov MH, Vanderah TW, Lai J, Dussor GO, Fields HL, Price TJ, Porreca F. Engagement of descending inhibition from the rostral ventromedial medulla protects against chronic neuropathic pain. Pain. 2011;152:2701–2709. doi: 10.1016/j.pain.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson LF, Humphrey PS, Oldfield S, Giblett S, Grubb BD. Expression and regulation of prostaglandin E receptor subtype mRNAs in rat sensory ganglia and spinal cord in response to peripheral inflammation. Prostaglandins Other Lipid Mediat. 2001;63:109–122. doi: 10.1016/S0090-6980(00)00101-5. [DOI] [PubMed] [Google Scholar]
- Eisenach JC, Curry R, Rauck R, Pan P, Yaksh TL. Role of spinal cyclooxygenase in human postoperative and chronic pain. Anesthesiology. 2010a;112:1225–1233. doi: 10.1097/ALN.0b013e3181d94dc0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenach JC, Curry R, Tong C, Houle TT, Yaksh TL. Effects of intrathecal ketorolac on human experimental pain. Anesthesiology. 2010b;112:1216–1224. doi: 10.1097/ALN.0b013e3181d94d8b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Bueno B, Serrats J, Sawchenko PE. Cerebrovascular cyclooxygenase-1 expression, regulation, and role in hypothalamic-pituitary-adrenal axis activation by inflammatory stimuli. J Neurosci. 2009;29:12970–12981. doi: 10.1523/JNEUROSCI.2373-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Géranton SM, Jiménez-Díaz L, Torsney C, Tochiki KK, Stuart SA, Leith JL, Lumb BM, Hunt SP. A rapamycin-sensitive signaling pathway is essential for the full expression of persistent pain states. J Neurosci. 2009;29:15017–15027. doi: 10.1523/JNEUROSCI.3451-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JY, Huo HR, Zhao BS, Liu HB, Li LF, Ma YY, Guo SY, Jiang TL. Cinnamaldehyde reduces IL-1beta-induced cyclooxygenase-2 activity in rat cerebral microvascular endothelial cells. Eur J Pharmacol. 2006;537:174–180. doi: 10.1016/j.ejphar.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Heinricher MM, Martenson ME, Neubert MJ. Prostaglandin E2 in the midbrain periaqueductal gray produces hyperalgesia and activates pain-modulating circuitry in the rostral ventromedial medulla. Pain. 2004;110:419–426. doi: 10.1016/j.pain.2004.04.026. [DOI] [PubMed] [Google Scholar]
- Heinricher MM, Tavares I, Leith JL, Lumb BM. Descending control of nociception: specificity, recruitment and plasticity. Brain Res Rev. 2009;60:214–225. doi: 10.1016/j.brainresrev.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrero JF, Cervero F. Changes in nociceptive reflex facilitation during carrageenan-induced arthritis. Brain Res. 1996;717:62–68. doi: 10.1016/0006-8993(95)01585-X. [DOI] [PubMed] [Google Scholar]
- Hsieh MT, Donaldson LF, Lumb BM. Differential contributions of A- and C-nociceptors to primary and secondary inflammatory hypersensitivity in the rat. Pain. 2015;156:1074–1083. doi: 10.1097/j.pain.0000000000000151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes SW, Hickey L, Hulse RP, Lumb BM, Pickering AE. Endogenous analgesic action of the pontospinal noradrenergic system spatially restricts and temporally delays the progression of neuropathic pain following tibial nerve injury. Pain. 2013;154:1680–1690. doi: 10.1016/j.pain.2013.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juteau H, Gareau Y, Labelle M, Sturino CF, Sawyer N, Tremblay N, Lamontagne S, Carrière MC, Denis D, Metters KM. Structure-activity relationship of cinnamic acylsulfonamide analogues on the human EP3 prostanoid receptor. Bioorg Med Chem. 2001;9:1977–1984. doi: 10.1016/S0968-0896(01)00110-9. [DOI] [PubMed] [Google Scholar]
- Kennedy I, Coleman RA, Humphrey PP, Levy GP, Lumley P. Studies on the characterisation of prostanoid receptors: a proposed classification. Prostaglandins. 1982;24:667–689. doi: 10.1016/0090-6980(82)90036-3. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Imaizumi R, Sumichika H, Tanaka H, Goda M, Fukunari A, Komatsu H. Sodium iodoacetate-induced experimental osteoarthritis and associated pain model in rats. J Vet Med Sci. 2003;65:1195–1199. doi: 10.1292/jvms.65.1195. [DOI] [PubMed] [Google Scholar]
- Koppert W, Wehrfritz A, Körber N, Sittl R, Albrecht S, Schüttler J, Schmelz M. The cyclooxygenase isozyme inhibitors parecoxib and paracetamol reduce central hyperalgesia in humans. Pain. 2004;108:148–153. doi: 10.1016/j.pain.2003.12.017. [DOI] [PubMed] [Google Scholar]
- Korotkova M, Jakobsson PJ. Persisting eicosanoid pathways in rheumatic diseases. Nat Rev Rheumatol. 2014;10:229–241. doi: 10.1038/nrrheum.2014.1. [DOI] [PubMed] [Google Scholar]
- Kosek E, Altawil R, Kadetoff D, Finn A, Westman M, Le Maître E, Andersson M, Jensen-Urstad M, Lampa J. Evidence of different mediators of central inflammation in dysfunctional and inflammatory pain–interleukin-8 in fibromyalgia and interleukin-1 beta in rheumatoid arthritis. J Neuroimmunol. 2015;280:49–55. doi: 10.1016/j.jneuroim.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutsikou S, Parry DM, MacMillan FM, Lumb BM. Laminar organization of spinal dorsal horn neurones activated by C- vs. A-heat nociceptors and their descending control from the periaqueductal grey in the rat. Eur J Neurosci. 2007;26:943–952. doi: 10.1111/j.1460-9568.2007.05716.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaMotte RH, Lundberg LE, Torebjörk HE. Pain, hyperalgesia and activity in nociceptive C units in humans after intradermal injection of capsaicin. J Physiol. 1992;448:749–764. doi: 10.1113/jphysiol.1992.sp019068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leith JL, Wilson AW, Donaldson LF, Lumb BM. Cyclooxygenase-1-derived prostaglandins in the periaqueductal gray differentially control C- versus A-fiber-evoked spinal nociception. J Neurosci. 2007;27:11296–11305. doi: 10.1523/JNEUROSCI.2586-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leith JL, Wilson AW, You HJ, Lumb BM, Donaldson LF. Periaqueductal grey cyclooxygenase-dependent facilitation of C-nociceptive drive and encoding in dorsal horn neurons in the rat. J Physiol. 2014;592:5093–5107. doi: 10.1113/jphysiol.2014.275909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtman AH, Cook SA, Martin BR. Investigation of brain sites mediating cannabinoid-induced antinociception in rats: evidence supporting periaqueductal gray involvement. J Pharmacol Exp Ther. 1996;276:585–593. [PubMed] [Google Scholar]
- Light AR, Perl ER. Spinal termination of functionally identified primary afferent neurons with slowly conducting myelinated fibers. J Comp Neurol. 1979;186:133–150. doi: 10.1002/cne.901860203. [DOI] [PubMed] [Google Scholar]
- Liu XG, Morton CR, Azkue JJ, Zimmermann M, Sandkühler J. Long-term depression of C-fibre-evoked spinal field potentials by stimulation of primary afferent A delta-fibres in the adult rat. Eur J Neurosci. 1998;10:3069–3075. doi: 10.1046/j.1460-9568.1998.00310.x. [DOI] [PubMed] [Google Scholar]
- Lu J, Xing J, Li J. Prostaglandin E2 (PGE2) inhibits glutamatergic synaptic transmission in dorsolateral periaqueductal gray (dl-PAG) Brain Res. 2007;1162:38–47. doi: 10.1016/j.brainres.2007.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magerl W, Fuchs PN, Meyer RA, Treede RD. Roles of capsaicin-insensitive nociceptors in cutaneous pain and secondary hyperalgesia. Brain. 2001;124:1754–1764. doi: 10.1093/brain/124.9.1754. [DOI] [PubMed] [Google Scholar]
- Mantyh PW, Hunt SP. Setting the tone: superficial dorsal horn projection neurons regulate pain sensitivity. Trends Neurosci. 2004;27:582–584. doi: 10.1016/j.tins.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Martindale JC, Wilson AW, Reeve AJ, Chessell IP, Headley PM. Chronic secondary hypersensitivity of dorsal horn neurones following inflammation of the knee joint. Pain. 2007;133:79–86. doi: 10.1016/j.pain.2007.03.006. [DOI] [PubMed] [Google Scholar]
- Matousek SB, Hein AM, Shaftel SS, Olschowka JA, Kyrkanides S, O'Banion MK. Cyclooxygenase-1 mediates prostaglandin E(2) elevation and contextual memory impairment in a model of sustained hippocampal interleukin-1beta expression. J Neurochem. 2010;114:247–258. doi: 10.1111/j.1471-4159.2010.06759.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMullan S, Lumb BM. Spinal dorsal horn neuronal responses to myelinated versus unmyelinated heat nociceptors and their modulation by activation of the periaqueductal grey in the rat. J Physiol. 2006a;576:547–556. doi: 10.1113/jphysiol.2006.117754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMullan S, Lumb BM. Midbrain control of spinal nociception discriminates between responses evoked by myelinated and unmyelinated heat nociceptors in the rat. Pain. 2006b;124:59–68. doi: 10.1016/j.pain.2006.03.015. [DOI] [PubMed] [Google Scholar]
- McMullan S, Simpson DA, Lumb BM. A reliable method for the preferential activation of C- or A-fibre heat nociceptors. J Neurosci Methods. 2004;138:133–139. doi: 10.1016/j.jneumeth.2004.03.020. [DOI] [PubMed] [Google Scholar]
- Menétrey D, Giesler GJ, Jr, Besson JM. An analysis of response properties of spinal cord dorsal horn neurones to nonnoxious and noxious stimuli in the spinal rat. Exp Brain Res. 1977;27:15–33. doi: 10.1007/BF00234822. [DOI] [PubMed] [Google Scholar]
- Menétrey D, Chaouch A, Besson JM. Responses of spinal cord dorsal horn neurones to non-noxious and noxious cutaneous temperature changes in the spinal rat. Pain. 1979;6:265–282. doi: 10.1016/0304-3959(79)90048-4. [DOI] [PubMed] [Google Scholar]
- Merrill EG, Ainsworth A. Glass-coated platinum-plated tungsten microelectrodes. Med Biol Engineer. 1972;10:662–672. doi: 10.1007/BF02476084. [DOI] [PubMed] [Google Scholar]
- Millan MJ. Descending control of pain. Prog Neurobiol. 2002;66:355–474. doi: 10.1016/S0301-0082(02)00009-6. [DOI] [PubMed] [Google Scholar]
- Mouton LJ, Holstege G. The periaqueductal gray in the cat projects to lamina VIII and the medial part of lamina VII throughout the length of the spinal cord. Exp Brain Res. 1994;101:253–264. doi: 10.1007/BF00228745. [DOI] [PubMed] [Google Scholar]
- Myren M, Baun M, Ploug KB, Jansen-Olesen I, Olesen J, Gupta S. Functional and molecular characterization of prostaglandin E2 dilatory receptors in the rat craniovascular system in relevance to migraine. Cephalalgia. 2010;30:1110–1122. doi: 10.1177/0333102409357957. [DOI] [PubMed] [Google Scholar]
- Myren M, Olesen J, Gupta S. Prostaglandin E2 receptor expression in the rat trigeminal-vascular system and other brain structures involved in pain. Neurosci Lett. 2012;506:64–69. doi: 10.1016/j.neulet.2011.10.050. [DOI] [PubMed] [Google Scholar]
- Naka A, Gruber-Schoffnegger D, Sandkühler J. Non-Hebbian plasticity at C-fiber synapses in rat spinal cord lamina I neurons. Pain. 2013;154:1333–1342. doi: 10.1016/j.pain.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natura G, Bär KJ, Eitner A, Boettger MK, Richter F, Hensellek S, Ebersberger A, Leuchtweis J, Maruyama T, Hofmann GO, Halbhuber KJ, Schaible HG. Neuronal prostaglandin E2 receptor subtype EP3 mediates antinociception during inflammation. Proc Natl Acad Sci U S A. 2013;110:13648–13653. doi: 10.1073/pnas.1300820110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olango WM, Roche M, Ford GK, Harhen B, Finn DP. The endocannabinoid system in the rat dorsolateral periaqueductal grey mediates fear-conditioned analgesia and controls fear expression in the presence of nociceptive tone. Br J Pharmacol. 2012;165:2549–2560. doi: 10.1111/j.1476-5381.2011.01478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva P, Berrino L, de Novellis V, Palazzo E, Marabese I, Siniscalco D, Scafuro M, Mariani L, Rossi F, Maione S. Role of periaqueductal grey prostaglandin receptors in formalin-induced hyperalgesia. Eur J Pharmacol. 2006;530:40–47. doi: 10.1016/j.ejphar.2005.11.025. [DOI] [PubMed] [Google Scholar]
- Palazzo E, Rossi F, Maione S. Role of TRPV1 receptors in descending modulation of pain. Mol Cell Endocrinol. 2008;286:S79–S83. doi: 10.1016/j.mce.2008.01.013. [DOI] [PubMed] [Google Scholar]
- Palazzo E, Guida F, Gatta L, Luongo L, Boccella S, Bellini G, Marabese I, de Novellis V, Rossi F, Maione S. EP1 receptor within the ventrolateral periaqueductal grey controls thermonociception and rostral ventromedial medulla cell activity in healthy and neuropathic rat. Mol Pain. 2011;7:82. doi: 10.1186/1744-8069-7-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. San Diego: Academic; 2006. [DOI] [PubMed] [Google Scholar]
- Pertovaara A. A neuronal correlate of secondary hyperalgesia in the rat spinal dorsal horn is submodality selective and facilitated by supraspinal influence. Exp Neurol. 1998;149:193–202. doi: 10.1006/exnr.1997.6688. [DOI] [PubMed] [Google Scholar]
- Petersen KL, Brennum J, Dahl JB. Experimental evaluation of the analgesic effect of ibuprofen on primary and secondary hyperalgesia. Pain. 1997;70:167–174. doi: 10.1016/S0304-3959(96)03316-7. [DOI] [PubMed] [Google Scholar]
- Raja SN, Campbell JN, Meyer RA. Evidence for different mechanisms of primary and secondary hyperalgesia following heat injury to the glabrous skin. Brain. 1984;107:1179–1188. doi: 10.1093/brain/107.4.1179. [DOI] [PubMed] [Google Scholar]
- Rees H, Sluka KA, Lu Y, Westlund KN, Willis WD. Dorsal root reflexes in articular afferents occur bilaterally in a chronic model of arthritis in rats. J Neurophysiol. 1996;76:4190–4193. doi: 10.1152/jn.1996.76.6.4190. [DOI] [PubMed] [Google Scholar]
- Samad TA, Moore KA, Sapirstein A, Billet S, Allchorne A, Poole S, Bonventre JV, Woolf CJ. Interleukin-1beta-mediated induction of Cox-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature. 2001;410:471–475. doi: 10.1038/35068566. [DOI] [PubMed] [Google Scholar]
- Sandkühler J. Models and mechanisms of hyperalgesia and allodynia. Physiol Rev. 2009;89:707–758. doi: 10.1152/physrev.00025.2008. [DOI] [PubMed] [Google Scholar]
- Schaible HG, Richter F, Ebersberger A, Boettger MK, Vanegas H, Natura G, Vazquez E, Segond von Banchet G. Joint pain. Exp Brain Res. 2009;196:153–162. doi: 10.1007/s00221-009-1782-9. [DOI] [PubMed] [Google Scholar]
- Serou MJ, DeCoster MA, Bazan NG. Interleukin-1 beta activates expression of cyclooxygenase-2 and inducible nitric oxide synthase in primary hippocampal neuronal culture: platelet-activating factor as a preferential mediator of cyclooxygenase-2 expression. J Neurosci Res. 1999;58:593–598. [PubMed] [Google Scholar]
- Serra J, Campero M, Bostock H, Ochoa J. Two types of C nociceptors in human skin and their behavior in areas of capsaicin-induced secondary hyperalgesia. J Neurophysiol. 2004;91:2770–2781. doi: 10.1152/jn.00565.2003. [DOI] [PubMed] [Google Scholar]
- Staud R. The important role of CNS facilitation and inhibition for chronic pain. Int J Clin Rheumtol. 2013;8:639–646. doi: 10.2217/ijr.13.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubhaug A, Romundstad L, Kaasa T, Breivik H. Methylprednisolone and ketorolac rapidly reduce hyperalgesia around a skin burn injury and increase pressure pain thresholds. Acta Anaesthesiol Scand. 2007;51:1138–1146. doi: 10.1111/j.1399-6576.2007.01415.x. [DOI] [PubMed] [Google Scholar]
- Su X, Leon LA, Wu CW, Morrow DM, Jaworski JP, Hieble JP, Lashinger ES, Jin J, Edwards RM, Laping NJ. Modulation of bladder function by prostaglandin EP3 receptors in the central nervous system. Am J Physiol Renal Physiol. 2008;295:F984–F994. doi: 10.1152/ajprenal.90373.2008. [DOI] [PubMed] [Google Scholar]
- Suzuki R, Morcuende S, Webber M, Hunt SP, Dickenson AH. Superficial NK1-expressing neurons control spinal excitability through activation of descending pathways. Nat Neurosci. 2002;5:1319–1326. doi: 10.1038/nn966. [DOI] [PubMed] [Google Scholar]
- Torebjörk E. Nociceptor activation and pain. Philos Trans R Soc Lond B Biol Sci. 1985;308:227–234. doi: 10.1098/rstb.1985.0023. [DOI] [PubMed] [Google Scholar]
- Torebjörk HE, Lundberg LE, LaMotte RH. Central changes in processing of mechanoreceptive input in capsaicin-induced secondary hyperalgesia in humans. J Physiol. 1992;448:765–780. doi: 10.1113/jphysiol.1992.sp019069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tortorici V, Vanegas H. Anti-nociception induced by systemic or PAG-microinjected lysine-acetylsalicylate in rats. Effects on tail-flick related activity of medullary off- and on-cells. Eur J Neurosci. 1995;7:1857–1865. doi: 10.1111/j.1460-9568.1995.tb00706.x. [DOI] [PubMed] [Google Scholar]
- Tracey I, Mantyh PW. The cerebral signature for pain perception and its modulation. Neuron. 2007;55:377–391. doi: 10.1016/j.neuron.2007.07.012. [DOI] [PubMed] [Google Scholar]
- Treede RD, Magerl W. Multiple mechanisms of secondary hyperalgesia. Prog Brain Res. 2000;129:331–341. doi: 10.1016/S0079-6123(00)29025-0. [DOI] [PubMed] [Google Scholar]
- Treede RD, Meyer RA, Raja SN, Campbell JN. Peripheral and central mechanisms of cutaneous hyperalgesia. Prog Neurobiol. 1992;38:397–421. doi: 10.1016/0301-0082(92)90027-C. [DOI] [PubMed] [Google Scholar]
- Urban MO, Gebhart GF. Supraspinal contributions to hyperalgesia. Proc Natl Acad Sci U S A. 1999;96:7687–7692. doi: 10.1073/pnas.96.14.7687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban MO, Smith DJ, Gebhart GF. Involvement of spinal cholecystokininB receptors in mediating neurotensin hyperalgesia from the medullary nucleus raphe magnus in the rat. J Pharmacol Exp Ther. 1996;278:90–96. [PubMed] [Google Scholar]
- Urban MO, Zahn PK, Gebhart GF. Descending facilitatory influences from the rostral medial medulla mediate secondary, but not primary hyperalgesia in the rat. Neuroscience. 1999;90:349–352. doi: 10.1016/S0306-4522(99)00002-0. [DOI] [PubMed] [Google Scholar]
- Vanegas H. To the descending pain-control system in rats, inflammation-induced primary and secondary hyperalgesia are two different things. Neurosci Lett. 2004;361:225–228. doi: 10.1016/j.neulet.2003.12.002. [DOI] [PubMed] [Google Scholar]
- Vanegas H, Schaible HG. Prostaglandins and cyclooxygenases in the spinal cord. Prog Neurobiol. 2001;64:327–363. doi: 10.1016/S0301-0082(00)00063-0. [DOI] [PubMed] [Google Scholar]
- Vanegas H, Schaible HG. Descending control of persistent pain: inhibitory or facilitatory? Brain Res Brain Res Rev. 2004;46:295–309. doi: 10.1016/j.brainresrev.2004.07.004. [DOI] [PubMed] [Google Scholar]
- Vanegas H, Vazquez E, Tortorici V. NSAIDs, opioids, cannabinoids and the control of pain by the central nervous system. Pharmaceuticals. 2010;3:1335–1347. doi: 10.3390/ph3051335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan CW. Enhancement of opioid inhibition of GABAergic synaptic transmission by cyclo-oxygenase inhibitors in rat periaqueductal grey neurones. Br J Pharmacol. 1998;123:1479–1481. doi: 10.1038/sj.bjp.0701818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan CW, Ingram SL, Connor MA, Christie MJ. How opioids inhibit GABA-mediated neurotransmission. Nature. 1997;390:611–614. doi: 10.1038/37610. [DOI] [PubMed] [Google Scholar]
- Vermeirsch H, Biermans R, Salmon PL, Meert TF. Evaluation of pain behavior and bone destruction in two arthritic models in guinea pig and rat. Pharmacol Biochem Behav. 2007;87:349–359. doi: 10.1016/j.pbb.2007.05.010. [DOI] [PubMed] [Google Scholar]
- Waschbisch A, Fiebich BL, Akundi RS, Schmitz ML, Hoozemans JJ, Candelario-Jalil E, Virtainen N, Veerhuis R, Slawik H, Yrjänheikki J, Hüll M. Interleukin-1 beta-induced expression of the prostaglandin E-receptor subtype EP3 in U373 astrocytoma cells depends on protein kinase C and nuclear factor-kappaB. J Neurochem. 2006;96:680–693. doi: 10.1111/j.1471-4159.2005.03599.x. [DOI] [PubMed] [Google Scholar]
- Woolf CJ. Evidence for a central component of post-injury pain hypersensitivity. Nature. 1983;306:686–688. doi: 10.1038/306686a0. [DOI] [PubMed] [Google Scholar]
- Yeomans DC, Proudfit HK. Nociceptive responses to high and low rates of noxious cutaneous heating are mediated by different nociceptors in the rat: electrophysiological evidence. Pain. 1996;68:141–150. doi: 10.1016/S0304-3959(96)03177-6. [DOI] [PubMed] [Google Scholar]
- You HJ, Dahl Morch C, Chen J, Arendt-Nielsen L. Simultaneous recordings of wind-up of paired spinal dorsal horn nociceptive neuron and nociceptive flexion reflex in rats. Brain Res. 2003;960:235–245. doi: 10.1016/S0006-8993(02)03895-7. [DOI] [PubMed] [Google Scholar]
- You HJ, Lei J, Sui MY, Huang L, Tan YX, Tjølsen A, Arendt-Nielsen L. Endogenous descending modulation: spatiotemporal effect of dynamic imbalance between descending facilitation and inhibition of nociception. J Physiol. 2010;588:4177–4188. doi: 10.1113/jphysiol.2010.196923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Lu Y, Chen Y, Westlund KN. Group I metabotropic glutamate receptor antagonists block secondary thermal hyperalgesia in rats with knee joint inflammation. J Pharmacol Exp Ther. 2002;300:149–156. doi: 10.1124/jpet.300.1.149. [DOI] [PubMed] [Google Scholar]
- Ziegler EA, Magerl W, Meyer RA, Treede RD. Secondary hyperalgesia to punctate mechanical stimuli. Central sensitization to A-fibre nociceptor input. Brain. 1999;122:2245–2257. doi: 10.1093/brain/122.12.2245. [DOI] [PubMed] [Google Scholar]