

Abstract
Microglial dysfunction is increasingly recognized as a key contributor to the pathogenesis of Alzheimer's disease (AD). Environmental enrichment (EE) is well documented to enhance neuronal form and function, but almost nothing is known about whether and how it alters the brain's innate immune system. Here we found that prolonged exposure of naive wild-type mice to EE significantly altered microglial density and branching complexity in the dentate gyrus of hippocampus. In wild-type mice injected intraventricularly with soluble Aβ oligomers (oAβ) from hAPP-expressing cultured cells, EE prevented several morphological features of microglial inflammation and consistently prevented oAβ-mediated mRNA changes in multiple inflammatory genes both in vivo and in primary microglia cultured from the mice. Microdialysis in behaving mice confirmed that EE normalized increases in the extracellular levels of the key cytokines (CCL3, CCL4, TNFα) identified by the mRNA analysis. Moreover, EE prevented the changes in microglial gene expression caused by ventricular injection of oAβ extracted directly from AD cerebral cortex. We conclude that EE potently alters the form and function of microglia in a way that prevents their inflammatory response to human oAβ, suggesting that prolonged environmental enrichment could protect against AD by modulating the brain's innate immune system.
SIGNIFICANCE STATEMENT Environmental enrichment (EE) is a potential therapy to delay Alzheimer's disease (AD). Microglial inflammation is associated with the progression of AD, but the influence of EE on microglial inflammation is unclear. Here we systematically applied in vivo methods to show that EE alters microglia in the dentate gyrus under physiological conditions and robustly prevents microglial inflammation induced by human Aβ oligomers, as shown by neutralized microglial inflammatory morphology, mRNA changes, and brain interstitial fluid cytokine levels. Our findings suggest that EE alters the innate immune system and could serve as a therapeutic approach to AD and provide new targets for drug discovery. Further, we propose that the therapeutic benefits of EE could extend to other neurodegenerative diseases involving microglial inflammation.
Keywords: Alzheimer's disease, brain inflammation, environmental enrichment, microglia, oligomeric amyloid beta
Introduction
Attempts to date to experimentally treat Alzheimer's disease (AD) with pharmacological agents have met with limited success, and a disease-modifying therapeutic remains to be unequivocally validated and approved. Given the potential side effects, complex delivery logistics, and expense of chronically administering drugs or biologics to the global AD population, nonpharmacological approaches, such as prolonged behavioral modification, are also highly attractive, but they are relatively understudied in AD and models thereof. Environmental enrichment (EE) in rodents can provide resistance to neuronal toxicity from soluble oligomers of amyloid β-protein (oAβ), the most widely validated pathogenic moiety in AD, and can effectively ameliorate multiple AD-like neuronal phenotypes in animal models, such as impaired cognition, decreased neurogenesis, and suppressed LTP (Lazarov et al., 2005; Cracchiolo et al., 2007; Herring et al., 2009; Li et al., 2013). EE has been extensively shown to confer benefits to synaptic plasticity and neuronal structure, with evidence that includes biochemical, electrophysiological, and behavioral read-outs (Nithianantharajah and Hannan, 2006; Olson et al., 2006; Li et al., 2013).
Over the past two decades, inflammatory mechanisms in AD have become a mainstream research direction, and the interactions between diffusible Aβ oligomers, the major cytotoxic species in AD (Walsh and Selkoe, 2007), and microglia are being studied intensively (Solito and Sastre, 2012; Wyss-Coray and Rogers, 2012). Emerging evidence suggests that neuroinflammation is a fundamental contributor to AD pathogenesis, with the brain's innate immune system, particularly microglia, playing a major role in generating chronic and self-sustaining inflammation, which contributes to neuronal dysfunction and death (Rubio-Perez and Morillas-Ruiz, 2012). A few reports suggest that the neuronal benefits of EE may require the presence of innate immune cells, including microglia (Ziv et al., 2006; Choi et al., 2008; Ziv and Schwartz, 2008; Vukovic et al., 2012), encouraging further study of the benefits of EE for non-neuronal cells. Limited work suggests that EE can modulate microglia quantity in cerebral cortex and hippocampus as well as microglial responses to lipopolysaccharide stimulation, such as cytokine elevation and phagocytic activity (Ehninger and Kempermann, 2003; Williamson et al., 2012). However, despite the emerging interest in the possible effects of EE on microglia, whether EE influences AD pathobiology at the level of microglia has barely been studied. Challenges here include the technical difficulty of separating endogenous microglia from CNS-entering peripheral monocytes and analyzing samples of extremely small quantity. We lack systematic, cell-type-specific, quantitative analyses of the microglial responses to EE in AD models.
Here, we have designed in vivo experimental paradigms that address qualitatively and quantitatively the question of whether EE modulates microglia and whether such modulation relates to EE's beneficial effects on AD phenotypes in a pathophysiologically relevant and reproducible manner. We performed intracerebroventricular microinjections in wild-type (wt) mice to expose the animals to human oAβ. We then selectively FACS-isolated and analyzed microglia from these animals, with no significant contamination from peripheral monocytes or other immune cells. Our results demonstrate that prolonged EE exposure alters microglia in the dentate gyrus of hippocampus and results in a prominent neutralization of the neuroinflammation induced by oAβ, including oAβ purified from AD brain tissue, as shown by both microglial morphometry and unbiased inflammatory gene expression profiles. We also identify several cytokines at both the mRNA and protein levels, the latter using in vivo microdialysis, which help mediate the protective benefits of EE. Our results directly link enriched environments to the innate immune response of microglia to oAβ and provide strong evidence for EE's protective effect on AD at the level of CNS immunology.
Materials and Methods
Animals.
The Harvard Medical School Standard Committee on Animals approved all experiments involving mice used for the study. All mice were male and contained a mixed background of C57BL/6 and 129 (http://www.taconic.com/mouse-model/b6129f1). Animals were housed in a temperature-controlled room on a 12 h light/12 h dark cycle and had free access to food and water.
EE.
Three week male BL6/129 mice were purchased from Taconic. The mice were housed either under standard housing condition (SH) or EE starting at 4 weeks for a total of 7–8 weeks. The EE paradigm allows 8 mice housed in one large cage (EE 38 × 60 cm vs SH 14 × 34 cm) consisting of running wheels, tunnels, and objects of various colors and shapes. The mice were housed in EE cages for 8 h per day and rotated daily through 4 different EE cages.
Electrophysiology.
We used weak stimulation protocol to induce LTP within hippocampus region, as described by Li et al. (2011).
Tissue section preparation and immunofluorescence staining.
The mice were perfused with ice-cold HBSS and then ice-cold 4% PFA. The brains were rapidly removed and immersed in 4% PFA for 2 h at 4°C, transferred to 30% sucrose solution for 48 h at 4°C, and embedded in OCT. The 14 μm coronal sections were prepared. The sections were washed for 5 min in 70% ethanol, 3 min ×2 in water, and 5 min in phosphate saline buffer with 0.1% Tween, then blocked (10% horse serum, 2% BSA, 1% glycine, 0.3% Triton-X) for 2 h in a humidity chamber at room temperature. Afterward the sections were incubated with primary antibody (anti-p2ry12 1:500, Butovsky laboratory; anti-CD68 1:200, Abcam catalog #ab53444 RRID:AB_869007; anti-Iba1 1:200 (Wako catalog #019-19741 RRID:AB_839504); anti-DCX 1:1000 (Abcam catalog #ab18723 RRID:AB_732011) overnight at 4°C and subsequently with secondary antibody (Invitrogen catalog #A11006 RRID:AB_10561520, 1:1000; Invitrogen catalog #A11010 RRID:AB_10584649, 1:1000) for 2 h at room temperature with adequate PBST wash in between. The sections were mounted with Vectashield H-1500.
5-Ethynyl-2-deoxyuridine labeling.
The mice were injected intraperitoneal with 5-ethynyl-2-deoxyuridine (EdU) solution (Invitrogen E10415) at 35 g/kg body weight every other day for the duration of EE. The brain sections were processed with Click-iT Plus EdU Alexa Fluor Flow Cytometry Assay Kit (Invitrogen C10632).
The 7PA2 conditioned medium preparation and immunodepletion.
Secreted human Aβ peptides were collected and prepared from the conditioned media (CM) of a CHO cell line (7PA2) that stably expresses human APP751 containing the V717F AD mutation (Podlisny et al., 1995), as described previously (Walsh et al., 2005). Immunodeleted 7PA2 CM was prepared by serial immunodepletion using 4G8 antibody (Biolegend RRID:AB_10175149). The antibody was crosslinked to agarose beads using Pierce CoImmunoprecipitation Kit (26149) before applying to 7PA2 CM. We analyzed the supernatant from the last round by Aβ ELISA (Yang et al., 2013) and used Western blot to confirm.
Human brain extract preparation and immunodepletion.
AD-TBS extracts were prepared as described before (Shankar et al., 2008). Immunodeleted ADTBS (ID-ADTBS) was prepared using the same method as with 7PA2 CM.
Confocal microscopy and image analysis.
All images were obtained under Zeiss LSM 510 confocal microscope with 20×/0.75 objective. Images obtained for the cell morphology analysis were set at 1024 × 1024 pixels and collected in Z-stacks for 3D reconstruction. Image analysis was completed in ImageJ.
Stereotactic intracerebroventricular injection.
Lateral ventricle was located by bregma −2.5 mm, midline 3.1 mm, and dura −3.7 mm. Then, 4 μl of injection material is slowly delivered over 5 min.
Microglia purification and mRNA extraction.
Mice were perfused with ice-cold HBSS. The brains were rapid removed, and an ∼3 mm cubic of contralateral hemisphere corresponding to the tissue surrounding injection site was cut out. Tissue blocks were homogenized manually. Microglia cells were purified using 37%/70% Percoll (GE Healthcare 17-5445-02) gradient followed by FACS. Antibodies: Anti-CD11b (BD Biosciences catalog #552850 RRID:AB_394491); anti-CD45 (eBioscience catalog #17-0451-82 RRID:AB_469392); anti-Ly6C (eBioscience catalog #12-5932-82 RRID:AB_10804510); and anti-Ly6G (Biolegend catalog #127605). mRNA samples were prepared from purified cells using mirVana mRNA isolation kit (AM1561).
Primary microglia culture.
Primary microglia were isolated as described above. The cells were plated at 20,000 cells/well in a poly-d-Lysine coated 96 well plate using DMEM/F12 + GlutaMAX medium supplemented with 10 ng/ml M-CSF (R&D Systems) and 50 ng/mL human TGF-β1 (Miltenyi Biotec). Cells were cultured at 37°C with 5% CO2 and controlled humidity.
Phagocytosis assay.
Primary microglia were isolated and plated as described above. The cells were incubated with 1 μm oligomeric FITC-Aβ42 (McCarthy et al., 2016) for 3 h at 37°C, placed on ice, and washed thoroughly with ice-cold PBS before their fluorescent signal was measured by a plate reader. Untreated cells were used to measure baseline fluorescence.
qPCR.
mRNA were converted to cDNA using Applied Biosystems High-capacity cDNA reverse transcription kit (#4368813). The samples were then mixed with Taqman fast universal PCR mix (#4352042) and ABI gene expression assay for qPCR analysis.
Nanostring analysis.
mRNA samples were concentrated to required concentration by Zymo RNA Clean and Concentrator (R1015). mRNA concentration was determined by Nanodrop. The samples were further processed with Nanostring nCounter Prep-station then scanned at high resolution with nCounter Digital Analyzer (Mouse Inflammation v2).
Microdialysis and cytokine ELISA.
The mice had intracerebral guide cannula implanted following the coordinates for left hippocampal placement (bregma −3.1 mm, midline −2.5 mm, dura −1.2 mm at 12° angle). After inserting probes with 1000 kDa molecular weight cutoff (MWCO) membranes (Eicom AtmosLM), 4% BSA in artificial CSF in mm as follows: 1.3 CaCl2, 1.2 MgSO4, 3 KCl, 0.4 KH2PO4, 25 NaHCO3, and 122 NaCl, pH 7.35, perfused through at flow rates 0.4 μl/min with an infusion syringe pump (Stoelting). Microdialysates were collected for 12 h. Mice were kept on 12 h light/dark cycles, and housed in a Raturn cage system (Bioanalytical Systems), which allowed normal activities. All samples were analyzed with Meso Scale Discovery customized HB Prototype Mouse SP 4-Plex ELISA.
Statistical analysis.
All quantification results were analyzed using two-tailed Student's t test and one-way ANOVA to determine statistical significance. N indicates total number of mice under each condition.
Results
EE increases microglial density and branching complexity in the dentate gyrus of naive wt mice
We initially assessed the possible effects of EE on hippocampal microglia by exposing 4-week-old wt mice to a well described and validated EE paradigm (Li et al., 2013) daily for 7–8 weeks. In each experiment, eight outbred wt mice (C57BL/6 × 129) were housed for 8 h/d, 7 d/week in a large cage (38 × 60 cm, threefold larger than a standard cage) with plastic toys of various shapes and colors and two different running wheels for voluntary physical exercise. To maximize novelty exposure, which our prior work showed to be more effective than physical exercise (Li et al., 2013), the mice were rotated each day among four large cages equipped with distinct sets of toys. The activity of the mice (i.e., exploring and running) was monitored throughout the EE period to confirm active participation in EE. Control littermate mice were housed within the same facility but in standard cages (SH, 14 × 34 cm) with only customary bedding and access to food pellets and water. Recording of long-term synaptic potentiation (LTP) in hippocampal slices using a weak electrical stimulus (Li et al., 2011) that cannot induce LTP in SH mice produced significant LTP in slices from the EE mice, confirming the effectiveness of the EE paradigm on neurons, in accord with our prior work (data not shown).
Following this EE training, we used confocal microscopy to quantify microglial density, defined as the number of microglia/mm2, in different regions of hippocampus. P2y12 receptor (P2ry12) has recently been established as a new and specific marker for resident microglia that does not recognize peripheral macrophages/monocytes (Butovsky et al., 2014). Although it shows superior specificity for microglia, it has not yet been used to quantify microglia number. We labeled microglia in situ with anti-P2ry12 monoclonal antibody, took confocal mosaic scans that covered the entire hippocampus, manually quantified the number of P2ry12+ cells in dentate gyrus (DG), CA1, and CA3, and calculated microglial density by normalizing the cell numbers to area (mm2), as determined by ImageJ. We analyzed 8 EE mice and 8 SH mice from each of 3 different cohorts, quantifying 10–12 sections/mouse brain. There was a highly significant 29% increase in microglial density in the DG of EE mice compared with SH mice (SH 95.22 ± 4.47 vs EE 123.10 ± 9.63, p < 0.0001, N = 8) (Fig. 1A,B), but not in the CA1 and CA3 regions (data not shown). To verify this P2ry12 finding, we applied a widely used macrophage marker, Iba-1, and repeated the quantification. We obtained densities (cells/mm2) of Iba-1+ cells in situ that were consistent with those of P2ry12+ cells above (SH 98.55 ± 6.17 vs EE 118.37 ± 5.36, p < 0.001, N = 8). We also observed a highly consistent ∼20% increase in DG microglial density in mice that were exposed to various durations of EE (4 week or longer) (Fig. 1C). Because Iba-1 marks both resident microglia and invading peripheral macrophages, we also analyzed the EE and SH brains by flow cytometry (FACS) to assess any potential confounding effects from macrophages. Resident microglia were labeled as CD11b+/CD45med/Ly6Clo/Ly6Glo, whereas peripheral macrophages were CD11b+/CD45high/Ly6Chigh (Butovsky et al., 2014). Flow cytometry demonstrated that the vast majority of CD11b+ cells are CD11b+/CD45med/Ly6Clo/Ly6Glo regardless of housing conditions (SH, 97.11 ± 0.22%; EE, 96.83 ± 0.35%) (Fig. 1D); thus, the CD11b+/CD45high/Ly6Chigh population was minimal within brain tissue, confirming that the Iba-1+ cells we observe are endogenous microglia.
Figure 1.

EE increases microglial density in the dentate gyrus of naive WT mice. A, Representative images from SH and EE mice DG showing difference in cellular density. P2ry12 antibody (red), together with DAPI (blue), was used to visualize microglia. Scale bar, 200 μm. Only cells showing P2ry12 and DAPI colocalization are included in cell counting. B, Quantification analysis of SH and EE mice DG microglia shows that EE significantly increases DG microglial density (*p < 0.0001, N = 8), which is calculated by dividing total cell count per DG with DG area (mm2). C, Quantitative analysis of Iba-1 labeling of microglia shows significant increase of DG microglial density in EE mice starting from 4 to 6 weeks of EE (*p < 0.05, **p < 0.01, ***p < 0.001; 7–8 weeks: N = 8; others: N = 2–4). ns, Not significant. D, Representative images from FACS gating show minimal invasion of cells other than resident microglia into brain parenchyma in naive mice. Quantitative data are mean ± SEM.
When we administrated EdU by intraperitoneal injection during the EE period, we observed no increase of EdU labeling among P2ry12+ microglia (Fig. 2A), suggesting that the increase in cell density was not a result of microglial division. As a simultaneous positive control, EdU+ neurons positive for DCX were substantially increased in the EE mice (EdU+/DCX+ neuron density: SH = 36.47 ± 3.123; EE = 64.41 ± 2.537, p < 0.02, N = 6; Fig. 2B,C), as expected from many studies in the literature (Kempermann et al., 1997; Nilsson et al., 1999).
Figure 2.

EdU labeling during EE shows minimal microglial division while significantly more neurogenesis by EE. A, Representative images showing minimal EdU (green)/P2ry12 (red) colocalization (blue, DAPI). Scale bar, 100 μm. B, Representative images showing majority of EdU (green) signals colocalize with DCX (red), a marker for newborn neurons. Scale bar, 100 μm. C, Quantification of EdU+/DCX+ cell counts (no. of cells/mm2) in the dentate gyrus of EE and SH mice indicates significantly higher level of neurogenesis in EE mice. *p < 0.02. N = 6. Data are mean ± SEM.
To assess further effects of EE on microglia beyond their increased density, we performed automated morphological analysis on individual cells using ImageJ. P2ry12 labeling, compared with Iba-1, provided outstanding resolution of fine processes, allowing detailed cell morphology analyses. We created Z-stack scan images of P2ry12+ microglia from three separate areas within the DG and imported images of individual microglia into ImageJ for automated, unbiased quantification of circularity (4π[area]/[perimeter]2, with 1.0 indicating a perfect circle) and solidity ([area]/[convex area], with a maximum solidity value of 1.0). Both of these parameters reflect the overall complexity of cell morphology and successfully distinguished microglia under different conditions, whereas roundness (4× [Area]/(π × [Major axis]2)), which focuses only on the major cellular axis, failed to do so and was excluded from further analyses. We also performed skeleton analysis (Morrison and Filosa, 2013) to measure the number of cellular processes and their branches on individual microglia (#branches/microglia) (Fig. 3A). Using these several measures, we analyzed all mice used for the above microglial density analyses, quantifying 30–40 cells from multiple brain sections per mouse. We found that, in addition to the higher microglial density, DG microglia in the EE mice possessed significantly more branches per cell on average (SH 93.52 ± 8.45 vs EE 124.40 ± 20.54, p < 0.05, N = 8) (Fig. 3B,C), indicating that, after EE, microglia are more ramified in their morphology under physiological conditions, suggesting a likely alteration of their function. No significant differences were observed in the measurements of circularity and solidity (data not shown).
Figure 3.
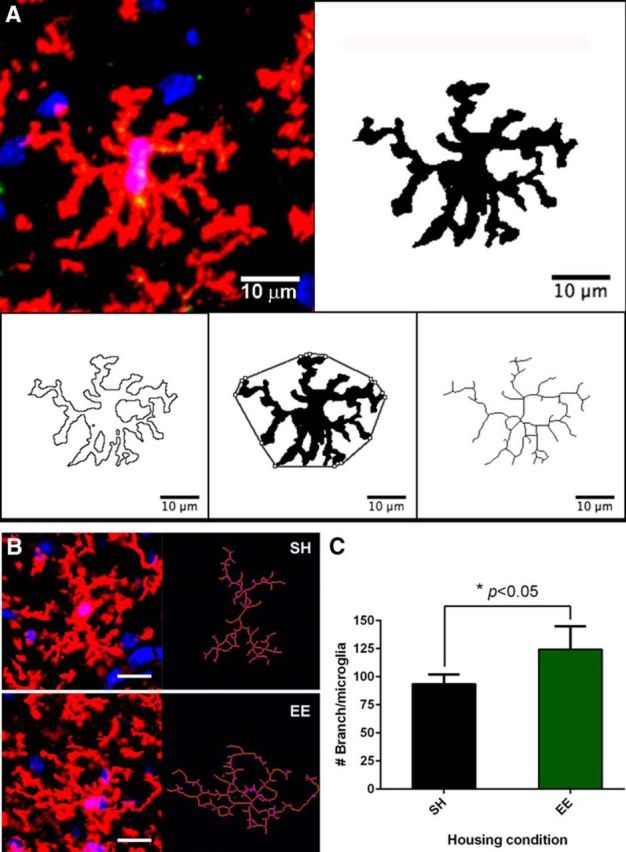
EE significantly increases individual microglia branching complexity in naive mice. A, Demonstration of microglia morphology analysis methods. Binary mask, perimeter, convex, and skeleton are generated using ImageJ software. Scale bar, 10 μm. B, Higher-magnification representative images of single microglia from each housing condition and their corresponding cell skeletons. Purple areas represent the branching points. Scale bar, 20 μm. C, Quantification of SH and EE mice DG branching complexity (#Branch/microglia) shows that EE significantly increases #Branch/microglia (*p < 0.05, N = 8). Data are mean ± SEM.
Oligomeric Aβ induces changes in microglial density and morphology in SH mice
Next, we systematically compared the effects of soluble human Aβ oligomers (oAβ) on microglia under SH conditions. The CM of CHO cells stably expressing the hAPP-Val717Phe FAD mutant (7PA2 CM) that is oAβ-rich (Podlisny et al., 1995; Walsh et al., 2002; Welzel et al., 2014) was delivered into the lateral ventricle (intracerebroventricularly) by stereotactic microinjection. After 48 h, the contralateral hemisphere was harvested to search for microglial changes induced by the diffusion of the soluble Aβ oligomers into the opposite lateral ventricle. To control for any possible alterations in microglia morphology and density caused by the surgical procedure or by non-Aβ substances in the cell medium, we microinjected CM from parental CHO cells expressing only the vector but not the hAPP-V717F AD mutant (called CHO− CM) as a negative control. To verify that the changes in microglial density and morphology upon 7PA2 CM injection are directly attributable to oAβ, we also repeated the experiments using 7PA2 CM that had been quantitatively immunodepleted of oAβ using anti-Aβ-conjugated beads while maintaining the same APPsα level (yielding ID-7PA2 CM) (Fig. 4A,B). Hippocampal images were acquired for microglia analysis (Fig. 5A). We observed a significant decrease in DG microglial density in the SH mice receiving 7PA2 CM and an almost full rescue from immunodepleting Aβ (SH CHO− CM 115.37 ± 11.04 vs SH 7PA2 CM 98.67 ± 11.43, p < 0.05; vs SH ID-7PA2 CM 118.68 ± 1.33, nonsignificant to SH CHO− CM, N = 6) (Fig. 5B).
Figure 4.

Serial immunodepletion with 4G8 antibody successfully removes oAβ from 7PA2 CM. A, Supernatant from the last immunodepletion was analyzed by ELISA for Aβ x-40 and Aβ x-42 and compared with original 7PA2 CM. Result shows >90% removal of overall oAβ species. Western blot shows no detectable oAβ after first immunodepletion, confirming the result. B, The supernatant was analyzed by ELISA for APPsα. Result shows no change of APPsα level from immunodepletion. Quantitative data are mean ± SEM.
Figure 5.
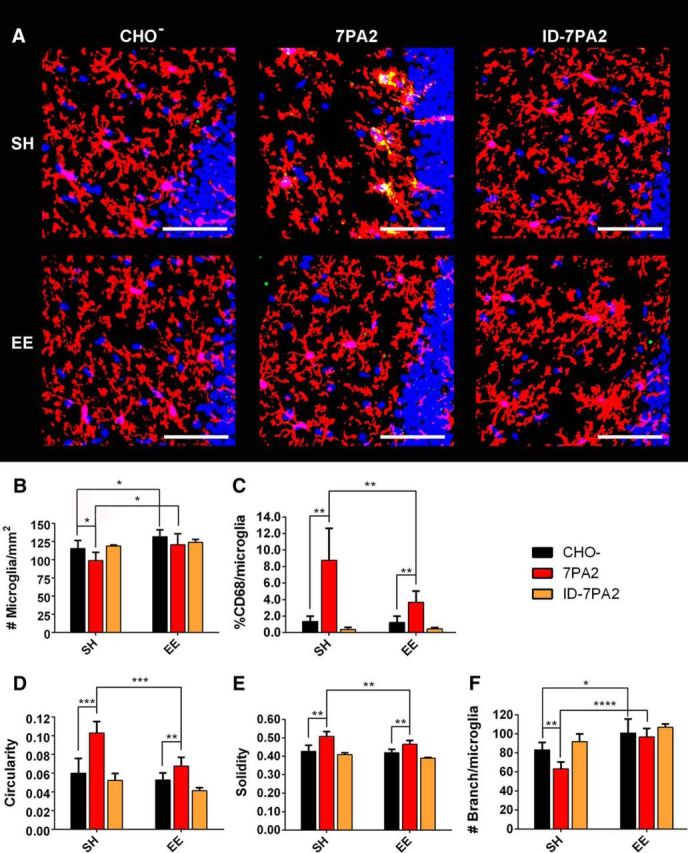
Oligomeric Aβ induces changes in microglial density and morphology in SH mice, and EE prevents such changes. A, Representative images from SH and EE mice receiving 7PA2 CM, CHO− CM, or immunodepleted 7PA2 CM (ID-7PA2) show strong microglial alteration in SH from 7PA2 CM, as reflected by microglial morphology (microglia labeled with P2ry12 antibody, red) and high level of CD68 (green). DAPI (blue) demonstrates that all images are from consistent DG area. EE potently neutralizes microglial alteration. Removing oAβ from 7PA2 CM also successfully abolishes microglial alteration. Scale bar, 50 μm. B–F, Quantification: 7PA2 CM significantly decreases DG microglial density, significantly increases % CD68/microglia, circularity, and solidity, and decreases #branch/microglia in SH mice. EE mice upon 7PA2 CM exposure have significantly higher microglial density than that of SH mice, and show significantly different morphological features: smaller value of % CD68/microglia, circularity, and solidity, as well as higher #branch/microglia. EE mice receiving CHO− CM show significantly higher DG microglial density and higher #branch/microglia than CHO− CM-receiving SH mice, just as in naive mice. ID-7PA2 CM shows no difference from CHO− CM in both SH and EE mice. *p < 0.05. **p < 0.01. ***p < 0.001. ****p < 0.0001. N = 6. Quantitative data are mean ± SEM.
Next, we colabeled microglia with anti-CD68 antibody. CD68 is a lysosomal protein marker such that % CD68 has been shown to be associated with microglial phagocytic activity and can be used to evaluate the degree of microglial inflammatory activity (Lee et al., 2010; Elmore et al., 2014). We measured the percentage of CD68+ area within each P2ry12+ microglia (% CD68) by dividing CD68+ area by P2ry12+ area, revealing a marked increase of this marker in SH mice after 7PA2 CM in vivo administration that is rescued by removing oAβ (SH CHO− CM 1.35 ± 0.63% vs SH 7PA2 CM 8.74 ± 3.89%, p < 0.01; vs SH ID-7PA2 CM 0.37 ± 0.25%, nonsignificant to SH CHO− CM, N = 6) (Fig. 5C). We then analyzed the morphology of individual microglia. Using the same analytical methods described in the previous section for the naive mice, we found that in vivo exposure to the oAβ-rich 7PA2 CM led to increased microglial circularity (SH CHO− CM: 0.0598 ± 0.0158 vs SH 7PA2 CM: 0.1029 ± 0.0122, p < 0.001, N = 6) (Fig. 5D) and increased solidity (SH CHO− CM: 0.4260 ± 0.0333 vs SH 7PA2 CM: 0.5076 ± 0.0268, p < 0.01, N = 6) (Fig. 5E), as well as to decreased #branches/microglia (SH CHO− CM: 83.04 ± 7.96 vs SH 7PA2 CM: 63.24 ± 7.24, p < 0.01, N = 6) (Fig. 5F). These significant changes all indicate a cellular morphology that is less ramified and more amoeboid, with shorter, thicker, fewer processes and a larger cell body after in vivo exposure to soluble oAβ. Such a shift in microglial morphology correlates with a more inflammatory microglial state (Kettenmann et al., 2011). Again, these three individual morphology measurements were almost 100% rescued by Aβ immunodepletion (Fig. 5D–F). No statistically significant morphological changes were observed for microglia in the cerebral cortex in the same brain sections.
EE prevents the changes in microglial density and morphology induced by oligomeric Aβ in SH mice
We applied the same experimental procedures and analyses as just described to EE mice, which were studied simultaneously with their littermate SH mice. In contrast to their SH littermate controls, EE mice showed no statistically significant change from oAβ in their microglial density (cells/mm2) (EE CHO− CM 131.62 ± 9.52 vs EE 7PA2 CM 120.48 ± 15.08, p = 0.16988, N = 6) (Fig. 5B). Overall, EE mice exposed to oAβ in vivo showed no lower microglial density than SH mice receiving CHO− CM and significantly higher microglial density than SH mice receiving oAβ (p < 0.05). Highly reproducible results were observed in three independent cohorts of EE and matched SH mice studied over the course of >9 months.
In accord with this finding for microglial density, EE exposure also produced a strong preventative effect on all of the aforementioned measurements of individual microglial morphology. EE mice receiving 7PA2 CM showed much smaller changes than did SH mice in % CD68, circularity and solidity, and no change in #branches/microglia at all. When considering the absolute values, we found highly significant differences between SH mice and EE mice receiving 7PA2 CM in all microglial measurements (% CD68 SH 8.74 ± 3.89% vs EE 3.66 ± 1.06%, p < 0.01; Circularity SH 0.1029 ± 0.0122 vs EE 0.0673 ± 0.0094, p < 0.001; Solidity SH 0.5076 ± 0.0268 vs EE 0.4645 ± 0.0214, p < 0.01; #branches/microglia SH 63.24 ± 7.24 vs EE 96.71 ± 9.08, p < 0.0001, N = 6) (Fig. 5C–F). These data indicate that EE-treated mice have microglial morphologies corresponding to a much less activated state than do SH mice. Even in the face of the intracerebroventricular microinjection procedure, the EE mice still showed significantly higher microglial density and higher #branch/microglia than SH mice upon exposure to oAβ, in agreement with the benefit of EE we observed in naive (uninjected) EE versus SH mice (Fig 5B,F).
Together, the above results suggest that in vivo exposure to soluble oAβ consistently causes a significant decrease in microglial density and changes microglial morphology in SH mice, suggesting significant microglial inflammatory alterations in the DG. Exposure to an EE effectively neutralizes this microglial alteration, indicating that EE can potently enhance microglial tolerance to oAβ and lower microglial inflammation in the CNS.
Aβ oligomers induce inflammatory shifts in microglial-specific mRNA profiles in SH mice
To seek further information about oAβ activity on microglia, we used the same intracerebroventricular oAβ microinjection model described above and isolated microglia by FACS from the contralateral hemisphere at 24 and 48 h after injection, to perform unbiased mRNA profiling. We extracted mRNA from the freshly FACS-purified microglia and analyzed total mRNA on the Nanostring platform with a specific Mouse Inflammation Code-set that measures a total of 253 inflammatory genes. The results indicated that, in SH mice, multiple proinflammatory genes (41 of 186 detectable genes at 24 h and 19 of 163 detectable genes at 48 h) were either activated or suppressed by exposure to oAβ-rich 7PA2 CM compared with CHO− CM (Fig. 6A,B), indicating that cell-secreted oAβ, which is present at low but pathophysiologically relevant levels of ∼1–3 nm in the CM, can induce a specific and quantifiable inflammatory response in microglia in vivo.
Figure 6.
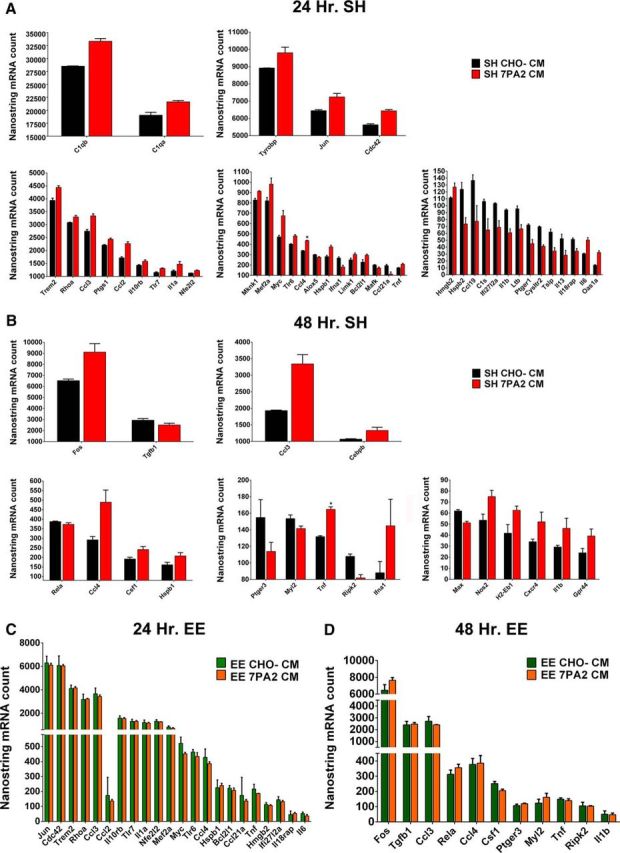
Oligomeric Aβ induces microglial inflammatory mRNA shifts in SH mice as shown by Nanostring mRNA counts, and EE prevents such shifts. A, At 24 h, 41 genes are significantly altered by oAβ in SH mice. Ccl4 is significant with correction (*p < 0.0001), whereas others are without. The genes are assigned to each panel according to exact mRNA counts from Nanostring, which reflects expression level. N = 5. B, At 48 h, 19 genes are significantly altered by oAβ in SH mice. Tnf is significant with correction (*p < 0.0001), whereas others are without. The genes are assigned to each panel according to exact mRNA counts from Nanostring, which reflects expression level. N = 5. C, At 24 h, 22 of 41 genes plotted in A are rescued by EE. Total mRNA counts for the 22 genes show no significant difference between EE mice receiving 7PA2 CM and receiving CHO− CM. N = 5. D, At 48 h, 11 of 19 genes plotted in B are rescued by EE. Total mRNA counts for the 11 genes show no significant difference between EE mice receiving 7PA2 CM and receiving CHO− CM. N = 5. Quantitative data are mean ± SEM.
EE prevents the inflammatory shift in microglial mRNA profile induced by oAβ in SH mice
When we applied the Nanostring mRNA profiling analysis just described in SH mice to littermate mice that had been exposed to EE simultaneously, we again observed a strong ameliorating effect of EE on oAβ microglial alteration. EE successfully neutralized changes in 22 of 41 genes at 24 h and 11 of 19 genes at 48 h that were altered by oAβ in the SH mice (Fig. 6C,D). There was a similar trend toward ameliorated responses with the remaining genes, but to lesser extent. Next, to directly compare SH animals with EE animals as regards oAβ-induced microglial inflammation, the central purpose of our study, we normalized each individual mRNA level in the 7PA2 CM-exposed microglia to the average corresponding level in CHO− CM-exposed microglia within each housing condition and compared the resultant ratios in SH versus EE animals. This additional analysis should ignore any effects of EE not related to oAβ exposure per se (e.g., wound healing; inflammatory responses to non-Aβ substances in the CHO− CM) and focus specifically on the environment-based differences in the microglial response to oAβ. The results of this analysis indicated that whereas in SH mice, multiple inflammatory genes had 7PA2:CHO− mRNA ratios that were significantly higher or lower than 1.0, mice chronically exposed to EE had 7PA2:CHO− mRNA ratios that were instead very close to 1.0. (Because of the many genes we quantified, the SH gene changes were no longer statistically significant after correction for multiplicity, except for the changes in Ccl4 at 24 h and in Tnf at 48 h, p < 0.0001). The consistent prevention by EE of the oAβ-mediated microglial inflammatory gene changes of SH mice that this ratio analysis demonstrated indicates minimal or no effect of oAβ on microglial inflammatory profile after EE (Fig. 7A,B). This molecular result supports our interpretation of the microglial morphology analysis that EE can successfully prevent oAβ-induced microglial alteration. Finally, when we microinjected 7PA2 CM selectively immunodepleted of Aβ as described earlier, we no longer observed any noticeable effect of the 7PA2 CM on SH mice 48 h after exposure in vivo (data not shown).
Figure 7.

EE rescues the shift in microglial inflammatory mRNA profile induced by oAβ in SH mice both in vivo and in vitro, with supportive data from qPCR. A, At 24 h, 22 of 186 inflammatory genes actively expressed by microglia are significantly different between SH and EE mice (uncorrected except for Ccl4). *p < 0.0001. N = 5. SH mice have significantly different expression levels from receiving 7PA2 CM than CHO− CM, whereas EE mice show no alteration, as measured by Nanostring nCounter platform. B, At 48 h, 11 of 163 inflammatory genes actively expressed by microglia are significantly different between SH and EE mice (uncorrected except for Tnf). *p < 0.0001. N = 5. SH mice have significantly different expression levels from receiving 7PA2 CM than CHO− CM, whereas EE mice show no alteration, as measured by Nanostring nCounter platform. C, qPCR analysis on Ccl3, Ccl4, and Tnf shows consistent results as Nanostring: that SH mice have significantly higher levels of Ccl3, Ccl4, and Tnf upon exposure to 7PA2 CM than CHO− CM. *p < 0.001. N = 5. EE neutralizes the responses. ID-7PA2 CM fails to induce any significant changes in mRNA in both SH and EE mice. D, In vitro exposure of 7PA2 CM versus ID-7PA2 CM to microglia isolated from naive SH and EE mice indicates that EE has consistent neutralizing effects on the three main cytokines in vitro as measured by Taqman qPCR. *p < 0.005. N = 8. E, Primary microglia isolated from EE mice show decreased phagocytosis activity toward FITC-Aβ42 oligomer. *p < 0.01. N = 12. Quantitative data are mean ± SEM.
Among the numerous microglial inflammatory genes that responded to in vivo soluble Aβ oligomers differently in SH and EE mice (22 of 186 detectable mRNAs at 24 h; 11 of 163 detectable at 48 h), many of them fell into two functional categories: cytokine signaling pathways and the JNK signaling pathway (Table 1). Three genes were of special interest because they responded to oAβ at both time points but more so at 48 h (thus less likely to be affected by acute effects of the intracerebroventricular injection) and because of their possible functional correlation: Ccl3, Ccl4, and Tnf (Glabinski et al., 2003). Additionally, the elevations of Ccl4 and Tnf in SH mice were still statistically significant after multiplicity correction. Several other of our hits are also thought to be involved in TNFα production and signaling in different model systems, such as Mef2a (Kain et al., 2015), Rhoa (Wang et al., 2015), and the JNK signaling pathway (McCoy and Tansey, 2008), but their functional interactions are not as well characterized as these three (Ccl3, Ccl4, and Tnf) in CNS inflammation. To incorporate a methodologically independent validation of our overall findings, we quantified the same FACS-isolated microglial samples (48 h after intracerebroventricular injection) on a Taqman qPCR platform to measure the expression levels of Ccl3, Ccl4, and Tnf. The results were highly consistent with those obtained by Nanostring: SH mice showed significant increases in Ccl3, Ccl4, and Tnf mRNA levels when exposed in vivo to oAβ (p < 0.001, N = 5), whereas both EE and oAβ immunodepletion of 7PA2 CM effectively diminished the effect (Fig. 7C).
Table 1.
Genes responding differently to oAβ in SH versus EE mice at 24 and 48 h that belong to either cytokine signaling pathways or the JNK signaling pathwaya
| Gene | Response to oAβ (7PA2:CHO− mRNA ratio) |
|||||
|---|---|---|---|---|---|---|
| SH 24 h | SH 48 h | EE 24 h | EE 48 h | p(SH vs EE) 24 h | p(SH vs EE) 48 h | |
| Ccl2 | 1.32 ± 0.04 | — | 1.00 ± 0.04 | — | <0.001 | — |
| Ccl3b | 1.21 ± 0.03 | 1.73 ± 0.14 | 0.94 ± 0.03 | 0.89 ± 0.01 | <0.001 | <0.001 |
| Ccl4b | 1.29 ± 0.01* | 1.68 ± 0.22 | 0.90 ± 0.03 | 1.02 ± 0.13 | <0.0001 | <0.05 |
| Csf1 | — | 1.25 ± 0.08 | — | 0.82 ± 0.04 | — | <0.01 |
| IL-1β | — | 1.58 ± 0.31 | — | 0.92 ± 0.22 | — | <0.05 |
| IL-6 | 1.65 ± 0.11 | — | 0.70 ± 0.17 | — | <0.01 | — |
| Mef2a | 1.20 ± 0.07 | — | 0.86 ± 0.08 | — | <0.01 | — |
| Ptger3 | — | 0.74 ± 0.07 | — | 1.12 ± 0.04 | — | <0.01 |
| Rhoa | 1.07 ± 0.02 | — | 1.01 ± 0.01 | — | — | <0.05 |
| Tgfb1 | — | 0.85 ± 0.05 | — | 1.02 ± 0.05 | — | <0.05 |
| Tnfb | 1.20 ± 0.05 | 1.25 ± 0.02* | 0.86 ± 0.00 | 0.94 ± 0.08 | <0.001 | <0.01 |
| Fos | — | 1.40 ± 0.12 | — | 1.18 ± 0.05 | — | <0.05 |
| Jun | 1.12 ± 0.03 | 1.15 ± 0.10 | 0.97 ± 0.02 | 1.29 ± 0.02** | <0.01 | — |
| Myc | 1.43 ± 0.11 | — | 0.87 ± 0.02** | — | <0.001 | — |
aAll genes in table are selected based on significant response to oAβ in SH mice without correction.
bGene selected for further analysis.
*Significance with correction.
**Significance without correction.
We also performed in vitro exposure of microglia freshly isolated (by FACS) from uninjected SH and EE mice to the 7PA2 CM or ID-7PA2 CM. This in vitro treatment with oAβ yielded results consistent with those observed in vivo: 7PA2 CM significantly increased the mRNA levels of Ccl3, Ccl4, and Tnf in SH microglia, whereas EE produced a significant neutralizing effect on all three genes (Fig. 7D). This confirmatory microglial culture experiment supports our interpretation that EE is directly changing microglial function in vivo in a way that induces tolerance toward the microglial effects of oAβ. Next, we performed experiments suggesting that EE-derived microglia in vitro also have lower phagocytic activity toward fluorescently tagged (FITC) synthetic Aβ oligomers (Fig. 7E), consistent with our data that EE is associated with a neutralized inflammatory response.
In all in vivo experiments throughout this study, we observed very few peripheral monocytes (<5% of the microglial cell counts) in the CD11b+ cell population, and in vivo exposure to oAβ did not increase or decrease this tiny peripheral monocyte population(Fig. 8). The absence of significant monocyte invasion suggests that microglia are the principal macrophage-type cells responding acutely to soluble oligomeric Aβ in the brain and that EE's ability to alter microglial responses to oAβ could have a significant influence at the tissue and organismal levels.
Figure 8.

Flow cytometry gating on postinjection brains indicates minimal invasion of peripheral monocytes. A, Representative gating images from SH/EE animals receiving intracerebroventricular injections of 7PA2 CM or CHO−. Little CD11b+ cells exist of CD45Med/Ly6CLow/Ly6GLow population, indicating minimal invasion of peripheral monocytes and neutrophils. B, Quantification of the percentage of CD45Med/Ly6CLow/Ly6GLow cells within CD11b+ population indicates that >96% CD11b+ cells are resident microglia. There were no significant difference observed among all conditions. N = 8.
EE neutralizes the elevation of extracellular cytokines in brain interstitial fluid induced by oAβ
To measure in vivo the three top cytokines identified in our unbiased mRNA profiling at the protein level, we developed a protocol to use hippocampal microdialysis with a 1000 kDa MWCO. In vivo microdialysis enabled us to measure protein concentrations in the brain interstitial fluid (ISF) of awake, behaving mice. The 1000 kDa MWCO should allow recovery of almost any kind of cytokine, making it an ideal system to analyze quantitatively the oAβ-induced inflammatory response in vivo. Mice again received an intracerebroventricular injection of 7PA2 CM or CHO− CM and then had a guide cannula for microdialysis implanted into the contralateral hippocampus immediately afterward on day 0. Microdialysis began 48 h after surgery, as specified in our intracerebroventricular injection model described above. The 48 h waiting time and contralateral locus for ISF collection allow inflammation caused by intracerebroventricular injection to largely resolve; the ISF collection then continues for 12 h (Fig. 9A). All mice were dissected afterward to confirm the placement of the microdialysis probes and observe the injection site. Mice with substantial bleeding at the injection site were excluded from analysis. We analyzed the ISF samples for our three top analytes: TNFα, CCL3, CCL4. We normalized each ISF sample from a mouse receiving 7PA2 CM to its paired ISF of the mouse in the same environment (SH or EE) receiving the control CHO− CM. Each such pair of mice shared the same microdialysis pumping system, removing any possible variation caused by the equipment. We observed significant elevations of CCL3, CCL4, and TNFα proteins in the ISF of SH mice receiving 7PA2 CM but not in the matched EE mice (7PA2 CM/ CHO− CM ratios: CCL3: SH 1.443 ± 0.335 vs EE 0.923 ± 0.188, p < 0.01; CCL4: SH 1.579 ± 0.168 vs EE 0.949 ± 0.298, p < 0.01; TNFα: SH 1.872 ± 0.663 vs EE 0.734 ± 0.213, p < 0.01. N = 6) (Fig. 9B). These ISF cytokine findings are entirely consistent with the neutralizing effects of EE at the mRNA level and by microglial morphometry.
Figure 9.

EE neutralizes the elevation of cytokines in brain interstitial fluid induced by oAβ. A, ISF collection timeline: the mice were intracerebroventricularly injected with 7PA2 CM or CHO− CM and had guide cannula implanted into contralateral hippocampus. After 48 h, ISF was collected for 12 h at 0.4 μl/min perfusion rate. B, The ISF samples were analyzed by customized ELISA from Meso Scale Discovery to measure CCL3, CCL4, and TNFα. Results show that SH mice have significantly higher ratios of 7PA2 CM/CHO− CM in all three molecules compared with EE mice (*p < 0.01, N = 6), with EE mice showing ratio very close to 1.0. Data are mean ± SEM.
EE neutralization of microglial mRNA changes is confirmed using human oAβ from AD brain
To extend this cumulative evidence of a preventive effect of EE against oAβ into the most pathobiologically relevant context, we examined saline (TBS) extracts from the brains of patients with clinically and neuropathologically diagnosed AD (AD-TBS) as a source of endogenous human Aβ oligomers. Previous work in our laboratory has established that such soluble extracts potently suppress hippocampal synaptic plasticity, decrease synapse density, and actually interfere with the memory of a learned behavior in rodents (Shankar et al., 2008). Moreover, the AD brain oligomers induce hyperphosphorylation of tau protein in primary neurons at AD-relevant epitopes, and this is followed by progressive neuritic dystrophy (Jin et al., 2011). As a key negative control, we performed selective immunodepletions of oAβ from the human AD-TBS with conjugated 4G8 antibody. The supernatant from the last of two serial immunodepletion steps was analyzed by ELISAs specific for either Aβ oligomers, Aβ x-40 monomers or Aβ x-42 monomers. The ELISAs, including the one specific for oligomers, had been developed here to accurately and sensitively measure Aβ species of different forms (Yang et al., 2013, 2015). Because the Aβ oligomers in the AD-TBS fraction are specifically responsible for inducing all of the aforementioned AD-relevant phenotypes, it was important that we were able to confirm by ELISA a marked decrease (>90%) of oAβ and almost complete removal of Aβ x-40 and Aβ x-42 species by the immunodepletion. Western blots of the immunoprecipitates confirmed almost complete removal of all Aβ species, consistent with the ELISA data (Fig. 10A).
Figure 10.

EE neutralization of microglial mRNA changes is confirmed using human oAβ from AD brain. A, Supernatant from the last immunodepletion was analyzed by ELISA for oligomeric Aβ, Aβ x-40, and Aβ x-42. Results show >90% removal of all Aβ species. Western blot shows no detectable oAβ after first immunodepletion, confirming the result. B, Taqman qPCR analysis on Ccl3, Ccl4, and Tnf shows results similar to those of 7PA2 CM: that SH mice have significantly higher levels of Ccl3, Ccl4, and Tnf upon exposure to human AD-TBS extract (ADTBS) than immunodepleted AD-TBS extract (ID-ADTBS). **p < 0.002. ***p < 0.001. ****p < 0.0001. N = 6. EE neutralizes the response. C, Nanostring analysis on the same samples shows a consistent overall neutralizing effect of EE. Among all genes significantly (uncorrected) changed in SH mice by oAβ and at the same time showing significantly (uncorrected) rescue effect from EE, 5 overlap with that of 7PA2 CM (marked by red dots). Quantitative data are mean ± SEM.
We applied the AD-TBS and immunodepleted AD-TBS (ID-ADTBS) to EE and SH mice by following the same intracerebroventricular injection procedure and microglial-selective mRNA analysis (48 h) as described above. The samples were first analyzed by Taqman qPCR for Ccl3, Ccl4, and Tnf mRNAs. We observed very consistent induction of the three molecules by the human AD-TBS in SH but not in EE mice, with the differences being significant, as we had seen by qPCR with the oAβ-rich 7PA2 CM. The values were expressed as a ratio of the mRNA level after AD-TBS versus that after ID-ADTBS injection, as follows: Ccl3: SH 1.65 ± 0.31 vs EE 0.86 ± 0.10, p < 0.0001; Ccl4: SH 1.52 ± 0.24 vs EE 0.96 ± 0.17, p < 0.001; Tnf: SH 1.93 ± 0.47 vs EE 1.07 ± 0.20, p < 0.002, N = 6) (Fig. 10B).
We then analyzed all microglial mRNA samples after the AD-TBS (or ID-ADTBS) microinjections on the Nanostring platform with the same Code-set used previously and saw results consistent with those of the 7PA2 CM. EE prevented the selective microglial gene changes seen in SH mice 48 h after injection of the AD-TBS. We found that 31 of 120 actively expressed microglial genes were significantly changed (uncorrected) by AD-TBS in SH mice, and 24 of these were rescued by the EE exposure (Fig. 10C) (raw values before normalization are plotted in Fig. 11A,B). Among these 24 hits, 5 of them, including Ccl3, Ccl4, and Tnf that we had tested by qPCR, are in common with the 11 hits we had identified at 48 h after the 7PA2 CM injection (Fig. 10C, red dots). AD-TBS also induced gene expression changes in several other cytokines and MAP kinase family members that are important for proper microglial signaling, the majority of which had shown comparable trends in the 7PA2 CM dataset, although not to as strong an extent as with the human brain Aβ oligomers.
Figure 11.

Human AD-TBS induces microglial inflammatory mRNA profile shift in SH mice as shown by Nanostring mRNA counts, and EE rescues such shift. A, At 48 h, 31 genes are significantly altered by oAβ in SH mice (uncorrected). The genes are assigned to each panel according to exact mRNA counts from Nanostring, which reflects expression level. N = 6. B, At 48 h, 24 of 31 genes plotted in A are rescued by EE. Total mRNA counts for the 24 genes show no significant difference between EE mice receiving AD-TBS and receiving ID-ADTBS. N = 6. Quantitative data are mean ± SEM.
Discussion
EE has been studied in animal models for its potential benefit in ameliorating the neuronal effects of factors implicated in AD. Our laboratory previously documented the benefits of EE in enhancing hippocampal LTP and increasing the levels of select synaptic markers and NMDA receptors in a way that decreased the adverse neuronal effects of oAβ (Li et al., 2013). However, whether EE can modify the brain's innate immune system and thereby lessen microglial abnormalities caused by oligomeric Aβ has not been addressed. Here, we systematically examined the possible benefits of EE on microglia from multiple perspectives because despite extensive knowledge of EE's neuronal benefits, whether and how EE may regulate microglial function in healthy animals and in those exposed selectively to pathogenic species of human oAβ is unstudied. A single very recent paper appearing as we completed this work examined certain aspects of this topic with a methodologically distinct approach (Rodríguez et al., 2015). Our multiple assays indicate that exposure to EE effectively modulates microglial form and function in a way that results in robust tolerance toward the effects of oAβ, resulting in significantly less proinflammatory microglial alteration.
We initially documented a significant increase in microglial density in the DG of naive mice exposed to EE, consistent with two recent studies using different methods (Williamson et al., 2012; Reshef et al., 2014). EdU labeling during EE showed minimal EdU+ microglia, suggesting a source of additional microglia other than cell division. We speculate that the extra microglia either migrate from nearby brain regions or mature from progenitor cells reported to develop into microglia (Elmore et al., 2014). Deeper morphological analyses revealed significant changes in microglial branching complexity by EE as measured by skeleton analysis, indicating that modulation of microglia by EE can occur without exposure to adverse stimuli. Next, we observed almost complete rescue by EE of microglial form in hippocampus (as judged by multiple morphological parameters) when animals were exposed in vivo to human oAβ. Unlike hippocampal microglia, cortical microglia exhibited minimal alteration with oAβ exposure. Such a difference could relate in part to significant regional differences in the microglial transcriptome, as recently reported (Grabert et al., 2016).
The molecular changes underlying the prominent EE rescue effect were next probed by unbiased examination of microglial inflammatory mRNA profiles. We found that EE suppressed the elevated expression of select inflammatory genes that occurred in response to oAβ in SH mice and similarly neutralized the depression of others. The highly consistent differences between the mRNA profiles of mice in the two environments and the fact that selective removal of oAβ from the 7PA2 CM prevented the mRNA profile shift in the SH animals together strongly suggest that EE can significantly mitigate the majority of inflammatory gene responses of microglia to oAβ, in accord with the results of our morphological phenotyping in situ. The preventive effect of EE was confirmed at the protein level in vivo by performing microdialysis in awake, behaving animals and quantifying the proteins of the 3 most altered mRNAs: TNFα, CCL3, and CCL4. All of these data support the hypothesis that EE effectively primes microglia before exposure to oAβ so that microglia are more resistant to challenges, resulting in a significant tolerance to human oAβ species previously shown to be highly synaptotoxic (Shankar et al., 2008; Jin et al., 2011) and thereby lowering inflammatory cytokines and potentially mitigating secondary neuronal toxicity. Upon establishing primary cultures of microglia from uninjected mice and exposing them to oAβ in vitro, we observed similar increases in mRNA levels of Ccl3, Ccl4, and Tnf in cells from SH but not EE animals, supporting our interpretation that EE ameliorates a direct response of microglia to Aβ per se.
We performed preliminary experiments to assess the phagocytic response of the cultured primary microglia to oAβ and found decreased uptake of synthetic oAβ by microglia from the EE mice. Establishing this in vitro assay will enable more extensive experiments to be done on this aspect of EE, but we interpret the apparent decrease in phagocytosis as suggesting that the sharply decreased proinflammatory reaction documented for EE in vivo and in vitro is associated with less phagocytic activity toward oAβ, as phagocytosis and inflammation can be related in macrophages (Aderem, 2003). In AD and models thereof, microglial function can be impaired by Aβ (Hickman et al., 2008; Krabbe et al., 2013) and by certain AD-linked genetic variants (Kleinberger et al., 2014; Heneka et al., 2015), allowing chronic overproduction of some cytokines that may then contribute to neuronal toxicity. We hypothesize that the ability of EE to neutralize overall microglial responses to oAβ could curb such cytokine release in the brain and help protect neurons. This idea is supported by evidence that eliminating microglia in vivo rescues neuronal injury (Spangenberg et al., 2016), whereas enhanced microglial activation exacerbates it (Cho et al., 2011), in each case without necessarily altering Aβ burden.
Our choice to use wt mice not overexpressing early-onset AD gene mutations sought to address whether environmental factors alone are adequate to generate immunomodulatory effects that could be beneficial in very early “sporadic” subjects lacking a specific genetic diathesis toward familial AD. We used an intracerebroventricular microinjection model to expose wt mice acutely to pathophysiologically relevant (low nanomolar) levels of human oAβ, including oligomers isolated directly from typical AD brains, to better model the earliest effects of Aβ dyshomeostasis in AD in the large majority of humans lacking deterministic FAD mutations and thereby quantify oAβ microglial activities without the potentially confounding effects of genetic manipulation.
It is notable that TNFα, CCL3, and CCL4, the top three hits from our unbiased mRNA screen among 253 inflammatory genes, were confirmed at the protein level upon in vivo microdialysis and were similarly elevated by the human oAβ oligomers in 7PA2 CM and in AD brain extracts. In all cases, the changes were significantly neutralized by EE, suggesting a key involvement of these cytokines (among others) in the microglial oAβ response and the benefits of EE. TNFα has been widely studied in AD and other neurodegenerative disorders as a key regulator of neuroinflammation (Fillit et al., 1991; Meda et al., 1995; Collins et al., 2000; Tan et al., 2007; Holmes et al., 2009) and can induce neuronal death (Bhaskar et al., 2014). TNFα is predominantly expressed by microglia in the CNS (Renno et al., 1995; Chabot et al., 1997) and interacts with CCL3 and CCL4 during inflammation (Glabinski et al., 2003). TNFα may also be involved in maintaining microglial inflammation and contribute to related morphological changes (Pearse et al., 2004). Based on these results, our finding regarding significant differences in microglial morphology between SH and EE mice may relate in part to the potent suppression of TNFα expression by EE that we document.
A previous study from our laboratory found that EE activates β2-adrenergic receptors (β2AR) primarily in neuronal synapses as an early part of its neuronal protective effect (Li et al., 2013). There is evidence that microglia express adrenergic receptors (Mori et al., 2002; Tanaka et al., 2002). The activation of such receptors, in particular by β2-AR agonists, leads to suppression of the proinflammatory response to LPS (Hetier et al., 1991; Markus et al., 2010; Qian et al., 2011). β2AR can also mediate the dynamics of microglial processes (Gyoneva and Traynelis, 2013). Our data demonstrate that EE modulates the complexity of microglial processes in naive mice and has immunosuppressive effects on the microglial response to Aβ oligomers. Therefore, we speculate that the molecular mechanism of EE's immunomodulating capacity is partially due to EE increasing the level and activity of β2AR on microglia, as it does in neurons, resulting in potential changes in the structure and function of microglial processes before challenge and the suppression of inflammation when challenged by oAβ.
In conclusion, our study reveals that EE has substantial immunomodulatory effects on innate microglia that are sufficient to increase their density and alter their morphology in healthy, unchallenged mice. EE significantly increases microglial resistance to the effects of oAβ and largely suppresses its proinflammatory activity. Intriguingly, two recent studies report immunosuppressive effects of EE on microglia in mouse models of depression (Chabry et al., 2015) or glioma (Garofalo et al., 2015). In humans, physical exercise and higher levels of cognitive activity are thought to be among the few environmental factors that may delay the onset and progression of clinical AD. For example, recent data in human subjects showed an inverse correlation between degree of intellectual involvement in one's occupation and the extent of cognitive decline (Then et al., 2015). Our study uncovers a new feature of the mechanism of EE's potential protective effect in AD by revealing its potent immunomodulation of microglia and identifying select cytokines, including CCL3, CCL4, and TNFα, which provide targets for future mechanistic and therapeutic studies. We recommend further intensive study of the molecular pathways linking EE to microglial biology, with attendant implications for the potential delay or prevention of brain dysfunction before clinical onset in AD and other neurodegenerative diseases.
Footnotes
This work was supported by National Institutes of Health Grants R01 AG06173 and R01 AG043975 to D.S. We thank Drs. Oleg Butovsky and Howard Weiner for providing anti-P2ry12 antibody and helpful advice and the members of the D.S. laboratory for many helpful discussions.
The authors declare no competing financial interests.
References
- Aderem A. Phagocytosis and the inflammatory response. J Infect Dis. 2003;187(Suppl 2):S340–S345. doi: 10.1086/374747. [DOI] [PubMed] [Google Scholar]
- Bhaskar K, Maphis N, Xu G, Varvel NH, Kokiko-Cochran ON, Weick JP, Staugaitis SM, Cardona A, Ransohoff RM, Herrup K, Lamb BT. Microglial derived tumor necrosis factor-alpha drives Alzheimer's disease-related neuronal cell cycle events. Neurobiol Dis. 2014;62:273–285. doi: 10.1016/j.nbd.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, Koeglsperger T, Dake B, Wu PM, Doykan CE, Fanek Z, Liu L, Chen Z, Rothstein JD, Ransohoff RM, Gygi SP, Antel JP, Weiner HL. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci. 2014;17:131–143. doi: 10.1038/nn.3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabot S, Williams G, Yong VW. Microglial production of TNF-alpha is induced by activated T lymphocytes: involvement of VLA-4 and inhibition by interferonbeta-1b. J Clin Invest. 1997;100:604–612. doi: 10.1172/JCI119571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabry J, Nicolas S, Cazareth J, Murris E, Guyon A, Glaichenhaus N, Heurteaux C, Petit-Paitel A. Enriched environment decreases microglia and brain macrophages inflammatory phenotypes through adiponectin-dependent mechanisms: relevance to depressive-like behavior. Brain Behav Immun. 2015;50:275–287. doi: 10.1016/j.bbi.2015.07.018. [DOI] [PubMed] [Google Scholar]
- Cho SH, Sun B, Zhou Y, Kauppinen TM, Halabisky B, Wes P, Ransohoff RM, Gan L. CX3CR1 protein signaling modulates microglial activation and protects against plaque-independent cognitive deficits in a mouse model of Alzheimer disease. J Biol Chem. 2011;286:32713–32722. doi: 10.1074/jbc.M111.254268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SH, Veeraraghavalu K, Lazarov O, Marler S, Ransohoff RM, Ramirez JM, Sisodia SS. Non-cell-autonomous effects of presenilin 1 variants on enrichment-mediated hippocampal progenitor cell proliferation and differentiation. Neuron. 2008;59:568–580. doi: 10.1016/j.neuron.2008.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins JS, Perry RT, Watson B, Jr, Harrell LE, Acton RT, Blacker D, Albert MS, Tanzi RE, Bassett SS, McInnis MG, Campbell RD, Go RC. Association of a haplotype for tumor necrosis factor in siblings with late-onset Alzheimer disease: the NIMH Alzheimer disease genetics initiative. Am J Med Genet. 2000;96:823–830. doi: 10.1002/1096-8628(20001204)96:6%3C823::AID-AJMG26%3E3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Cracchiolo JR, Mori T, Nazian SJ, Tan J, Potter H, Arendash GW. Enhanced cognitive activity-over and above social or physical activity-is required to protect Alzheimer's mice against cognitive impairment, reduce Abeta deposition, and increase synaptic immunoreactivity. Neurobiol Learn Mem. 2007;88:277–294. doi: 10.1016/j.nlm.2007.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehninger D, Kempermann G. Regional effects of wheel running and environmental enrichment on cell genesis and microglia proliferation in the adult murine neocortex. Cereb Cortex. 2003;13:7. doi: 10.1093/cercor/13.8.845. [DOI] [PubMed] [Google Scholar]
- Elmore MR, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, Kitazawa M, Matusow B, Nguyen H, West BL, Green KN. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron. 2014;82:380–397. doi: 10.1016/j.neuron.2014.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fillit H, Ding WH, Buee L, Kalman J, Altstiel L, Lawlor B, Wolf-Klein G. Elevated circulating tumor necrosis factor levels in Alzheimer's disease. Neurosci Lett. 1991;129:318–320. doi: 10.1016/0304-3940(91)90490-K. [DOI] [PubMed] [Google Scholar]
- Garofalo S, D'Alessandro G, Chece G, Brau F, Maggi L, Rosa A, Porzia A, Mainiero F, Esposito V, Lauro C, Benigni G, Bernardini G, Santoni A, Limatola C. Enriched environment reduces glioma growth through immune and non-immune mechanisms in mice. Nat Commun. 2015;6:6623. doi: 10.1038/ncomms7623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glabinski AR, Bielecki B, Kolodziejski P, Han Y, Selmaj K, Ransohoff RM. TNF-α microinjection upregulates chemokines and chemokine receptors in the central nervous system without inducing leukocyte infiltration. J Interferon Cytokine Res. 2003;23:457–466. doi: 10.1089/107999003322277874. [DOI] [PubMed] [Google Scholar]
- Grabert K, Michoel T, Karavolos MH, Clohisey S, Baillie JK, Stevens MP, Freeman TC, Summers KM, McColl BW. Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat Neurosci. 2016;19:504–516. doi: 10.1038/nn.4222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyoneva S, Traynelis SF. Norepinephrine modulates the motility of resting and activated microglia via different adrenergic receptors. J Biol Chem. 2013;288:15291–15302. doi: 10.1074/jbc.M113.458901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss-Coray T, Vitorica J, Ransohoff RM, Herrup K, Frautschy SA, Finsen B, Brown GC, Verkhratsky A, Yamanaka K, Koistinaho J, Latz E, Halle A, Petzold GC, et al. Neuroinflammation in Alzheimer's disease. Lancet Neurol. 2015;14:388–405. doi: 10.1016/S1474-4422(15)70016-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herring A, Ambrée O, Tomm M, Habermann H, Sachser N, Paulus W, Keyvani K. Environmental enrichment enhances cellular plasticity in transgenic mice with Alzheimer-like pathology. Exp Neurol. 2009;216:184–192. doi: 10.1016/j.expneurol.2008.11.027. [DOI] [PubMed] [Google Scholar]
- Hetier E, Ayala J, Bousseau A, Prochiantz A. Modulation of interleukin-1 and tumor necrosis factor expression by β-adrenergic agonists in mouse ameboid microglial cells. Exp Brain Res. 1991;86:407–413. doi: 10.1007/BF00228965. [DOI] [PubMed] [Google Scholar]
- Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer's disease mice. J Neurosci. 2008;28:8354–8360. doi: 10.1523/JNEUROSCI.0616-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes C, Cunningham C, Zotova E, Woolford J, Dean C, Kerr S, Culliford D, Perry VH. Systemic inflammation and disease progression in Alzheimer disease. Neurology. 2009;73:768–774. doi: 10.1212/WNL.0b013e3181b6bb95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin M, Shepardson N, Yang T, Chen G, Walsh D, Selkoe DJ. Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc Natl Acad Sci U S A. 2011;108:5819–5824. doi: 10.1073/pnas.1017033108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kain V, Kapadia B, Viswakarma N, Seshadri S, Prajapati B, Jena PK, Teja Meda CL, Subramanian M, Kaimal Suraj S, Kumar ST, Prakash Babu P, Thimmapaya B, Reddy JK, Parsa KV, Misra P. Co-activator binding protein PIMT mediates TNF-alpha induced insulin resistance in skeletal muscle via the transcriptional down-regulation of MEF2A and GLUT4. Sci Rep. 2015;5:15197. doi: 10.1038/srep15197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempermann G, Kuhn HG, Gage FH. More hippocampal neurons in adult mice living in an enriched environment. Nature. 1997;386:493–495. doi: 10.1038/386493a0. [DOI] [PubMed] [Google Scholar]
- Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. 2011;91:461–553. doi: 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- Kleinberger G, Yamanishi Y, Suárez-Calvet M, Wyss-Coray T, Hardy J, Colonna M, Haass C. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med. 2014;6:14. doi: 10.1126/scitranslmed.3009093. [DOI] [PubMed] [Google Scholar]
- Krabbe G, Halle A, Matyash V, Rinnenthal JL, Eom GD, Bernhardt U, Miller KR, Prokop S, Kettenmann H, Heppner FL. Functional impairment of microglia coincides with Beta-amyloid deposition in mice with Alzheimer-like pathology. PLoS One. 2013;8:e60921. doi: 10.1371/journal.pone.0060921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarov O, Robinson J, Tang YP, Hairston IS, Korade-Mirnics Z, Lee VM, Hersh LB, Sapolsky RM, Mirnics K, Sisodia SS. Environmental enrichment reduces Abeta levels and amyloid deposition in transgenic mice. Cell. 2005;120:701–713. doi: 10.1016/j.cell.2005.01.015. [DOI] [PubMed] [Google Scholar]
- Lee S, Varvel NH, Konerth ME, Xu G, Cardona AE, Ransohoff RM, Lamb BT. CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer's disease mouse models. Am J Pathol. 2010;177:2549–2562. doi: 10.2353/ajpath.2010.100265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Jin M, Koeglsperger T, Shepardson NE, Shankar GM, Selkoe DJ. Soluble Abeta oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J Neurosci. 2011;31:6627–6638. doi: 10.1523/JNEUROSCI.0203-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Jin M, Zhang D, Yang T, Koeglsperger T, Fu H, Selkoe DJ. Environmental novelty activates beta2-adrenergic signaling to prevent the impairment of hippocampal LTP by Abeta oligomers. Neuron. 2013;77:929–941. doi: 10.1016/j.neuron.2012.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markus T, Hansson SR, Cronberg T, Cilio C, Wieloch T, Ley D. beta-Adrenoceptor activation depresses brain inflammation and is neuroprotective in lipopolysaccharide-induced sensitization to oxygen-glucose deprivation in organotypic hippocampal slices. J Neuroinflammation. 2010;7:94. doi: 10.1186/1742-2094-7-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy RC, Lu DY, Alkhateeb A, Gardeck AM, Lee CH, Wessling-Resnick M. Characterization of a novel adult murine immortalized microglial cell line and its activation by amyloid-beta. J Neuroinflammation. 2016;13:21. doi: 10.1186/s12974-016-0484-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy MK, Tansey MG. TNF signaling inhibition in the CNS: implications for normal brain function and neurodegenerative disease. J Neuroinflammation. 2008;5:45. doi: 10.1186/1742-2094-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meda L, Cassatella MA, Szendrei GI, Otvos L, Jr, Baron P, Villalba M, Ferrari D, Rossi F. Activation of microglial cells by [beta]-amyloid protein and interferon-[gamma] Nature. 1995;374:647–650. doi: 10.1038/374647a0. [DOI] [PubMed] [Google Scholar]
- Mori K, Ozaki E, Zhang B, Yang L, Yokoyama A, Takeda I, Maeda N, Sakanaka M, Tanaka J. Effects of norepinephrine on rat cultured microglial cells that express α1, α2, β1 and β2 adrenergic receptors. Neuropharmacology. 2002;43:1026–1034. doi: 10.1016/S0028-3908(02)00211-3. [DOI] [PubMed] [Google Scholar]
- Morrison HW, Filosa JA. A quantitative spatiotemporal analysis of microglia morphology during ischemic stroke and reperfusion. J Neuroinflammation. 2013;10:4. doi: 10.1186/1742-2094-10-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson M, Perfilieva E, Johansson U, Orwar O, Eriksson PS. Enriched environment increases neurogenesis in the adult rat dentate gyrus and improves spatial memory. J Neurobiol. 1999;39:569–578. doi: 10.1002/(SICI)1097-4695(19990615)39:4%3C569::AID-NEU10%3E3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Nithianantharajah J, Hannan AJ. Enriched environments, experience-dependent plasticity and disorders of the nervous system. Nat Rev Neurosci. 2006;7:697–709. doi: 10.1038/nrn1970. [DOI] [PubMed] [Google Scholar]
- Olson AK, Eadie BD, Ernst C, Christie BR. Environmental enrichment and voluntary exercise massively increase neurogenesis in the adult hippocampus via dissociable pathways. Hippocampus. 2006;16:250–260. doi: 10.1002/hipo.20157. [DOI] [PubMed] [Google Scholar]
- Pearse DD, Pereira FC, Stolyarova A, Barakat DJ, Bunge MB. Inhibition of tumour necrosis factor-alpha by antisense targeting produces immunophenotypical and morphological changes in injury-activated microglia and macrophages. Eur J Neurosci. 2004;20:3387–3396. doi: 10.1111/j.1460-9568.2004.03799.x. [DOI] [PubMed] [Google Scholar]
- Podlisny MB, Ostaszewski BL, Squazzo SL, Koo EH, Rydell RE, Teplow DB, Selkoe DJ. Aggregation of secreted amyloid-protein into sodium dodecyl sulfate-stable oligomers in cell culture. J Biol Chem. 1995;270:9564–9570. doi: 10.1074/jbc.270.16.9564. [DOI] [PubMed] [Google Scholar]
- Qian L, Wu HM, Chen SH, Zhang D, Ali SF, Peterson L, Wilson B, Lu RB, Hong JS, Flood PM. beta2-adrenergic receptor activation prevents rodent dopaminergic neurotoxicity by inhibiting microglia via a novel signaling pathway. J Immunol. 2011;186:4443–4454. doi: 10.4049/jimmunol.1002449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renno T, Krakowski M, Piccirillo C, Lin JY, Owens T. TNF-alpha expression by resident microglia and infiltrating leukocytes in the central nervous system of mice with experimental allergic encephalomyelitis: regulation by Th1 cytokines. J Immunol. 1995;154:944–953. [PubMed] [Google Scholar]
- Reshef R, Kreisel T, Beroukhim Kay D, Yirmiya R. Microglia and their CX3CR1 signaling are involved in hippocampal, but not olfactory, bulb-related memory and neurogenesis. Brain Behav Immun. 2014;41:239–250. doi: 10.1016/j.bbi.2014.04.009. [DOI] [PubMed] [Google Scholar]
- Rodríguez JJ, Noristani HN, Verkhratsky A. Microglial response to Alzheimer's disease is differentially modulated by voluntary wheel running and enriched environments. Brain Struct Funct. 2015;220:941–953. doi: 10.1007/s00429-013-0693-5. [DOI] [PubMed] [Google Scholar]
- Rubio-Perez JM, Morillas-Ruiz JM. A review: inflammatory process in Alzheimer's disease, role of cytokines. Sci World J. 2012;2012:756357. doi: 10.1100/2012/756357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solito E, Sastre M. Microglia function in Alzheimer's disease. Front Pharmacol. 2012;3:14. doi: 10.3389/fphar.2012.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spangenberg EE, Lee RJ, Najafi AR, Rice RA, Elmore MR, Blurton-Jones M, West BL, Green KN. Eliminating microglia in Alzheimer's mice prevents neuronal loss without modulating amyloid-beta pathology. Brain. 2016;139:1265–1281. doi: 10.1093/brain/aww016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka KF, Kashima H, Suzuki H, Ono K, Sawada M. Existence of functional beta1- and beta2-adrenergic receptors on microglia. J Neurosci Res. 2002;70:232–237. doi: 10.1002/jnr.10399. [DOI] [PubMed] [Google Scholar]
- Tan ZS, Beiser AS, Vasan RS, Roubenoff R, Dinarello CA, Harris TB, Benjamin EJ, Au R, Kiel DP, Wolf PA, Seshadri S. Inflammatory markers and the risk of Alzheimer disease: the Framingham Study. Neurology. 2007;68:1902–1908. doi: 10.1212/01.wnl.0000263217.36439.da. [DOI] [PubMed] [Google Scholar]
- Then FS, Luck T, Luppa M, König HH, Angermeyer MC, Riedel-Heller SG. Differential effects of enriched environment at work on cognitive decline in old age. Neurology. 2015;84:2169–2176. doi: 10.1212/WNL.0000000000001605. [DOI] [PubMed] [Google Scholar]
- Vukovic J, Colditz MJ, Blackmore DG, Ruitenberg MJ, Bartlett PF. Microglia modulate hippocampal neural precursor activity in response to exercise and aging. J Neurosci. 2012;32:6435–6443. doi: 10.1523/JNEUROSCI.5925-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ. A beta oligomers: a decade of discovery. J Neurochem. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid b protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Townsend M, Podlisny MB, Shankar GM, Fadeeva JV, El Agnaf O, Hartley DM, Selkoe DJ. Certain inhibitors of synthetic amyloid beta-peptide (Abeta) fibrillogenesis block oligomerization of natural Abeta and thereby rescue long-term potentiation. J Neurosci. 2005;25:2455–2462. doi: 10.1523/JNEUROSCI.4391-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Song S, Zhang Y, Ge Y, Fang X, Huang T, Du J, Gao J. Inhibition of the Rho/Rho kinase pathway prevents lipopolysaccharide-induced hyperalgesia and the release of TNF-alpha and IL-1beta in the mouse spinal cord. Sci Rep. 2015;5:14553. doi: 10.1038/srep14553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welzel AT, Maggio JE, Shankar GM, Walker DE, Ostaszewski BL, Li S, Klyubin I, Rowan MJ, Seubert P, Walsh DM, Selkoe DJ. Secreted amyloid beta-proteins in a cell culture model include N-terminally extended peptides that impair synaptic plasticity. Biochemistry. 2014;53:3908–3921. doi: 10.1021/bi5003053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson LL, Chao A, Bilbo SD. Environmental enrichment alters glial antigen expression and neuroimmune function in the adult rat hippocampus. Brain Behav Immun. 2012;26:500–510. doi: 10.1016/j.bbi.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T, Rogers J. Inflammation in Alzheimer disease: a brief review of the basic science and clinical literature. Cold Spring Harbor Perspect Med. 2012;2:a006346. doi: 10.1101/cshperspect.a006346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Hong S, O'Malley T, Sperling RA, Walsh DM, Selkoe DJ. New ELISAs with high specificity for soluble oligomers of amyloid beta-protein detect natural Abeta oligomers in human brain but not CSF. Alzheimers Dement. 2013;9:99–112. doi: 10.1016/j.jalz.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, O'Malley TT, Kanmert D, Jerecic J, Zieske LR, Zetterberg H, Hyman BT, Walsh DM, Selkoe DJ. A highly sensitive novel immunoassay specifically detects low levels of soluble Abeta oligomers in human cerebrospinal fluid. Alzheimers Res Ther. 2015;7:14. doi: 10.1186/s13195-015-0100-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziv Y, Schwartz M. Immune-based regulation of adult neurogenesis: implications for learning and memory. Brain Behav Immun. 2008;22:167–176. doi: 10.1016/j.bbi.2007.08.006. [DOI] [PubMed] [Google Scholar]
- Ziv Y, Ron N, Butovsky O, Landa G, Sudai E, Greenberg N, Cohen H, Kipnis J, Schwartz M. Immune cells contribute to the maintenance of neurogenesis and spatial learning abilities in adulthood. Nat Neurosci. 2006;9:268–275. doi: 10.1038/nn1629. [DOI] [PubMed] [Google Scholar]