

Abstract
Neuroligins are postsynaptic cell-adhesion molecules that contribute to synapse specification. However, many other postsynaptic cell-adhesion molecules are known and the relative contributions of neuroligins versus other such molecules in different types of synapses and neurons remains largely unknown. Here, we have studied the role of neuroligins in cerebellar stellate interneurons that participate in a well defined circuit that converges on Purkinje cells as the major output neurons of cerebellar cortex. By crossing triple conditional knock-out (cKO) mice targeting all three major neuroligins [neuroligin-1 to neuroligin-3 (NL123)] with parvalbumin-Cre (PV-Cre) transgenic mice, we deleted neuroligins from inhibitory cerebellar interneurons and Purkinje cells, allowing us to study the effects of neuroligin deletions on cerebellar stellate cell synapses by electrophysiology in acute slices. PV-Cre/NL123 cKO mice did not exhibit gross alterations of cerebellar structure or cerebellar interneuron morphology. Strikingly, electrophysiological recordings in stellate cells from these PV-Cre/NL123 cKO mice revealed a large decrease in NMDAR-mediated excitatory synaptic responses, which, in stellate cells, are largely extrasynaptic, without a change in AMPA-receptor-mediated responses. Parallel analyses in PV-Cre/NL1 mice that are single NL1 cKO mice uncovered the same phenotype, demonstrating that NL1 is responsible for recruiting extrasynaptic NMDARs. Moreover, we observed only a modest impairment in inhibitory synaptic responses in stellate cells lacking NL123 despite a nearly complete suppression of inhibitory synaptic transmission in Purkinje cells by the same genetic manipulation. Our results suggest that, unlike other types of neurons investigated, neuroligins are selectively essential in cerebellar stellate interneurons for enabling the function of extrasynaptic NMDARs.
SIGNIFICANCE STATEMENT Neuroligins are postsynaptic cell-adhesion molecules genetically linked to autism. However, the contributions of neuroligins to interneuron functions remain largely unknown. Here, we analyzed the role of neuroligins in cerebellar stellate interneurons. We deleted neuroligin-1, neuroligin-2, and neuroligin-3, the major cerebellar neuroligin isoforms, from stellate cells in triple NL123 conditional knock-out mice and analyzed synaptic responses by acute slice electrophysiology. We find that neuroligins are selectively essential for extrasynaptic NMDAR-mediated signaling, but dispensable for both AMPAR-mediated and inhibitory synaptic transmission. Our results reveal a critical and selective role for neuroligins in the regulation of NMDAR responses in cerebellar stellate interneurons.
Keywords: cerebellum, neuroligin, stellate cell, synaptic transmission
Introduction
Knowledge of how neurons form synapses and how synapses function is critical to our understanding of the brain and of neuropsychiatric disorders. Neuroligins are a family of four postsynaptic cell-adhesion molecules (NL1, NL2, NL3, and NL4) that contribute to synapse specification via binding to presynaptic neurexins (Ichtchenko et al., 1995, 1996; Song et al., 1999; Varoqueaux et al., 2004; Chih et al., 2005; Budreck and Scheiffele, 2007; Craig and Kang, 2007; Südhof, 2008; Hoon et al., 2011; McMahon et al., 2011; Krueger et al., 2012). Conditional knock-out (cKO) of neuroligins in projection neurons in different circuits demonstrated an important role of neuroligins in synapse specification (Rothwell et al., 2014; Liang et al., 2015; Zhang et al., 2015; Jiang et al., 2016). However, the function of neuroligins in local interneurons and the relative importance of neuroligins vis-à-vis other postsynaptic cell-adhesion molecules such as leucine-rich repeat transmembrane proteins (LRRTMs) (Ko et al., 2009; de Wit et al., 2009; Siddiqui et al., 2010) remains unclear.
Cerebellar stellate cells are interneurons in the molecular layer of the cerebellar cortex that receive glutamatergic inputs from parallel fibers and inhibitory inputs from stellate cells and other cerebellar interneurons (Sultan and Bower, 1998). Low-frequency parallel fiber stimulation triggers EPSCs in stellate cells that are mediated solely by AMPARs (Carter and Regehr, 2000; Clark and Cull-Candy, 2002; Malagon et al., 2016), whereas high-frequency parallel fiber stimulation induces glutamate “spillover” at stellate cell synapses and activates NMDARs that are thought to be largely extrasynaptic (Carter and Regehr, 2000; Clark and Cull-Candy, 2002; Nahir and Jahr, 2013). Extrasynaptic NMDARs in stellate cells are critical for multiple functions, including modulation of synaptic plasticity via endocannabinoids (Beierlein and Regehr, 2006) and mGluR1α signaling (Karakossian and Otis, 2004), regulation of the insertion of GluA2-containing AMPARs (Sun and June Liu, 2007), and activation of axonal Ca2+-channels (Christie and Jahr, 2008). In parallel fiber synapses of stellate cells, postsynaptic GluRδ1 and GluRδ2, which act as cell-adhesion molecules instead of ionotropic glutamate receptors (Hollmann and Heinemann, 1994; Yuzaki, 2003; Collingridge et al., 2009), are indispensable for synapse formation or maintenance and for AMPAR-mediated synaptic transmission (Landsend et al., 1997; Takeuchi et al., 2005; Yamasaki et al., 2011; Konno et al., 2014). GluRδ1 and GluRδ2 act via binding presynaptic neurexin/cerebellin complexes (Matsuda et al., 2010; Uemura et al., 2010; Yamasaki et al., 2011; Ryu et al., 2012; Konno et al., 2014). Moreover, several other transmembrane proteins, such as TARPs and GSGL1, modulate the function and cell-surface expression of AMPARs in cerebellar stellate cells (Soto et al., 2007; Bats et al., 2012; McGee et al., 2015).
In the present study, we examined the role of neuroligins in cerebellar stellate cell synapses. Using mouse genetics, immunohistochemistry, and electrophysiological recordings, we demonstrate that in cerebellar stellate cells neuroligins selectively control NMDAR-mediated transmission, whereas AMPAR-mediated excitatory and GABAR-mediated inhibitory synaptic transmission were not massively altered. Together with previous studies, our results suggest that neuroligins perform profound but context-specific functions that differ greatly between distinct types of neurons.
Materials and Methods
Animal handling.
The following mouse lines were used for the current study: mice in which cre-recombinase is expressed under control of the parvalbumin (PV) gene promoter (PV-Cre mice; Hippenmeyer et al., 2005), Ai14 Cre reporter mice (Jackson Laboratory, stock no. 007908), and neuroligin triple NL123 cKO mice (Zhang et al., 2015). All analyses were performed on littermate mice at postnatal day 21 (P21) to P25 of either sex, with the experimenter blinded to the mouse genotype. All mice were analyzed on a hybrid genetic background to avoid the confounding effects of contaminating homozygous mutations in inbred mouse strains. All procedures conformed to National Institutes of Health's Guidelines for the Care and Use of Laboratory Animals and were approved by the Stanford University Administrative Panel on Laboratory Animal Care.
Electrophysiology.
Sagittal slices (250 μm thick) of the cerebellum were made according to standard procedures with a vibratome (Leica, VT1200S) using PV-NL123 mice or PV-NL1 mice and their control littermate mice at P21–P23, as described previously (Dugué et al., 2005; Zhang et al., 2015). To preserve best cell quality, different cutting solutions were used. For stellate cell recordings, the solution contained the following (in mm): 130 K-gluconate, 15 KCl, 20 HEPES, 25 glucose, 0.05 EGTA, and 0.05 D-AP5, pH 7.4 with NaOH. For Purkinje cell recordings, the solution contained the following (in mm): 125 NaCl, 25 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 25 glucose, 0.4 ascorbic acid, 3 myo-inositol, 2 Na-pyruvate, 0.1 CaCl2, and 3 MgCl2, pH 7.4, when aerated with 95% O2/5% CO2. The extracellular artificial CSF (aCSF) recording solutions contained the following (in mm): 125 NaCl, 25 NaHCO3,2.5 KCl, 1.25 NaH2PO4, 25 glucose, 0.4 ascorbic acid, 3 myo-inositol, 2 Na-pyruvate, 2 CaCl2, and 1 MgCl2, pH 7.4, when aerated with 95% O2/5% CO2. For recordings of spontaneous EPSCs, picrotoxin (50 μm) and strychnine (2 μm) were added to the extracellular solution. For recordings of spontaneous IPSCs (sIPSCs), CNQX (20 μm) and D-AP5 (50 μm) were added. Tetrodotoxin (TTX, 1 μm) was also added for recordings of miniature IPSCs (mIPSCs). For recordings of AMPAR-mediated sEPSCs or EPSCs in stellate cells, picrotoxin (50 μm), strychnine (2 μm), and D-AP5 (50 μm) were added. For recordings of NMDAR-mediated EPSCs in stellate cells, picrotoxin (50 μm), strychnine (2 μm), and CNQX (20 μm) were added. Internal solutions in the pipette contained the following (in mm): 140 Cs-gluconate, 10 HEPES, 5 Na2-phosphocreatine, 4 MgATP, 0.3 Na2GTP, 0.5 Cs-EGTA, and 0.1 spermine, pH 7.2. Whole-cell recordings in voltage-clamp mode were made with an Axon amplifier, under visualization of neurons with an upright microscope (BX51Wil; Olympus) equipped with a 40× water-immersion objective (Zeiss). For stellate cell whole-cell recording, patch pipettes had resistances of 4–5 MΩ and the series resistance (15–20 MΩ) was comparable between genotypes and was not compensated. For Purkinje cell whole-cell recording, patch pipettes had resistances of 2–3 MΩ, and the series resistance (8–9 MΩ) was comparable between genotypes and was not compensated. Measurement of current transient elicited by a 10 mV hyperpolarizing voltage step at regular intervals was used to monitor series resistance, input capacitance, and input resistance. Recordings were rejected if the series resistance altered by >20% for single recording. Stellate cells were held at −60 mV in voltage-clamp mode and Purkinje cells were held at −70 mV; membrane potentials were not corrected for liquid junction potentials. Parallel fiber-mediated EPSCs were evoked using a bipolar stimulator and placed ∼100 μm away of the recording cell in the upper one-third molecular layer. Stimuli consisted of paired pulses (10–40 V; 20 μs duration) separated by different intervals. The paired-pulse ratio (PPR) of evoked EPSCs was determined by dividing the amplitude of the second EPSC by that of the first. Amplitude and PPR of EPSCs were determined from individual sweeps before numerical averaging. For the glutamate puff experiments, the glutamate-containing pipette, with pipette resistance ∼2 MΩ, was positioned <5 μm from soma of voltage-clamped stellate cells to induce maximal current. Glutamate was puffed onto the cells with a Picospritzer III (Parker Hannifin) at a pressure of 8 psi for 5 ms. Responses are the average of two to three traces per cell with an intertrace time of 25 s. For recording glutamate-puff-induced, NMDAR-mediated EPSCs, a mixture of drugs (1 μm TTX, 20 μm CNQX, 50 μm picrotoxin, and 2 μm strychnine) was added in MgCl2-free aCSF. Glass pipettes also contained 50 mm glutamate in the aCSF with the mixture of drugs. For recording glutamate-puff-induced, AMPAR-mediated EPSCs, a mixture of drugs (1 μm TTX, 50 μm D-AP5, 50 μm picrotoxin, and 2 μm strychnine) was added in normal aCSF. Glass pipettes also contained 50 mm glutamate in the aCSF with the mixture of drugs. Charge transfer was calculated by integration of between 0 and 680 ms after 100 Hz train stimulation (the start point of stimulation as set as 0 s) or by integration of between 0 and 8 s after glutamate puffing.
Immunohistochemistry experiments were performed as described previously (Zhang et al., 2015). Cryostat sections (30 μm thickness) of mouse brain were processed on Superfrost Plus slides. Mice at P21–P25 were transcardially perfused and then brains were dissected and postfixed in 4% paraformaldehyde in PBS overnight at room temperature. Permeabilization and blocking of the sections were done in PBS with 0.3% Triton ×100 (Sigma-Aldrich) and 1% GSA. The following primary antibodies were used: mouse anti-PV (dilution 1:2000) and rabbit anti-calbindin (1:1000; Millipore) and were incubated overnight at 4°C. Secondary antibodies (Invitrogen) were goat anti-mouse Alexa Fluor 488 (1:500), goat anti-rabbit Alexa Fluor 554 (1:500) and were incubated overnight at 4°C. Confocal images were acquired with an inverted Nikon IR+ microscope equipped with a 63× objective. Images were taken at 1024 × 1024 pixels. Imaging analysis was performed with software from Nikon IR+.
Immunoblotting was performed as described previously (Zhang et al., 2015), with fluorescently labeled secondary antibodies and LI-COR detection (LI-COR Bioscience). Littermate mice (P21–P25) with the indicated genotypes were deeply anesthetized with isoflurane and cerebella were dissected and lysed in lysis buffer containing the following (in mm): 20 Tris-HCl, pH 7.5, 100 NaCl, 4 KCl, 2 MgCl2, and 2 CaCl2 with 1% Triton X-100 and protease inhibitor mixture (Roche) for 1 h at 4°C. The samples were then centrifuged for 20 min at 20,000 × g at 4°C (3 times) to remove insoluble materials. Proteins (10 μg) in the lysate were separated on 4–20% Tris-HCl Gel (Bio-Rad) and blotted onto nitrocellulose membranes following the manufacturer's instructions. Membranes were blocked in 5% milk powder prepared in 0.05% TBS-Tween for 30 min at room temperature. Primary antibodies were diluted in blocking buffer and incubated overnight at 4°C. Membranes then were washed with 0.05% TBS-Tween and then incubated with a fluorescence-labeled secondary antibodies (donkey anti-rabbit IR dye 680CW, 1:5000; donkey anti-mouse IR dye 800CW, 1:5000, 1;5000; LI-COR Bioscience) were used and signals were detected with an Odyssey Infrared Imager and Odyssey software (LI-COR Biosciences). The total intensity values for calculated by Odyssey software and each values normalized to actin first then normalized to control. The antibodies used were as follows: NL1 (1:500, mouse monoclonal antibody 4C12); NL2 (rabbit #129203; Synaptic Systems); NL3 (1:500, rabbit polyclonal antibody 639B); GluR1 (1:1000, rabbit polyclonal antibody AB1504, Millipore); Synaptotagmin-2 (1:1000, rabbit monoclonal antibody A320); NR1 (1:1000, 54.2, Synaptic Systems); β-actin (1:5000, mouse monoclonal antibody AC-15; Sigma-Aldrich).
Data analysis.
Data analysis was performed using IgorPro software. Both mIPSCs and sEPSCs were analyzed with MiniAnalysis software (Synaptosoft). Data are reported as average ± SEM values and statistical significance was calculated using unpaired, two-tailed Student's t test, Kolmogorov–Smirnov test, or one-way ANOVA as follows: *p < 0.05, **p < 0.01, ***p < 0.001. A bracket without a symbol indicates that the value is not significant.
Results
Normal stellate cell morphology in PV-Cre NL123 mice
To address whether neuroligins play a role in cerebellar interneurons, we aimed to delete neuroligins in stellate cells of the cerebellar cortex. However, constitutive NL123 triple KO mice die at birth (Varoqueaux et al., 2006), which precludes analysis in cerebellar interneurons, and deleting individual neuroligin isoforms might produce only a partial loss-of-function phenotype because of possible functional redundancy. Therefore, we aimed to remove neuroligins conditionally in stellate cells using recently produced floxed mouse lines in which the expression of the three major neuroligins expressed in the cerebellum (NL1, NL2, and NL3) can be abolished by expression of Cre-recombinase (Zhang et al., 2015). Parvalbumin is a marker protein of inhibitory neurons that, in the cerebellum, is expressed in interneurons of the molecular layer in addition to Purkinje cells (Collin et al., 2005; Alcami and Marty, 2013); all of these cell types are readily identified by light microscopy because of their size and the location of their cell bodies (Fig. 1A).
Figure 1.

PV-Cre-mediated conditional KO of NL123 does not alter the overall morphology of the cerebellar cortex or of cerebellar stellate cells. A, Single focal plane showing triple immunostaining tdTomato (red fluorescence), parvalbumin (Pv, green fluorescence), and calbindin (Cal, purple fluorescence) of the cerebellum. Data are from a 21-day-old tdTomato-Cre-reporter mouse crossed with PV-Cre transgenic mice. For the current study, the interneurons in the upper third territory of the molecular layer (above dashed line) were classified as stellate cells; cells expressing both Pv and tdTomato appear yellow. Note the robust Cre activity in Pv-positive stellate cells, even though Cre activity is higher in basket cells. B, Illustration of the breeding strategy for conditional ablation of neuroligins in PV-expressing neurons of the cerebellum. PV-Cre mice were crossed with NL123 triple cKO mice; littermate NL123 triple cKO mice lacking PV-Cre were used as controls. C, Single focal plane showing parvalbumin (green fluorescence) and calbindin (red fluorescence) double immunostaining of the cerebellum in control and PV-Cre-NL123 cKO mice. D, Summary data of cell density and soma size of control and neuroligin-deficient interneurons in the molecular layer of the cerebellum. Data are means ± SEM. Numbers in bars represent the number of slices (or cells)/number of mice used. Statistical significance was determined by Student's t test.
We found that, in PV-Cre mice, Cre-recombinase activity is present at P21 in cerebellar interneurons as well as in Purkinje cells, as revealed after crossing PV-Cre mice with tdTomato reporter mice (Fig. 1A). All examined Purkinje cells (41 cells) and almost all examined stellate cells (104 of 105 cells) were both tdTomato-positive and PV-positive. We noted that those stellate cells from distal part of cerebellar molecular layer tend to have a lower tdTomato reporter signal. For simplicity, we generally designated interneurons from the upper third of the cerebellar molecular layer as stellate cells that were used for morphological and electrophysiological studies in all subsequent experiments.
We crossed PV-Cre and NL123 triple-floxed cKO mice to generate PV-Cre NL123 mice and used littermate PV-Cre-negative NL123 triple-floxed mice as controls (Fig. 1B). To identify possible changes in the overall development and survival of stellate cells after deletion of neuroligins, we quantified the density and soma size of stellate cells. Compared with littermate control mice, PV-positive stellate cells from PV-Cre NL123 mice exhibited no major changes in density or soma size (Fig. 1C,D). We also measured membrane capacitance and input resistance of stellate cells using patch-clamp recordings. We found no significant changes of either of these two parameters (membrane capacitance: control, 20.2 ± 0.5 pF, n = 37; PV-NL123, 20.4 ± 0.6 pF, n = 35; input resistance: control, 533 ± 49 MΩ, n = 37; PV-NL123, 614 ± 58 MΩ, n = 35). These results suggest that neuroligins are dispensable for the differentiation and/or survival of stellate cells, in contrast to the significant role of GluRδ1 in these processes (Konno et al., 2014).
NL123 triple cKO causes only a mild inhibitory synaptic transmission deficit in cerebellar stellate cells
To determine whether neuroligins are required for synaptic transmission in cerebellar stellate cells, similar to their essential role in Purkinje cells (Zhang et al., 2015), we first examined inhibitory transmission. Each connection among cerebellar stellate cells exhibits a large quantal size of ∼100 pA at resting potential under symmetrical Cl− conditions (Llano and Gerschenfeld, 1993; Auger and Marty, 1997; Nusser et al., 1997; Trigo et al., 2012). We compared the characteristics of spontaneous mIPSCs (note that all events were included; Trigo et al., 2010) between control and PV-Cre NL123 mice at P21–P23 using whole-cell recordings from stellate cells in sagittal cerebellar slices in the presence of CNQX, D-AP5, and TTX. We identified a modest but significant reduction in the amplitude of mIPSCs in PV-Cre NL123 mice (Fig. 2A,B). Recording at a holding potential of −60 mV, control stellate cells exhibited an average mIPSC amplitude of 104.1 ± 7.1 pA (n = 15), whereas PV-Cre NL123 stellate cells displayed a lower average amplitude of 83.1 ± 3.9 pA (n = 17; p < 0.05; Fig. 2B). In contrast to the mIPSC amplitude, the mIPSC frequency was not significantly reduced (controls: 2.93 ± 0.68 Hz, n = 15; PV-Cre NL123 stellate cells: 2.51 ± 0.4 Hz, n = 17). In addition, we assessed the role of neuroligins in the kinetics of mIPSCs (Fig. 2B). We calculated a half-height duration from averaged mIPSC waveforms and found it to be comparable in PV-Cre NL123 (7.55 ± 0.22 ms; n = 15) and control mice (7.57 ± 0.29 ms; n = 17), indicating that the mIPSC kinetics were normal after ablation of NL123.
Figure 2.

Neuroligin deletion causes only mild impairments of synaptic transmission at inhibitory synapses of stellate cells, in contrast to a severe impairment of synaptic transmission at inhibitory synapses of Purkinje cells. A, Sample traces of mIPSC in stellate cells from control (black traces) and PV-Cre NL123 littermate mice (red traces) monitored at P21–P23. Averaged mIPSCs are displayed on the right. B, C, Summary data of mIPSCs (amplitude distribution and average and frequency distribution and average) recorded from stellate cells as described in A. The half-height duration (HHD) of mIPSCs was 7.55 ± 0.22 ms (n = 15) in PV-Cre/NL123 mice and 7.57 ± 0.29 ms (n = 17) in control littermates. C, Sample traces of sIPSCs recorded from Purkinje cells from control (black traces) and PV-NL123 (red traces) littermate mice at P21–P23. Averaged sIPSC traces are displayed on the right. D, Summary data of sIPSCs in C. HHD of sIPSCs was 5.38 ± 1.1 ms (n = 3) in PV-Cre NL123 mice and 5.97 ± 0.65 ms (n = 14) in control littermates. The amplitude of sIPSCs was set at 0 pA in 12 silent cells. Data are means ± SEM. Numbers in bars represent the number of cells/mice used. Statistical significance was determined by Student's t test.
Together, these data suggested that stellate cells from PV-Cre NL123 mice exhibit an overall mild reduction in inhibitory synaptic transmission that is likely due to a slight impairment in postsynaptic GABA/glycine receptors, but not in presynaptic release probability. We were concerned that the modest inhibitory transmission phenotype in neuroligin-deficient stellate cells may be due to an incomplete activity of the PV-Cre transgenic mice, although the reporter mice suggested otherwise (Fig. 1). Because PV is also expressed in Purkinje cells, we were able to validate the efficiency of the PV-Cre manipulation using recordings of sIPSCs from Purkinje cells with the same genetic manipulation. In contrast to the modest stellate cell phenotype in inhibitory synaptic transmission, we observed a massive suppression of sIPSCs in Purkinje cells of PV-Cre NL123 mice (frequency: control, 1.26 ± 0.16 Hz; PV-Cre NL123, 0.02 ± 0.01 Hz, p < 0.001; amplitude: control, 42.7 ± 6.5 pA; PV-Cre NL123, 2.9 ± 1.7 pA, p < 0.001; Fig. 2C,D). The difference in the impairment of inhibitory synaptic transmission between stellate and Purkinje cells likely reflects a synapse- and neuron-specific role of neuroligins.
NL123 triple cKO induces no phenotype in AMPAR-mediated excitatory synaptic transmission in stellate cells
We next aimed to determine whether neuroligins are required for excitatory synapse function in stellate cells. We examined the characteristics of sEPSCs, which we measured instead of mEPSCs because of the low mEPSC frequency in stellate cells (Crowley et al., 2007). We generated whole-cell recordings from stellate cells in cerebellar slices in the presence of picrotoxin, strychnine, and D-AP5. These conditions ensured that we selectively monitored AMPAR-mediated synaptic responses. At a holding potential of −60 mV, control stellate cells exhibited an average frequency of 0.32 ± 0.08 Hz (n = 9), whereas PV-Cre NL123 stellate cells had an average frequency of 0.27 ± 0.07 Hz (n = 9), representing a normal value (Fig. 3A,B). The amplitude of sEPSCs was also indistinguishable because control sEPSCs had an average amplitude of 38.5 ± 1.4 pA (n = 9), whereas sEPSCs from PV-Cre NL123 stellate cells had an average amplitude of 39.4 ± 2 pA (n = 9; Fig. 3B). Together, these data show that stellate cells from PV-Cre NL123 mice exhibit an overall normal basal AMPAR-mediated synaptic transmission.
Figure 3.
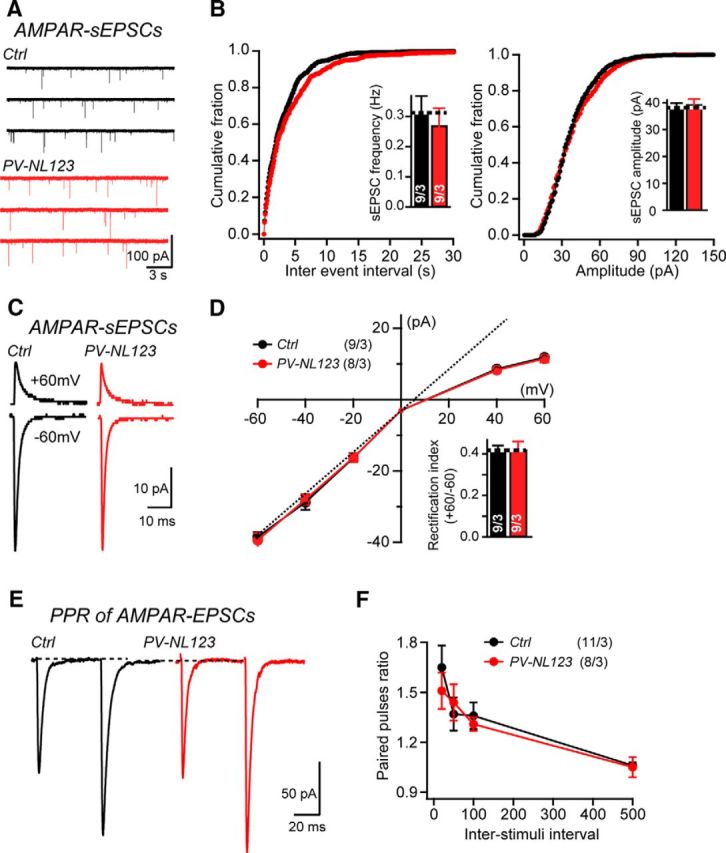
Neuroligin deletion does not alter AMPAR-mediated synaptic responses at parallel fiber synapses in stellate cells. A, B, Sample traces and summary data of sEPSCs recorded from stellate cells from control mice (black traces) and PV-NL123 (red traces) littermates. C, D, Sample traces and summary data of average sEPSCs with different holding potentials recorded from stellate cells from control mice (black traces) and PV-Cre NL123 littermate mice (red traces) at P21–P23. E, F, Sample traces and summary data of EPSC PPRs recorded with different interstimulus intervals from control cells (black traces) and PV-Cre NL123 stellate cells (red traces) at P21–P23. Stimulation artifacts are removed for cosmetic purposes. Data are means ± SEM. Numbers in bars represent the number of cells/mice used. Statistical significance was determined using Student's t test (pairwise comparison), Kolmogorov–Smirnov test (cumulative distributions), or one-way ANOVA.
We next investigated whether neuroligins are involved in determining the default subunit composition of synaptic AMPARs in stellate cells, because it has been suggested that neuroligins may determine the AMPAR subunit composition of excitatory synapses via preferential interactions with particular GluA subunits (Choquet et al., 2008). An informative measure of the AMPAR subunit composition is the rectification index, which takes advantage of the fact that GluA2-lacking AMPARs exhibit a voltage-dependent block by endogenous intracellular polyamines and thus display inwardly rectifying I–V curves (Kamboj et al., 1995). The rectification index of sEPSCs, recorded with an internal solution containing 0.1 mm spermine, could be calculated from the I–V curve and the content of GluA2-containing AMPARs could then be inferred from rectification index.
Under basal conditions, synaptic AMPARs at parallel fiber–stellate cell synapses are moderately inwardly rectifying with average RI of ∼0.4 (Liu and Cull-Candy, 2000; Liu et al., 2010), suggesting a mixture of GluA2-lacking and GluA2-containing synaptic AMPARs. Recording in the presence of picrotoxin, strychnine, and D-AP5, we found that, under basal conditions, synaptic AMPARs on stellate cells are indeed moderately inwardly rectifying, with an average rectification index of 0.42 ± 0.02 (n = 9), which is broadly consistent with previously reported values. Parallel fiber–stellate cell synapses from PV-Cre NL123 mice showed a similar inward rectification, with an average index of 0.42 ± 0.04 (n = 9), suggesting that parallel fiber–stellate cell synapses in PV-Cre NL123 mice have a normal complement of GluA2-lacking AMPARs similar to control mice (Fig. 3C,D).
Finally, to assess the possibility that neuroligins might have a direct or indirect presynaptic role in stellate cells (Wittenmayer et al., 2009), we examined the PPR of evoked EPSCs, which is thought to be inversely proportional to the presynaptic release probability. Parallel fiber–stellate cell synapses have been shown to exhibit prominent paired-pulse facilitation (Atluri and Regehr, 1996). We used pairs of stimuli separated by a serial interval and found no significant difference in paired-pulse facilitation (at 50 ms interval control: 1.38 ± 0.1; n = 11; PV-NL123: 1.44 ± 0.11; n = 8; Fig. 3E,F), indicating a normal presynaptic release probability at parallel fiber–stellate cell synapses in PV-NL123 mice.
Together, these results indicate that neuroligins are not required for any aspect of AMPAR-mediated synaptic transmission at parallel fiber–stellate cell synapses. This conclusion agrees well with previous observations that all excitatory inputs onto stellate cells derive from parallel fibers and that, in Purkinje cells, neuroligins are also dispensable for excitatory parallel fiber synapses (Zhang et al., 2015). Moreover, this conclusion nicely conforms to the notion that parallel fiber synapses use cerebellins in a complex with Gluδ1 or Gluδ2 as neurexin ligands instead of neuroligins (Matsuda et al., 2010; Uemura et al., 2010; Yamasaki et al., 2011; Ryu et al., 2012; Konno et al., 2014) and thus do not need neuroligins as a neurexin ligand for synapse specification and maintenance.
NL123 triple cKO in stellate cells dramatically impairs the delayed phase of EPSCs that is mediated by extrasynaptic NMDARs
The lack of a major phenotype in GABAR- and AMPAR-mediated synaptic transmission in neuroligin-deficient stellate cells was surprising given the prominent role of neuroligins in excitatory and inhibitory synaptic transmission in many other neurons (Varoqueaux et al., 2006; Chubykin et al., 2007; Gibson et al., 2009; Poulopoulos et al., 2009; Radyushkin et al., 2009; Földy et al., 2013; Jaramillo et al., 2014; Rothwell et al., 2014; Espinosa et al., 2015; Liang et al., 2015; Zhang et al., 2015; Jiang et al., 2016). Interestingly, excitatory synapses formed by parallel fibers on stellate cells exhibit an unusual feature in that synaptic transmission is nearly exclusively mediated by AMPARs, similar to excitatory synapses formed by parallel fibers on Purkinje cells. However, stimulation of parallel fiber–stellate cell synapses by high-frequency action potential trains causes spillover of synaptic glutamate, which then activates perisynaptic or extrasynaptic NMDARs (Clark and Cull-Candy, 2002). Therefore, at parallel fiber–stellate cell synapses, NMDARs are thought to be primarily nonsynaptic (different from other synapses), to be localized to both dendrites and the soma of the stellate cells, and to be activated only by glutamate spillover during massive release induced by bursts of presynaptic action potentials. This conserved feature of parallel fiber–stellate cell synapes is not present in parallel fiber synapses on Purkinje cells and may enable parallel fiber synapses on stellate cells to perform signaling functions that are not available at parallel fiber-Purkinje cell synapses.
In the experiments on AMPAR-mediated EPSCs described above, we evoked EPSCs by low-frequency activation of only a small number of parallel fibers and recorded EPSCs in the presence of AP5, which blocks NMDARs (Fig. 3). To test whether neuroligins might be involved in organizing NMDARs that are activated by glutamate spillover during repetitive stimulation (Fig. 4A), we stimulated multiple parallel fibers in acute cerebellar slices with a burst of five action potentials applied at 100 Hz. We recorded EPSCs in the absence of AP5, but in the presence of picrotoxin and strychnine to block inhibitory transmission, and voltage clamped the patched cells at +60 mV to enlarge NMDAR-mediated responses that are normally blocked by Mg2+ at more negative holding potentials. In these experiments, we adjusted the stimulation intensity in control and PV-Cre NL123 cells to evoke EPSCs of similar initial amplitudes, which primarily reflected AMPAR-mediated currents even at +60 mV (control: 45.6 ± 8 pA, n = 9; PV-NL123: 49.8 ± 7.4 pA, n = 8; Fig. 4B–D). We then quantified the EPSC amplitude at 100 ms after the peak of the last EPSC because, at that time point, the EPSCs should be composed primarily of NMDAR-mediated currents. Finally, we calculated the ratios of the delayed EPSC amplitude to the first EPSC amplitude and to the cumulative peak EPSC amplitudes to quantify the relative strength of NMDAR-mediated perisynaptic or extrasynaptic signals compared with synaptic AMPAR-mediated signals. Because we have shown above that AMPAR-mediated EPSCs are not changed by the neuroligin triple KO in PV-Cre NL123 stellate cells, any changes in the ratios of the delayed EPSC to the peak AMPAR-mediated EPSC likely reflects a change of NMDAR-mediated responses in stellate cells.
Figure 4.
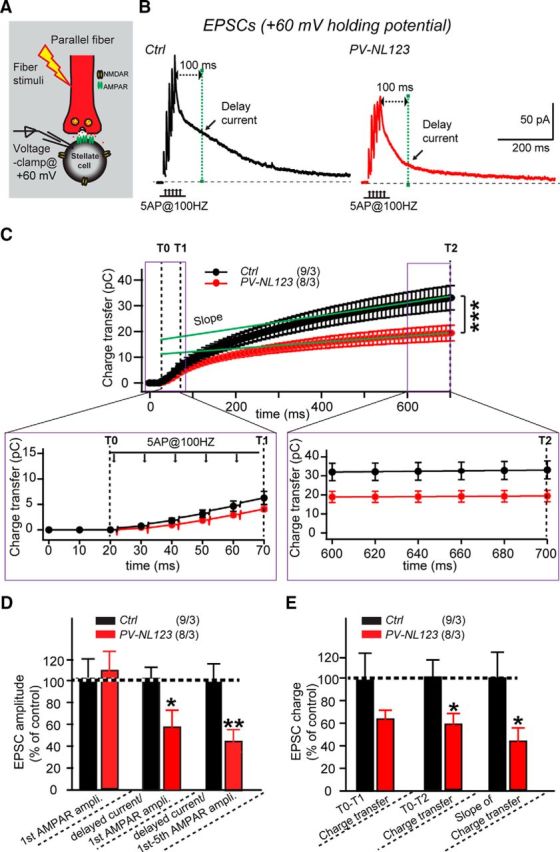
Neuroligin deletion from stellate cells dramatically impairs the late phase of EPSCs elicited by short stimulus trains. A, Illustration of the experimental strategy for monitoring AMPAR- and NMDAR-mediated EPSCs in stellate cells (Carter and Regehr, 2000; Clark and Cull-Candy, 2002; Sun and June Liu, 2007; Nahir and Jahr, 2013; Bidoret et al., 2015). B, Sample traces of EPSCs in response to five APs at 100 Hz recorded at a +60 mV holding potential in cerebellar stellate cells from littermate control mice (black traces) and PV-Cre NL123 mice (red traces) at P21–P23. The peak EPSC amplitude is thought to represent primarily AMPAR-mediated currents, whereas the delayed EPSC (calculated at 100 ms after the fifth EPSC peak in the train) is thought to be primarily due to NMDAR-mediated currents. C–E, Plot of the cumulative EPSC charge and summary graphs of EPSCs induced by short high-frequency trains and recorded as described in B. Data are means ± SEM. Numbers in bars represent the number of cells/mice used. Statistical significance was determined by Student's t test or one-way ANOVA.
Strikingly, we found that both ways of calculating the delayed EPSC/AMPAR-mediated ratio revealed a large, ∼50% decrease in the delayed EPSC in PV-Cre NL123 stellate cells (Fig. 4D). The average ratio of the delayed EPSC/first EPSC amplitude in control stellate cells was 1.19 ± 0.1 (n = 9), whereas that in PV-Cre NL123 mice was 0.65 ± 0.16 (n = 8). The average ratio of delayed EPSC/first–fifth EPSC amplitudes in control stellate cells was 0.29 ± 0.04 (n = 9), whereas that in PV-Cre NL123 mice was 0.13 ± 0.03 (n = 8). We also measured the charge transfer of EPSCs recorded in +60 mV and observed a trend toward a reduction at 50 ms after stimulation (control: 6.5 ± 1.3 pC, n = 9; PV-Cre NL123: 4.2 ± 0.4 pC, n = 8, p > 0.05) and a significant reduction at 680 ms after stimulation (control: 34.3 ± 5.5 pC, n = 9; PV-Cre NL123: 19.4 ± 3.0 pC, n = 8, p < 0.05; Fig. 4C–E). The slope after linear regression of the charge transfer, which may reflect the continual activation of NMDARs during the later stimulation period, also showed a reduced value (control: 24.7 ± 5.4 pC/s, n = 9; PV-Cre NL123: 11.2 ± 2.7 pC/s, n = 8, p < 0.05; Fig. 4C–E). These data indicate that NMDAR-mediated transmission is impaired in PV-Cre NL123 mice.
NL123 triple cKO in stellate cells dramatically impairs NMDAR-mediated synaptic transmission
Because recordings of peak and delayed EPSCs are only indirect measurements of AMPAR- and NMDAR-mediated EPSCs, we ensured the validity of the conclusions from these indirect measurements using pharmacological isolation of NMDAR-mediate EPSCs. Specifically, we measured EPSCs evoked by bursts of five action potentials at 100 Hz using whole-cell voltage-clamp recordings in stellate cells held at +60 mV before and after the addition of the AMPAR blocker CNQX. By subtracting the EPSCs recorded in CNQX, which represent pure NMDAR-mediated currents from the EPSCs recorded in the absence of CNQX, we could calculate the AMPAR-mediated current. With this subtraction method, we found that the delayed EPSC measured at 100 ms after the peak of the last peak EPSC is produced by a mixture of both AMPAR- and NMDAR-mediated currents (Fig. 5A). The delayed AMPAR-mediated current could be carried by extrasynaptic AMPARs and/or produced by synaptic AMPARs that are activated by asynchronous release from parallel fibers, whereas the delayed NMDAR-mediated current likely reflects the activity of extrasynaptic NMDARs.
Figure 5.
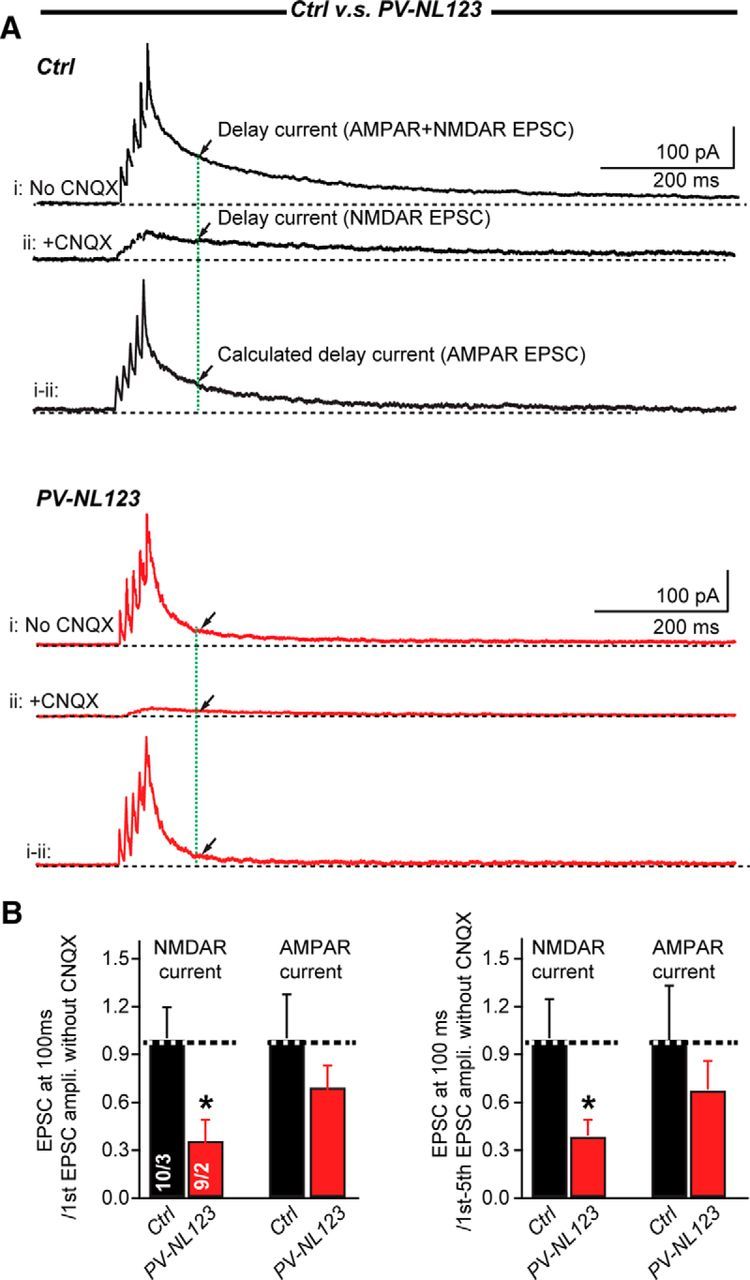
Neuroligins deletion impairs NMDAR-mediated EPSCs elicited by short-stimulus trains in stellate cells. A, Sample traces of EPSCs in response to 5 APs at 100 Hz recorded without (trace i) or with (trace ii) CNQX at a +60 mV holding potential in cerebellar stellate cells from P21–P23 littermate control mice (black traces) and PV-Cre NL123 mice (red traces). The current without CNQX (trace i) is mixture of AMPAR- and NMDAR-mediated currents, whereas the current in the presence of CNQX (trace ii) is mediated exclusively by NMDARs. AMPAR-mediated currents were calculated by subtracting trace ii from trace i. Arrows indicated the current at 100 ms after the last EPSC peak. B, Summary graphs of NMDAR- and calculated AMPAR-mediated EPSCs. Data are means ± SEM. Numbers in bars represent the number of cells/mice used. Statistical significance was determined by Student's t test.
When we examined the effect of the neuroligin ablation on NMDAR- and AMPAR- mediated EPSCs in stellate cells, we found that the amplitude of NMDAR-mediated EPSCs was significantly reduced in PV-Cre NL123 mice (normalized to the first EPSC amplitude, control: 1 ± 0.19, n = 9; PV-NL123: 0.37 ± 0.12, n = 8; p < 0.05; normalized to the cumulative EPSC amplitude, control: 1 ± 0.25, n = 9; PV-NL123: 0.39 ± 0.1, n = 9; p < 0.05). In contrast, delayed AMPAR-mediated EPSCs did not significantly change (normalized to the first EPSC amplitude, control: 1 ± 0.27, n = 9; PV-NL123: 0.7 ± 0.13, n = 9; normalized to the cumulative EPSCs amplitude, control: 1 ± 0.34, n = 9; PV-NL123: 0.81 ± 0.21, n = 9) (Fig. 5B). These data demonstrate that NMDAR-mediated transmission was selectively impaired in PV-Cre NL123 mice.
NL1 single cKO in stellate cells dramatically impairs NMDAR-mediated synaptic transmission similar to the NL123 triple cKO
Because NL1 is essential for NMDAR-mediated synaptic EPSCs (Soler-Llavina et al., 2011; Shipman and Nicoll, 2012; Jiang et al., 2016), we tested whether the deletion of NL1 alone may impair extrasynaptic NMDAR-mediated EPSCs in stellate cells. We performed the same experiment with PV-Cre/NL1 mice that we performed with PV-Cre/NL123 mice. Strikingly, we found that the amplitude of NMDAR-mediated EPSCs was significantly reduced in PV-NL1 mice (normalized to the first EPSC amplitude, control: 1 ± 0.17, n = 10; PV-NL1: 0.34 ± 0.08, n = 9; p < 0.05; normalized to the cumulative EPSC amplitude, control: 1 ± 0.24, n = 10; PV-NL1: 0.21 ± 0.04, n = 9; p < 0.05), whereas the amplitude of the delayed AMPAR-mediated EPSCs did not change in PV-NL1 mice (normalized to the first EPSC amplitude, control: 1 ± 0.32, n = 10. PV-NL1: 0.83 ± 0.14, n = 9; normalized to the cumulative EPSCs amplitude, control: 1 ± 0.22, n = 10; PV-NL123: 0.9 ± 0.14, n = 9; Fig. 6A,B). These data demonstrate that NL1 is selectively essential for extrasynaptic NMDAR-mediated transmission in stellate cells.
Figure 6.
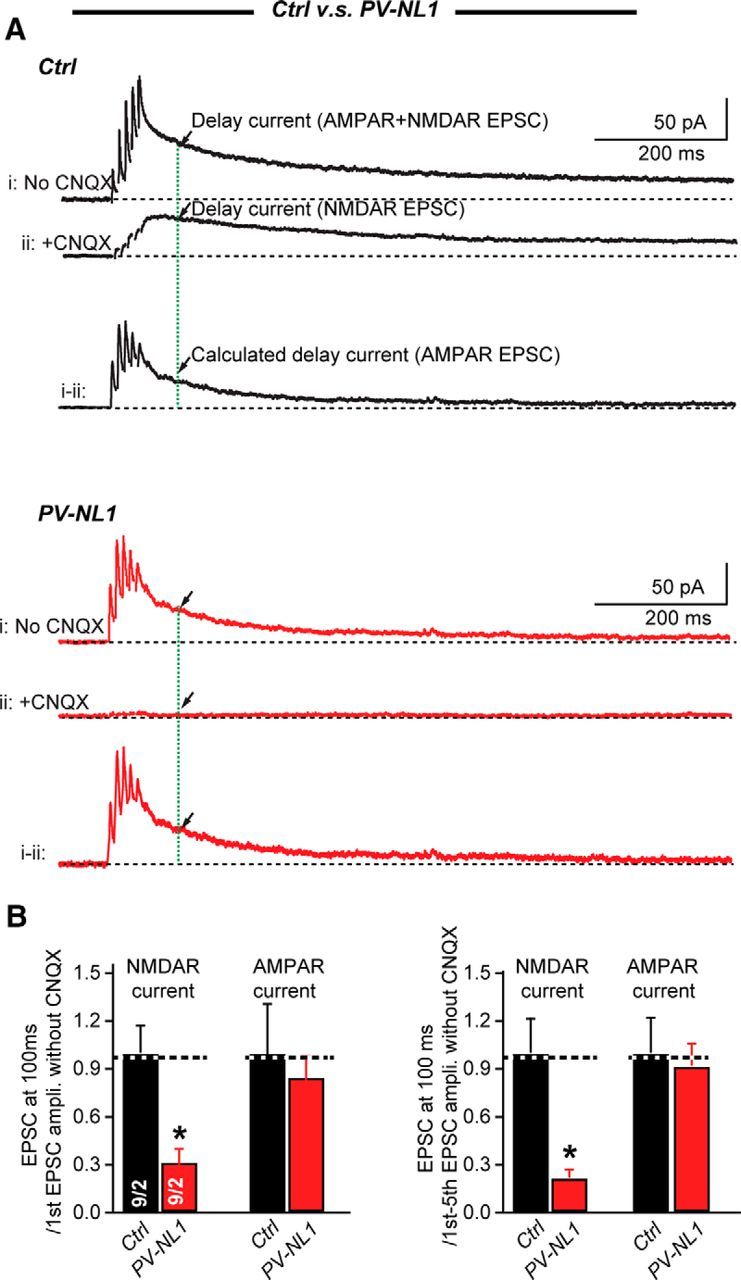
NL1 deletion impairs NMDAR-mediated EPSCs elicited by short stimulus trains in stellate cells. A, Sample traces of EPSCs in response to five APs at 100 Hz recorded without (trace i) or with (trace ii) CNQX at a +60 mV holding potential in cerebellar stellate cells from P21–P23 littermate control (black traces) and PV-Cre NL1 mice (red traces). The current without CNQX (trace i) is mixture of AMPAR- and NMDAR-mediated currents, whereas the current in the presence of CNQX (trace ii) is mediated exclusively by NMDARs. AMPAR-mediated currents were calculated by subtracting trace ii from trace i. Arrows indicate the current at 100 ms after the last EPSC peak. B, Summary graphs of NMDAR- and calculated AMPAR-mediated EPSCs. Data are means ± SEM. Numbers in bars represent the number of cells/mice used. Statistical significance was determined by Student's t test.
PV-Cre-induced NL123 deletion reduces the total surface levels of NMDARs in cerebellar stellate cells and total NMDAR protein levels in cerebellar lysates
The decrease in NMDAR-mediated synaptic responses after stimulus trains at parallel fiber synapses of stellate cells is most plausibly explained by a reduction in the pool of NMDAR receptors in the absence of neuroligins. To test this hypothesis independently, we puffed glutamate onto stellate cells in the presence of TTX, CNQX, and picrotoxin (Fig. 7A). In the presence of these drugs, glutamate-induced currents are primarily mediated by extrasynaptic/somatic NMDARs. We found that, in PV-Cre NL123 mice, the neuroligins deletion significantly reduced the amplitude and charge transfer of NMDAR-mediated currents. The average amplitude of glutamate-puff-induced currents in control stellate cells was 2.2 ± 0.2 nA (n = 12), whereas that in PV-Cre NL123 stellate cells was 1.5 ± 0.1 nA (n = 12); the average charge transfer of the induced currents in control stellate cells was 3.4 ± 0.5 nC (n = 12), whereas that in PV-Cre NL123 stellate cells was 2.2 ± 0.3 nC (n = 12) (Fig. 7B–D). As a control, we performed similar experiments for AMPAR-mediated currents induced by glutamate puffs. In contrast to NMDAR-mediated currents, the amplitude (control: 2.64 ± 0.15 nA, n = 9; PV-Cre NL123: 2.78 ± 0.19 nA, n = 8) and charge transfer (control: 4.79 ± 0.39 nC, n = 9; PV-Cre NL123: 4.22 ± 0.57 nC, n = 8) of AMPAR-mediated currents induced by glutamate puffs in the presence of TTX, D-AP5, and picrotoxin was similar in control and PV-NL123 stellate cells (Fig. 7E–G). This result independently confirms the observation of normal AMPAR-mediated synaptic transmission in neuroligin-deficient stellate cells (Fig. 3). Together, these data suggest that the NL123 triple KO causes a reduction in the content of extrasynaptic and/or somatic NMDARs.
Figure 7.

Neuroligin deletion significantly decreases NMDAR-mediated currents induced by puffing glutamate onto cerebellar stellate cells, but has no effect on AMPAR-mediated currents. A, Illustration of the recording configuration. B–D, Sample traces and summary graph of NMDAR-mediated EPSCs induced by local puffing of glutamate (50 mm, injected at 8 psi for 5 ms) onto cerebellar stellate cells from littermate control (black traces) and PV-Cre NL123 mice (red traces) at P21–P23. CNQX (20 μm), picrotoxin (50 μm), and TTX (1 μm) were added both to the extracellular and the puffing solutions to block AMPAR-mediated and inhibitory currents. E–G, Same as B–D, but for AMPAR-mediated EPSCs induced by local puffing of glutamate. D-AP5 (50 μm), picrotoxin (50 μm), and TTX (1 μm) were added both to the extracellular and the puffing solutions to block NMDAR-mediated and inhibitory currents. Data are means ± SEM. Numbers in bars represent the number of cells/mice used. Statistical significance was determined by Student's t test or one-way ANOVA.
Last, we measured the levels of NMDAR proteins, neuroligins, and synaptic control proteins in lysates of total cerebellum from control and PV-Cre NL123 mice by quantitative immunoblotting. As expected, PV-Cre NL123 mice showed a significant reduction in NL1, NL2, and NL3 protein levels (Fig. 8). NL2 and NL3 levels were robustly decreased, but NL1 levels were only modestly suppressed, suggesting that most NL1 in the cerebellum is primarily present in cells that do not express parvalbumin. Consistent with the decreased NMDAR-mediated EPSCs in PV-Cre NL123 mice, the PV-Cre NL123 mice also exhibited an ∼30% reduction of total content of the NMDAR subunit NR1 (Fig. 8A,B). We observed no change in PV-Cre NL123 mice in the levels of the AMPAR subunit GluA1 or the synaptic vesicle protein synaptotagmin-2 that functions as a presynaptic Ca2+-sensor for release (Sun et al., 2007). Because NMDARs are expressed in the cerebellum primarily in molecular layer interneurons (basket cells and stellate cells), but not in Purkinje cells at this young adult age (P21–P25) (Kano et al., 1995; Casado et al., 2002; Piochon et al., 2007), we believe that this reduction in NR1 levels might be largely due to the impairment of NMDARs in molecular layer interneurons. Therefore, both electrophysiological and biochemical results support the notion that deletion of neuroligins in PV-Cre NL123 mice at least partially decreases the cerebellar content of NMDARs and that the ensuing reduction in NMDAR-mediated synaptic responses is associated with a lowering of NR1 levels in these interneurons.
Figure 8.

PV-Cre induced neuroligin deletion removes only a fraction of neuroligins from cerebellum consistent with restricted PV expression in the cerebellum, but significantly decreases the total levels of NMDARs in the cerebellum. A, B, Representative quantitative immunoblots (A) and summary graphs (B) of protein levels in cerebellum from P21–P25 PV-NL123 triple cKO mice and littermate control. Proteins were solubilized from cerebellum and protein levels were measured by quantitative immunoblotting using fluorescently labeled secondary antibodies and LI-COR detection. Protein levels in PV-NL123 mice were normalized to those of control mice. Actin was used as an internal standard. Data are means ± SEM. Numbers in bars represent the number of cells/mice analyzed. Statistical significance was determined by Student's t test.
Discussion
Trans-synaptic cell-adhesion molecules are thought to control the formation and specification of different types of synapses and to regulate synapse maintenance and synaptic pruning. Many different types of trans-synaptic cell-adhesion molecules have been described (Bukalo and Dityatev, 2012; Krueger et al., 2012; Um and Ko, 2013; Bourgeron, 2015; Friedman et al., 2015). Among these, neuroligins arguably are particularly relevant because of their evolutionary conservation and their central role in synaptic transmission (Ichtchenko et al., 1996; Song et al., 1999; Varoqueaux et al., 2004; Budreck and Scheiffele, 2007; Hoon et al., 2011; Rothwell et al., 2014; Hammer et al., 2015; Liang et al., 2015; Zhang et al., 2015; Jiang et al., 2016). Moreover, neuroligins have been repeatedly implicated in autism pathogenesis (Jamain et al., 2003, 2008; Ylisaukko-oja et al., 2005; Südhof, 2008; Etherton et al., 2011; Rothwell et al., 2014), suggesting that this study is important, not only for insight into how neural circuits are formed via synaptic connections, but also for an understanding of how neural circuit formation is impaired in neuropsychiatric disorders. In the present study, we have extended previous reports on the role of neuroligins in constructing synaptic connections on cerebellar Purkinje cells (Zhang et al., 2015) and specifically examined neuroligin functions in one particular type of cerebellar neuron, namely stellate interneurons of the molecular layer. Our data demonstrate that, in stellate cells, neuroligins are surprisingly not essential for most forms of synaptic transmission, but are required for NMDAR responses (Fig. 9). This role of neuroligins is dramatically different from their role in Purkinje cells, in which they are required for the normal function of inhibitory synapses and of excitatory climbing-fiber synapses, although not for the function of parallel fiber synapses.
Figure 9.

Diagram model of the proposed functions of neuroligins in cerebellar stellate cell. Illustration for neuroligins as synaptic or extrasynaptic organizers in stellate cells. In the parallel fiber–stellate cell synapse (synapse “1” in the model), neuroligins play indispensable role for extrasynaptic NMDAR function (arrow), but a dispensable role for the synapse formation and AMPAR-mediated synaptic transmission, possibly because of paramount important role of Cbln1/GluRδ1/2 complex for AMPAR-mediated synaptic transmission (Konno et al., 2014). For basket cell/stellate cell to stellate cell inhibitory synapse (synapse “2” in the model), neuroligins play a minor role for inhibitory synaptic transmission, indicating that an additional unidentified protein “x” might play a critical role for stellate cell to stellate cell synaptic transmission.
Specifically, we found that deletion of neuroligins caused only a small decrease in inhibitory synaptic transmission (<20%) and no change in AMPAR-mediated excitatory synaptic transmission in stellate cells, but did produce a large decrease of NMDAR-mediated synaptic currents. In stellate cells, NMDARs are thought to be largely, if not exclusively, perisynaptic and extrasynaptic and are activated only by massive release induced by bursts of action potentials (Carter and Regehr, 2000; Clark and Cull-Candy, 2002; Nahir and Jahr, 2013). Our data demonstrate that the neuroligin deletion significantly decreases the total surface levels of functional NMDARs in stellate cells, suggesting that, in these cells, neuroligins are selectively essential for maintaining postsynaptic NMDAR levels. Moreover, we showed that deletion of only NL1 replicated this phenotype, suggesting that postsynaptic NMDARs are maintained primarily by NL1 alone. Our results thus extend and elaborate the emerging concept that neuroligins, and possibly other trans-synaptic cell-adhesion molecules, act in a functional mosaic to which many different cell-adhesion proteins contribute. In that functional mosaic, neuroligins provide different components in various types of neurons and different classes of synapses, such that their contribution in stellate cells is largely restricted to regulating perisynaptic and extrasynaptic NMDARs, whereas in Purkinje cells, their contribution extends to the overall maintenance of inhibitory synaptic function and to the specification of excitatory climbing-fiber synapses.
Neuroligin function in inhibitory transmission in stellate cells
Our data identify only a modest contribution of neuroligins to inhibitory synapse function in cerebellar stellate cells (Fig. 2). We speculate that the small observed inhibitory synaptic transmission phenotype in neuroligin-deficient stellate cells is attributable to NL2 and/or NL3, as reported with cell-type-specific neuroligin cKO mice in impaired inhibitory transmission recorded from Purkinje cell in cerebellum, medium spiny neurons in the striatum, and pyramidal cells in the cortex (Rothwell et al., 2014; Liang et al., 2015; Zhang et al., 2015). The reduction of inhibitory synaptic transmission onto stellate cells is small, at ∼20%, compared with an ∼85% reduction in interneuron/Purkinje cell synapses in Purkinje-cell-specific NL123 cKO mice (Zhang et al., 2015), an ∼70% reduction in PV interneuron to pyramidal cell synapse in hippocampus with NL3 KO mice (Földy et al., 2013), and an ∼50% reduction in PV interneuron to pyramidal cell synapse in cortex with NL2 KO mice (Gibson et al., 2009). The differences in the contribution of neuroligins to inhibitory synaptic strength observed between these different types of synapses and neurons could be due to synapse-specific factors (projection neurons vs interneurons) and/or context-dependent circuit features (e.g., cerebellum vs cortex vs hippocampus). Unraveling the molecular mechanisms of these differences will require dissecting the contributions of multiple candidate synaptic cell-adhesion molecules, which may constitute the next step in this overall project.
Neuroligins are not essential for AMPAR-mediated EPSCs in stellate cells
We did not find significant changes in the gross structure of stellate cells or in their AMPAR-mediated excitatory transmission in PV-Cre NL123 mice (Figs. 1, 3, 5, 6, 7). Because excitatory synapses on stellate cells are formed by parallel fibers, this finding agrees well with our previous observation that parallel fiber synapses on Purkinje cells are also not altered by deletion of neuroligins (Zhang et al., 2015). Accumulating evidence demonstrates an essential role of the neurexin/cerebellin-1/GluRδ2 complex in the formation and maintenance of parallel fiber synapses on Purkinje cells (Landsend et al., 1997; Hirai et al., 2005; Miura et al., 2009; Matsuda et al., 2010; Uemura et al., 2010; Konno et al., 2014). Importantly, GluRδ1, which is closely related to GluRδ2, was reported to be essential for formation of parallel fiber–stellate cell synapses (Konno et al., 2014), possibly via binding to cerebellin-1 (Matsuda and Yuzaki, 2011; Ryu et al., 2012). Because the binding affinity of cerebellin-1 for neurexins may be higher than that of neuroligins (Matsuda and Yuzaki, 2011), the synaptic role of neuroligins may be outcompeted by the synaptic cerebellin-1/GluRδ1 or cerebellin-1/GluRδ2 complex in parallel fiber synapses of the cerebellum. Gluδ2 KO mice exhibit an ∼50% parallel fiber Purkinje cell synapse loss, whereas Gluδ1 KO mice display an ∼30% loss of parallel fiber synapses on basket and stellate cells (Konno et al., 2014), implying that additional cell-adhesion molecules other than Gluδ1/2 function at the remaining parallel fiber synapses in cerebellum. LRRTMs, which are also neurexin-binding partners that may function in synapse specification (Ko et al., 2009; de Wit et al., 2009; Siddiqui et al., 2010), might be one of the potential candidates family. Whether and how these other cell-adhesion molecules work at parallel fiber synapses in the cerebellum, however, still needs to be determined.
Neuroligins are essential for maintaining perisynaptic and extrasynaptic NMDARs in stellate cells
We found that the triple NL123 deletion caused a substantial impairment of NMDAR-mediated synaptic transmission in cerebellar stellate cells (Figs. 4, 5, 6, 7). Consistent with this finding, we observed a reduction in the protein levels of the NMDAR subunit NR1 in cerebellar lysates (Fig. 8). Therefore, neuroligins are partially essential for only one feature of parallel fiber synaptic transmission on stellate cells, namely extrasynaptic NMDAR-mediated responses. Extrasynaptic NMDARs in stellate cells could also be activated by volume transmission from climbing fiber synapses (Nishiyama and Linden, 2007). Note that Purkinje cells do not express NMDARs in juvenile mice, so no such phenotype could be observed there.
The reduction in extrasynaptic NMDARs in stellate cells could affect transmission mediated by glutamate spillover both from climbing fibers and parallel fibers. We demonstrated here that NL1 is essential for extrasynaptic NMDARs in stellate cells (Fig. 6), significantly extending previous findings of NL1 being essential for the synaptic NMDAR function in hippocampal pyramidal neurons and amygdala principal neurons (Chubykin et al., 2007; Kim et al., 2008; Soler-Llavina et al., 2011; Jiang et al., 2016). Due to a lack of suitably specific antibodies for NL1 that can be used for immunohistochemistry, we do not know the exact location of NL1 at stellate cells. Therefore, the interaction between neuroligins and NMDARs could be direct or indirect through adaptor protein(s) (Dresbach et al., 2004; Meyer et al., 2004; Budreck et al., 2013). Considering the important role of extrasynaptic NMDARs in endocannabinoid (Beierlein and Regehr, 2006) and mGluR1α signaling (Karakossian and Otis, 2004), it is tempting to speculate that the essential but indirect role of neuroligins on synaptic strength via second message could be found in stellate cells, a similar mechanism as was reported for NL3 in hippocampus (Földy et al., 2013). Therefore, it will be important in the future to test whether neuroligins modulate short- and/or long-term plasticity in interneurons.
Footnotes
This work was supported by the Simons Foundation (307762, to T.C.S.) and the National Institute of Mental Health–National Institutes of Health (R37MH052804, to T.C.S.). We thank the members of the Südhof laboratory for invaluable discussions.
The authors declare no competing financial interests.
References
- Alcami P, Marty A. Estimating functional connectivity in an electrically coupled interneuron network. Proc Natl Acad Sci U S A. 2013;110:E4798–E4807. doi: 10.1073/pnas.1310983110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atluri PP, Regehr WG. Determinants of the time course of facilitation at the granule cell to Purkinje cell synapse. J Neurosci. 1996;16:5661–5671. doi: 10.1523/JNEUROSCI.16-18-05661.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auger C, Marty A. Heterogeneity of functional synaptic parameters among single release sites. Neuron. 1997;19:139–150. doi: 10.1016/S0896-6273(00)80354-2. [DOI] [PubMed] [Google Scholar]
- Bats C, Soto D, Studniarczyk D, Farrant M, Cull-Candy SG. Channel properties reveal differential expression of TARPed and TARPless AMPARs in stargazer neurons. Nat Neurosci. 2012;15:853–861. doi: 10.1038/nn.3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beierlein M, Regehr WG. Local interneurons regulate synaptic strength by retrograde release of endocannabinoids. J Neurosci. 2006;26:9935–9943. doi: 10.1523/JNEUROSCI.0958-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidoret C, Bouvier G, Ayon A, Szapiro G, Casado M. Properties and molecular identity of NMDA receptors at synaptic and non-synaptic inputs in cerebellar molecular layer interneurons. Front Synaptic Neurosci. 2015:7. doi: 10.3389/fnsyn.2015.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgeron T. From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat Rev Neurosci. 2015;16:551–563. doi: 10.1038/nrn3992. [DOI] [PubMed] [Google Scholar]
- Budreck EC, Scheiffele P. Neuroligin-3 is a neuronal adhesion protein at GABAergic and glutamatergic synapses. Eur J Neurosci. 2007;26:1738–1748. doi: 10.1111/j.1460-9568.2007.05842.x. [DOI] [PubMed] [Google Scholar]
- Budreck EC, Kwon OB, Jung JH, Baudouin S, Thommen A, Kim HS, Fukazawa Y, Harada H, Tabuchi K, Shigemoto R, Scheiffele P, Kim JH. Neuroligin-1 controls synaptic abundance of NMDA-type glutamate receptors through extracellular coupling. Proc Natl Acad Sci U S A. 2013;110:725–730. doi: 10.1073/pnas.1214718110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukalo O, Dityatev A. Synaptic cell adhesion molecules. Adv Exp Med Biol. 2012;970:97–128. doi: 10.1007/978-3-7091-0932-8_5. [DOI] [PubMed] [Google Scholar]
- Carter AG, Regehr WG. Prolonged synaptic currents and glutamate spillover at the parallel fiber to stellate cell synapse. J Neurosci. 2000;20:4423–4434. doi: 10.1523/JNEUROSCI.20-12-04423.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casado M, Isope P, Ascher P. Involvement of presynaptic N-methyl-D-aspartate receptors in cerebellar long-term depression. Neuron. 2002;33:123–130. doi: 10.1016/S0896-6273(01)00568-2. [DOI] [PubMed] [Google Scholar]
- Chih B, Engelman H, Scheiffele P. Control of excitatory and inhibitory synapse formation by neuroligins. Science. 2005;307:1324–1328. doi: 10.1126/science.1107470. [DOI] [PubMed] [Google Scholar]
- Heine M, Thoumine O, Mondin M, Tessier B, Giannone G, Choquet D. Activity-independent and subunit-specific recruitment of functional AMPA receptors at neurexin/neuroligin contacts. Proc Natl Acad Sci U S A. 2008;105:20947–20952. doi: 10.1073/pnas.0804007106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie JM, Jahr CE. Dendritic NMDA receptors activate axonal calcium channels. Neuron. 2008;60:298–307. doi: 10.1016/j.neuron.2008.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chubykin AA, Atasoy D, Etherton MR, Brose N, Kavalali ET, Gibson JR, Südhof TC. Activity-dependent validation of excitatory versus inhibitory synapses by neuroligin-1 versus neuroligin-2. Neuron. 2007;54:919–931. doi: 10.1016/j.neuron.2007.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark BA, Cull-Candy SG. Activity-dependent recruitment of extrasynaptic NMDA receptor activation at an AMPA receptor-only synapse. J Neurosci. 2002;22:4428–4436. doi: 10.1523/JNEUROSCI.22-11-04428.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collin T, Chat M, Lucas MG, Moreno H, Racay P, Schwaller B, Marty A, Llano I. Developmental changes in parvalbumin regulate presynaptic Ca2+ signaling. J Neurosci. 2005;25:96–107. doi: 10.1523/JNEUROSCI.3748-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collingridge GL, Olsen RW, Peters J, Spedding M. A nomenclature for ligand-gated ion channels. Neuropharmacology. 2009;56:2–5. doi: 10.1016/j.neuropharm.2008.06.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig AM, Kang Y. Neurexin-neuroligin signaling in synapse development. Curr Opin Neurobiol. 2007;17:43–52. doi: 10.1016/j.conb.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley JJ, Carter AG, Regehr WG. Fast vesicle replenishment and rapid recovery from desensitization at a single synaptic release site. J Neurosci. 2007;27:5448–5460. doi: 10.1523/JNEUROSCI.1186-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit J, Sylwestrak E, O'Sullivan ML, Otto S, Tiglio K, Savas JN, Yates JR, 3rd, Comoletti D, Taylor P, Ghosh A. LRRTM2 interacts with neurexin1 and regulates excitatory synapse formation. Neuron. 2009;64:799–806. doi: 10.1016/j.neuron.2009.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dresbach T, Neeb A, Meyer G, Gundelfinger ED, Brose N. Synaptic targeting of neuroligin is independent of neurexin and SAP90/PSD95 binding. Mol Cell Neurosci. 2004;27:227–235. doi: 10.1016/j.mcn.2004.06.013. [DOI] [PubMed] [Google Scholar]
- Dugué GP, Dumoulin A, Triller A, Dieudonné S. Target-dependent use of co-released inhibitory transmitters at central synapses. J Neurosci. 2005;25:6490–6498. doi: 10.1523/JNEUROSCI.1500-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa F, Xuan Z, Liu S, Powell CM. Neuroligin 1 modulates striatal glutamatergic neurotransmission in a pathway and NMDAR subunit-specific manner. Front Synaptic Neurosci. 2015:7. doi: 10.3389/fnsyn.2015.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etherton M, Földy C, Sharma M, Tabuchi K, Liu X, Shamloo M, Malenka RC, Südhof TC. Autism-linked neuroligin-3 R451C mutation differentially alters hippocampal and cortical synaptic function. Proc Natl Acad Sci U S A. 2011;108:13764–13769. doi: 10.1073/pnas.1111093108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Földy C, Malenka RC, Südhof TC. Autism-associated neuroligin-3 mutations commonly disrupt tonic endocannabinoid signaling. Neuron. 2013;78:498–509. doi: 10.1016/j.neuron.2013.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman LG, Benson DL, Huntley GW. Cadherin-based transsynaptic networks in establishing and modifying neural connectivity. Curr Top Dev Biol. 2015;112:415–465. doi: 10.1016/bs.ctdb.2014.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson JR, Huber KM, Südhof TC. Neuroligin-2 deletion selectively decreases inhibitory synaptic transmission originating from fast-spiking but not from somatostatin-positive interneurons. J Neurosci. 2009;29:13883–13897. doi: 10.1523/JNEUROSCI.2457-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer M, Krueger-Burg D, Tuffy LP, Cooper BH, Taschenberger H, Goswami SP, Ehrenreich H, Jonas P, Varoqueaux F, Rhee JS, Brose N. Perturbed hippocampal synaptic inhibition and gamma-oscillations in a neuroligin-4 knock-out mouse model of autism. Cell Rep. 2015;13:516–523. doi: 10.1016/j.celrep.2015.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hippenmeyer S, Vrieseling E, Sigrist M, Portmann T, Laengle C, Ladle DR, Arber S. A developmental switch in the response of DRG neurons to ETS transcription factor signaling. PLoS Biol. 2005;3:e159. doi: 10.1371/journal.pbio.0030159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai H, Pang Z, Bao D, Miyazaki T, Li L, Miura E, Parris J, Rong Y, Watanabe M, Yuzaki M, Morgan JI. Cbln1 is essential for synaptic integrity and plasticity in the cerebellum. Nat Neurosci. 2005;8:1534–1541. doi: 10.1038/nn1576. [DOI] [PubMed] [Google Scholar]
- Hollmann M, Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- Hoon M, Soykan T, Falkenburger B, Hammer M, Patrizi A, Schmidt KF, Sassoè-Pognetto M, Löwel S, Moser T, Taschenberger H, Brose N, Varoqueaux F. Neuroligin-4 is localized to glycinergic postsynapses and regulates inhibition in the retina. Proc Natl Acad Sci U S A. 2011;108:3053–3058. doi: 10.1073/pnas.1006946108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichtchenko K, Hata Y, Nguyen T, Ullrich B, Missler M, Moomaw C, Südhof TC. Neuroligin 1: a splice site-specific ligand for beta-neurexins. Cell. 1995;81:435–443. doi: 10.1016/0092-8674(95)90396-8. [DOI] [PubMed] [Google Scholar]
- Ichtchenko K, Nguyen T, Südhof TC. Structures, alternative splicing, and neurexin binding of multiple neuroligins. J Biol Chem. 1996;271:2676–2682. doi: 10.1074/jbc.271.5.2676. [DOI] [PubMed] [Google Scholar]
- Jamain S, Quach H, Betancur C, Råstam M, Colineaux C, Gillberg IC, Soderstrom H, Giros B, Leboyer M, Gillberg C, Bourgeron T, Paris Autism Research International Sibpair Study Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet. 2003;34:27–29. doi: 10.1038/ng1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamain S, Radyushkin K, Hammerschmidt K, Granon S, Boretius S, Varoqueaux F, Ramanantsoa N, Gallego J, Ronnenberg A, Winter D, Frahm J, Fischer J, Bourgeron T, Ehrenreich H, Brose N. Reduced social interaction and ultrasonic communication in a mouse model of monogenic heritable autism. Proc Natl Acad Sci U S A. 2008;105:1710–1715. doi: 10.1073/pnas.0711555105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaramillo TC, Liu S, Pettersen A, Birnbaum SG, Powell CM. Autism-related neuroligin-3 mutation alters social behavior and spatial learning. Autism Res. 2014;7:264–272. doi: 10.1002/aur.1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Polepalli J, Chen LY, Zhang B, Südhof TC, Malenka RC. Conditional ablation of neuroligin-1 in CA1 pyramidal neurons blocks LTP by a cell-autonomous NMDA receptor-independent mechanism. Mol Psychiatry. 2016 doi: 10.1038/mp.2016.80. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamboj SK, Swanson GT, Cull-Candy SG. Intracellular spermine confers rectification on rat calcium-permeable AMPA and kainate receptors. J Physiol. 1995;486:297–303. doi: 10.1113/jphysiol.1995.sp020812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano M, Hashimoto K, Chen C, Abeliovich A, Aiba A, Kurihara H, Watanabe M, Inoue Y, Tonegawa S. Impaired synapse elimination during cerebellar development in PKCy mutant mice. Cell. 1995;83:1223–1231. doi: 10.1016/0092-8674(95)90147-7. [DOI] [PubMed] [Google Scholar]
- Karakossian MH, Otis TS. Excitation of cerebellar interneurons by group I metabotropic glutamate receptors. J Neurophysiol. 2004;92:1558–1565. doi: 10.1152/jn.00300.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Jung SY, Lee YK, Park S, Choi JS, Lee CJ, Kim HS, Choi YB, Scheiffele P, Bailey CH, Kandel ER, Kim JH. Neuroligin-1 is required for normal expression of LTP and associative fear memory in the amygdala of adult animals. Proc Natl Acad Sci U S A. 2008;105:9087–9092. doi: 10.1073/pnas.0803448105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko J, Fuccillo MV, Malenka RC, Südhof TC. LRRTM2 functions as a neurexin ligand in promoting excitatory synapse formation. Neuron. 2009;64:791–798. doi: 10.1016/j.neuron.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno K, Matsuda K, Nakamoto C, Uchigashima M, Miyazaki T, Yamasaki M, Sakimura K, Yuzaki M, Watanabe M. Enriched expression of GluD1 in higher brain regions and its involvement in parallel fiber-interneuron synapse formation in the cerebellum. J Neurosci. 2014;34:7412–7424. doi: 10.1523/JNEUROSCI.0628-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger DD, Tuffy LP, Papadopoulos T, Brose N. The role of neurexins and neuroligins in the formation, maturation, and function of vertebrate synapses. Curr Opin Neurobiol. 2012;22:412–422. doi: 10.1016/j.conb.2012.02.012. [DOI] [PubMed] [Google Scholar]
- Landsend AS, Amiry-Moghaddam M, Matsubara A, Bergersen L, Usami S, Wenthold RJ, Ottersen OP. Differential localization of delta glutamate receptors in the rat cerebellum: coexpression with AMPA receptors in parallel fiber-spine synapses and absence from climbing fiber-spine synapses. J Neurosci. 1997;17:834–842. doi: 10.1523/JNEUROSCI.17-02-00834.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J, Xu W, Hsu YT, Yee A, Chen L, Südhof TC. Conditional neuroligin-2 knockout in adult medial prefrontal cortex links chronic changes in synaptic inhibition to cognitive impairments. Mol Psychiatry. 2015;20:850–859. doi: 10.1038/mp.2015.31. [DOI] [PubMed] [Google Scholar]
- Liu SQ, Cull-Candy SG. Synaptic activity at calcium-permeable AMPA receptors induces a switch in receptor subtype. Nature. 2000;405:454–458. doi: 10.1038/35013064. [DOI] [PubMed] [Google Scholar]
- Liu Y, Formisano L, Savtchouk I, Takayasu Y, Szabó G, Zukin RS, Liu SJ. A single fear-inducing stimulus induces a transcription-dependent switch in synaptic AMPAR phenotype. Nat Neurosci. 2010;13:223–231. doi: 10.1038/nn.2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llano I, Gerschenfeld HM. Inhibitory synaptic currents in stellate cells of rat cerebellar slices. J Physiol. 1993;468:177–200. doi: 10.1113/jphysiol.1993.sp019766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malagon G, Miki T, Llano I, Neher E, Marty A. Counting vesicular release events reveals binomial release statistics at single glutamatergic synapses. J Neurosci. 2016;36:4010–4025. doi: 10.1523/JNEUROSCI.4352-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda K, Yuzaki M. Cbln family proteins promote synapse formation by regulating distinct neurexin signaling pathways in various brain regions. Eur J Neurosci. 2011;33:1447–1461. doi: 10.1111/j.1460-9568.2011.07638.x. [DOI] [PubMed] [Google Scholar]
- Matsuda K, Miura E, Miyazaki T, Kakegawa W, Emi K, Narumi S, Fukazawa Y, Ito-Ishida A, Kondo T, Shigemoto R, Watanabe M, Yuzaki M. Cbln1 is a ligand for an orphan glutamate receptor delta2, a bidirectional synapse organizer. Science. 2010;328:363–368. doi: 10.1126/science.1185152. [DOI] [PubMed] [Google Scholar]
- McGee TP, Bats C, Farrant M, Cull-Candy SG. Auxiliary subunit GSG1L acts to suppress calcium-permeable AMPA receptor function. J Neurosci. 2015;35:16171–16179. doi: 10.1523/JNEUROSCI.2152-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon SA, Díaz E. Mechanisms of excitatory synapse maturation by trans-synaptic organizing complexes. Curr Opin Neurobiol. 2011;21:221–227. doi: 10.1016/j.conb.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer G, Varoqueaux F, Neeb A, Oschlies M, Brose N. The complexity of PDZ domain-mediated interactions at glutamatergic synapses: a case study on neuroligin. Neuropharmacology. 2004;47:724–733. doi: 10.1016/j.neuropharm.2004.06.023. [DOI] [PubMed] [Google Scholar]
- Miura E, Matsuda K, Morgan JI, Yuzaki M, Watanabe M. Cbln1 accumulates and colocalizes with Cbln3 and GluRδ2 at parallel fiber-Purkinje cell synapses in the mouse cerebellum. Eur J Neurosci. 2009;29:693–706. doi: 10.1111/j.1460-9568.2009.06632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahir B, Jahr CE. Activation of extrasynaptic NMDARs at individual parallel fiber-molecular layer interneuron synapses in cerebellum. J Neurosci. 2013;33:16323–16333. doi: 10.1523/JNEUROSCI.1971-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama H, Linden DJ. Pure spillover transmission between neurons. Nat Neurosci. 2007;10:675–677. doi: 10.1038/nn0607-675. [DOI] [PubMed] [Google Scholar]
- Nusser Z, Cull-Candy S, Farrant M. Differences in synaptic GABA(A) receptor number underlie variation in GABA mini amplitude. Neuron. 1997;19:697–709. doi: 10.1016/S0896-6273(00)80382-7. [DOI] [PubMed] [Google Scholar]
- Piochon C, Irinopoulou T, Brusciano D, Bailly Y, Mariani J, Levenes C. NMDA receptor contribution to the climbing fiber response in the adult mouse Purkinje cell. J Neurosci. 2007;27:10797–10809. doi: 10.1523/JNEUROSCI.2422-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulopoulos A, Aramuni G, Meyer G, Soykan T, Hoon M, Papadopoulos T, Zhang M, Paarmann I, Fuchs C, Harvey K, Jedlicka P, Schwarzacher SW, Betz H, Harvey RJ, Brose N, Zhang W, Varoqueaux F. Neuroligin 2 drives postsynaptic assembly at perisomatic inhibitory synapses through gephyrin and collybistin. Neuron. 2009;63:628–642. doi: 10.1016/j.neuron.2009.08.023. [DOI] [PubMed] [Google Scholar]
- Radyushkin K, Hammerschmidt K, Boretius S, Varoqueaux F, El-Kordi A, Ronnenberg A, Winter D, Frahm J, Fischer J, Brose N, Ehrenreich H. Neuroligin-3-deficient mice: model of a monogenic heritable form of autism with an olfactory deficit. Genes Brain Behav. 2009;8:416–425. doi: 10.1111/j.1601-183X.2009.00487.x. [DOI] [PubMed] [Google Scholar]
- Rothwell PE, Fuccillo MV, Maxeiner S, Hayton SJ, Gokce O, Lim BK, Fowler SC, Malenka RC, Südhof TC. Autism-associated neuroligin-3 mutations commonly impair striatal circuits to boost repetitive behaviors. Cell. 2014;158:198–212. doi: 10.1016/j.cell.2014.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu K, Yokoyama M, Yamashita M, Hirano T. Induction of excitatory and inhibitory presynaptic differentiation by GluD1. Biochem Biophys Res Commun. 2012;417:157–161. doi: 10.1016/j.bbrc.2011.11.075. [DOI] [PubMed] [Google Scholar]
- Shipman SL, Nicoll RA. A subtype-specific function for the extracellular domain of neuroligin 1 in hippocampal LTP. Neuron. 2012;76:309–316. doi: 10.1016/j.neuron.2012.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui TJ, Pancaroglu R, Kang Y, Rooyakkers A, Craig AM. LRRTMs and neuroligins bind neurexins with a differential code to cooperate in glutamate synapse development. J Neurosci. 2010;30:7495–7506. doi: 10.1523/JNEUROSCI.0470-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soler-Llavina GJ, Fuccillo MV, Ko J, Südhof TC, Malenka RC. Inaugural article: the neurexin ligands, neuroligins and leucine-rich repeat transmembrane proteins, perform convergent and divergent synaptic functions in vivo. Proc Natl Acad Sci U S A. 2011;108:16502–16509. doi: 10.1073/pnas.1114028108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song JY, Ichtchenko K, Südhof TC, Brose N. Neuroligin 1 is a postsynaptic cell-adhesion molecule of excitatory synapses. Proc Natl Acad Sci U S A. 1999;96:1100–1105. doi: 10.1073/pnas.96.3.1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto D, Coombs ID, Kelly L, Farrant M, Cull-Candy SG. Stargazin attenuates intracellular polyamine block of calcium-permeable AMPA receptors. Nat Neurosci. 2007;10:1260–1267. doi: 10.1038/nn1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature. 2008;455:903–911. doi: 10.1038/nature07456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultan F, Bower JM. Quantitative Golgi study of the rat cerebellar molecular layer interneurons using principal component analysis. J Comp Neurol. 1998;393:353–373. [PubMed] [Google Scholar]
- Sun J, Pang ZP, Qin D, Fahim AT, Adachi R, Südhof TC. A dual-Ca2+-sensor model for neurotransmitter release in a central synapse. Nature. 2007;450:676–682. doi: 10.1038/nature06308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, June Liu S. Activation of extrasynaptic NMDA receptors induces a PKC-dependent switch in AMPA receptor subtypes in mouse cerebellar stellate cells. J Physiol. 2007;583:537–553. doi: 10.1113/jphysiol.2007.136788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi T, Miyazaki T, Watanabe M, Mori H, Sakimura K, Mishina M. Control of synaptic connection by glutamate receptor delta2 in the adult cerebellum. J Neurosci. 2005;25:2146–2156. doi: 10.1523/JNEUROSCI.4740-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trigo FF, Bouhours B, Rostaing P, Papageorgiou G, Corrie JE, Triller A, Ogden D, Marty A. Presynaptic miniature gabaergic currents in developing interneurons. Neuron. 2010;66:235–247. doi: 10.1016/j.neuron.2010.03.030. [DOI] [PubMed] [Google Scholar]
- Trigo FF, Sakaba T, Ogden D, Marty A. Readily releasable pool of synaptic vesicles measured at single synaptic contacts. Proc Natl Acad Sci U S A. 2012;109:18138–18143. doi: 10.1073/pnas.1209798109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uemura T, Lee SJ, Yasumura M, Takeuchi T, Yoshida T, Ra M, Taguchi R, Sakimura K, Mishina M. Trans-synaptic interaction of GluRδ2 and neurexin through Cbln1 mediates synapse formation in the cerebellum. Cell. 2010;141:1068–1079. doi: 10.1016/j.cell.2010.04.035. [DOI] [PubMed] [Google Scholar]
- Um JW, Ko J. LAR-RPTPs: Synaptic adhesion molecules that shape synapse development. Trends Cell Biol. 2013;23:465–475. doi: 10.1016/j.tcb.2013.07.004. [DOI] [PubMed] [Google Scholar]
- Varoqueaux F, Jamain S, Brose N. Neuroligin 2 is exclusively localized to inhibitory synapses. Eur J Cell Biol. 2004;83:449–456. doi: 10.1078/0171-9335-00410. [DOI] [PubMed] [Google Scholar]
- Varoqueaux F, Aramuni G, Rawson RL, Mohrmann R, Missler M, Gottmann K, Zhang W, Südhof TC, Brose N. Neuroligins determine synapse maturation and function. Neuron. 2006;51:741–754. doi: 10.1016/j.neuron.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Wittenmayer N, Körber C, Liu H, Kremer T, Varoqueaux F, Chapman ER, Brose N, Kuner T, Dresbach T. Postsynaptic Neuroligin1 regulates presynaptic maturation. Proc Natl Acad Sci U S A. 2009;106:13564–13569. doi: 10.1073/pnas.0905819106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki M, Miyazaki T, Azechi H, Abe M, Natsume R, Hagiwara T, Aiba A, Mishina M, Sakimura K, Watanabe M. Glutamate receptor δ2 is essential for input pathway-dependent regulation of synaptic AMPAR contents in cerebellar Purkinje cells. J Neurosci. 2011;31:3362–3374. doi: 10.1523/JNEUROSCI.5601-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ylisaukko-oja T, Rehnström K, Auranen M, Vanhala R, Alen R, Kempas E, Ellonen P, Turunen JA, Makkonen I, Riikonen R, Nieminen-von Wendt T, von Wendt L, Peltonen L, Järvelä I. Analysis of four neuroligin genes as candidates for autism. Eur J Hum Genet. 2005;13:1285–1292. doi: 10.1038/sj.ejhg.5201474. [DOI] [PubMed] [Google Scholar]
- Yuzaki M. The delta2 glutamate receptor: 10 years later. Neurosci Res. 2003;46:11–22. doi: 10.1016/S0168-0102(03)00036-1. [DOI] [PubMed] [Google Scholar]
- Zhang B, Chen LY, Liu X, Maxeiner S, Lee SJ, Gokce O, Südhof TC. Neuroligins sculpt cerebellar Purkinje cell circuits by differential control of distinct classes of synapses. Neuron. 2015;87:781–796. doi: 10.1016/j.neuron.2015.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]