

Abstract
Neuroplastin (Nptn) is a member of the Ig superfamily and is expressed in two isoforms, Np55 and Np65. Np65 regulates synaptic transmission but the function of Np55 is unknown. In an N-ethyl-N-nitrosaurea mutagenesis screen, we have now generated a mouse line with an Nptn mutation that causes deafness. We show that Np55 is expressed in stereocilia of outer hair cells (OHCs) but not inner hair cells and affects interactions of stereocilia with the tectorial membrane. In vivo vibrometry demonstrates that cochlear amplification is absent in Nptn mutant mice, which is consistent with the failure of OHC stereocilia to maintain stable interactions with the tectorial membrane. Hair bundles show morphological defects as the mutant mice age and while mechanotransduction currents can be evoked in early postnatal hair cells, cochlea microphonics recordings indicate that mechanontransduction is affected as the mutant mice age. We thus conclude that differential splicing leads to functional diversification of Nptn, where Np55 is essential for OHC function, while Np65 is implicated in the regulation of synaptic function.
SIGNIFICANCE STATEMENT Amplification of input sound signals, which is needed for the auditory sense organ to detect sounds over a wide intensity range, depends on mechanical coupling of outer hair cells to the tectorial membrane. The current study shows that neuroplastin, a member of the Ig superfamily, which has previously been linked to the regulation of synaptic plasticity, is critical to maintain a stable mechanical link of outer hair cells with the tectorial membrane. In vivo recordings demonstrate that neuroplastin is essential for sound amplification and that mutation in neuroplastin leads to auditory impairment in mice.
Keywords: amplification, deafness, hair cells, inner ear, neuroplastin, tectorial membrane
Introduction
The organ of Corti of the mammalian cochlea contains the sensory epithelium for the perception of sound. The mechanosensory cells of the cochlea are the hair cells, which are patterned in one row of inner hair cells (IHCs) and three rows of outer hair cells (OHCs). Hair cells are surrounded by supporting cells and sandwiched between two specialized extracellular matrix assemblies, the basilar membrane (BM) and the tectorial membrane (TM; Kazmierczak and Müller, 2012). Sound-induced vibrations of the organ of Corti lead to deflections of the stereociliary bundles of hair cells, which cause the opening of mechanically gated ion channels in stereocilia and hair cell depolarization. IHCs transmit the sound information along afferent neurons to the CNS. In contrast, OHCs act as amplifiers that increase the motion of the organ of Corti (Fettiplace and Hackney, 2006). Somatic electromotility, length changes in the soma of OHCs driven by hair cell depolarization (Fettiplace and Hackney, 2006; Ashmore, 2008), and hair bundle motility, driven by the gating of the transducer channels (Fettiplace and Hackney, 2006; Richardson et al., 2011), are both thought to contribute to amplification. Cochlear amplification shows an impressive nonlinearity, where low sounds are amplified more strongly than loud sounds, thus conferring remarkable sensitivity to the cochlea and a large dynamic range. Since amplification is frequency selective, OHCs also contribute to the sharp tuning of the cochlea (Robles and Ruggero, 2001; Schwander et al., 2010).
Coherent vibrational motions within the organ of Corti during sound stimulation requires mechanical coupling between the different components of the organ of Corti, including the hair cells, support cells, the BM, and the TM. Remarkably, only the longest stereocilia of OHCs are coupled to the TM while the hair bundles of IHCs are freestanding and deflected by endolymph flow created by organ of Corti movement (Hudspeth, 1997; Tsuprun and Santi, 1998; Guinan, 2012). Thus, the coupling of OHCs to the TM is critical for them to be properly stimulated to create the forces of cochlear amplification necessary for the proper transmission of sound information to IHCs (Legan et al., 2000). However, little is known about the molecules and mechanisms that mechanically couple OHCs to the TM. One protein that has been implicated in this process is stereocilin, which may be a secreted protein without a noticeable transmembrane domain (Verpy et al., 2008, 2011; Richardson et al., 2011), although it might have a glycosylphosphatidylinositol (GPI) anchor (Jovine et al., 2002). In mature hair cells, stereocilin localizes to the horizontal top connectors that link adjacent stereocilia to each other and to the tips of stereocilia near the TM (Verpy et al., 2008, 2011). Stereocilin can also be detected within TMs that have been mechanically detached from hair cells, suggesting that the protein might be secreted and deposited into the TM (Verpy et al., 2011). Genetic studies in mice have demonstrated that stereocilin is required for the maintenance of hair bundle integrity and the coupling of stereocilia to the TM (Verpy et al., 2008, 2011). However, the mechanism by which stereocilin mediates interactions between stereocilia and the TM and the extent to which it acts alone or as part of a larger protein complex is not known.
In a N-ethyl-N-nitrosaurea (ENU) mutagenesis screen, we have now generated a deaf mouse line, audio-1, which carries a recessive mutation in the neuroplastin gene (Nptn), a member of the Ig superfamily that is expressed in two isoforms (Beesley et al., 2014). The longer Np65 isoform contains three extracellular Ig domains and is expressed in the CNS (Beesley et al., 2014). Recent studies suggest that Np65 may regulate the properties of synapses connecting IHCs with spiral ganglion neurons (SGNs; Carrott et al., 2016). We now demonstrate that the shorter Np55 isoform, which lacks the N-terminal Ig1 domain, is strongly expressed in the stereocilia of OHCs but not IHCs. In OHCs, Np55 is required for coupling of their stereocilia to the TM and for proper sound amplification. In addition, hair bundles are maintained in the mutant mice but they accumulate morphological changes as mice age. Cochlear microphonics recordings suggest that mechanotransduction is also affected in hair cells of adult mice, at least in the basal part of the cochlea. We therefore conclude that defects in OHC function likely contribute to the auditory impairment in audio-1 mice, where alternative splicing leads to Nptn isoforms with distinct functions in OHCs and at synaptic sites.
Materials and Methods
Ethics statement.
Institutional Animal Care and Use Committee Institutional Review Boards at the Scripps Research Institute, La Jolla, California, and at Stanford Medical School approved all animal procedures.
ENU mutagenesis, auditory brainstem response and distortion product otoacoustic emission measurement, and mapping of the audio-1 mutation.
ENU mutagenesis, auditory brainstem response (ABR) and distortion product otoacoustic emission (DPOAE) measurements, vestibular function tests, and single nucleotide polymorphism (SNP) mapping were performed as described previously (Schwander et al., 2007). All phenotypic analysis subsequent to positional cloning was performed with ENU mutant mice on a C57BL/6J background. For ABR and DPOAE measurements and data analysis, we used a TDT workstation (Tucker-Davis Technologies). Speakers were calibrated to minimize harmonic distortions. Exome sequencing of two affected audio-1 mice was performed by Australian Phenomics Facility. Libraries were prepared and captured using an Agilent SureSelectXT2 mouse All Exon kit. An Illumina HiSeq2500 was used for 100 bp paired-end sequencing. The bioinformatics analysis was processed through a custom SNP/indel analysis pipeline. To confirm the presence of the mutation in transcripts, RNA was prepared from the inner ear of P7 wild-type and audio-1 mice, reverse transcribed using Moloney murine leukemia virus reverse transcriptase, and amplified by RT-PCR using random primers and JumpStart Accu TaqLA DNA polymerase (Sigma-Aldrich). Nptn was amplified using gene-specific primers as follows: forward, 5′-GGTAAAGTGAAAGTCCCAGTGTAGTCC-3′; reverse, 5′-CATTCTTACGGGTGGCAGTGAGTT-3′. Amplification reactions were cycled using a standard protocol on a GeneMate Genius thermocycler (ISC BioExpress). Bidirectional sequencing of transcripts as well as exons and flanking regions was completed with a BigDye v3.1 Terminator Cycle Sequencing Kit (Applied Biosystems), according to the manufacturer's instructions. Sequencing products were resolved using an ABI 3730s Sequencer (PerkinElmer). All sequencing chromatograms were compared with published cDNA sequences; nucleotide changes were detected using Sequencher v4.5 (Gene Code).
Nptn−/− mice.
Nptn knock-out mice were generated using ES cells obtained from the European Mouse Mutagenesis Program (EUCOMM). The mice were originally constructed on a C57BL/6N background but in subsequent breedings were also crossed to C57BL/6J mice and CBA/J mice.
RT-PCR and in situ hybridization.
RT-PCR analysis with total RNA from mouse inner ear tissue was performed. Inner ears of postnatal day (P) 7, P14, and P28 mice were dissected from temporal bones. Total RNA was isolated using the RNeasy Mini kit (Qiagen) and reverse-transcribed into cDNA using the SuperScript III First-Strand Synthesis System (Thermo Fisher Scientific) from tissue of the cerebral cortex and cochlea. Nptn55/65-specific cDNA were amplified by PCR and separated by gel electrophoresis on a 1% agarose gel. The primers used are as follows: Np65 forward (Np65F), 5′-GAAGCGCCGTGTCACCGTAAAC-3′; Np55 forward (Np55F), 5′-CGCTGCTCAGAACGAACCAAGAA-3′; Np65 and Np55 common reverse, 5′-TTATGGCCAGTGATGTCAGGA-3′. The size of Np65 amplification is 480 bp and, for Np55, 303 bp. Sequences were cloned in pGEM-T (Promega) and sequenced.
In situ hybridization was performed as described previously (Grillet et al., 2009). Probes for Np55 and Np65 mRNA (GenBank FJ50876) were amplified from the murine P7 cochlea using Phusion (New England Biolabs) and cloned into pGEM-T (Promega). The Np55/65 probe was generated from the 341 bp sequence using the following primers: 5′-TTGTCACCAGTGAAG-3′ and 5′-GTAGCCAACTGACTTGCAGTA-3′. The Nptn65 probe was generated similarly (353 bp) using the following primers: 5′-GAACGCTGGGTTTGTCAAGTCGCCCAT-3′ and 5′-TCTGAAGGACGCTTATGGTGGC-3′.
Scanning electron microscopy.
Scanning electron microscopy was performed as described previously (Xiong et al., 2012). Briefly, cochlear tissue was fixed and the TM was removed for hair cell scanning. Samples were dehydrated, processed to the critical drying point, mounted, coated with iridium, and imaged. For TM scanning, the membrane was kept on the organ of Corti until mounting. The TM was further dissected and flat mounted on the carbon tape followed by coating and imaging.
Immunolocalization studies.
For immunolocalization studies, we used the following antibodies: sheep anti-mouse neuroplastin (NPTN) 55/65 (R&D Systems, AF7818), goat anti-human/mouse NPTN 65 (R&D Systems, AF5360), and rat anti-HA High Affinity (Roche). F-actin was visualized in whole mounts by staining with FITC-phalloidin (Thermo Fisher Scientific). Staining of histological sections and whole mounts were performed as described previously (Senften et al., 2006; Schwander et al., 2007). For immunogold localization, animals were perfused with the following: 4% PFA, 0.025% glutaraldehyde in Na-cacodylate 0.1 m, pH 7.4, 0.025% picric acid. The cochlear shell was opened and incubated in the same fixative for 1 h. Tissue was washed with Tris-buffered saline (TBS; 150 mm NaCl, 10 mm Tris-HCl), pH 7.6, and the cochlear shell, Reissner's membrane, and TM were removed. Tissue was blocked for 1 h at room temperature in TBS containing 4% BSA and 0.02% Triton X-100, and incubated overnight at 4°C with sheep anti-mouse Np55/65 antibody (1:150) in TBS containing 1% BSA and 0.02% Triton X-100. Tissue was washed in TBS and incubated for 2 d at 4°C with donkey anti-sheep antibody conjugated with 6 nm colloidal gold beads. Tissue was washed in TBS and 0.1 m Na-cacodylate, and postfixed for 24 h at 4°C with 2.5% glutaraldehyde in 0.1 m Na-cacodylate. Decalcification of the modiolus was performed by adding 1:3 v/v 0.5 m EDTA, pH 8.0, to fixative and incubating for 3 h at 4°C. Tissue was washed with 0.1 m Na-cacodylate and postfixed for 1.5 h in 1% OsO4 in 0.1 m Na-cacodylate, washed, dehydrated, and cleared in propylene oxide. Tissue was impregnated in Epon-Araldite resin and the organ of Corti further microdissected and samples polymerized at 60°C. Thick sections were initially taken to assess the orientation. Thin sections were cut and poststained for 30 min with uranyl acetate followed by 20 min incubation in lead citrate. Grids were examined on a Philips CM100 electron microscope (FEI).
Injectoporations.
Injectoporations were performed as described previously (Xiong et al., 2014). In brief, cochlear explants prepared from P3 animals were place under an inverted microscope (BX51WI, Olympus) equipped with a pair of micromanipulators (MP-285, Sutter Instrument) holding a pair of home-made electroporation electrodes and an injection electrode. Plasmid was purified using an Endo-free Maxiprep kit (Qiagen) and diluted with Endo-free water to a final working concentration of 1 μg/μl. The osmolarity of the plasmid solution was adjusted to 330–350 mOsm with 10× HBSS (14185-052, Invitrogen). The desirable injection location, position of electroporation electrodes, and position of injection electrode was assessed with a 60× water-immersed objective. The tip of injection electrode was pushed through the apical surface of the sensory epithelium and positioned next to the cell bodies of hair cells. Once the injection electrode was positioned, positive air pressure was used to perfuse 0.1 ml of plasmid solution over a time interval of 10 s. The electroporation electrodes were connected to a square-wave electroporator (ECM 830, Harvard Apparatus). Explants were electroporated with three pulses at 60 V amplitude, 15 ms duration, and 1 s interval. After electroporation, half of the medium was changed to fresh DMEM/F12 with 1% FBS and the tissues were transferred back in the incubator for culturing.
Electrophysiology.
Mechanotransduction currents were recorded following our published procedure (Xiong et al., 2012). Cells were whole-cell patched for recording mechanotransduction currents at −70 mV holding potential with a patch-clamp amplifier (EPC 10 USB, HEKA). For mechanical stimulation, hair bundles were deflected with a glass probe mounted on a piezoelectric stack actuator (P-885, Physik Instrument). The actuator was driven with voltage steps that were low-pass filtered at 10 kHz frequency to diminish the resonance of the piezo stack.
Nonlinear capacitance (NLC) was measured by a two-sinusoidal method and fitted to the first derivative of a two-state Boltzmann function with SigmaPlot (Zhu et al., 2013) as follows: Cm = NLC + Clin= Qmax * (ze/kT) * exp[−ze(Vm − Vpk)/kT)/(1 + exp(−ze(Vm − Vpk)/kT)] ∧ 2 + Clin, where Qmax is the maximum charge transferred, Vpk is the peak of NLC, z is the number of elementary charge (e), k is Boltzmann's constant, and T is the absolute temperature. Membrane potential (Vm) was corrected for electrode access resistance.
Analysis of purified hair bundles.
Hair bundles were isolated from mouse utricles at P4–P6 (P5) and P21–P25 (P23) developmental ages as described previously (Gillespie and Hudspeth, 1991; Dumont et al., 2002; Krey et al., 2015). Protein mass spectrometry, peptide and protein identification, protein quantification, and technical validation of this dataset (available from ProteomeXchange with the identifier PXD002167) were described in depth previously (Krey et al., 2015). Peptide mapping was performed as described previously (Shin et al., 2010; Francis et al., 2015), using the Np55 (ENSMUSP00000082793) and Np65 (ENSMUSP00000135199) amino acid sequences.
Protein immunoblotting used methods similar to those previously described (Shin et al., 2013). Utricles and brains were dissected from C57BL/6 mice and homogenized in reducing SDS-PAGE sample buffer, boiled for 5 min, and loaded into a 4–12% Bis-Tris gel (1.5 mm × 10 wells; Life Technologies). Proteins were transferred to PVDF membranes and blocked for 1 h with GE Healthcare ECL Prime Blocking reagent (GE Healthcare) and probed overnight with 0.05 μg/ml sheep anti-mouse NPTN 55/65 (R&D Systems, AF7818) diluted in blocking reagent. The membrane was washed in PBS/0.1% Tween, incubated for 2 h in 0.1 μg/ml HRP-conjugated anti-sheep IgG secondary antibody (R&D Systems), washed, and then developed with GE Healthcare ECL Prime Western Blotting Detection reagent (GE Healthcare).
In vivo vibrometry.
In vivo measurements of BM and TM vibrations in response to sound stimulation were obtained using volumetric optical coherence tomography vibrometry (VOCTV), as described previously (Lee et al., 2015). Briefly, mice (P26–P81 males and females) were anesthetized with ketamine/xylazine and the left middle ear bulla was surgically exposed. Widely opening the bulla then allowed optical access to the cochlear apex, from which images and vibration data could be obtained noninvasively without opening the otic capsule bone. Vibrations were measured from the malleus (near the orbicular apophysis). The displacement magnitudes and phases of BM and TM vibrations were first obtained in response to 200 ms tones that ranged in frequency from 2 to 14 kHz (0.5 kHz steps) at stimulus levels of 10–80 dB SPL (10 dB steps). Next, the vibration of the middle ear ossicular chain was then measured at the same frequencies using a stimulus level of 80 dB SPL, and used to normalize the responses of the BM and TM. Vibration data with magnitudes <3 SDs above the noise floor or <0.15 nm were not analyzed.
Cochlear microphonic recordings.
The animals were anesthetized using ketamine (100 mg/ml) and xylazine (10 mg/ml), and their head secured in a head holder. The left tympanic bulla was surgically uncovered and opened to expose the round window. The cochlear microphonic (CM) was measured from the ball-ended tip of a Teflon-coated silver wire (0.003 inch diameter; A-M Systems) advanced onto the round window membrane with a micromanipulator. The signal was referenced to a silver wire inserted under the skin near the vertex of the skull. The ground electrode was placed in the hind leg. A bioamplifier was used (DP-311 differential amplifier, Warner Instruments) to amplify the signals 100 times and bandpass filtered between 1 Hz and 10 kHz. The sound stimuli were 20 ms 6 kHz tones and the intensity ranged from 10 to 100 dB in 5 dB steps. The stimuli were synthesized in software and output by a speaker (MDR EX37B, Sony) inserted into the ear canal. We calibrated the intensity using a probe-tip microphone in the ear bar as previously described (Xia et al., 2010). The CM signal measured by the bioamplifier was digitized at 1 MHz and the magnitude of the response at 6 kHz was determined by fast Fourier transform. All stimulus harmonics and noise at all other frequencies were ≥50 dB below the primary signal at all stimulus intensities.
Data analysis.
Data analysis was performed using Excel (Microsoft), OriginLab, and Matlab (MathWorks). All data are mean ± SEM. Student's two-tailed paired or unpaired t tests were used to determine statistical significance between measurements in the same cohort or different cohorts, respectively (*p < 0.05, **p < 0.01, ***p < 0.001).
Results
Hearing loss in audio-1 mice
In an ENU mutagenesis screen, we generated a mouse line that we named audio-1 (Fig. 1A). In our initial phenotypic analysis of 8-week-old mice using ABR measurements as a screening tool, homozygous audio-1 mice manifested with profound deafness. When deaf audio-1 mice were crossed with wild-type mice, all F1 offspring showed normal hearing function (data not shown), indicating that the audio-1 mutation was inherited recessively. Consistent with this finding, when the F1 mice were intercrossed ∼25% of their F2 offspring were hearing impaired. We also observed occasional head tossing, suggestive of mild vestibular impairment. Measurements of the ABR to broadband click stimuli in offspring at P21 and P60 demonstrated that homozygous audio-1 mice were profoundly deaf already by 3 weeks of age (Fig. 1B,C). Analysis of pure-tone ABRs confirmed that hearing was impaired across the entire analyzed frequency spectrum (Fig. 1D).
Figure 1.

Hearing loss in audio-1 mice. A, Diagram of the ENU-induced forward genetics screen in C57BL/6J mice. G3 animals were tested for hearing impairment. Red asterisks indicate mutation originating from the G0 male, the blue asterisks from the G0 females. Animals with putative homozygous phenotype are represented with a black-filled square or circle, for male and female, respectively. B, Representative ABR traces to click stimuli in the indicated control and mutant mice at 8 weeks of age. C, Statistic results of ABR thresholds to click stimuli at P21 and P60 of wild-type (control) and homozygous audio-1 mutant mice. n = 15 mice for each group. D, ABR thresholds to pure tones at P21 and P60 of wild-type (control) and homozygous audio-1 mutant mice. n = 15 mice for each group.
Audio-1 mice carry a missense mutation in Nptn
To identify the affected gene in audio-1 mice, we backcrossed them with WT 129S1/SvlmJ mice. The F1 offspring were intercrossed and hearing function of F2 progeny was analyzed by ABR measurements to identify affected and unaffected offspring. Ten affected and 13 unaffected littermates were used for genome-wide SNP mapping. An interval of 20 Mb on chromosome 9 [56,586,960–77,090,797] was found to be homozygous for the C57BL/6J background in all affected animals but not in the unaffected ones (Fig. 2A). Exome sequencing using genomic DNA from two affected mice identified putative homozygous point mutations in a total of 18 autosomal genes (Fig. 2B). Mutations in three of the genes were shared between the two affected mice and only two of the mutations were localized within the affected genomic interval on chromosome 9 identified by SNP mapping. These mutations (T-to-A change) were located within coding exons of Nptn and the casein-kinase-1-gamma-1 gene (Csnk1g1; Fig. 2B). Primers were designed for the two genes and used to generate by PCR cDNA fragments from mRNA isolated from the P7 organ of Corti of seven affected and seven unaffected animals. The amplicons were directly sequenced. We detected a T-to-A change in the coding sequence of both Nptn and Csnk1g1 (Fig. 2C). All sequenced Nptn transcripts from affected audio-1 mice carried the mutation. In contrast, only some Csnk1g1 transcripts carried the mutation in affected audio-1 animals, while others encoded the wild-type protein. At the genomic level, only if the mutation in Nptn was present in both Nptn alleles were audio-1 mice hearing impaired, while the mutation in Csnk1g1 did not segregate with the hearing phenotype (Fig. 2C). We thus conclude that the auditory phenotype in audio-1 mice is likely caused by a recessive mutation in Nptn.
Figure 2.
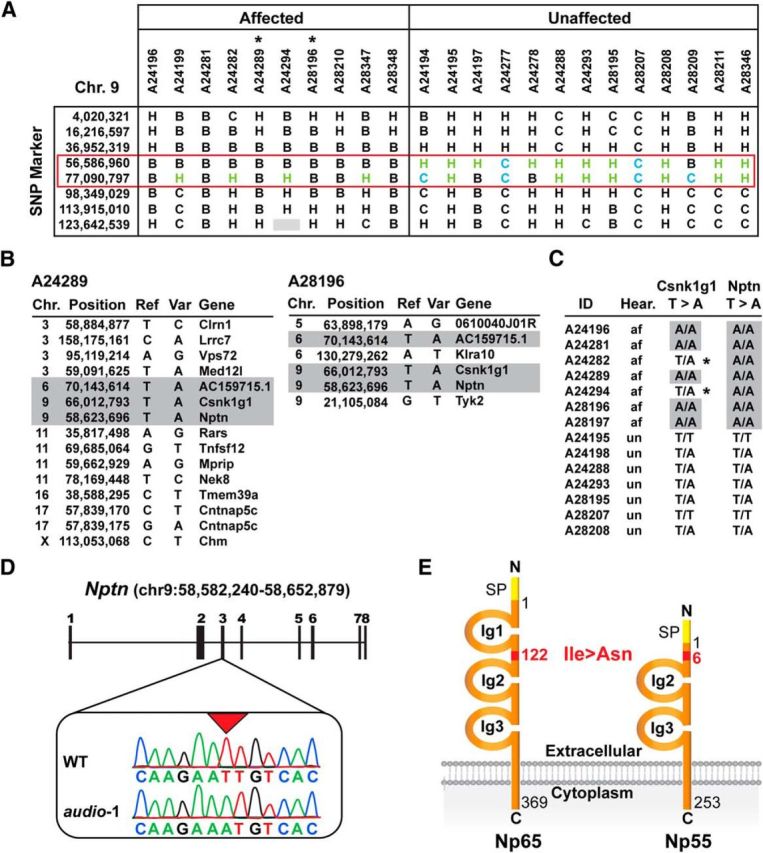
Positional cloning of the audio-1 mutation. A, Mapping of the audio-1 locus. The diagram shows the results from fine mapping of the region of chromosome 9 (Chr 9) linked to the mutant phenotype. Audio-1 mice (generated on a C57BL/6J background) were crossed to 129S1/SvlmJ mice. The F1 offspring were intercrossed to obtain F2 progeny for ABR phenotyping. DNA from affected and unaffected F2 mice and control mice was analyzed with SNP markers that distinguish between C57BL/6J and 129S1/SvlmJ alleles to detect linkage of a genomic region to the audio-1 phenotype. The locations of SNP markers on chromosome 9 are listed in the first column (the kilobase position 56,586,960–77,090,797 is indicated). The mouse identifiers and phenotypes are shown in the two top rows. The genotype of each mouse for a given SNP marker is represented as follows: B, homozygous C57BL/6J; C, homozygous 129S1/SvlmJ; H, heterozygous. The chromosomal interval that segregates with the mutant phenotype is highlighted with a red box. Two affected animals selected for the exome sequencing are indicated with an asterisk. B, Homozygous mutation found in the two animals subjected to exome sequencing (A24289 and A28196). The chromosomal position, the reference nucleotide (Ref), the nucleotide variation identified (Var), and the gene concerned are indicated. C, Verification of the nucleotide variation on a large mouse cohort for the two homozygous mutations lying in the identified locus. The mouse identifier (ID), the hearing ability (af, affected; un, unaffected), and the nucleotide variation observed are indicated. Outliers are indicated with an asterisk. D, Exon structure of the NPTN gene. Exons are shown as black squares. Coding exons are numbered. Sequence chromatograms of wild-type (WT) and homozygous audio-1 mice reveal a T-to-A transversion in exon 3 (box). E, Schematic representation of the two splicing isoform of Nptn. Np65 contains three Ig domains. Np55 contains only two. The missense mutation, indicted in red, is found in the amino acid #122 for Np65 and amino acid #6 for Np55. It leads to the replacement of an isoleucine by an asparagine. SP, Signal peptide.
Nptn encodes a transmembrane protein that is a member of the Ig superfamily and is expressed in two isoforms generated by alternative splicing of the primary transcript (Beesley et al., 2014). The longer Np65 isoform contains three extracellular Ig domains, while the shorter Np55 isoform lacks the N-terminal Ig1 domain (Fig. 2E) due to alternative splicing of exon 2. However, the T-to-A change is present in exon 3 and thus present in transcripts for both Nptn isoforms (Fig. 2E).
To confirm that Nptn is required for normal hearing function, we generated an Nptn knock-out mouse using ES cells obtained from EUCOMM (Fig. 3A,B). In this mouse a premature stop is inserted downstream of the third exon leading to a protein predicted to be truncated in the middle of IgG2. Mice homozygous for the Nptn mutation survived to adulthood but were smaller than their heterozygous littermates (Fig. 3C). Protein expression of Nptn was abolished in the mutant mice (Fig. 3D), indicating that the genetic modification led to an Nptn-null allele (and hence referred to as Nptn knock-out mice or Nptn−/−). Nptn expression was also drastically reduced in homozygous audio-1 mutant mice, although low levels of the mutant proteins appeared to persist (Fig. 3E). However, the remaining protein only encodes part of the extracellular domain, suggesting that the audio-1 mutation also leads to a functional null allele. Measurements of the ABR in Nptn knock-out mice demonstrated that hearing function was impaired at P21 and P60 across the entire analyzed frequency spectrum (Fig. 3F,G). We thus conclude that Nptn is essential for normal hearing.
Figure 3.

Nptn knock-out mice are deaf. A, Diagram of the Nptn allele used in the current study. The “knock-out-first” allele (Nptnnull) contains an SA:lacZ trapping cassette and a floxed promoter-driven neo cassette inserted into the intron the Nptn gene, disrupting gene function. B, PCR analysis of wild-type (+/+), heterozygous (+/−), and homozygous mutant (−/−) mice obtained after crossing the Nptnnull mice. C, Nptn−/− mice show reduced size during development when compared with Nptn+/− littermates. D, Immunoblots of brain extracts from Nptn+/+, Nptn+/−, and Nptn−/− mice probed with either Np55/65 or Np65-specific antibody. Immunoreactive bands of the expected molecular mass are detected in Nptn+/+ and Nptn+/− extracts, but not in Nptn−/− extracts. As a control, anti-α tubulin antibody was used to confirm equal loading of total protein lysate between groups. E, Immunoblots of brain extracts from +/+ and homozygous audio-1 mutant mice probed with Np55/65 antibody. Immunoreactive bands of the expected molecular mass are detected in +/+ but not in homozygous audio-1 mutant extracts. As a control, anti-α tubulin antibody was used to confirm equal loading of total protein lysate between groups. F, Statistic results of ABR thresholds to click stimuli at P21 and P60 of wild-type (control) and Nptn−/− mice. n = 15 mice for each group. G, ABR thresholds to pure tones at P21 and P60 of wild-type (control) and Nptn−/− mice. n = 15 mice for each group.
Nptn transcripts are expressed in hair cells and SGNs
Previous studies have shown that Np65 is expressed in the CNS, while Np55 is more widely expressed inside and outside the nervous system (Empson et al., 2006; Owczarek et al., 2010; Beesley et al., 2014). Consistent with the earlier studies, we detected by RT-PCR with isoform-specific primers (Fig. 4A) prominent expression of both Np55 and Np65 in the cerebral cortex of mice between P7 and P28 (Fig. 4B). In contrast, Np55 levels were much higher than Np65 levels in mRNA isolated from the organ of Corti at all time points analyzed (Fig. 4B).
Figure 4.

Nptn transcripts are expressed in the inner ear. A, Graphical representation of cDNA/protein composition of the Nptn isoforms. The position of the primer used in RT-PCR and in situ hybridization probes to amplify the sequence of each isoform is indicated. SP, Signal peptide; TM, Transmembrane domain; Cyto, cytoplasmic domain. B, Isoform-specific RT-PCR on cerebral cortex and cochlea from P7, P14, and P28. C–V, Analysis of Nptn expression by in situ hybridization on P4 inner ear sections. Comparable probes specific for both isoform (Nptn 55/65) and for the long isoform (Nptn 65) were used. A probe specific for hair cell expression (Loxhd1) was also used as positive control. AS, Antisense; S, sense. C–G, Sections of the inner ear showing three turns and expression of Np55 and Np65 in the SGNs (black arrowheads). H–L, Expression of Np55 in the organ of Corti and in hair cells (yellow dotted lines). M–Q, hair cells close up. R–V, Np55 is also expressed in the vestibular hair cells (white arrowheads). R'–V', Close-up of the vestibular sensory epithelium. Note that the Nptn 55/65 probe revealed expression in hair cells and support cells (R'). Scale bars: C–G, 200 μm; H–L, 50 μm; M–Q, 20 μm; R–V, 100 μm; R'–V', 50 μm.
To further analyze the expression pattern of Nptn in the inner ear, we generated a probe for in situ hybridization that detects both the Nptn55 and Nptn65 transcripts as well as a probe that recognizes the Nptn65-specific exon (Fig. 4A). As a control, we used a probe specific for Loxhd1, which is expressed in IHCs and OHCs (Grillet et al., 2009; Fig. 4G,L,Q). At P4, the Np55/65 probe detected strong expression in SGNs as well as the organ of Corti, where both hair and support cells were positive for Np55/65 transcripts (Fig. 4C,H,M). The Np65-specific probe detected weak expression in SGNs but no clear signal was observed in hair cells or support cells of the organ of Corti (Fig. 4E,J,O). In the vestibule, hair cells and support cells were positive with the Np55/65 probe, but not with the Np65-specific probe (Fig. 4R,R',T,T'). No signal was observed in hair cells, support cells, or SGNs with sense control probes (Fig. 4D,I,N,S,S',F,K,P,U,U'). These data agree with our analysis of gene expression by RT-PCR and suggest that Np55 is the predominant Nptn isoform in hair cells, while both Np55 and Np65 appear to be expressed in SGNs.
Np55 is expressed in SGNs and in the stereocilia of OHCs but not IHCs
We next analyzed the expression pattern of Nptn at the protein level using an antibody that specifically recognizes both the Np55 and Np65 isoforms. The specificity of the antibody was confirmed by expressing Np55 and Np65 in heterologous cells followed by immunocytochemistry and Western blotting (data not shown). In histological sections of animals at P7, we observed expression of Np55/65 at the cell surface of SGNs (Fig. 5A). In whole-mount staining of the organ of Corti at P7, we observed strong expression of Np55/65 in the stereocilia of OHCs but not IHCs and weak expression along the basolateral membrane (Fig. 5B,C; data not shown). Expression was also observed in the stereocilia of vestibular hair cells (Fig. 5D). No Np55/65 signal was detected in homozygous audio-1 mice (Fig. 5C, middle row). Staining appeared to be present in all rows of stereocilia and was most prominent toward the distal ends of stereocilia with low or undetectable signals toward their base (Fig. 5C, top row). Similar observations were made by immunogold staining of OHCs at P7, which revealed Np55/65 expression at the cell membrane surrounding the stereocilia of OHCs but not IHCs (Fig. 5E). A similar expression pattern in stereocilia was observed at P60 (Fig. 5F), indicating that Np55/65 expression was maintained into adulthood.
Figure 5.

Np55 is expressed in SGNs and in the stereocilia of OHCs but not IHCs. A, Sagittal inner ear sections of SGNs from wild-type (WT) mice at P7 were stained for Np55/65 (red) and Tuj1 (green). Note the localization of Np55/65 at the cell surface of SGN cell bodies. B, Cochlear whole mounts from C57BL/6 mice at P7 were stained for Np55/65 (red) and phalloidin (green). Note the localization of Np55/65 only in stereocilia OHCs but not IHCs. C, Higher-magnification view of hair cells showing Np55/65 immunoreactivity near the distal ends of stereocilia (top). No staining was observed in homozygous audio-1 mutant OHCs (middle) and IHCs of WT (bottom). D, Expression of Nptn in vestibular hair cells. E, F, Immunogold localization of Nptn in stereocilia at P7 and P60. Nptn was distributed along the length of stereocilia but with higher density toward their upper part and less staining in their lower part. Note IHC shows negative Nptn immune-reactivity. Scale bars: A, 10 μm; B, 2 μm; C, D, 10 μm; E, 500 nm.
We also evaluated Nptn expression with a commercial antibody to Np65 that has recently been used to evaluate Np65 expression in the inner ear (Carrott et al., 2016).
Unfortunately, the antibody gave a positive signal in hair cells not only in wild-type mice but also in Nptn knock-out animals (data not shown), confirming that the staining signal in hair cells was nonspecific. However, staining appeared reduced in SGNs from knock-out animals, indicating that in neurons it specifically detected Np65 (data not shown). Similar results were obtained with several other commercial antibodies to Np65 (data not shown).
Unfortunately, it is not possible to generate Np55-specific antibodies because all epitopes of Np55 are contained within Np65. Because Np55/65 is also expressed in vestibular hair cells, we examined NPTN in purified hair bundles and whole epithelium of mouse utricles using protein mass spectrometry (Krey et al., 2015). NPTN accounts for ∼0.2% of the total bundle protein at P5 and P23, and is enriched 50-fold and 20-fold at those time points compared with its concentration in whole epithelium (Fig. 6A).
Figure 6.

NPTN in purified vestibular hair bundles. A, Relative molar abundance of NPTN (summed isoforms) from purified hair bundles and whole epithelium from mouse utricles. NPTN is highly enriched in bundles. B, Protein immunoblotting of purified bundles using Np55/Np65 antibody. BUN, 18 ear-equivalents of P21–P24 C57BL/6 mouse utricle hair bundles; Agar, equivalent amount of agarose in saline; UTR, two P22 mouse utricles; Brain, 2 μg of C57BL/6 mouse brain extract. While the Np55 and Np65 isoforms are clearly identified in brain, whole mouse utricle has a complex isoform pattern. Bundles predominantly contain an NPTN isoform that migrates between Np55 and Np65. C, D, Peptide mapping of NPTN isoforms from utricle bundles and epithelium, using pooled P5 and P23 peptide identifications. The frequency of peptide identification is plotted as a function of the position of the peptide in the amino acid sequence. The exon structures of the Np55 and Np65 isoforms are indicated. No peptides in the Np65-specific exon (#2) were detected in bundles (C) or epithelium (D), despite frequent detection in other experiments.
Protein immunoblotting of purified hair bundles predominantly identified a single band, which ran in SDS-PAGE more slowly than brain Np55 but faster than brain Np65 (Fig. 6B). The NPTN band pattern in the whole utricle was more complex, with the majority of the immunoreactive protein again migrating between Np55 and Np65 (Fig. 6B). We also used peptide mapping to determine which NPTN isoforms were identified in isolated hair bundles (Fig. 6C,D). Most peptides identified in bundles mapped to exons 3, 4, and 5, which are shared between Np55 and Np65. In addition, one peptide was identified (KRPDEVPDDDEPAGPMK) that spans exons 6, 7, and 8. No peptides mapped to exon 1, but examination of the GPMdb database (http://gpmdb.thegpm.org) indicates that the large tryptic peptides predicted to span either exons 1 and 2 or exons 1 and 3 are very rarely detected. Finally, despite their frequent occurrence in the GPMdb, no peptides from bundles (Fig. 6C) or epithelium (Fig. 6D) mapped to exon 2, the Np65-specific exon.
Together, the in situ hybridization data and biochemical analysis show that vestibular hair bundles express an NPTN isoform that is likely the Np55 amino acid sequence with a glycosylation pattern that distinguishes it from brain Np55. There is no evidence that vestibular bundles possess significant amounts of an NPTN isoform with exon 2, specific to Np65.
Trafficking of Np55 to hair cell stereocilia of OHCs but not IHCs
Our in situ hybridization data suggested that Np55 is expressed in IHCs and OHCs, yet we observed protein expression only in the stereocilia of OHCs. To further confirm the immunolocalization data, we used our recently described injectoporation procedure (Xiong et al., 2014) to express Np55 modified to carry an HA tag at its C terminus in hair cells. In brief, the organ of Corti was dissected from mice at P3 and a plasmid expression vector for Np55-HA was injected into sensory epithelia followed by a brief electric pulse (Fig. 7A). The sensory epithelia were then cultured for 2 days. Np55-HA expression was monitored by staining with antibodies to HA. F-actin in hair cell stereocilia was visualized with phalloidin-FITC. Consistent with our immunolocalization data, Np55-HA was targeted to the stereocilia of OHCs with weak expression along the plasma membrane surrounding the cell body (Fig. 7B,C). Expression of Np55-HA was also observed in the cell bodies of IHCs, but the protein was not transported into the stereocilia of IHCs (Fig. 7E). An Np55-HA construct carrying the audio-1 mutation also failed to be transported to the stereocilia of OHCs (Fig. 7D). Our data thus lead to the surprising finding that OHCs but not IHCs shuttle Np55 into stereocilia, suggesting differences in the transport machinery for some proteins between the two hair cell types. The audio-1 mutation affects protein transport, although the predominant effect of the audio-1 mutation is likely to destabilize the protein because the Np55 and Np65 protein could no longer be detected in Nptn-deficient animals (Fig. 3E).
Figure 7.

Trafficking of Np55 to hair cell stereocilia of OHCs but not IHCs. A, Graphical representation of the injectoporation setup used for the transfection of hair cells of the organ of Corti. The HA-tagged Np55 plasmid for delivering (left), as well as the intercellular space for plasmid injection (right), are indicated. B, Organs of Corti from P3 mice were injectoporated with Np55-HA constructs and stained with phalloidin (Phal, F-actin) and anti-HA antibody after 24 h in vitro. C–E, Higher-magnification view of hair cells overexpressed with Np55-WT or audio-1 mutant (Np55-mut). Note the expression of Np55-HA in stereocilia of OHCs (D), not in IHCs (E). Np55-HA construct carrying the audio-1 mutation failed to be transported to the stereocilia of OHCs (D). Scale bars: B, 10 μm; C–E, 5 μm. Number of transfected cells: C, n = 36; D, n = 16; E, n = 12.
Hair cell morphology, survival, and mechanotransduction
To define the mechanism by which mutations in Nptn cause deafness, we analyzed the morphology and maintenance of hair cells by scanning electron microscopy. We did not observe obvious loss of hair bundles between P14 and P40 along the entire length of the cochlear duct in hair cells from audio-1 mutant mice and from Nptn knock-outs (Fig. 8A,B; data not shown). To obtain quantitative data, we determined the number of hair cells with hair bundles at P60 in the medial-apical part of the cochlea, where tissue was best preserved during sample preparation. Hair cell numbers appeared unaffected in audio-1 mutant mice (Fig. 8D), although hair bundle morphology appeared altered in some hair cells. This was more obvious at P120, when scanning electron microscopy analysis revealed not only a loss of hair bundles in nearly 25% of all OHCs (Fig. 8C,D), but also clear structural abnormalities in the remaining hair bundles (Fig. 8C), indicating that they were in the process of degeneration. The loss of hair bundles was likely not the primary cause of deafness in audio-1 mutant mice because hearing loss was already obvious at P21, which is before mutant animals experience a significant loss of hair bundles.
Figure 8.
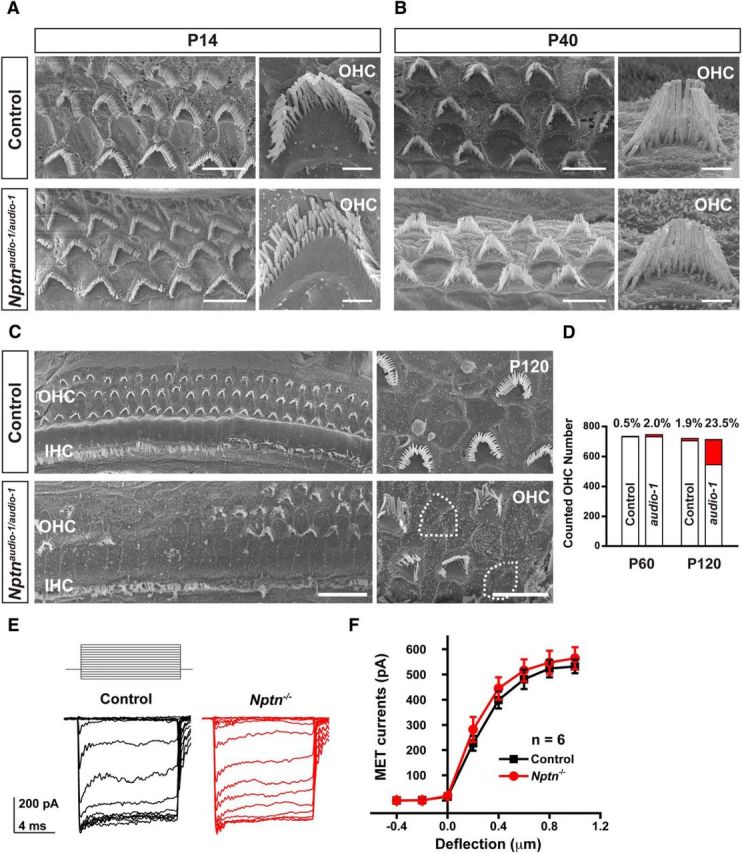
Hair cell survival and mechanotransduction. A, B, Scanning electron microscopy analysis of hair bundles from wild-type (control) and homozygous audio-1 mutant mice in the midapical cochlea at P14 and P40. Hair bundles from OHCs at higher magnification are shown. C, Severe degeneration of the OHCs was evident at P120 in homozygous audio-1 mutant mice. D, The average number of OHCs per sample of the entire organ of Corti was calculated at P60 and P120. Cell losses in each group are marked as red. Number of cell loss/survival from each group: 4/730 (P60, Control), 15/730 (P60, audio-1), 14/706 (P120, Control), and 167/545 (P120, audio-1). E, Traces of transduction currents in OHCs from wild-type (control) and Nptn−/− mice at P7 in response to a set of 10 ms hair bundle deflections ranging from −400 to 1000 nm (100 nm steps) at a holding potential of −70 mV. F, Current displacement plots obtained from similar data as shown in E. All values are mean ± SEM by Student's t test.
Next we analyzed the extent to which mechanotransduction was affected in Nptn-deficient OHCs. We stimulated hair bundles from P7 mice with a stiff glass probe and recorded mechanotransduction currents in the whole-cell configuration. As previously reported, control OHCs had rapidly activating transducer currents, which were subsequently adapted (Xiong et al., 2012; Fig. 8E). There was no significant difference in mechanically evoked currents between wild-type and Nptn-deficient hair cells (Fig. 8E,F). The amplitude of saturated mechanotransduction currents at maximal deflection was at 532 ± 27 pA (mean ± SEM) for wild-type OHCs and 564 ± 44 pA (mean ± SEM) for OHCs from knock-out mice (Fig. 8F). We thus conclude that the deafness phenotype in Nptn-deficient mice is likely not caused by defects in mechanotransduction.
In vivo vibrometry reveals defects in cochlear amplification
Through deflection of their stereociliary bundle, OHCs detect the mechanical vibrations of the cochlear traveling wave. This stimulates the OHCs to produce forces that amplify and sharpen the frequency tuning of the vibrations of the organ of Corti (Xia et al., 2013; Gao et al., 2014; Song et al., 2015). To assess the intracochlear anatomy and the function of the cochlear amplifier, we used VOCTV to study Nptn knock-out mice and wild-type littermates in vivo at P21. We first imaged the cochlea approximately one-half of a turn down from the apical end (Fig. 9A,B). While the anatomic imaging provided by VOCTV has limited resolution, the intracochlear tissues appeared normal in Nptn-knock-out mice.
Figure 9.

VOCTV reveals absence of cochlear amplification. A, B, In vivo cochlear cross-sectional images from the apical cochlear turn of control (Nptn+/+) and Nptn−/− mice revealed no gross anatomical differences in the TM and its relationship to the BM and the intervening hair cell epithelium. Also, Reissner's membrane (RM) appeared normal in Nptn−/− mice. C, D, Sound-evoked BM and TM vibrations in representative control and Nptn−/− mice. Thicker lines were used to represent the response to higher stimulus intensities. E, BM sensitivity was calculated by normalizing BM displacement to middle ear displacement. In control mice, BM sensitivity increased and tuning became sharper with decreasing stimulus level, a hallmark of cochlear amplification. For Nptn−/− mice, BM sensitivity was broad and did not change with stimulus level, indicating a lack of cochlear amplification. F, Measurements of cochlear gain at both the BM and TM were normal in control mice but absent in Nptn−/− mice (p = 0.02 for both comparisons). G, The phase of the TM relative to the BM was not significantly different between Nptn−/− and control mice (p > 0.7 at both 5 and 9 kHz, the frequencies of largest vibration to low-intensity stimuli in Nptn−/− and control mice; gray arrows). All values are mean ± SEM.
We then selected measurement points on the images corresponding to the midpoint of the BM and the tip of the TM, and recorded vibrations in response to sound stimuli. The frequency of maximal vibration of the BM to 10 dB SPL stimuli (i.e., the characteristic frequency) in wild-type mice was ∼9 kHz. At this frequency, both BM and TM vibrations in wild-type mice were larger than those in Nptn knock-out mice (Fig. 9C,D). In particular, while wild-type mice demonstrated a compressive nonlinear amplification centered at this frequency, Nptn-knock-out mice had purely linear responses centered at ∼4 kHz, identical to the passive responses seen in the wild-type mice after death (data not shown). The phases of BM and TM vibration in both genotypes were similar, demonstrating a progressive phase lag as the frequency was increased. This is consistent with traveling wave propagation (Xia et al., 2007).
Cochlear amplification can be appreciated more clearly by normalizing the magnitude of BM and TM vibrations to those of the middle ear, yielding a sensitivity ratio. Nonlinear amplification in wild-type mice is evident as increasing BM sensitivity with decreasing stimulus levels (Fig. 9E,F). In contrast, there was no amplification in Nptn knock-out mice because BM sensitivity did not change with stimulus level. We calculated the gain of amplification by dividing the peak sensitivity of a quiet stimulus (20 dB SPL for wild type; 40 dB SPL for Nptn knock-outs) by the peak sensitivity of the loudest stimulus (80 dB SPL for both). In the wild-type mice, the frequency of peak sensitivity shifted from ∼4 to ∼9 kHz as the stimulus intensity was reduced, and the cochlear gains measured at the TM were consistent with previously published data in CBA mice (Lee et al., 2015). In contrast, the frequency of peak sensitivity was ∼4 kHz regardless of the stimulus intensity and there was no evidence of cochlear gain measured at either the BM or the TM (Fig. 9E,F).
We also examined the phase of TM motion relative to that of the BM in the two genotypes (Fig. 9G). Over the frequencies tested, the vibrational pattern was similar for control and Nptn knock-out mice. There was in-phase motion of the TM and BM at lower frequencies (<6 kHz) with a gradually increasing phase lag at higher frequencies. For quantification, we compared the phase difference between the TM and BM at 4 and 9 kHz, and there were no significant differences between the genotypes. Together these data indicate that while the TM is near the OHC stereociliary bundles, OHCs in Nptn knock-outs do not produce force and thus there is no cochlear amplification.
Otoacoustic emissions and hair cell electromotility
VOCTV measurements demonstrated that cochlear amplification was lacking in Nptn-deficient mice, indicating that OHC function was affected. To confirm this, we recorded DPOAEs, which are mechanical distortions generated in the inner ear when two primary tones (f1 and f2) are presented. OHCs amplify the distortions, which then propagate back through the middle ear and ear canal and can be measured as sounds emitted by the ear (Ashmore, 2008). Consistent with the lack of cochlear amplification, DPOAEs were absent in audio-1 homozygous mice at P21 and P60 at all frequencies tested (Fig. 10A,B). Similar observations were made in Nptn knock-out mice (Fig. 10C). As these emissions depend on the mechanical activity of OHCs, the deafness in audio-1 mice must be due, at least in part, to defects in OHC function.
Figure 10.
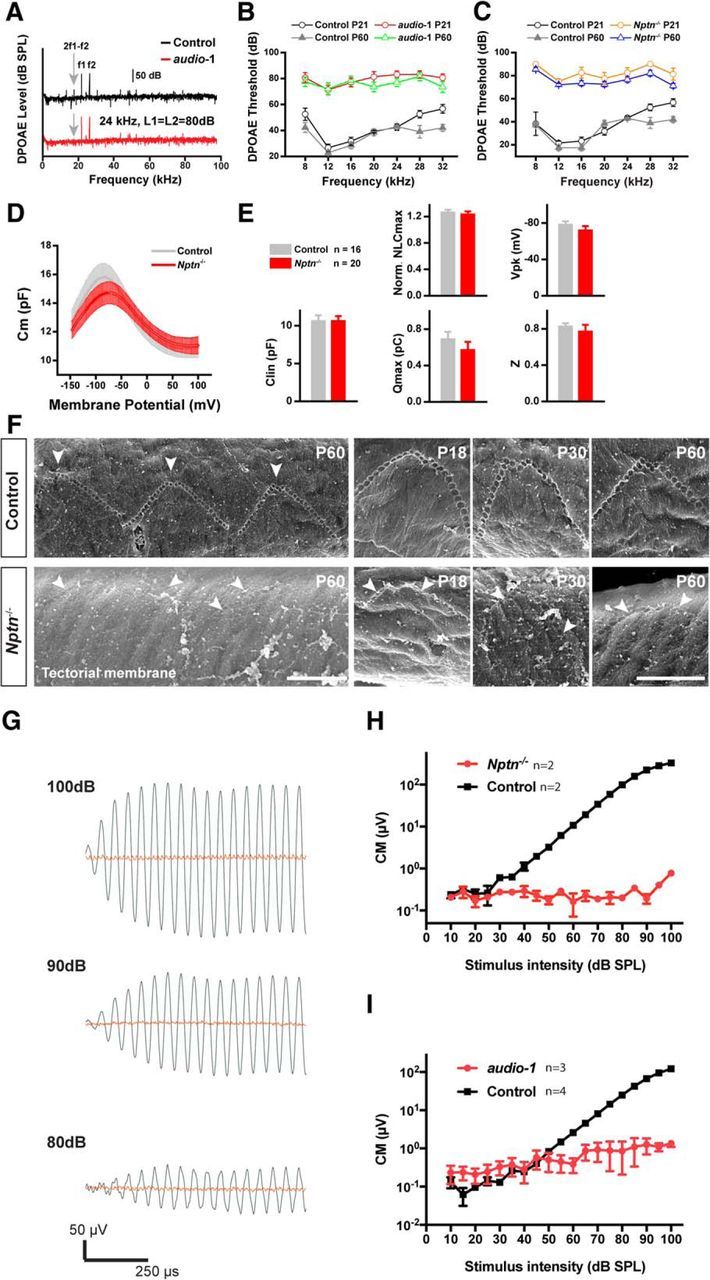
OHC amplification, electromotility, and coupling of OHC stereocilia to the TM. A, Representative DPOAE response spectra from wild-type (control) and homozygous audio-1 mutant mice at a single stimulus condition. Note the 2f1–f2 peak (gray arrow), which is absent in mutant mice. B, DPOAE thresholds at different frequencies at P21 and P60 of wild-type (control) and homozygous audio-1 mutant mice. n = 15 mice for each group. C, DPOAE thresholds at different frequencies at P21 and P60 of wild-type (control) and Nptn−/− mice. n = 15 mice for each group. D, NLC recorded from wild-type (control) and Nptn−/− mice at P21. E, Parameters of NLC fitting. Normalized NLC peak (Norm. NLCmax) was performed using the peak value of NLC normalized with the base line. Clin, Linear capacitance. F, Scanning electron micrographs of V-shaped TM imprints from wild-type (control) and Nptn−/− mice of different ages. These imprints (arrows) are formed by the notches remaining after the tallest row of OHC stereocilia was pulled away from the TM. Such imprints are not found or are disorganized in the TM of Nptn−/− mice. Scale bars, 2 μm. All values are mean ± SEM. ***p < 0.001, by two-way ANOVA in B–D and Student's t test in E. G–I, CM. Representative CM recordings measured from a P28 Nptn−/− mouse (in red) and a WT littermate (in black) in response to a 6 kHz stimulus at 80, 90, and 100 dB SPL. The peak-to-peak amplitude of the CM versus sound intensity measured in P28 Nptn−/− mice (n = 2) and WT littermates (n = 2) in H, and measured in P30 audio-1 mice (n = 3) and littermate WT (n = 4) in I.
Np55 was prominently expressed in the stereocilia of OHCs (Figs. 5–67) but also at lower levels in the plasma membrane of their cell bodies (data not shown). Amplification by OHCs depends on electromotility, length changes of the cell bodies of OHCs. Electromotility is accompanied by a voltage-dependent gating charge movement within the lateral hair cell membrane, manifesting as NLC in OHCs (Fettiplace and Hackney, 2006; Ashmore, 2008). Measurements of the NLC did not reveal any significant difference between OHCs from wild-type and Nptn knock-out mice (Fig. 10D,E). We thus conclude that defects in electromotility likely did not cause the observed defects in amplification in Nptn-deficient mice.
Defect in the coupling of OHC stereocilia to the TM
The strong expression of Np55 toward the upper part of OHC stereocilia suggested that Np55 might couple stereocilia to the TM. We therefore wondered whether defects in amplification in Nptn-deficient mice might be caused by perturbations in the coupling of the stereocilia of OHCs to the TM. Previous studies have shown that the longest stereocilia of OHCs are tightly connected to the TM, forming indentations that correspond to these anchoring points (Tsuprun and Santi, 1998). In wild-type mice, scanning electron microscopy analysis revealed these indentations on the lower surface of the microdissected TM with the characteristic V-shape of the hair bundles of OHCs (Fig. 10F). These indentations were prominently visible in the TM from wild-type mice already by P18 and were maintained into adulthood (Fig. 10F). In contrast, indentations in Nptn knock-out mice were nearly absent along the entire length of the cochlear duct at P18, the first time point analyzed (Fig. 10F). Small remnants of the indentations could occasionally be observed (Fig. 10F, bottom, arrows), but they were much less well defined in the mutant animals compared with wild type. We therefore conclude that Np55 is critical for stably linking the stereocilia of OHCs to the TM. In the absence of efficient mechanical coupling between OHCs and the TM, OHC stimulation is inadequate, thus affecting cochlear amplification and normal hearing.
Defects in cochlear microphonics
The lack of DPOAEs, the lack of ABRs, and the lack of cochlear amplification in mutant mice suggest a lack of OHC function. Given the localization of NPTN protein to the stereociliary bundles of the OHCs, we sought to assess the ability of OHCs to be stimulated in vivo. We therefore recorded CM potentials in P28–P30 animals from the round window, which measures the flow of transducer currents across the cochlear partition in the basal turn of the cochlea (Dallos, 1975; Patuzzi et al., 1989; Cheatham et al., 2011). Control littermates from both mutant strains, Nptn−/− and audio-1, demonstrated normal responses, although the magnitude of the response differed between the two lines, likely because they were on different genetic backgrounds (Fig. 10G–I). Audio-1 mice were on a C57BL/6J background and we used as controls littermates that were also on a pure C57BL/6J background. In contrast, Nptn−/− mutants were originally generated on a C57BL/6N background but intercrossed both with C57BL/6N and CBA/J mice; as controls for Nptn−/− mice we therefore used littermates with a similarly mixed genetic background (Fig. 10G–I). Indeed previous reports have demonstrated effects of genetic background on CM (Henry and Lepkowski, 1978; Henry, 1984). Importantly, we could not detect CM potentials above the noise floor in either Nptn−/− and audio-1 homozygous mutants, indicating the lack of transduction currents. This finding argues that the Nptn mutations either affects the ability of the TM to deflect OHC stereociliary bundles or that it affects the ability of bundle deflection to elicit mechanoelectrical transduction currents at least in the basal part of the cochlea, where our measurements were performed.
Discussion
OHCs are critical for cochlear amplification, a process responsible for the remarkable sensitivity and sharp frequency tuning of the cochlea (Fettiplace and Hackney, 2006; Schwander et al., 2010). Cochlear amplification is driven by somatic electromotility and hair bundle motility (Fettiplace and Hackney, 2006; Ashmore, 2008) and requires mechanical coupling of OHCs to the TM to feed back motion into the organ of Corti. The mechanisms that mediate this coupling between OHCs and the TM are not well understood. We now show that Np55 is expressed in the stereocilia of OHCs but not IHCs and is essential for mediating stable interactions between the stereocilia with the TM, thus allowing for proper sound amplification. Intriguingly, Np55 is specifically transported in the stereocilia of OHCs but not IHCs, leading to the unexpected finding that the transport machinery of OHCs and IHCs are not identical. While mechanotransduction currents could still be recorded at early postnatal ages, measurements of the CM indicated an absence of transducer currents in the basal part of the cochlea of adult mice. This could be a consequence of degenerative changes in hair cells, such as disruption in the normal morphology of the hair bundle. Our findings thus identify Np55 as an essential component of the molecular machinery that stably couples OHCs to the TM and suggests that mutations in NPTN might lead to hearing impairment not only in mice but also in humans.
Previous studies have shown that coupling of the TM to OHC stereocilia is defective in mice with mutations in stereocilin. Stereocilin immunoreactivity is observed in association with the tips of OHC stereocilia and with horizontal top connectors that link the stereocilia to each other (Verpy et al., 2011). Stereocilin is also expressed in the kinocilium of hair cells, a structural specialization of hair cells that is present only during the development of cochlear hair cells (Verpy et al., 2008, 2011). Surprisingly, stereocilin lacks an obvious transmembrane domain and can be detected within the TM, suggesting that it is a secreted molecule that is associated but not tightly embedded into the plasma membrane of OHCs, although it might have a GPI anchor (Jovine et al., 2002). Other molecules are likely critical to contribute to the coupling of OHCs to the TM to resist mechanical forces during sound stimulation and amplification. Our findings suggest that Np55 in the stereocilia of OHC is critical for mechanical coupling. RT-PCR data show that Np55 is more prominently expressed in the inner ear than Np65 and our in situ hybridization data detect Np55 but not Np65 in hair cells. Immunolocalization studies with an antibody that detects Np55 and Np65 stained the sterocilia of OHCs. Unfortunately, all epitopes of Np55 are contained in Np65, thus preventing the specific analysis of Np55 expression by immunohistochemistry. For an independent confirmation, we therefore analyzed Np55/65 expression by tandem mass spectrometry. Peptides representing Np55 were prominently present in the proteome of hair bundles but peptides for Np65 were absent. Np65 peptides were readily recovered following immunoprecipitation of Np55/65 from brain extracts, indicating that Np65-specific peptides can be detected by tandem mass spectroscopy. As one caveat, we cannot exclude the possibility that the N terminus of Np65 might carry an inner ear-specific modification that prevents its detection. However, in combination with our RNA expression data, it seems likely that Np55 is the predominant Nptn isoform in the stereocilia of OHCs.
The mechanism by which Np55 contributes to establishing a stable mechanical link of OHC stereocilia with the TM remains to be determined. Np65 has been shown to act as a homophilic cell-adhesion molecule but no ligands have so far been identified for Np55 (Beesley et al., 2014). The first Ig domain of Np65 that is absent in Np55 is required for homophilic binding of Np65 and it is tempting to speculate that the alternative-splicing event unmasks a new binding surface at the N terminus of Np55, which most likely binds to an extracellular matrix ligand within the TM. The TM consists of several ECM proteins, including collagens, tectorin-α, tectorin-β, CEACAM16, and otogelin, and stereocilin also appears to be deposited into the TM (Richardson et al., 2008, 2011). We failed to identify convincing interactions between Np55 and stereocilin following their expression in heterologous cells. Our initial attempts to express other components of the TM in heterologous cells have so far also failed since the proteins tend to form intracellular aggregates. Thus, further studies will be necessary to identify the putative Np55 ligand within the TM.
We were surprised that Np55 is transported into the stereocilia of OHCs but not IHCs even after ectopic overexpression of Np55 in hair cells. These findings suggest molecular differences in the composition of the protein transport machinery in hair cells. Stereocilia are densely packed with actin filaments (Gillespie and Müller, 2009; Schwander et al., 2010) and no transport vesicles for membrane proteins have been observed in stereocilia. Cell-surface receptors, such as Np55, must therefore be transported into stereocilia following their insertion into the plasma membrane near the cell body. Myosin motor proteins are thought to be instrumental in protein transport (Nambiar et al., 2010) but no motor protein has so far been shown to be differentially expressed between OHCs and IHCs. Perhaps, OHCs and IHCs express distinct adaptor proteins that link cargo molecules to motor proteins. Current efforts in our laboratory are directed toward the identification of interaction partners for Np55 that might be critical for the transport of Np55, and possibly of other cargo proteins, into the stereocilia of OHCs.
OHCs amplify input sound signals while IHCs transmit sound information via afferent neurons to the CNS. Defects in OHCs are expected to lead to a decrease in hearing sensitivity but not necessarily to complete deafness. Notably, Nptn-deficient mice are completely deaf, suggesting that Nptn might have additional functions in the auditory system. We observed that cochlear microphonics was nearly absent in Nptn-deficient mice. This indicates that the flow of transducer current across the cochlear partition was affected at least in the basal part of the cochlea where our recordings were performed.
This does not necessarily mean that the transduction channel is dysfunctional after P7, particularly given our patch-clamp results, which reveal normal transduction ex vivo at P7. Perhaps, changes in transduction were a secondary consequence of degenerative changes in the hair bundle that manifested in a slow progressive form in adult mice, which could affect tip links and the transduction complex of hair cells secondarily. In addition, it is currently not entirely clear how defects in the interactions of stereocilia with the TM regulate signal transmission to the transduction machinery by affecting direct coupling and fluid flow within the cochlea. In vitro experiments highlight the complex response of hair bundles to different stimulation protocols. For example, different forms of bundle deflection ex vivo elicit key aspects of transduction in distinct ways (i.e., the differences in adaptation found in stiff probe vs fluid jet stimulation; Peng et al., 2013; Corns et al., 2014). The “natural” way that the TM stimulates the bundle in vivo is not yet known, and may be altered in the absence of Nptn. While we cannot exclude a more direct role for NPTN in the mechanotransduction process in OHCs, we favor the hypothesis that in the mutant mice BM vibrations fail to produce transducer currents because of a failure of BM and TM movements to stimulate OHC sterociliary bundles appropriately. This would be significantly different from results obtained in TectA mutant mice, where connections between OHCs and the TM are also perturbed but cochlear microphonics, albeit with changes in phase and symmetry, can still be recorded (Legan et al., 2000). Nevertheless, further studies will be necessary to address more directly potential effects of Np55 on the transduction machinery in adult hair cells.
Importantly, our in situ hybridization data suggest that SGN express Nptn, including the Np65 isoform, which has been implicated in the regulation of synaptic plasticity in the hippocampus (Smalla et al., 2000; Empson et al., 2006; Owczarek et al., 2011). It is therefore tempting to speculate that Nptn might also regulate synaptic transmission between ribbon synapses and SGNs. In fact, a recent study reported defects in synaptic transmission at ribbon synapses in an Nptn mutant mouse generated in an independent ENU mutagenesis screen (Carrott et al., 2016). However, the defects in synaptic function were relatively small and alone can likely not explain the deafness phenotype associated with mutations in Nptn. Deafness caused by mutations in Nptn is thus likely a consequence of perturbations of several processes in the inner ear, including the function of hair cells and SGNs.
Footnotes
This work was supported by National Institute on Deafness and Other Communication Disorders Grants DC005965, DC007704, and DC014713 (U.M.); DC014450, DC013774, and DC010363 (J.S.O.); the Dorris Neuroscience Center; the Skaggs Institute for Chemical Biology (U.M.); the Dean's Postdoctoral Fellowship at the Stanford University School of Medicine (J.B.D.); and a fellowship from the George E. Hewitt Foundation for Medical Research (W.-Z.Z.).
The authors declare no competing financial interests.
References
- Ashmore J. Cochlear outer hair cell motility. Physiol Rev. 2008;88:173–210. doi: 10.1152/physrev.00044.2006. [DOI] [PubMed] [Google Scholar]
- Beesley PW, Herrera-Molina R, Smalla KH, Seidenbecher C. The neuroplastin adhesion molecules: key regulators of neuronal plasticity and synaptic function. J Neurochem. 2014;131:268–283. doi: 10.1111/jnc.12816. [DOI] [PubMed] [Google Scholar]
- Carrott L, Bowl MR, Aguilar C, Johnson SL, Chessum L, West M, Morse S, Dorning J, Smart E, Hardisty-Hughes R, Ball G, Parker A, Barnard AR, MacLaren RE, Wells S, Marcotti W, Brown SD. Absence of neuroplastin-65 affects synaptogenesis in mouse inner hair cells and causes profound hearing loss. J Neurosci. 2016;36:222–234. doi: 10.1523/JNEUROSCI.1808-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheatham MA, Naik K, Dallos P. Using the cochlear microphonic as a tool to evaluate cochlear function in mouse models of hearing. J Assoc Res Otolaryngol. 2011;12:113–125. doi: 10.1007/s10162-010-0240-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corns LF, Johnson SL, Kros CJ, Marcotti W. Calcium entry into stereocilia drives adaptation of the mechanoelectrical transducer current of mammalian cochlear hair cells. Proc Natl Acad Sci U S A. 2014;111:14918–14923. doi: 10.1073/pnas.1409920111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallos P. Electrical correlates of mechanical events in the cochlea. Audiology. 1975;14:408–418. doi: 10.3109/00206097509071753. [DOI] [PubMed] [Google Scholar]
- Dumont RA, Zhao YD, Holt JR, Bähler M, Gillespie PG. Myosin-I isozymes in neonatal rodent auditory and vestibular epithelia. J Assoc Res Otolaryngol. 2002;3:375–389. doi: 10.1007/s101620020049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Empson RM, Buckby LE, Kraus M, Bates KJ, Crompton MR, Gundelfinger ED, Beesley PW. The cell adhesion molecule neuroplastin-65 inhibits hippocampal long-term potentiation via a mitogen-activated protein kinase p38-dependent reduction in surface expression of GluR1-containing glutamate receptors. J Neurochem. 2006;99:850–860. doi: 10.1111/j.1471-4159.2006.04123.x. [DOI] [PubMed] [Google Scholar]
- Fettiplace R, Hackney CM. The sensory and motor roles of auditory hair cells. Nat Rev Neurosci. 2006;7:19–29. doi: 10.1038/nrn1828. [DOI] [PubMed] [Google Scholar]
- Francis SP, Krey JF, Krystofiak ES, Cui R, Nanda S, Xu W, Kachar B, Barr-Gillespie PG, Shin JB. A short splice form of Xin-actin binding repeat containing 2 (XIRP2) lacking the Xin repeats is required for maintenance of stereocilia morphology and hearing function. J Neurosci. 2015;35:1999–2014. doi: 10.1523/JNEUROSCI.3449-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao SS, Wang R, Raphael PD, Moayedi Y, Groves AK, Zuo J, Applegate BE, Oghalai JS. Vibration of the organ of Corti within the cochlear apex in mice. J Neurophysiol. 2014;112:1192–1204. doi: 10.1152/jn.00306.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie PG, Hudspeth AJ. High-purity isolation of bullfrog hair bundles and subcellular and topological localization of constituent proteins. J Cell Biol. 1991;112:625–640. doi: 10.1083/jcb.112.4.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie PG, Müller U. Mechanotransduction by hair cells: models, molecules, and mechanisms. Cell. 2009;139:33–44. doi: 10.1016/j.cell.2009.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillet N, Schwander M, Hildebrand MS, Sczaniecka A, Kolatkar A, Velasco J, Webster JA, Kahrizi K, Najmabadi H, Kimberling WJ, Stephan D, Bahlo M, Wiltshire T, Tarantino LM, Kuhn P, Smith RJ, Müller U. Mutations in LOXHD1, an evolutionarily conserved stereociliary protein, disrupt hair cell function in mice and cause progressive hearing loss in humans. Am J Hum Genet. 2009;85:328–337. doi: 10.1016/j.ajhg.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guinan JJ., Jr How are inner hair cells stimulated? Evidence for multiple mechanical drives. Hear Res. 2012;292:35–50. doi: 10.1016/j.heares.2012.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry KR. Noise and the young mouse: genotype modifies the sensitive period for effects on cochlear physiology and audiogenic seizures. Behav Neurosci. 1984;98:1073–1082. doi: 10.1037/0735-7044.98.6.1073. [DOI] [PubMed] [Google Scholar]
- Henry KR, Lepkowski CM. Evoked potential correlates of genetic progressive hearing loss. Age-related changes from the ear to the inferior colliculus of C57BL/6 and CBA/J mice. Acta Otolaryngol. 1978;86:366–374. doi: 10.3109/00016487809107515. [DOI] [PubMed] [Google Scholar]
- Hudspeth A. Mechanical amplification of stimuli by hair cells. Curr Opin Neurobiol. 1997;7:480–486. doi: 10.1016/S0959-4388(97)80026-8. [DOI] [PubMed] [Google Scholar]
- Jovine L, Park J, Wassarman PM. Sequence similarity between stereocilin and otoancorin points to a unified mechanism for mechanotransduction in the mammalian inner ear. BMC Cell Biol. 2002;3:28. doi: 10.1186/1471-2121-3-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazmierczak P, Müller U. Sensing sound: molecules that orchestrate mechanotransduction by hair cells. Trends Neurosci. 2012;35:220–229. doi: 10.1016/j.tins.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krey JF, Sherman NE, Jeffery ED, Choi D, Barr-Gillespie PG. The proteome of mouse vestibular hair bundles over development. Sci Data. 2015;2:150047. doi: 10.1038/sdata.2015.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HY, Raphael PD, Park J, Ellerbee AK, Applegate BE, Oghalai JS. Noninvasive in vivo imaging reveals differences between tectorial membrane and basilar membrane traveling waves in the mouse cochlea. Proc Natl Acad Sci U S A. 2015;112:3128–3133. doi: 10.1073/pnas.1500038112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legan PK, Lukashkina VA, Goodyear RJ, Kössi M, Russell IJ, Richardson GP. A targeted deletion in alpha-tectorin reveals that the tectorial membrane is required for the gain and timing of cochlear feedback. Neuron. 2000;28:273–285. doi: 10.1016/S0896-6273(00)00102-1. [DOI] [PubMed] [Google Scholar]
- Nambiar R, McConnell RE, Tyska MJ. Myosin motor function: the ins and outs of actin-based membrane protrusions. Cell Mol Life Sci. 2010;67:1239–1254. doi: 10.1007/s00018-009-0254-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owczarek S, Kiryushko D, Larsen MH, Kastrup JS, Gajhede M, Sandi C, Berezin V, Bock E, Soroka V. Neuroplastin-55 binds to and signals through the fibroblast growth factor receptor. FASEB J. 2010;24:1139–1150. doi: 10.1096/fj.09-140509. [DOI] [PubMed] [Google Scholar]
- Owczarek S, Soroka V, Kiryushko D, Larsen MH, Yuan Q, Sandi C, Berezin V, Bock E. Neuroplastin-65 and a mimetic peptide derived from its homophilic binding site modulate neuritogenesis and neuronal plasticity. J Neurochem. 2011;117:984–994. doi: 10.1111/j.1471-4159.2011.07269.x. [DOI] [PubMed] [Google Scholar]
- Patuzzi RB, Yates GK, Johnstone BM. The origin of the low-frequency microphonic in the first cochlear turn of guinea-pig. Hear Res. 1989;39:177–188. doi: 10.1016/0378-5955(89)90089-0. [DOI] [PubMed] [Google Scholar]
- Peng AW, Effertz T, Ricci AJ. Adaptation of mammalian auditory hair cell mechanotransduction is independent of calcium entry. Neuron. 2013;80:960–972. doi: 10.1016/j.neuron.2013.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson GP, Lukashkin AN, Russell IJ. The tectorial membrane: one slice of a complex cochlear sandwich. Curr Opin Otolaryngol Head Neck Surg. 2008;16:458–464. doi: 10.1097/MOO.0b013e32830e20c4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson GP, de Monvel JB, Petit C. How the genetics of deafness illuminates auditory physiology. Annu Rev Physiol. 2011;73:311–334. doi: 10.1146/annurev-physiol-012110-142228. [DOI] [PubMed] [Google Scholar]
- Robles L, Ruggero MA. Mechanics of the mammalian cochlea. Physiol Rev. 2001;81:1305–1352. doi: 10.1152/physrev.2001.81.3.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwander M, Sczaniecka A, Grillet N, Bailey JS, Avenarius M, Najmabadi H, Steffy BM, Federe GC, Lagler EA, Banan R, Hice R, Grabowski-Boase L, Keithley EM, Ryan AF, Housley GD, Wiltshire T, Smith RJ, Tarantino LM, Müller U. A forward genetics screen in mice identifies recessive deafness traits and reveals that pejvakin is essential for outer hair cell function. J Neurosci. 2007;27:2163–2175. doi: 10.1523/JNEUROSCI.4975-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwander M, Kachar B, Müller U. Review series: the cell biology of hearing. J Cell Biol. 2010;190:9–20. doi: 10.1083/jcb.201001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senften M, Schwander M, Kazmierczak P, Lillo C, Shin JB, Hasson T, Géléoc GS, Gillespie PG, Williams D, Holt JR, Müller U. Physical and functional interaction between protocadherin 15 and myosin VIIa in mechanosensory hair cells. J Neurosci. 2006;26:2060–2071. doi: 10.1523/JNEUROSCI.4251-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JB, Longo-Guess CM, Gagnon LH, Saylor KW, Dumont RA, Spinelli KJ, Pagana JM, Wilmarth PA, David LL, Gillespie PG, Johnson KR. The R109H variant of fascin-2, a developmentally regulated actin crosslinker in hair-cell stereocilia, underlies early-onset hearing loss of DBA/2J mice. J Neurosci. 2010;30:9683–9694. doi: 10.1523/JNEUROSCI.1541-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JB, Krey JF, Hassan A, Metlagel Z, Tauscher AN, Pagana JM, Sherman NE, Jeffery ED, Spinelli KJ, Zhao H, Wilmarth PA, Choi D, David LL, Auer M, Barr-Gillespie PG. Molecular architecture of the chick vestibular hair bundle. Nat Neurosci. 2013;16:365–374. doi: 10.1038/nn.3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smalla KH, Matthies H, Langnäse K, Shabir S, Böckers TM, Wyneken U, Staak S, Krug M, Beesley PW, Gundelfinger ED. The synaptic glycoprotein neuroplastin is involved in long-term potentiation at hippocampal CA1 synapses. Proc Natl Acad Sci U S A. 2000;97:4327–4332. doi: 10.1073/pnas.080389297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Xia A, Lee HY, Wang R, Ricci AJ, Oghalai JS. Activity-dependent regulation of prestin expression in mouse outer hair cells. J Neurophysiol. 2015;113:3531–3542. doi: 10.1152/jn.00869.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuprun V, Santi P. Structure of outer hair cell stereocilia links in the chinchilla. J Neurocytol. 1998;27:517–528. doi: 10.1023/A:1006903926571. [DOI] [PubMed] [Google Scholar]
- Verpy E, Weil D, Leibovici M, Goodyear RJ, Hamard G, Houdon C, Lefèvre GM, Hardelin JP, Richardson GP, Avan P, Petit C. Stereocilin-deficient mice reveal the origin of cochlear waveform distortions. Nature. 2008;456:255–258. doi: 10.1038/nature07380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verpy E, Leibovici M, Michalski N, Goodyear RJ, Houdon C, Weil D, Richardson GP, Petit C. Stereocilin connects outer hair cell stereocilia to one another and to the tectorial membrane. J Comp Neurol. 2011;519:194–210. doi: 10.1002/cne.22509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia A, Visosky AM, Cho JH, Tsai MJ, Pereira FA, Oghalai JS. Altered traveling wave propagation and reduced endocochlear potential associated with cochlear dysplasia in the BETA2/NeuroD1 null mouse. J Assoc Res Otolaryngol. 2007;8:447–463. doi: 10.1007/s10162-007-0092-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia A, Gao SS, Yuan T, Osborn A, Bress A, Pfister M, Maricich SM, Pereira FA, Oghalai JS. Deficient forward transduction and enhanced reverse transduction in the alpha tectorin C1509G human hearing loss mutation. Dis Model Mech. 2010;3:209–223. doi: 10.1242/dmm.004135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia A, Song Y, Wang R, Gao SS, Clifton W, Raphael P, Chao SI, Pereira FA, Groves AK, Oghalai JS. Prestin regulation and function in residual outer hair cells after noise-induced hearing loss. PLoS One. 2013;8:e82602. doi: 10.1371/journal.pone.0082602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong W, Grillet N, Elledge HM, Wagner TF, Zhao B, Johnson KR, Kazmierczak P, Müller U. TMHS is an integral component of the mechanotransduction machinery of cochlear hair cells. Cell. 2012;151:1283–1295. doi: 10.1016/j.cell.2012.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong W, Wagner T, Yan L, Grillet N, Müller U. Using injectoporation to deliver genes to mechanosensory hair cells. Nat Protocols. 2014;9:2438–2449. doi: 10.1038/nprot.2014.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Liang C, Chen J, Zong L, Chen GD, Zhao HB. Active cochlear amplification is dependent on supporting cell gap junctions. Nat Commun. 2013;4:1786. doi: 10.1038/ncomms2806. [DOI] [PMC free article] [PubMed] [Google Scholar]