

Abstract
Background : Based on epidemiological commonalities, multiple sclerosis (MS) and Hodgkin lymphoma (HL), two clinically distinct conditions, have long been suspected to be aetiologically related. MS and HL occur in roughly the same age groups, both are associated with Epstein-Barr virus infection and ultraviolet (UV) light exposure, and they cluster mutually in families (though not in individuals). We speculated if in addition to sharing environmental risk factors, MS and HL were also genetically related. Using data from genome-wide association studies (GWAS) of 1816 HL patients, 9772 MS patients and 25 255 controls, we therefore investigated the genetic overlap between the two diseases.
Methods: From among a common denominator of 404 K single nucleotide polymorphisms (SNPs) studied, we identified SNPs and human leukocyte antigen (HLA) alleles independently associated with both diseases. Next, we assessed the cumulative genome-wide effect of MS-associated SNPs on HL and of HL-associated SNPs on MS. To provide an interpretational frame of reference, we used data from published GWAS to create a genetic network of diseases within which we analysed proximity of HL and MS to autoimmune diseases and haematological and non-haematological malignancies.
Results: SNP analyses revealed genome-wide overlap between HL and MS, most prominently in the HLA region. Polygenic HL risk scores explained 4.44% of HL risk (Nagelkerke R 2 ), but also 2.36% of MS risk. Conversely, polygenic MS risk scores explained 8.08% of MS risk and 1.94% of HL risk. In the genetic disease network, HL was closer to autoimmune diseases than to solid cancers.
Conclusions: HL displays considerable genetic overlap with MS and other autoimmune diseases.
Key Messages
Epidemiological similarities have suggested common aetiologies for Hodgkin lymphoma and multiple sclerosis.
Consistent with this hypothesis, detailed analyses reveal considerable genetic overlap between Hodgkin lymphoma and multiple sclerosis.
Genetically, Hodgkin lymphoma lies closer to autoimmune diseases than to solid cancers.
Introduction
Hodgkin lymphoma (HL) is an immunologically active malignant neoplasm of B cells in a heterogeneous reactive cellular infiltrate. 1 HL is roughly equally common in men and women, and in socioeconomically affluent populations HL occurrence displays a bimodal age distribution, with separate peaks in younger adults (15-34 years old) and older adults (over 50 years old). 2–4 In socioeconomically deprived settings, in contrast, there is no young adult HL incidence peak, but rather one among children. 5
Multiple sclerosis (MS) is a debilitating disease of the central nervous system (CNS) characterized by chronic polycellular inflammation (including T cells, monocytes and B cells), myelin loss, gliosis, axonal and oligodendrocyte pathology and accumulation of progressive neurological disability. 6 Onset is typically between the ages of 20 to 40 years, with a female to male ratio of three to one. 6
Based on conspicuous epidemiological similarities between the two conditions, e.g. regarding age patterns and geographical distributions, Newell in 1970 proposed that HL and MS were somehow aetiologically related. 7 Subsequent epidemiological studies have tested this hypothesis by assessing clustering of HL with MS in individuals and in families. Whereas previous studies generally suggest that patients suffering from either condition are not at increased risk of the other, 8–16 two partially overlapping investigations have reported mutual clustering of the two diseases among first-degree relatives. 17,18
Familial clustering of HL and MS may reflect shared environmental and genetic risk factors. Evidence implicates infection with the Epstein-Barr virus (EBV) 19–22 and levels of ultraviolet light exposure 23,24 in the pathogenesis of the roughly one-third of HL cases that harbour EBV in the malignant cells (EBV-positive HL), as well as in the pathogenesis of MS. Moreover, common genetic risk factors have emerged in HL and MS. 25–28 For example, HLA-A*02 appears to be associated with a decreased risk of both MS 29 and EBV-positive HL, 30,31 and DNA variants in the Relish oncogene ( REL , a member of the NF-kappaB transcription factor family) have been associated with both MS 32 and the EBV-negative subset of HL. 25 This suggests that the relationship between HL and MS is not limited to either EBV-positive or EBV-negative HL.
Unveiling of aetiological commonalities for HL and MS could contribute to the understanding of their pathogenesis and might even have clinical implications. We therefore combined data from previous genome-wide association studies (GWAS) of MS 28 and HL 26,27,33,34 and evaluated the genetic overlap between the two diseases.
Methods
Overview
Analysis was performed on a total of 1816 HL patients, 9772 MS patients and 25 255 controls, using 404 K single nucleotide polymorphisms ( Figure 1 ). We first sought to identify single nucleotide polymorphisms (SNPs) and HLA alleles that associated independently with both diseases. Next, we calculated polygenic risk scores to assess the cumulative genome-wide effect of MS-associated SNPs on HL, and of HL-associated SNPs on MS. To describe the overlap between HL and MS, we performed a protein interaction network-based pathway analysis (PINBPA) on associated genes from each disease, and then investigated the intersection of networks for biological relevance. To place the genetic similarity between HL and MS in context, we used data from previously reported GWAS to create a diseasome: a network of diseases. Within the diseasome, we analysed proximity of HL and MS to autoimmune diseases and haematological and non-haematological malignancies (‘solid’ cancers).
Figure 1.

Study design and data analysis procedures. Results from previously reported genome-wide associations studies (GWAS) of Hodgkin lymphoma (HL) and multiple sclerosis (MS) were used to assess genetic overlap between the two diseases. Single nucleotide polymorphisms (SNPs) independently associated with both HL and MS were identified, and disease-specific polygenic risk scores were compared in HL cases, MS cases and healthy controls. Protein-interaction network-based pathway analysis (PINBPA) was performed on the intersection of nominally associated ( P < 0.05) SNPs in HL and MS and gene ontology (GO) analysis was performed to identify common genetic pathways. Genetic similarity between HL and MS was further evaluated in the context of other immune diseases, haematological malignancies and solid cancers by constructing a diseasome using data from previously reported GWAS.
HL and MS dataset characteristics
The HL 26,27,33,34 and MS 28 cohorts have previously been described in detail. For MS, data from the Wellcome Trust Case Control Consortium 2 meta-analysis project totalled 9772 cases and 17 376 controls. Individuals in this dataset were of European descent and originated from 15 geographical regions, including the USA, Australia, New Zealand and numerous European countries. Included in this dataset were summary-level association results for a total of 464 434 SNPs.
For HL, data from a recent meta-analysis of three GWAS studies consisted of 1816 cases and 7879 controls. 34 Individuals in this dataset were of European descent and originated from locations in the USA and Europe. Summary-level meta-association results were included for a total of 1 036 304 SNPs. The cases were subdivided into nodular lymphocytic predominant and classical HL, and classical HL further divided into subgroups by EBV tumour status (EBV-positive and EBV-negative) as determined by immunohistochemistry or in situ hybridization as previously described 27 and histology [mixed cellularity (MC), nodular sclerosis (NS) and other or unspecified], when such data were available. Summary characteristics are shown in Supplementary Data (available as Supplementary data at IJE online).
The HL and MS datasets were merged (by rsID), giving a final dataset containing summary-level results for 404 069 overlapping SNPs.
Overlap between diseases: SNP-level
We followed a procedure similar to that used in other meta-analyses of complex genetic diseases. 35 To assess genetic overlap between HL and MS, we sought SNPs that associated independently with both diseases. We identified top ( P -value < 5 x 10 −8 ) independent (r 2 < 0.1 in CEU) MS-susceptibility SNPs, and determined how many of these SNPs are associated with HL after correction by Benjamini-Hochberg method (corrected P -value < 0.05). The process was repeated for increasingly liberal values of the P -value threshold for defining MS susceptibility SNPs (ranging from P -value < 5 x 10 −8 to P -value < 5 x 10 −2 ). Corresponding analysis was repeated after switching roles for HL and MS. The HLA region was analysed in further detail using imputed classical HLA alleles (see Supplementary methods for details, available as Supplementary data at IJE online).
Supplementary analysis considered MS SNPs on subsets of the HL dataset as defined by tumour EBV status (EBV-positive HL and EBV-negative HL), tumour histology [nodular sclerosis (NS), mixed cellularity (MC)], or tumour histology combined with age (NS among 15-35 year olds), in order to explore possible heterogeneity among the HL samples.
Overlap between diseases: polygenic risk
Polygenic risk scores were calculated to test the cumulative effect of SNPs associated with HL on MS and vice versa, as described in detail in other complex genetic diseases. 32,35–37 For each trait (HL and MS), sets of top independent SNPs were chosen as described above. Multiple sclerosis genetic burden (MSGB) and Hodgkin lymphoma genetic burden (HLGB) were calculated for each individual: the weighted sum of the number of risk alleles at each SNP in the set, weighted by the log-odds ratio of association for each SNP. We assessed the ability of MSGB to distinguish HL cases from controls and the ability of HLGB to distinguish MS cases from controls using the Nagelkerke’s R 2 (note that the P -values of the linear regression models will largely be driven by large sample sizes, and the biological significance lies in the R2 value rather than the P -value). This analysis was repeated for subgroups of HL (EBV-positive, EBV-negative, MC and NS).
Protein interaction network-based pathway analysis (PINBPA)
To visualize the sets of interacting genes found to associate with both HL and MS, a protein interaction network-based pathway analysis (PINBPA) was performed using methods described previously. 32 Sub-networks of aggregate score of three or greater were chosen as associated. Network discovery was performed independently in HL and MS, networks of score three or greater being chosen as associated. The intersection of the HL and MS networks was visualized. Gene ontology analysis was performed on genes in this intersection.
Diseasome analysis
To further assess genetic similarity between HL and MS, a representation of the human diseasome (network of diseases) 38,39 was constructed in which diseases (nodes) were connected by the extent of their shared genetic aetiology (edges) 40,41 as reported by the GWAS catalogue 42 . This network is termed the diseasome. Diseases were manually classified as haematological malignancies, solid cancers or autoimmune diseases. Pairwise proximity measures between diseases were calculated as described in Supplementary methods . Relative distances between haematological malignancies, solid cancers, and auto-immune diseases were tested by t-test.
Results
SNP and HLA allele overlap between HL and MS
We identified SNPs associated with MS across multiple P -value thresholds ranging from P -value < 5 x 10 −8 to P -value < 5 x 10 −2 . Among these SNPs, we then identified those that were also associated with HL (FDR < 0.05; false discovery rate with Benjamini-Hochberg adjustment of P -values for the total number of SNPs tested at each threshold) ( Table 1 ).
Table 1.
Overlap of associated SNPs in HL and MS at increasing thresholds
|
Top MS-associated SNPs in HL
| |||
|---|---|---|---|
| MS P -value threshold | Number of SNPs associated with MS | Number of independent MS SNPs | Number of independent MS SNPs also associated with HL (FDR < 0.05) |
| 5 x 10 −8 | 429 | 36 | 3 |
| 5 x 10 −7 | 497 | 50 | 4 |
| 5 x 10 −6 | 601 | 76 | 6 |
| 5 x 10 −5 | 825 | 138 | 4 |
| 5 x 10 −4 | 1422 | 386 | 3 |
| 0.005 | 4317 | 1715 | 3 |
| 0.05 | 24225 | 9107 | 2 |
|
| |||
| Top HL-associated SNPs in MS | |||
|
| |||
| HL P -value threshold | Number of SNPs associated with HL | Number of independent HL SNPs | Number of independent HL SNPs also associated with MS (FDR < 0.05) |
|
| |||
| 5 x 10 −8 | 11 | 6 | 5 |
| 5 x 10 −7 | 23 | 12 | 9 |
| 5 x 10 −6 | 37 | 17 | 12 |
| 5 x 10 −5 | 60 | 30 | 15 |
| 5 x 10 −4 | 291 | 165 | 19 |
| 0.005 | 2053 | 1155 | 32 |
| 0.05 | 17541 | 7196 | 36 |
In the upper panel, top MS-associated SNPs at a given P -value threshold (column 1) are counted (column 2), thinned to include only independent SNPs (column 3). Independent MS SNPs are tested in HL for association; the number of independent SNPs which pass FDR < 0.05 in HL is shown (column 4). In the lower panel, the top HL SNPs are counted, thinned and tested for association with MS.
LD, linkage disequilibrium; FDR, Benjamini-Hochberg false discovery rate, adjusted for the total number of independent SNPs tested.
At a threshold of P -value < 5 x 10 −8 , 429 SNPs were associated with MS; 36 of these 429 were independent (r 2 < 0.1), and three of these 36 were associated with HL at an FDR < 0.05 (Benjamini-Hochberg correction for 36 tests), summarized in row 1 of Table 1 . Panel 1 of Table 1 shows results for other P -value thresholds and panel 2 shows results when top HL SNPs were tested for association in MS. SNPs found to be overlapping (final column of Table 1 ) are described in Table 2 (after HLA is removed). While the actual number and proportion of overlapping SNPs varied by the P -value threshold, the majority of overlapping SNPs belonged to genes in the HLA region of chromosome 6; however, several mutually associated non-HLA SNPs were also detected ( Table 2 ; Supplementary Data , Supplementary Data , available as Supplementary data at IJE online). It should be noted that the direction of association was not taken into account in this analysis, which reveals only shared genetic risk loci (see genetic burden analysis below, which accounts for direction of association).
Table 2.
Non-HLA SNPs associated with both HL and MS at decreasing thresholds

|
Top: a grey box indicates that an SNP was associated with MS (at the P- value threshold shown in the top row), and was also associated with HL (FDR < 0.05; adjusted for the total number of SNPs that were tested in HL at the given MS threshold). Bottom: a grey box indicates that an SNP was associated with HL (at the P -value threshold shown in the top row), and was also associated with MS (FDR < 0.05; adjusted for the total number of SNPs that were tested in MS at the given HL threshold). Only independent SNPs are shown (r 2 < 0.1). The HLA region is omitted.
CHR, chromosome.
The SNP-level overlap between diseases was repeated for each HL subgroup: MS versus EBV-positive HL, MS versus EBV-negative HL, MS versus NS-HL, MS versus NS-HL in 15 to 35-year-olds and MS versus MC-HL. These analyses revealed no major differences between HL subgroups.
Given the strong genetic overlap at HLA, HLA alleles were imputed from SNP information via the HIBAG algorithm (see Supplementary methods , available as Supplementary data at IJE online). Figure 2 demonstrates overlap between risk alleles for EBV-negative HL and risk alleles for MS, as well as overlap between protective alleles for EBV-negative HL and protective alleles for MS. In contrast, there is no overlap between risk alleles for EBV-positive HL and risk alleles for MS, whereas there is overlap between protective alleles for EBV-positive HL and protective alleles for MS. Supplementary Data (available as Supplementary data at IJE online) provides details of HLA allelic P -values and odds ratios in each disease. Analysis of NS-HL revealed a pattern similar to EBV-negative HL.
Figure 2.

Legend. Classical HLA alleles were imputed in each disease using SNP data. Each point in each plot represents a classical HLA allele. Axes represent the odds ratio of association for each allele in the designated disease. Protective alleles have odds ratios less than 1 (lower values on each axis) and risk alleles have odds ratios greater than 1 (high values on each axis). (A) HLA risk alleles for EBV-positive HL tend to be neutral for EBV-negative HL, while HLA risk alleles for EBV-negative HL are neutral to protective for EBV-positive HL. Some HLA alleles are protective for both diseases. (B) HLA risk alleles for EBV-positive HL are neutral or protective for MS, and HLA risk alleles for MS are neutral or protective for EBV-positive HL. There are a large number of HLA alleles which are protective for both MS and EBV-positive HL. (C) There is an overlap between HLA risk alleles for MS and EBV-negative HL, and overlap between protective alleles for MS and EBV-negative HL.
Polygenic risk overlap between diseases
To assess the extent of genetic risk overlap between HL and MS at the genome-wide level (including HLA), polygenic risk scores, termed MS genetic burden (MSGB) and HL genetic burden (HLGB) were calculated in all cases of MS, all cases of HL and all controls used in the MS study. Figure 3A shows the MSGB (y-axis) in each population. As expected, the MSGB was higher in MS cases than in controls ( P -value < 1.0 x 10 −200 ) and explained 8.08% of the risk for MS (Nagelkerke's R 2 ). However, the MSGB was also higher in HL cases than in controls ( P -value < 2.8 x 10 −35 ) and explained 1.94% of the risk for HL. Figure 3B shows the HLGB (y-axis) in each population. As expected, the HLGB was higher in HL cases than in controls ( P -value < 2.8 x 10 −81 ) and explained 4.44% of the risk for HL. However, HLGB was also higher in MS cases compared with controls ( P -value < 2.0 x 10 −121 ) and explained 2.36% of the MS risk. Results shown here use a threshold of P -value < 5 x 10 −6 for including SNPs in the polygenic risk score, which results in 76 independent SNPs used for MSGB and 17 independent SNPs used for HLGB. Similar results held true at other P -value thresholds. We observed no major differences among HL subgroups.
Figure 3.

Polygenic risk scores demonstrate overlap between diseases. Hodgkin lymphoma (HL) and multiple sclerosis (MS) polygenic risk scores in HL cases, MS cases and healthy controls. A. MS genetic burden (MSGB) on y-axis, an aggregate measure of MS genetic risk across the genome of a given individual (includes human leukocyte antigen region of chromosome 6). MSGB is higher in HL cases than controls, indicating genetic overlap between HL and MS. B. HL genetic burden (HLGB) on y-axis, an aggregate measure of HL genetic risk across the genome of a given individual (includes human leukocyte antigen region of chromosome 6). HLGB is higher in MS cases than controls, indicating genetic overlap between HL and MS.
Pathway analysis
To generate hypotheses about potential functional pathways that are common to HL and MS, we carried out PINBPA in each independent dataset based on the nominally associated SNPs. We found 100 associated modules (with threshold of score > 3) in MS comprising 1404 genes and 4050 edges, and 100 associated modules in HL comprising 1049 genes and 3652 edges. The intersection of the HL and MS networks yielded a network of 430 genes and 1066 edges. A gene ontology (GO) analysis of this intersection network using the software binGO revealed enrichment in JUN kinase activity, antigen processing and presentation, peptidyl-tyrosine phosphorylation and lymphocyte-mediated immunity ( Figure 4 ; Supplementary Data , available as Supplementary data at IJE online). When the analysis was repeated after a seven mega-base region surrounding the HLA was masked, the antigen processing and presentation pathway was no longer associated but JUN kinase activity, peptidyl-tyrosine phosphorylation and lymphocyte-mediated immunity remained.
Figure 4.

Protein-interaction network-based pathway analysis (PINBPA) and gene ontology (GO). Four top pathways identified using GO analysis on PINBPA networks discovered in both Hodgkin lymphoma (HL) and multiple sclerosis (MS). A. Positive regulation of JUN kinase activity. B. Antigen processing and presentation of peptide antigen. C. Peptidyl-tyrosine phosphorylation. D. Lymphocyte-mediated immunity. Individual gene P -values for MS and HL are indicated when P < 0.05 (*) or when P < 0.1 (‡).
Diseasome analysis
To assess the relative position of HL and MS among other autoimmune diseases and cancers (in terms of shared genetic risk), pairwise proximities were calculated among 37 complex autoimmune diseases, solid cancers and haematological malignancies ( Table 3 ), where proximity is a network-based relatedness measure derived from shared GWAS loci between diseases. Autoimmune diseases showed more genetic similarity to other autoimmune diseases than to solid cancers ( P = 3.5 x 10 −38 , Figure 5A ), and analogously, the individual solid cancers showed more genetic similarity to other solid cancers than to autoimmune diseases ( P = 5.4 x 10 −9 , Figure 5C ). MS was closer to other autoimmune diseases than to solid cancers ( P = 9.2 x 10 −6 , Figure 5D ).
Table 3.
Classification of immune and neoplastic diseases from the diseasome
| Autoimmune diseases | Solid cancers |
|
| |
| Alopecia areata (AR) | Basal cell carcinoma (BCC) |
| Ankylosing spondylitis (AS) | Bladder carcinoma (BLC) |
| Behcet's disease (Beh) | Breast carcinoma (BRC) |
| Coeliac disease (Cel) | Central nervous system cancer (CNS) |
| Crohn's disease (CD) | Oesophageal carcinoma (OESC) |
| Graves' disease (GD) | Lung adenocarcinoma (LUA) |
| IGa glomerulonephritis (IGA) | Lung carcinoma (LUC) |
| Kawasaki disease (KAW) | Melanoma (MEl) |
| Multiple sclerosis (MS) | Ovarian carcinoma (OVC) |
| Primary biliary cirrhosis (PBC) | Pancreatic carcinoma (PAC) |
| Psoriasis (PS) | Prostate carcinoma (PRC) |
| Psoriatic arthritis (PSA) | Renal cell carcinoma (RCC) |
| Rheumatoid arthritis (RA) | Squamous cell carcinoma (SCC) |
| Sclerosing cholangitis (PSC) | Stomach carcinoma (STC) |
| Systemic lupus erythematosus (SLE) | Thyroid carcinoma (THC) |
| Systemic scleroderma (SS) | |
| Type 1 diabetes mellitus (T1D) | |
| Ulcerative colitis (UC) | |
| Vitiligo (Vit) | |
| Haematological cancers | |
| Chronic lymphocytic leukemia (CLL) | |
| Hodgkin lymphoma (HL) | |
| Multiple myeloma (MM) | |
Figure 5.
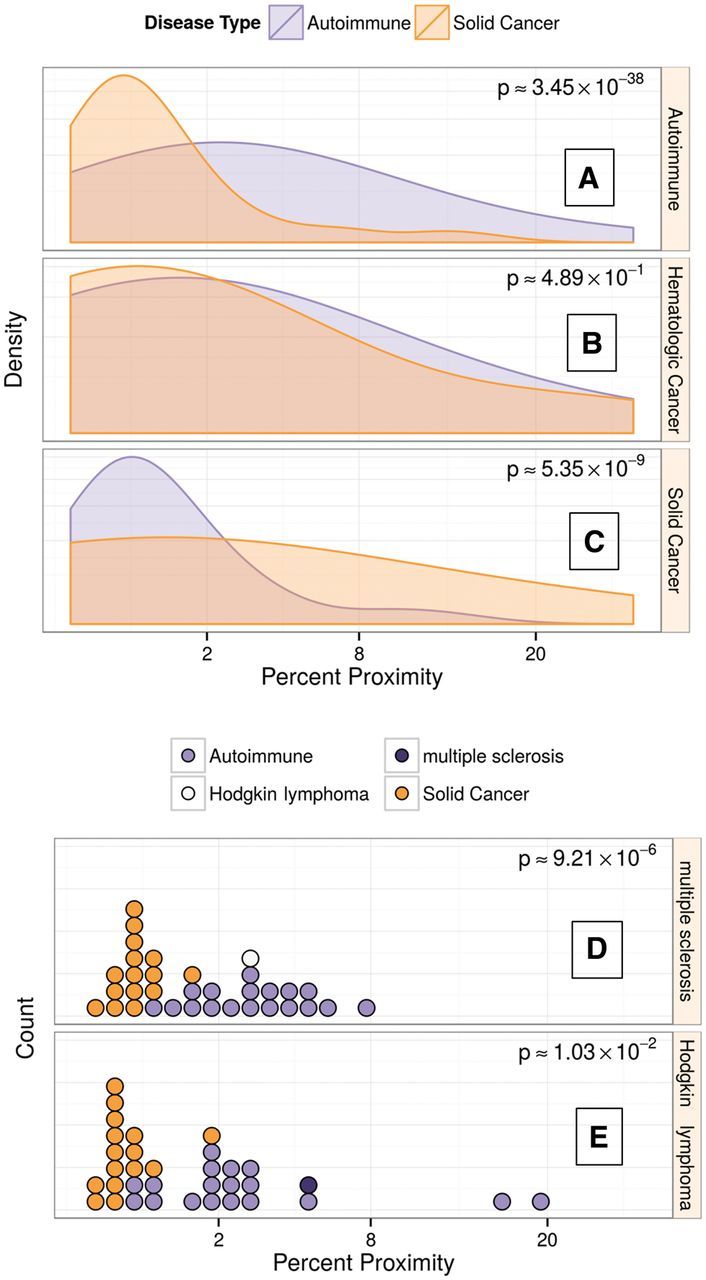
Diseasome analysis reveals that haematological malignancies lie somewhere between autoimmune diseases and solid cancer. A. Proximity of autoimmune diseases to other diseases. Density plots represent all possible pair-wise proximities between autoimmune diseases and solid cancers (orange), and all pair-wise proximities between autoimmune diseases and other autoimmune disease (purple). Higher degree of proximity (higher values on the x-axis) indicates more genetic similarity to autoimmune diseases. The P -value indicates that autoimmune diseases are closer to other autoimmune diseases than to solid cancers. B. Proximity of haematological malignancies to solid cancers (orange) and to autoimmune diseases (purple). Haematological malignancies show genetic overlap with both solid cancers and autoimmune diseases.C. Proximity of solid cancers to other solid cancers (orange) and to autoimmune diseases (purple).Solid cancers are closer to other solid cancers than to autoimmune diseases. D. Proximity of MS to all diseases. Each circle represents a disease in the diseasome. Higher degrees of proximity (higher values on x-axis) represent more genetic similarity with MS. Solid cancers are orange, autoimmune diseases are purple, HL is white. The P -value indicates MS is closer to autoimmune diseases than to solid cancers. E. Proximity of HL to all diseases. HL is closer to autoimmune diseases than to solid cancers.
Haematological malignancies, as a group, displayed approximately equal proximity to solid cancers and autoimmune disorders ( P = 0.49 for a difference, Figure 5B ). However, when the haematological malignancies were considered individually, HL was closer to autoimmune diseases than to solid cancers ( P = 0.01, Figure 5E ). Chronic lymphocytic leukaemia (CLL) was also closer to autoimmune diseases ( P = 0.038, Supplementary Data available as Supplementary data at IJE online) but also shared some loci with solid cancers. In contrast, multiple myeloma (MM) was closer to solid cancers than to autoimmune diseases ( P = 0.08, Supplementary Data ). Figure 6 is a graphical representation of the proximity network between diseases.
Figure 6.

Human disease network shows distinct autoimmune and solid cancer clusters and places hematologic cancers in context. In a network of disease proximity, constructed using systematic GWAS data, autoimmune diseases (purple) tightly cluster. Solid cancers (orange) also form a distinct cluster, but exhibit less relatedness in terms of genetic etiology than autoimmune diseases. Hematologic cancers (white) do not form a cohesive cluster and instead ranged from autoimmune related to solid cancer related. Hodgkin lymphoma (HL), in particular, appeared strongly autoimmune. See table 3 for a list of abbreviations.
Discussion
In this study, we undertook a series of analyses exploring genetic overlap between HL and MS. We found that top HL-associated SNPs were associated with MS, and conversely that top MS-associated SNPs were associated with HL. Overlap was particularly prominent in the HLA region of chromosome 6 but also applied to non-HLA loci. Genetic overlap between HL and MS was also observed in analyses of disease-specific polygenic risk scores (HLGB and MSGB) which included the HLA region. The HLGB explained 4.44% of the risk of HL and the MSGB explained 1.94% of the risk of HL. Similarly, MSGB explained 8.08% of the risk of MS and HLGB explained 2.36% of the risk for MS. Thus, the MSGB captured approximately 40% of the genetic susceptibility to HL measured by the HLGB and the HLGB captured approximately 30% of the genetic susceptibility to MS measured by the MSGB. Additionally, pathway analysis suggested shared biological pathways between HL and MS, involving a common theme of immune activation and cell proliferation, including lymphocyte-mediated immunity, positive regulation of JUN kinase activity (which plays roles in cellular response to stress, T cell differentiation, inflammation and apoptosis), peptidyl-tyrosine-phosphorylation (a non-specific intermediate step in multiple tyrosine pathways) and antigen processing and presentation.
The shared genetic element between HL and MS suggested by the present investigation is consistent with the original hypothesis of shared associations between the two conditions and with their previously observed mutual clustering within families. 17,18 Therefore further investigation of pathogenic pathways shared by these two clinically distinct conditions, similar to those being conducted for other neurodegenerative diseases associated with cancer risk, 43 is warranted.
Both EBV-positive and EBV-negative HL 44–46 and MS 47 have been consistently and strongly associated with HLA alleles. Our study confirms that this locus confers the highest known effect for the three phenotypes. Interestingly, in stratified analyses the patterns of overlap with MS clearly differed between EBV-positive HL and EBV-negative HL. Risk alleles were shared between EBV-negative HL and MS, but not between EBV-positive HL and MS. These findings suggest that EBV-positive HL and EBV-negative HL may each be independently associated with MS, but via different genes.
The precise mechanisms underlying the HLA associations have not been firmly established either for HL or MS. In MS, one immune model holds that T cells, activated peripherally by an infectious agent, cross the blood-brain barrier and induce MS lesions upon reactivation by myelin fragment antigens presented in the context of HLA. 48 For HL, speculation has centred primarily on its presumed infectious aetiology, 30,31,44 with EBV present and expressing antigens with plausible pathogenic functions in all tumour cells and therefore likely playing a causative role in the EBV-positive subset of HL. 49 Moreover, like MS, 22 EBV-positive HL has been associated with infectious mononucleosis caused by primary EBV infection 20 and with aberrant anti-EBV nuclear antigen antibody patterns, albeit different from those associated with MS. 19,21 Accordingly, HLA-specific variation in immune response to EBV infection may mediate the effects of the HLA associations shared by EBV-positive HL and MS. Although an analogous scenario involving an HLA-specific immune response directed against an infectious organism different from EBV may be envisioned for EBV-negative HL, no such agent has been linked to this HL subgroup 50 or to MS as yet.
Construction of a diseasome based on disease-gene associations 42 showed that the close genetic relationship between HL and the autoimmune disease MS applied to a broad spectrum of autoimmune conditions and that HL was in general closer to autoimmune diseases than to solid cancers. Importantly, the observation was not entirely explained by HLA, and the close relationship between HL and autoimmune conditions remained when the diseasome analysis was repeated after masking a seven mega-base region surrounding the HLA region. In line with this, there is evidence to suggest that the risk of HL is increased in patients with autoimmune diseases such as rheumatoid arthritis and systemic lupus erythematosus. 51,52 In the absence of evidence of familial clustering of HL and autoimmune diseases, 53 mechanisms such as chronic immune stimulation and/or immune-modulating treatment have been considered the most plausible explanations for the association. However, the present analyses suggest that shared genetic constitution may also contribute to the increased prevalence of HL among patients with autoimmune diseases (though an interaction between genetics and immune-modulating treatment is also a possibility). Indeed, our approach of combining GWAS data may prove more efficient in demonstrating overlapping pathogenic pathways between diseases than traditional epidemiological analytical designs, which may suffer from inadequate statistical power.
Besides HL, the diseasome analysis also included two other haematological malignancies. Among these, CLL was also closer to autoimmune diseases whereas multiple myeloma showed similarity to solid cancers. CLL belongs to the group of non-Hodgkin lymphomas 54 and, although strong associations between autoimmune disorders and CLL per se have not been noted, 55 an increased risk of the combined group of non-Hodgkin lymphomas among patients with various autoimmune diseases is well documented in the literature. 56
The main limitation of the present study was the uneven distribution of HL and MS patients with GWAS data and the lack of independent validation datasets. These limitations likely do not affect the diseasome results which are based on multiple GWAS in each disease and are robust to the removal of any single GWAS. However, SNP-level summary data for other autoimmune diseases and other cancers would have allowed assessment of polygenic risk scores and specific genetic overlap between other pairs of diseases that were closely associated in the diseasome, i.e. in the same way that the genetic overlap between HL and MS was assessed. Perhaps the diseasome analysis will provide impetus for further collaborative meta-analyses of haematologicalmalignancies and autoimmune diseases.
In summary, this study demonstrated commonalities in the genetic susceptibility to HL and MS as evidenced by analyses of individual SNPs, polygenic risk scores and protein-interaction networks. Diseasome analysis further suggested that HL shares a genetic architecture more similar to that of autoimmune diseases than to solid cancers. We speculate that autoimmune diseases and HL are different manifestations of a shared underlying genetic syndrome.
Funding
Funding for this project was obtained from the Nordic Cancer Union (16-02-D to H.H.), the Lundbeck Foundation (R19-A2364 to H.H.), the Danish Cancer Research Foundation (41-08 to H.H.), the National Institutes of Health (CA110836 to W.C.; HD0433871, CA129045, and CA40046 to K.O.; CA55727 to L.L.R.; CA58839 to T.M.M.), the United States Army Medical Research and Materiel Command (Department of Defense PR054600 to W.C.), the American Cancer Society Illinois Division (to K.O.), the American Lebanese Syrian Associated Charities (to L.L.R.), the Leukemia & Lymphoma Society (TR6137-07 to W.C.), the Cancer Research Foundation (to K.O.), the National Cancer Institute Surveillance Epidemiology and End Results Population-based Registry Program at the National Institutes of Health in the Department of Health and Human Services (R01-NS-026799 to J.R.O., N01-PC-35139 to W.C. and N01-PC-35136 to the Cancer Prevention Institute of California), the National Cancer Institute (263-MQ-417755 to S.L.G.), the Leukemia Lymphoma Research and Kay Kendall Leukemia fund (grant 12022 to R.F.J.), the California Cancer Registry and the Dutch Cancer Society (KWF grants RUG 2009-4313 and RUG 2014-6698 to A.v.d.B.), the National Science Foundation (Graduate Research Fellowship Grant 1144247 to D.S.H.) and the Center for Neuroengineering and Therapeutics at the University of Pennsylvania (P.K.).
Supplementary Material
Acknowledgments
We thank the Childhood Cancer Survivor Study (National Cancer Institute grant U24 CA 55727) for the Hodgkin Lymphoma data, and the International Multiple Sclerosis Genetics Consortium and the Wellcome Trust Case-Control Consortium for the multiple sclerosis data. We would like to thank Hans-Olov Adami, David Conti, Arjan Diepstra, Bengt Glimelius, Annette Lake, Dorothy Montgomery, Les Robison, Malcolm Taylor, and Maria Timofeeva for data accrual, sample handling and analysis of Hodgkin lymphoma data. We would like to thank Stacy J. Caillier and Sarah Beth Hill for assistance with figure creation.
Author contributions
Contributions: P.K., W.C., D.S.H., S.E.B., J.D.M., J.R.O. and H.H. designed the experiment. P.K., D.S.H., L.M., L.D., T.M., J.M.M., A.L., P.A.G., M.D.S., D.L. and J.D.M. analysed the data. A.v d B., S.L.G., K.E.S., T.M.M., S.d.S., A.N., S.L.H., P.C., M.M., L.F., A.S., M.D.S., S.B., M.M., K.O., R.J., J.D.M., J.R.O. and H.H. contributed reagents, materials and analysis tools. P.K., W.C., D.S.H., S.E.B., P.A.G., R.J., J.D.M., J.R.O. and H.H. wrote the paper. All authors read and approved the final manuscript.
Conflict of interest : None declared.
References
- 1. Stein H. Hodgkin lymphoma - Introduction . In: Swerdlow SH, Campo E, Harris NLet al. . (eds). Tumours of Haematopoietic and Lymphoid Tissues . Lyon, France: : IARC Press; , 2008. . [Google Scholar]
- 2. MacMahon B. Epidemiology of Hodgkin's disease . Cancer Res 1966. ; 26:1189 –. [PubMed] [Google Scholar]
- 3. Mueller N, Grufferman S. Hodgkin lymphoma . In: Schottenfeld D, Fraumeni J., Jr (eds). Cancer Epidemiology and Prevention . 3rd edn.Oxford, UK: : Oxford University Press; , 2006. . [Google Scholar]
- 4. MacMahon B. Epidemiological evidence of the nature of Hodgkin's disease . Cancer 1957. ; 10:1045 – 13 . [DOI] [PubMed] [Google Scholar]
- 5. Correa P, O'Conor GT. Epidemiologic patterns of Hodgkin's disease . Int J Cancer 1971. ; 8:192 – 201 . [DOI] [PubMed] [Google Scholar]
- 6. Hauser S, Goodin DS. Multiple sclerosis and other demyelinating diseases . In: Longo DL, Fauci AS, Kasper DL, Hauser SL, Jameson L, Loscalzo J. (eds). Harrison's Principles of Internal Medicine . 18th edn.New York, NY: : McGraw-Hill; , 2012. . [Google Scholar]
- 7. Newell GR. Etiology of multiple sclerosis and Hodgkin's disease . Am J Epidemiol 1970. ; 91:11 – 22 . [DOI] [PubMed] [Google Scholar]
- 8. Bahmanyar S, Montgomery SM, Hillert J, Ekbom A, Olsson T. Cancer risk among patients with multiple sclerosis and their parents . Neurology 2009. ; 72:1170 – 77 . [DOI] [PubMed] [Google Scholar]
- 9. Bernard SM, Cartwright RA, Darwin CM. et al. . Hodgkin's disease: case control epidemiological study in Yorkshire . Br J Cancer 1987. ; 55:85 – 90 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Koch-Henriksen N, Bronnum-Hansen H, Stenager E. Underlying cause of death in Danish patients with multiple sclerosis: results from the Danish Multiple Sclerosis Registry . J Neurol Neurosurg Psychiatry 1998. ; 65:56 – 59 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lebrun C, Debouverie M, Vermersch P. et al. . Cancer risk and impact of disease-modifying treatments in patients with multiple sclerosis . Mult Scler 2008. ; 14:399 – 405 . [DOI] [PubMed] [Google Scholar]
- 12. Midgard R, Glattre E, Gronning M, Riise T, Edland A, Nyland H. Multiple sclerosis and cancer in Norway. A retrospective cohort study . Acta Neurol Scand 1996. ; 93:411 – 15 . [DOI] [PubMed] [Google Scholar]
- 13. Moller H, Kneller RW, Boice JD, Jr, Olsen JH. Cancer incidence following hospitalization for multiple sclerosis in Denmark . Acta Neurol Scand 1991. ; 84:214 – 20 . [DOI] [PubMed] [Google Scholar]
- 14. Palo J, Duchesne J, Wikstrom J. Malignant diseases among patients with multiple sclerosis . J Neurol 1977. ; 216:217 – 22 . [DOI] [PubMed] [Google Scholar]
- 15. Sadovnick AD, Eisen K, Ebers GC, Paty DW. Cause of death in patients attending multiple sclerosis clinics . Neurology 1991. ; 41:1193 – 96 . [DOI] [PubMed] [Google Scholar]
- 16. Vineis P, Crosignani P, Vigano C. et al. . Lymphomas and multiple sclerosis in a multicenter case-control study . Epidemiology 2001. ; 12:134 – 35 . [DOI] [PubMed] [Google Scholar]
- 17. Hjalgrim H, Rasmussen S, Rostgaard K. et al. . Familial clustering of Hodgkin lymphoma and multiple sclerosis . J Natl Cancer Inst 2004. ; 96:780 – 84 . [DOI] [PubMed] [Google Scholar]
- 18. Landgren O, Kerstann KF, Gridley G. et al. . Re: Familial clustering of Hodgkin lymphoma and multiple sclerosis . J Natl Cancer Inst 2005. ; 97:543 – 44 . [DOI] [PubMed] [Google Scholar]
- 19. Levin LI, Munger KL, Rubertone MV. et al. . Temporal relationship between elevation of Epstein-Barr virus antibody titers and initial onset of neurological symptoms in multiple sclerosis . JAMA 2005. ; 293:2496 – 500 . [DOI] [PubMed] [Google Scholar]
- 20. Hjalgrim H, Askling J, Rostgaard K. et al. . Characteristics of Hodgkin's lymphoma after infectious mononucleosis . N Engl J Med 2003. ; 349:1324 – 32 . [DOI] [PubMed] [Google Scholar]
- 21. Levin LI, Chang ET, Ambinder RF. et al. . Atypical prediagnosis Epstein-Barr virus serology restricted to EBV-positive Hodgkin lymphoma . Blood 2012. ; 120:3750 – 55 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nielsen TR, Rostgaard K, Nielsen NM. et al. . Multiple sclerosis after infectious mononucleosis . Arch Neurol 2007. ; 64:72 – 75 . [DOI] [PubMed] [Google Scholar]
- 23. Monnereau A, Glaser SL, Schupp CW. et al . Exposure to UV radiation and risk of Hodgkin lymphoma: a pooled analysis . Blood 2013. ; 122:3492 – 99 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Beretich BD, Beretich TM. Explaining multiple sclerosis prevalence by ultraviolet exposure: a geospatial analysis . Mult Scler 2009. ; 15:891 – 98 . [DOI] [PubMed] [Google Scholar]
- 25. Enciso-Mora V, Broderick P, Ma Y. et al. . A genome-wide association study of Hodgkin's lymphoma identifies new susceptibility loci at 2p16.1 (REL), 8q24.21 and 10p14 (GATA3) . Nat Genet 2010. ; 42:1126 – 30 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cozen W, Li D, Best T. et al. . A genome-wide meta-analysis of nodular sclerosing Hodgkin . Blood 2012. ; 119:469 – 75 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Urayama KY, Jarrett RF, Hjalgrim H. et al. . Genome-wide association study of classical Hodgkin lymphoma and Epstein-Barr virus status-defined subgroups . J Natl Cancer Inst 2012. ; 104:240 – 53 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sawcer S, Hellenthal G, Pirinen M. et al. . Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis . Nature 2011. ; 476:214 – 19 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Isobe N, Gourraud PA, Harbo HF. et al. . Genetic risk variants in African Americans with multiple sclerosis . Neurology 2013 July 16. ; 81 ( 3 ): 219 – 27 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Niens M, Jarrett RF, Hepkema B. et al. . HLA-A*02 is associated with a reduced risk and HLA-A*01 with an increased risk of developing EBV+ Hodgkin lymphoma . Blood 2007. ; 11:3310 – 15 . [DOI] [PubMed] [Google Scholar]
- 31. Hjalgrim H, Rostgaard K, Johnson PC. et al. . HLA-A alleles and infectious mononucleosis suggest a critical role for cytotoxic T cell response in EBV-related Hodgkin lymphoma . Proc Natl Acad Sci U S A 2010. ; 107:6400 – 05 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Baranzini SE, Khankhanian P, Patsopoulos NA. et al . Network-based multiple sclerosis pathway analysis with GWAS data from 15,000 cases and 30,000 controls . Am J Hum Genet 2013. ; 92:854 – 65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Best T, Li D, Skol AD. et al . Variants at 6q21 implicate PRDM1 in the etiology of therapy-induced second malignancies after Hodgkin's lymphoma . Nat Med 2011. ; 17:941 – 43 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cozen W, Timofeeva MN, Li D. et al. . A meta-analysis of Hodgkin lymphoma reveals 19p13.3 TCF3 as a novel susceptibility locus . Nat Commun 2014. ; 5:3856 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Goris A, van SJ, Diekstra F. et al. . No evidence for shared genetic basis of common variants in multiple sclerosis and amyotrophic lateral sclerosis . Hum Mol Genet 2014. ; 23:1916 – 22 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gourraud PA, McElroy JP, Caillier SJ. et al. . Aggregation of multiple sclerosis genetic risk variants in multiple and single case families . Ann Neurol 2011. ; 69:65 – 74 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Isobe N, Damotte V, Lo R. et al. . Genetic burden in multiple sclerosis families . Genes Immun 2013. ; 14:434 – 40 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Goh KI, Cusick ME, Valle D, Childs B, Vidal M, Barabasi AL. The human disease network . Proc Natl Acad Sci U S A 2007. ; 104:8685 – 90 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang K, Ng SK, McLachlan GJ. Clustering of time-course gene expression profiles using normal mixture models with autoregressive random effects . BMC Bioinformatics 2012. ; 13:300 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Himmelstein D, Khankhanian P, Baranzini SE . A human disease network from GWAS loci . Zenodo 2015. . [Google Scholar]
- 41. Himmelstein D. Extracting disease-gene associations from the GWAS Catalog . ThinkLab 2015. . [Google Scholar]
- 42. Welter D, MacArthur J, Morales J. et al. . The NHGRI GWAS Catalog, a curated resource of SNP-trait associations . Nucleic Acids Res 2014. ; 42(Database issue):D1001 – D1006 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Plun-Favreau H, Lewis PA, Hardy J, Martins LM, Wood NW. Cancer and neurodegeneration: between the devil and the deep blue sea . PLoS Genet 2010. ; 6:e1001257 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Huang X, Kushekhar K, Nolte I. et al. . HLA associations in classical Hodgkin lymphoma: EBV status matters . PLoS One 2012. ; 7:e39986 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Niens M, van den Berg A, Diepstra A. et al. . The human leukocyte antigen class I region is associated with EBV-positive Hodgkin's lymphoma: HLA-A and HLA complex group 9 are putative candidate genes . Cancer Epidemiol Biomarkers Prev 2006. ; 15:2280 – 84 . [DOI] [PubMed] [Google Scholar]
- 46. Johnson PC, McAulay KA, Montgomery D. et al. . Modeling HLA associations with EBV-positive and -negative Hodgkin lymphoma suggests distinct mechanisms in disease pathogenesis . Int J Cancer 2015. ; 137:1066 – 75 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Beecham AH, Patsopoulos NA, Xifara DK. et al. . Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis . Nat Genet 2013. ; 45:1353 – 60 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lang HL, Jacobsen H, Ikemizu S. et al. . A functional and structural basis for TCR cross-reactivity in multiple sclerosis . Nat Immunol 2002. ; 3:940 – 43 . [DOI] [PubMed] [Google Scholar]
- 49. Kuppers R. The biology of Hodgkin's lymphoma . Nat Rev Cancer 2009. ; 9:15 – 27 . [DOI] [PubMed] [Google Scholar]
- 50. Jarrett RF. Viruses and Hodgkin's lymphoma . Ann Oncol 2002. ; 13(Suppl 1):23 – 29 . [DOI] [PubMed] [Google Scholar]
- 51. Fallah M, Liu X, Ji J, Forsti A, Sundquist K, Hemminki K. Hodgkin lymphoma after autoimmune diseases by age at diagnosis and histological subtype . Ann Oncol 2014. ; 25:1397 – 404 . [DOI] [PubMed] [Google Scholar]
- 52. Landgren O, Caporaso NE. New aspects in descriptive, etiologic, and molecular epidemiology of Hodgkin's lymphoma . Hematol Oncol Clin North Am 2007. ; 21:825 – 40 . [DOI] [PubMed] [Google Scholar]
- 53. Landgren O, Engels EA, Pfeiffer RM. et al. . Autoimmunity and susceptibility to Hodgkin lymphoma: a population-based case-control study in Scandinavia . J Natl Cancer Inst 2006. ; 98:1321 – 30 . [DOI] [PubMed] [Google Scholar]
- 54. World Health Organization . Classification of Tumours of Haematopoietic and Lymphoid Tissues . Lyon, France: : IARC; , 2008. . [Google Scholar]
- 55. Landgren O, Engels EA, Caporaso NE. et al. . Patterns of autoimmunity and subsequent chronic lymphocytic leukemia in Nordic countries . Blood 2006. ; 108:292 – 96 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Baecklund E, Smedby KE, Sutton LA, Askling J, Rosenquist R. Lymphoma development in patients with autoimmune and inflammatory disorder - what are the driving forces? Semin Cancer Biol 2014. ; 24:61 – 70 . [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.