

Abstract
In order to describe the physical characteristics, medical complications, and natural history of classic 7q11.23 duplication syndrome [hereafter Dup7 (MIM 609757)], reciprocal duplication of the region deleted in Williams syndrome [hereafter WS (MIM 194050)], we systematically evaluated 53 individuals aged 1.25–21.25 years and 11 affected adult relatives identified in cascade testing. In this series, 27% of probands with Dup7 had an affected parent. Seven of the 26 de novo duplications that were examined for inversions were inverted; in all 7 cases one of the parents had the common inversion polymorphism of the WS region. We documented the craniofacial features of Dup7: brachycephaly, broad forehead, straight eyebrows, broad nasal tip, low insertion of the columella, short philtrum, thin upper lip, minor ear anomalies, and facial asymmetry. Approximately 30% of newborns and 50% of older children and adults had macrocephaly. Abnormalities were noted on neurological examination in 88.7% of children, while 81.6% of MRI studies showed structural abnormalities such as decreased cerebral white matter volume, cerebellar vermis hypoplasia, and ventriculomegaly. Signs of cerebellar dysfunction were found in 62.3%, hypotonia in 58.5%, Developmental Coordination Disorder in 74.2%, and Speech Sound Disorder in 82.6%. Behavior problems included anxiety disorders, ADHD, and oppositional disorders. Medical problems included seizures, 19%; growth hormone deficiency, 9.4%; patent ductus arteriosus, 15%; aortic dilation, 46.2%; chronic constipation, 66%; and structural renal anomalies, 18%. We compare these results to the WS phenotype and offer initial recommendations for medical evaluation and surveillance of individuals who have Dup7.
Keywords: 7q11.23 duplication syndrome, Williams syndrome, aortic dilation, macrocephaly, cerebellar vermis hypoplasia, anxiety, psychopathology, developmental coordination disorder, speech sound disorder
INTRODUCTION
Williams syndrome (WS, MIM 194050) is caused by a 1.5Mb microdeletion of chromosome 7q11.23 mediated by nonallelic homologous recombination (NAHR). Due to the NAHR mechanism, it was known that reciprocal duplication of the WS region must exist, but the first case was not reported until 2005 when a boy being tested for 22q11 deletion using real time PCR was found to have duplication of the elastin gene; subsequent analysis revealed the microduplication of the WS region (7q11.23 duplication syndrome, hereafter Dup7, MIM 609757) [Somerville et al., 2005]. Since that time, we have had the opportunity to evaluate 64 individuals with classic Dup7 in order to characterize the clinical features, medical problems, phenotypic variability, and natural history of the syndrome.
In the past 10 years, 46 children and 15 adults with classic Dup7 have been reported in the literature in case reports [Somerville et al., 2005; Kriek et al., 2006; Kirchhoff et al., 2007; Depienne et al.,2007; Torniero et al., 2007, 2008; Merritt and Lindor, 2008; Orellana et al., 2008; Malenfant et al., 2011; Degerliyurt et al., 2012; McGrew et al., 2012; Prontera et al, 2014; Zarate et al., 2014] or in clinical case series [Berg et al., 2007; van der Aa et al., 2009; Dixit et al., 2013; Parrott et al., 2015]. Some of these individuals were identified when special populations were screened by microarray for copy number variants (Table I). While affected individuals have been identified in populations of persons with autism spectrum disorder (ASD), intellectual disability, schizophrenia, and epilepsy, and Dup7 has been reported to be a risk factor for both ASD [Sanders et al., 2011] and schizophrenia [Mulle et al., 2014], the conclusion of these studies is that Dup7 is not a common cause of any of these conditions. Most of the probands reported were identified in the course of routine clinical evaluation for developmental delay, dysmorphic features, or behavior problems. Six transmitting parents, two affected half siblings, and three affected siblings were identified by cascade testing. The prevalence of Dup 7 has been estimated between 1/7,500 – 20,000 [Van der Aa et al., 2009; Velleman and Mervis, 2011].
Table I.
Cases of Dup7 Identified When Screening Special Populations for Copy Number Variants
| Report | Population Screened | N | Dup7 cases |
|---|---|---|---|
| Depienne et al., 2007 | Autism spectrum disorder | 206 | 1 |
| Kirchhoff et al., 2007 | Intellectual disability | 258 | 1 |
| Torniero et al., 2008 | Epilepsy/neuronal migration defects | 134 | 2 |
| van der Aa et al., 2009 | Intellectual disability | 300 | 2 |
| Sanders et al., 2011 | Autism spectrum disorder | 1,124 | 4 |
| Malenfant et al., 2011 | Autism spectrum disorder | 1,142 | 1 |
| McGrew et al., 2012 | Autism spectrum disorder | 97 | 1 |
| Mulle et al., 2014 | Schizophrenia Cohort 1 Schizophrenia Cohort 2 (meta-analysis)a |
554 14,387 |
2 11b |
Samples analyzed only for Dup7.
Includes 9 cases of classic Dup7 and 2 cases in which the duplication extended at least 1 Mb beyond the classic telomeric breakpoint.
These case reports and clinical case series have provided a preliminary phenotypic description of Dup7 and highlighted its variability. Craniofacial features commonly noted in the literature include macrocephaly, broad forehead, straight eyebrows, deep set eyes, short philtrum, thin upper lip, micrognathia, and minor ear anomalies leading to a recognizable facial gestalt [Van der Aa et al., 2009; Dixit et al., 2013]. Various malformations have been reported in single patients including congenital glaucoma, choanal atresia, submucous cleft palate, congenital diaphragmatic hernia, vertebral anomalies, and talipes equinovarus. Two children have been reported with trigonocephaly, two with exotropia, two with cleft lip and palate, and four with cryptorchidism. Abnormal MRI findings were noted in 13 of the 16 individuals with classic Dup7 for whom MRI results were reported. The MRI abnormalities varied and included cerebral atrophy, increased cortical thickness, ventriculomegaly, hyperintensities in the white matter, simplified gyral pattern, dilated perivascular spaces, and cerebellar vermis hypoplasia [Berg et al. 2007; Depienne et al., 2007; Torniero et al., 2007, 2008; van der Aa et al., 2009; Orellana et al., 2009; Dixit et al., 2013; Prontera et al., 2014].
Medical problems noted in previous reports include hypotonia (18), seizure disorder (8), constipation (3), and short stature (1). Congenital heart disease has been reported in 6 individuals (4 patent ductus arteriosus, 1 atrial septal defect, 1 supravalvar aortic stenosis with post stenotic dilation). Two recent studies drew attention to the occurrence of aortic dilation in classic Dup7. Zarate et al. [2014] reported a 2.5-year-old with dilation of the ascending aorta. Parrott et al. [2015] described the cardiovascular findings in 8 individuals with classic Dup7: one had aortic dilation in infancy and was treated with a beta blocker at age 5 years due to worsening dilation. Two children had dilation of the aorta that normalized as they got older, one had moderate dilation, and four other individuals (the oldest age 34 years) had mild aortic dilation.
Developmental delay or intellectual disability, ASD or features of ASD, language delay or disorder, and/or severe speech delay have been noted in almost all case reports of individuals with Dup7. These important aspects of the phenotype, along with anxiety and behavioral problems, were analyzed for a group of 63 children aged 4 – 17 years and 16 toddlers and young preschoolers by Mervis et al. [2015]. In both Mervis et al. [2015] and the present study, only individuals with classic Dup7 are reported. This decision was made because both shorter [Morris et al., 2003] and longer [Stock et al., 2003] deletions of the WS region result in phenotypes that differ from classic WS in important ways and therefore it was expected that shorter or longer duplications of the WS region would also result in phenotypes that include important differences from classic Dup7.
MATERIALS AND METHODS
Participants
Sixty four individuals who had a classic 1.5 Mb microduplication of 7q11.23 participated in the study. Group 1 consisted of 53 individuals (28 females, 25 males) aged 1.25 – 21.25 years (median: 7.09 years, mean: 8.12 years, SD: 4.87 years). Forty eight were probands and 5 were siblings of probands identified in cascade testing. All probands had had a chromosome microarray (using various commercially available platforms) demonstrating a classic duplication of the WS region. The classic size of the duplication was confirmed by FISH and/or PCR for all participants, including siblings diagnosed in cascade testing. One individual (#20) was previously described in a case report [Somerville et al., 2005] and 48 of the 53 were included in our article on psychological characteristics of children with classic Dup7 [Mervis et al., 2015]. Group 2 was comprised of 11 adult relatives of probands, all diagnosed by cascade testing. Four mothers, five fathers, and two grandmothers were identified in nine families. FISH and/or PCR was used to confirm that all individuals in Group 2 had a classic duplication. Eight of the 11 were included in the adult group reported in Mervis et al. [2015]. Informed consent/assent was obtained from all study participants and/or their parents. Research protocols were prospectively reviewed and approved by the Institutional Review Boards of the University of Nevada School of Medicine, the University of Louisville, and the University of Toronto.
Genetic Studies
FISH or qPCR was used to confirm the classic size of the duplication in all participants. Chromosome preparations were made using our laboratory’s variation of standard cytogenetic protocols for G-banded staining of metaphase cells. FISH was performed by co-denaturation of target DNA and our research FISH probe DNAs produced from BACs located in the regions of interest. The following probes from 7q11.23 were used: RP11-815K3 (815), BCL7 (BCL), RP5-1186P10 (1186), CTB-139P11 (139). 815 and 139 were used as anchor probes, since both are located outside the Williams syndrome critical region. For hybridization, probes were suspended in denHyb-2 hybridization solution (Insitus, Albuquerque, NM). Overnight hybridization at 37°C followed co-denaturation of the probe and the target metaphases at 77.5°C for 6 minutes. Counterstain was DAPI. Observation was performed with a Zeiss Axioscop and documented on a Metasystems (Altlussheim, Germany) imaging system. Direct duplication versus inverted duplication was determined as previously described [Hobart et al., 2010].
DNA was extracted directly from participant blood samples and the extent of the duplication confirmed using real-time PCR with existing validated primer sets as previously described [Somerville et al., 2005; Tam et al., 2008]. Wherever possible, parent-of-origin of the duplication was determined for each individual using analysis of existing highly polymorphic microsatellite markers from the 7q11.23 region in DNA from the participants and their parents as described previously [Somerville et al., 2005].
Clinical Assessments
Physical examination of all participants, including documentation of craniofacial features, was completed by the same dysmorphologist (C.A.M.), and all available medical records were reviewed. Interviews to obtain family, developmental, and medical history were conducted. Detailed neurological examinations were completed on Group 1; the adult relatives were not assessed because there were too many potentially complicating factors in the group (e.g., closed head injury, neurofibromatosis I, untreated hypothyroidism). Children aged 5.00 – 17.99 years (N = 31) were evaluated for Developmental Coordination Disorder (DCD) using DSM-5 criteria. Diagnosis of DCD was based upon the synthesis of medical and developmental history, medical records, neurological examination, and school and physical therapy reports. Most of the child participants also were evaluated for Speech Sound Disorder (SSD) according to DSM-5 criteria and for anxiety disorders, Attention Deficit Hyperactivity Disorder (ADHD), and Oppositional Defiant Disorder (ODD) or Disruptive Behavior Disorder – Not Otherwise Specified (DBD-NOS) according to DSM-IV criteria as detailed in Mervis et al. [2015]. MRI images were available for 15 participants and CT images were available for one participant; all were reviewed by the same neurologist (A.R.P.).
RESULTS
Genetic Studies
Examination of the indications for genetic testing for the probands revealed that the most common was developmental delay (21 of 48; 43.8%). The second most common indicator was autism spectrum disorder (ASD) (9 of 48; 18.8%). Seven of the nine were later assessed for ASD by one of the authors (B.P.K.-T.) using the gold-standard procedure (ADI-R and ADOS-2, plus clinical judgment). Four of the seven met criteria for ASD. Other indicators listed for at least 10% of the probands were speech and/or language delay/disorder, dysmorphic facial features, macrocephaly, and hypotonia. Indicators that were less commonly provided but that correspond to phenotypic characteristics of Dup7 were hydrocephalus, decreased pain sensitivity, and anxiety. Only one individual was specifically evaluated for possible Dup7 (patient #19). This 7-year-old-boy previously had been diagnosed with Childhood Apraxia of Speech and his speech therapist referred him to the study after attending a presentation regarding Dup7 by one of the authors (S.L.V.).
Twenty of the probands were born in 2005 or later. For this group, age at diagnosis ranged from 0.02 years – 5.46 years, with a median of 2.08 years (mean: 2.50 years, SD: 1.58). Three children were diagnosed before their first birthday, six as 1-year-olds, five as 2-year-olds, three as 3-year-olds, and three as 5-year-olds.
Thirty nine of the probands were the only members of their family known to have Dup7. For 29, both parents were available for testing and the proband’s duplication was determined to be de novo. For four probands, only one parent was available for testing; that parent was determined not to have Dup7. For the remaining six probands, neither parent was tested; four of these probands were adopted. Parent of origin was determined for 15 of the 39; it was maternal for eight and paternal for seven. Inversion status was determined for 26 of the 39. Seven (26.9%) had inverted duplications; in all cases one of the parents had the common inversion polymorphism of the WS region (5 maternal 2 paternal). Parent of origin was known for only one of the seven children; for this child, the father, who was the parent of origin, had an inversion. Nineteen children had direct duplications. In 10 of these cases neither parent had an inversion; in the remaining nine cases one or both parents were not available for testing.
The remaining 25 individuals were members of nine families. In two families, Dup7 was present in at least three generations and in seven families Dup7 was present in at least two generations. Five of the families had a direct duplication, three families had an inverted duplication (Figure 1), and one family did not have FISH studies performed. In one of the families with both grandparents available for testing, the grandfather had the WS region inversion polymorphism and the mother with Dup7 had a de novo inverted duplication. Therefore, in this series, 27% of probands with Dup7 had an affected parent.
Figure 1.
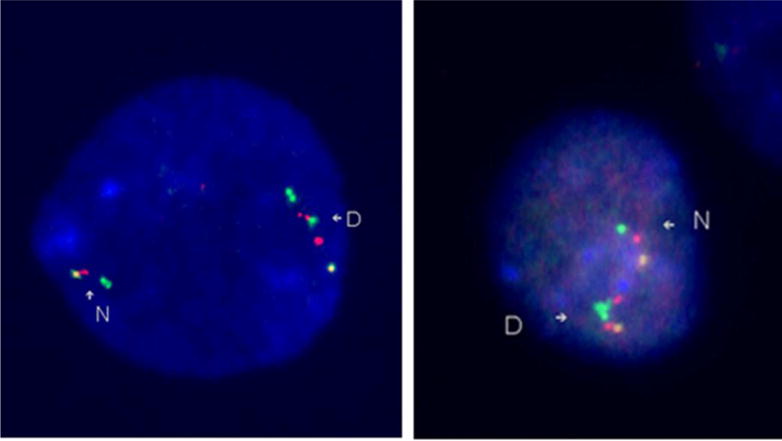
FISH images of a direct duplication and an inverted duplication of the WS region. The left image demonstrates a direct duplication. The N indicates the normal, non-duplicated, chromosome, with the probe order being yellow-red-green. The yellow is an anchor probe (RP11-815K3 (815)), and the red (BCL7 (BCL)) and green (RP5-1186P10 (1186)) probes are inside the WS region. The D indicates the duplicated chromosome. The probe order is yellow-red-green-red-green, or 815-BCL-1186-BCL-1186. Since the duplicated region, the extra red and green signals, is in the same orientation as the original region, the duplicated segment has been inserted in the normal orientation. The right image demonstrates an inverted duplication. The N indicates the normal chromosome and the D indicates the duplicated chromosome. The probe color order is yellow-red-green-green-red, or 815-BCL-1186-1186-BCL. Since the duplicated segment is in the opposite orientation, green-red, instead of the normal orientation, red-green, the duplicated segment has been inserted in the opposite direction, giving an inverted duplication.
Four individuals had a copy number variant reported on chromosome microarray in addition to the 7q11.23 duplication. Patient #36, a female, had a 10kb deletion of Xq28 disrupting IDS, the gene responsible for Hunter syndrome in males. Three other individuals had deletions that have not been reported with a phenotype; each deleted a single gene. Patient #45 had a 31kb deletion of 4q26 that included ANK2, Patient #25 had a 180 kb deletion of 10p14 that included CELF2, and patient #41 had a maternally-inherited 70kb deletion of 3p26.1 that included LRRN1. Siblings #41 and #42 had inherited the 7q11.23 duplication from their father, #43. Their mother, who did not have Dup7, did have a history of intellectual disability and a seizure disorder and had dysmorphic facial features shared by her daughter #42 but not her other daughter #41 (Figure 2). It is possible that patient #42 who did not inherit the maternal LRRN1 deletion has an autosomal dominant disorder inherited from her mother as well as Dup7.
Figure 2.

Family with Dup7. The father, patient #43, transmitted the duplication to his daughters, patients #41 and 42. The mother of the children had a history of intellectual disability and died secondary to complications of a seizure disorder, but did not have Dup7. Patient #42 (the girl on the left) resembles her mother.
Natural History
Prenatal and Neonatal
Birth records were available for 53 participants. Prenatal problems were noted in eight pregnancies: two had oligohydramnios, one had decreased fetal movement, two had maternal seizures and prenatal exposure to phenobarbital, and two had prenatal exposure to alcohol (timing and amount unspecified). One pregnancy was complicated by hyperemesis gravidarum; fetal ventriculomegaly and abnormal kidney were noted on prenatal ultrasound at 20 weeks gestation. Microarray obtained on the newborn demonstrated Dup7. As indicated in Supplementary Table I, 9 of the 53 (17.0%) were born preterm (range: 27 – 36 weeks gestation, median: 34 weeks). Eight children (four preterm, four full term) required NICU treatment for respiratory distress. Three term infants, all males, had prolonged hospital nursery stays due to feeding difficulty (poor latching on).
Nine newborns (6 males, 3 females) were noted to be large for gestational age, while two (1 male, 1 female) were small for gestational age. Birth weights were available for 50 individuals. Median birth weight was at the 75th centile; for three newborns (6.0%) birth weight was ≤ 5th centile and for 14 (28.0%) birth weight was ≥ 95th centile. Birth length was available for 45 participants. Median birth length was at the 80th centile, with birth length for three newborns (6.7%) ≤ 5th centile and for 13 (28.9%) ≥ 95th centile. Head circumference measurement was noted in 37 newborns. Median head circumference was at the 75th centile with no newborn below the 25th centile; 11 (29.7%) newborns had an OFC ≥ 95th centile (Supplementary Table I).
Malformations or dysmorphic features were noted in the newborn record in 29 of 53 participants (54.7%). Four of the infants had multiple congenital anomalies. A 39-week gestation male (#29) had right diaphragmatic hernia, cubitus valgus, and postaxial polydactyly. A 40-week gestation female (#36) had 11 ribs, unilateral renal agenesis, and Mullerian agenesis. A 40-week gestation male (#9) had hydrocephalus, sagittal craniosynostosis, ventricular septal defect, bowel malrotation, and hydronephrosis. A 39-week gestation female (#64) had ventriculomegaly, left renal agenesis and uterus didelphys. Macrocephaly was the most common physical anomaly, noted in 11 of the 37 newborns who had a head circumference measurement recorded (29.7%). One infant had isolated communicating hydrocephalus. Cardiovascular problems included atrial septal defect and ventricular septal defect in one, subvalvar aortic stenosis in one, and patent ductus arteriosus in four (two requiring surgical intervention and one treated with prostaglandin). Musculoskeletal abnormalities included hemivertebra in one, club foot in three, pectus excavatum in one, and sacral dimple in one. Cryptorchidism was found in three, hypospadias in one, inguinal hernia in one, hydronephrosis in one, and anterior-placed anus in one. One infant had tracheomalacia, two had gastroesophageal reflux, and one had salt wasting congenital adrenal hyperplasia. Dysmorphic craniofacial features were noted in eight newborns (15.1%): frontal bossing (one), hypertelorism (one), flat midface (one), thin upper lip (one), and abnormal ears (four; overfolded helices, low set, posteriorly rotated.)
Craniofacial
Craniofacial features were assessed in both Group 1 and Group 2 (Table II). Macrocephaly (OFC ≥ 95th centile) was present in 32 of 64 (50%). The median head circumference was at the 90th centile. Features present in more than 50% included brachycephaly, broad forehead, straight eyebrows, broad nasal tip, low insertion of the columella, short philtrum, thin upper lip, minor ear anomalies, and facial asymmetry. These salient features comprise a recognizable facial gestalt (Figures 3–6). Review of family photographs indicated that the broad nasal tip, short philtrum, and minor ear anomalies were evident in infancy, while the appearance of the columella changed with age (Figure 7). Columella abnormalities, evidenced by 50 of 64 patients (78.1%), were noted as follows: 26 (40.6%) had low insertion of the columella alone, 5 (7.8%) had low hanging columella alone, and 19 (29.7%) had both low insertion of the columella and low hanging columella. In the individuals > 18 years, 9 of 14 (64.3%) had a low hanging columella as compared to 15 of the 50 individuals < 18 years (30%).
Table II.
Craniofacial features of Classic Dup7 (N = 64)
| Feature | N | % |
|---|---|---|
| Macrocephaly | 32 | 50.0 |
| Microcephaly | 2 | 3.1 |
| Brachycephaly | 42 | 65.6 |
| Dolichocephaly | 3 | 4.7 |
| Plagiocephaly | 2 | 3.1 |
| Normal cranial shape | 17 | 26.6 |
| Broad forehead | 40 | 62.5 |
| High forehead | 13 | 20.3 |
| Straight eyebrows | 33 | 51.6 |
| Deep set eyes | 29 | 45.3 |
| Long eyelashes | 29 | 45.3 |
| Broad nasal tip | 49 | 76.6 |
| Any columella anomaly | 50 | 78.1 |
| Low hanging columella | 24 | 37.5 |
| Low insertion of columella | 35 | 54.7 |
| Short philtrum <3rd% | 12 | 18.8 |
| Short philtrum >3rd%-<25% | 25 | 39.0 |
| Thin upper lip | 52 | 81.3 |
| Diastema | 20 | 31.3 |
| High arched palate | 28 | 43.8 |
| Micrognathia | 19 | 29.7 |
| Ears normal | 24 | 37.5 |
| Ears overfolded helix | 27 | 42.2 |
| Ears lateral protrusion | 11 | 17.2 |
| Ears posterior rotation | 7 | 10.9 |
| Facial asymmetry | 54 | 84.4 |
| Facial asymmetry-right small | 31 | 48.4 |
| Facial asymmetry-left small | 23 | 35.9 |
Figure 3.

Facial features of Dup7 in young children. Top row left to right: 1.25 years, 1.5 years, 2.17 years, 2.5 years. Bottom row left to right: 3.9 years, 4.25 years, 4.25 years, 4.29 years.
Figure 6.

Facial features of adults with Dup7. Top row left to right: 30 years, 31 years (front and profile), 33 years. Bottom row left to right: 39 years, 44 years, 49 years, 60 years.
Figure 7.

Facial features over time. Top row: a boy at ages 1 day, 4 years, and 5 years. Bottom row: a boy at ages 6 months, 7 years, 11.6 years.
Neurological, Neurodevelopmental, and Behavioral
Age at attainment of two developmental milestones was examined, based on parental report. Parents of 49 of the 51 individuals who were 21 years or younger and were walking at the time of the study indicated that they remembered the age at which their child started to walk independently. Median age of independent walking was 1.33 years (mean: 1.48, SD: 0.51), with a range from 0.75 – 3.75 years. Two children (one aged 1.25 years, one aged 1.33 years) had not yet started to walk. Parents of 48 of the 52 individuals who had started to talk at the time of the study indicated that they remembered the age at which their child first produced single words. One child (aged 4.75 years) had not yet started to talk. Median age at onset of single words was 2.00 years (mean: 2.25 years, SD: 0.99) with a range from 1.00 – > 4.75 years. Of the 48 children whose parents recalled both the age at which they started to walk and the age at which they started to talk, 39 children walked first, 8 children began to use single words first, and 1 child reached both milestones at the same age. Parents also were asked how old their child was when he or she was referred for developmental delay; 49 of 53 were able to answer this question. Median age at referral for developmental delay was 1.25 years (mean: 1.46 years, SD: 1.06), with a range from 0.10 – 5.00 years. One adolescent (aged 15.28 years) had never been referred for developmental delay. Of the 47 individuals whose parents recalled both when they began to use single words and when they were referred for developmental delay, 32 were referred before they began to talk, 10 were referred after they had begun to talk, and 5 were referred at about the time that they began to talk.
Neurological problems were common. Seizure disorder was present in 10 of 53 (18.9%). One participant died in her sleep at age 15 years, thought to be related to a nocturnal seizure. Four children were diagnosed with hydrocephalus. Two had ventriculo-peritoneal shunts (one at age 4 months, one at 18 months) and one had a ventriculo-atrial shunt when overproduction of CSF resulting in hydrocephalus was diagnosed at age 3 months. One child had communicating hydrocephalus that was stable over time. In Group 1, 38 participants had neuroimaging studies. MRI or CT images were available for review for 16 participants; all but one was abnormal (Table III). Common MRI findings included varying degrees of ventriculomegaly, thin corpus callosum, increased extra-axial spaces, white matter immaturity and underdevelopment, posterior fossa cysts, and cerebellar vermis hypoplasia. Representative MRI findings are illustrated in Figure 8. For the remaining 22 participants who had had MRI scans only medical records were available (Table IV). Sixteen were reported to be abnormal and 6 normal. Combining the two groups, 31 of 38 studies (81.6%) were abnormal.
Table III.
Participant Characteristics: Age, Sex, OFC Percentile, Height Percentile, Reviewed MRI/CT Findings, and History of Seizures
| # | Age | Sex | OFC% | Ht% | Reviewed MRI Findings | Seizures | |||
|---|---|---|---|---|---|---|---|---|---|
| Cerebellar Vermis | Ventricles | White Matter | Other | ||||||
| 1 | 1.3 | f | 50 | 50 | normal | moderate dilation | thin white matter, thin CC, increased T2 signal | small midbrain and pons | no |
| 39 | 2.2 | f | >98 | 55 | hypoplasia | mild dilation | NAa | increased extra-axial space | no |
| 4 | 3.9 | f | >98 | 95 | normal | asymmetric, mild dilation | normal | large posterior fossa with increased extra-axial space | no |
| 5 | 4.0 | m | >98 | 75 | hypoplasia | mild dilation | thin CC, thin white matter, increased T2 signal occipital lobes | prominent extra axial spaces, posterior fossa cyst | yes |
| 56 | 4.3 | m | >98 | 90 | mild hypoplasia | moderate dilation | rounded CC, thin white matter | Increased extra-axial space | yes |
| 9 | 4.6 | m | 35 | 15 | hypoplasia | shunted hydrocephalus | collapsed CC (secondary to shunt) | large frontal lobes, small pons, increased extra-axial space posterior fossa | yes |
| 12 | 5.3 | m | 50 | 35 | normal | normal | increased T2 signal occipital lobes | increased extra-axial space posterior fossa | yes |
| 13 | 5.6 | f | 95 | 90 | mild hypoplasia | asymmetric, mild dilation | increased T2 signal, thin white matter, rounded CC | increased extra-axial fluid | no |
| 15 | 5.9 | f | >98 | 5 | normal | moderate dilationb | increased T2 signal, thin rounded CC, thin white matter | cerebellar tonsil ectopia, increased extra-axial space | no |
| 17 | 6.8 | f | 45 | 15 | mild anterior atrophy | normal | increased T2 signal frontal lobes | cavum septum pellucidum | no |
| 22 | 7.9 | m | 60 | 10 | normal | Mild asymmetry lateral ventricles | mildly thin white matter | no | |
| 23 | 9.7 | f | >98 | 95 | normal | mild dilation | Immature white matter, increased perivascular space | increased extra-axial space | yes |
| 26 | 10.9 | m | >98 | 5 | mild hypoplasia | asymmetric, mild dilation | normal | posterior fossa cyst | no |
| 27 | 11.0 | f | 80 | 70 | mild anterior atrophy | mild dilation | mildly thin posterior white matter | no | |
| 29 | 12.2 | m | 45 | 20 | hypoplasia | mild dilation | thin CC, thin white matter | Posterior fossa cyst | no |
| 30 | 13.7 | f | >98 | 95 | small, mild atrophy, prominent folia | mild dilation | increased T2 signal frontal, parietal, occipital lobes | no | |
Abbreviations: OFC% = head circumference percentile; Ht% = height percentile; CC = corpus callosum
Only CT scan available for review.
MRI done at age 17 months. At 18 months had VP shunt placed for hydrocephalus.
Figure 8.
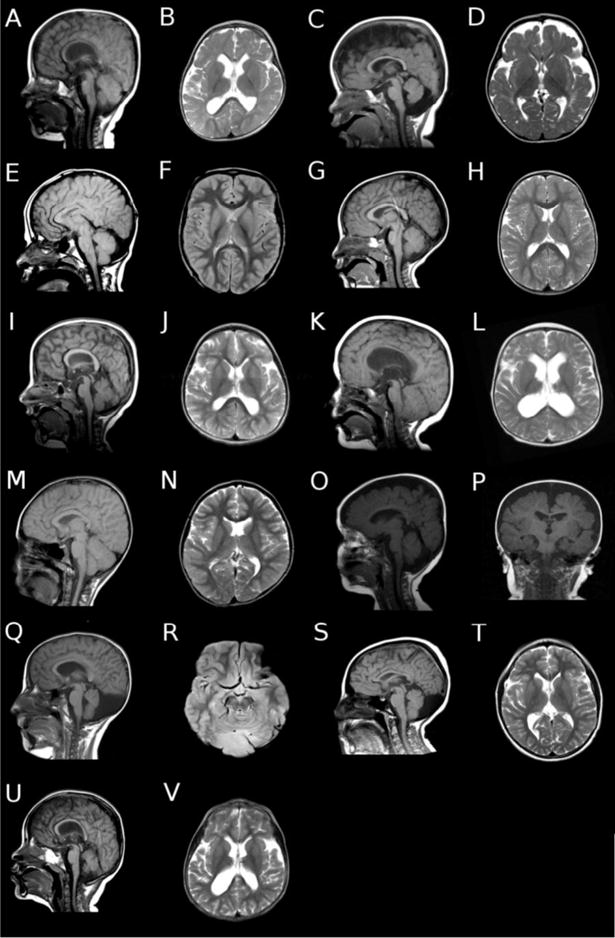
Representative brain MRI findings in individuals with Dup7. (A) Sagittal and (B) axial images of patient #1 showing thin corpus callosum and white matter, with mildly dilated lateral ventricles. (C) Sagittal and (D) axial images of patient #5 showing cerebellar vermis hypoplasia, thin corpus callosum and white matter, and increased cortical extra-axial space. (E) Sagittal and (F) axial images of patient #9 showing collapsed ventricles status post ventricular peritoneal shunting for hydrocephalus, and mildly increased extra-axial space in the posterior fossa. (G) Sagittal and (H) axial images of patient #12 showing normal brain morphology. (I) Sagittal and (J) axial images of patient #13 showing rounded corpus callosum, thin white matter, mild dilation of the lateral ventricles, and mild cerebellar vermis hypoplasia. (K) Sagittal and (L) axial images of patient #15 showing rounded corpus callosum, moderate dilation of the lateral ventricles, and thin white matter. There is also mild cerebellar tonsillar ectopia. (M) Sagittal and (N) axial images of patient #22 showing mild asymmetry of the lateral ventricles, probably due to mildly thin white matter, but otherwise normal brain morphology. (O) Sagittal and (P) coronal images of patient #23 showing increased extra axial spaces around both the cortex and in the posterior fossa. (Q) Sagittal and (R) axial images of patient #26, showing cerebellar vermis hypoplasia, and extra axial fluid collection in the posterior fossa. (S) Sagittal and (T) axial images of patient #29 showing mildly thin white matter, mildly dilated lateral ventricles, and cerebellar vermis hypoplasia with an extra axial fluid collection in the posterior fossa. (U) Sagittal and (V) axial images of patient #56 showing rounded corpus callosum, thin white matter, and moderately dilated lateral ventricles. The cerebellar vermis is mildly hypoplastic.
Table IV.
Participant Characteristics: Age, Sex, OFC Percentile, Height Percentile, MRI Findings from Medical Records, and History of Seizures
| # | Age | Sex | OFC% | Ht% | MRI Findings | Seizures | ||
|---|---|---|---|---|---|---|---|---|
| Overall | Ventricles | Other | ||||||
| 64 | 1.25 | f | 98 | 45 | abnormal | enlarged | white matter intensities, thin corpus callosum, prominent perivascular spaces | no |
| 52 | 1.5 | f | 40 | 50 | abnormal | white matter lesions | no | |
| 41 | 2.0 | f | 50 | 25 | NA | no | ||
| 59 | 2.2 | f | 50 | 3 | abnormal | mild volume loss, delayed myelination | no | |
| 2 | 2.5 | m | 50 | 35 | normal | no | ||
| 3 | 2.8 | f | >98 | 5 | normal | no | ||
| 6 | 4.1 | f | >98 | 90 | abnormal | enlarged | increased signal white matter | no |
| 53 | 4.3 | m | 50 | 75 | abnormal | volume loss on right (postoperative stroke) | yes | |
| 7 | 4.3 | m | >98 | 25 | abnormal | enlarged | prominent sulci, thin corpus callosum | no |
| 8 | 4.3 | m | >98 | 55 | abnormal | communicating hydrocephalus | no | |
| 42 | 4.3 | f | 5 | 15 | NA | no | ||
| 10 | 4.7 | m | >98 | 90 | abnormal | subcortical hyperintensities frontal lobe | no | |
| 11 | 4.9 | f | 95 | 90 | NA | no | ||
| 14 | 5.7 | f | >98 | 50 | normal | no | ||
| 16 | 6.4 | m | >98 | 60 | abnormal | optic nerve tortuosity, arachnoid cyst | yes | |
| 18 | 6.9 | f | 60 | 90 | abnormal | enlarged | posterior fossa cyst | no |
| 19 | 7.1 | m | >98 | 75 | NA | no | ||
| 47 | 7.3 | m | 50 | 50 | abnormal | enlarged | delayed myelinization | no |
| 20 | 8.8 | m | 15 | 50 | normal | yes | ||
| 21 | 8.8 | m | 2 | 25 | NA | no | ||
| 48 | 9.0 | f | 50 | 10 | NA | no | ||
| 57 | 9.8 | f | >98 | 95 | NA | no | ||
| 24 | 9.9 | f | 50 | 30 | NA | no | ||
| 25 | 10.0 | m | 50 | 65 | abnormal | hydrocephalusa | no | |
| 45 | 10.3 | m | 20 | <3 | NA | no | ||
| 61 | 10.5 | m | 90 | 95 | NA | no | ||
| 50 | 10.9 | m | >98 | <3 | abnormal | small pituitary | no | |
| 28 | 11.6 | m | 90 | 50 | abnormal | decreased white matter volume, thin corpus callosum | no | |
| 62 | 13.3 | f | >98 | 50 | NA | yes | ||
| 31 | 13.9 | m | 90 | 75 | NA | no | ||
| 32 | 13.9 | m | >98 | 75 | abnormal | prominent perivascular spaces | no | |
| 33 | 14.8 | f | 40 | 3 | abnormal | enlarged | no | |
| 34 | 15.3 | f | 95 | 5 | normal | no | ||
| 35 | 15.3 | f | 95 | 75 | NA | no | ||
| 38 | 18.2 | m | 95 | 80 | NA | no | ||
| 36 | 18.9 | f | <2 | <3 | NA | yes | ||
| 37 | 21.3 | f | >98 | 95 | normal | no | ||
Abbreviations: OFC% = head circumference percentile; Ht% = height percentile; NA = Not applicable (no MRI performed).
Hydrocephalus due to overproduction of cerebral spinal fluid, treated with a ventricular-atrial shunt.
Abnormalities were noted on neurological examination in 47 of 53 (88.7%) participants in Group 1 (Table V). One of the most common findings was hypotonia, present in 31 of 53 (58.5%). Abnormalities of gait and station suggested cerebellar dysfunction in 33 of 53 (62.3%). While gait and station tasks such as hopping, balancing, dysmetria and stressed gait reach ceiling at age 7 years in typically developing children [Gidley Larson et al., 2007], 14 of the 27 (51.8%) individuals over age 7 in this series demonstrated deficits. Involuntary overflow movements occurring with an intentional movement were common (83% of those older than 14 years in the Dup7 group) as compared to the typically developing population in which the movements gradually disappear between ages 7 – 14 years, thought to be related to maturity of the cortical spinal tracts [Gidley Larson et al., 2007]. Of the 31 children assessed for Developmental Coordination Disorder, 23 (74.2%) met DSM-5 diagnostic criteria. The children were often described as clumsy, were slow to develop coordinated motor skills, and had difficulty participating in physical activities with peers. Of the 46 children we evaluated for Speech Sound Disorder (almost all as part of Mervis et al. [2015]), 38 (82.6%) met DSM-5 diagnostic criteria. The affected children had problems with articulation and phonology that interfered with verbal communication. All of the children except the two youngest, including those who did not have a current SSD diagnosis, were receiving or had previously received speech therapy. Excluding two 4-year-old boys with ASD and severe developmental delay, mean GCA (similar to IQ) on the Differential Ability Scales-II [Elliott, 2007] was 80.54 (SD: 14.93, range: 42 – 107) for the children with SSD and 94.50 (SD: 11.90, range: 79 – 118) for the children who did not have SSD.
Table V.
Participant Characteristics: Neurological Examination Findings, Developmental Coordination Disorder, and Speech Sound Disorder
| # | Age | Neurological Examinationa | DCD | SSD | ||||
|---|---|---|---|---|---|---|---|---|
| Overall | Cranial Nerves | Adventitious Movements | Gait and Station | Hypotonia | ||||
| 64 | 1.2 | abnormal | asymmetric crying face | yes | NEL | |||
| 1 | 1.3 | abnormal | asymmetric crying face | yes | NEL | |||
| 52 | 1.5 | normal | no | yes* | ||||
| 41 | 2.0 | normal | no | NEL | ||||
| 39 | 2.2 | abnormal | yes | yes* | ||||
| 59 | 2.2 | abnormal | yes | NEL | ||||
| 2 | 2.5 | normal | no | yes* | ||||
| 3 | 2.8 | abnormal | yes | yes* | ||||
| 4 | 3.9 | abnormal | wide based gait | no | yes* | |||
| 5 | 4.0 | abnormal | hearing loss aided | wide based gait | no | yes* | ||
| 6 | 4.1 | abnormal | not able to jump, walk on toes or heels | yes | yes | |||
| 53 | 4.3 | abnormal | no | yes | ||||
| 56 | 4.3 | abnormal | esotropia | no | yes | |||
| 7 | 4.3 | abnormal | repetitive movements | wide based gait | yes | yes | ||
| 8 | 4.3 | abnormal | esotropia | wide based gait | yes | yes | ||
| 42 | 4.3 | abnormal | esotropia, ptosis | wide based gait | yes | not eval. | ||
| 9 | 4.6 | abnormal | esotropia | wide based gait | yes | yes* | ||
| 10 | 4.7 | abnormal | tremor with excitement | wide based gait | yes | yes | ||
| 11 | 4.9 | abnormal | unable to hop | yes | yes | |||
| 12 | 5.3 | abnormal | high pitched voice | tics | unable to hop | yes | yes | yes |
| 13 | 5.6 | abnormal | unable to jump, dysmetria | no | yes | yes* | ||
| 14 | 5.7 | abnormal | hearing loss aided | abnormal unipedal stance | yes | yes | yes | |
| 15 | 5.9 | abnormal | difficulty jumping, heel walking | yes | yes | yes | ||
| 16 | 6.4 | abnormal | high pitched voice | abnormal unipedal stance | yes | yes | yes | |
| 17 | 6.8 | abnormal | high pitched voice | abnormal tandem gait | no | no | yes | |
| 18 | 6.9 | abnormal | hypernasal voice, drooling | abnormal left unipedal stance, dysmetria, abnormal tandem gait | yes | yes | no | |
| 19 | 7.1 | abnormal | intention tremor | awkward run | no | yes | yes | |
| 47 | 7.3 | abnormal | tics | abnormal unipedal stance, abnormal tandem gait | yes | yes | yes | |
| 22 | 7.9 | abnormal | high pitched voice | dysmetria, abnormal tandem gait, abnormal unipedal stance | no | yes | yes | |
| 20 | 8.8 | abnormal | asymmetric crying face | abnormal unipedal stance | no | no | yes | |
| 21 | 8.8 | abnormal | dysmetria, abnormal unipedal stance | no | no | yes | ||
| 48 | 9.0 | abnormal | pressured speech | motor overflow | yes | no | no | |
| 23 | 9.7 | abnormal | abnormal unipedal stance | yes | yes | yes | ||
| 57 | 9.8 | normal | no | no | no | |||
| 24 | 9.9 | abnormal | facial grimacing, hypernasal voice | poor unipedal stance left | yes | yes | yes | |
| 25 | 10.0 | abnormal | intention tremor, motor overflow | dysmetria, abnormal tandem gait and unipedal stance | yes | yes | yes | |
| 45 | 10.3 | abnormal | awkward run | yes | no | yes | ||
| 61 | 10.5 | abnormal | hearing loss, abnormal oral motor movement | no | no | yes | ||
| 50 | 10.9 | abnormal | difficult oral motor movements, facial grimacing | synkinesia, motor overflow | yes | yes | yes | |
| 26 | 10.9 | abnormal | motor overflow | abnormal unipedal stance left, | yes | yes | no | |
| 27 | 11.0 | abnormal | whispered speech | dysmetria, abnormal unipedal stance | no | yes | yes | |
| 28 | 11.6 | abnormal | esotropia, high pitched voice | motor overflow | abnormal unipedal stance | yes | yes | yes |
| 29 | 12.2 | abnormal | awkward gait | no | yes | no | ||
| 62 | 13.3 | normal | no | yes | no | |||
| 30 | 13.7 | abnormal | asymmetric crying face, soft voice | no | yes | no | ||
| 31 | 13.9 | abnormal | hypernasal voice | abnormal tandem gait, | yes | yes | no | |
| 32 | 13.9 | abnormal | exotropia, soft voice | abnormal unipedal stance | yes | yes | yes | |
| 33 | 14.8 | abnormal | exotropia, hypernasal voice | right motor overflow | awkward running, skipping, abnormal unipedal stance | no | yes | yes |
| 34 | 15.3 | abnormal | high pitched voice | motor overflow | yes | no | yes | |
| 35 | 15.3 | abnormal | esotropia, soft voice | motor overflow | abnormal tandem gait | yes | yes | yes |
| 38 | 18.2 | abnormal | difficulty lifting tongue tip, pressured speech | motor overflow left hand | yes | yes | ||
| 36 | 18.9 | abnormal | synkinesia | no | not eval. | |||
| 37 | 21.3 | normal | no | no | ||||
Abbreviations: DCD = Developmental Coordination Disorder, SSD = Speech Sound Disorder, NEL: Not enough language to evaluate for SSD, yes* = SSD evaluation took place at an older age and child was found to have SSD, not eval. = not evaluated.
Note: SSD evaluations were conducted as part of Mervis et al. [2015].
Additional findings: #1: left side muscle weakness; #6: pronator drift; # 25: Left lower extremity muscle weakness; #26: poor fine motor coordination; #30: hyperreflexia lower extremities; # 38: clonus at ankles; #47: mild diffuse weakness; #53: left hemiparesis.
The parents of the 53 individuals aged 21 years or younger were asked to describe any behavioral concerns they had. The most common concern was anxiety, which was raised by parents of 38 individuals (71.7%). The most frequently mentioned types were separation anxiety (especially for young children) and social anxiety; obsessive compulsive behavior was mentioned by parents of five individuals. Of the 46 children whose parents completed the Anxiety Disorders Interview Schedule for DSM-IV: Parent [ADIS-P; Silverman and Albano, 1996], 7 (15.2%) met criteria for separation anxiety disorder, 22 (47.8%) met criteria for social phobia, and 1 (2.2%) met criteria for obsessive compulsive disorder. Most of these interviews were conducted as part of Mervis et al. [2015]. Mood swings were mentioned by parents of three individuals. Nine individuals (17.0%) were being treated with SSRIs (8 for anxiety, 1 for mood swings). Another frequent concern was attention problems, which were mentioned by parents of 17 individuals (32.1%, 13 of whom also had anxiety problems or mood swings). Based on the ADIS-P interview, 17 of 46 children (37.0%) met criteria for ADHD. Eleven children (18.9%) were medicated for attention problems, primarily with stimulants. Parents of seven children (13.2%, five of whom also had attention problems) mentioned concerns with aggression or defiance. Based on the ADIS-P interview, 12 of 46 children (26.1%) met criteria for either ODD or DBD-NOS. Seven individuals (13.2%, most of whose parents had not mentioned aggression or defiance) were treated or had been treated in the past with atypical antipsychotics, and three children were treated with a mood stabilizer (valproic acid). Based on parental report, 10 of the children (18.9%) had previously been diagnosed with ASD. Parents of 14 children (26.4%, the oldest of whom was 13½ years) indicated that their child had a high pain tolerance. Examples provided of situations in which a strong reaction to pain would have been expected but in which the child showed little or no reaction included slamming a finger in a car door, fracturing a foot or arm, severe lacerations resulting from a fall, and severe suppurative otitis media.
The 11 adults in Group 2 were interviewed regarding past school and behavioral history. Seven reported difficulties with learning, and four remembered having speech therapy in elementary school. Five had dropped out of high school, three had graduated from high school, one had graduated from community college with an associate degree, and two had graduated from college with a bachelor degree. The two who had a bachelor degree received services for students with learning disabilities while in college. Occupations included roofer, cook, newspaper deliverer, day care teacher, laborer, computer technician, and homemaker. Six noted that they were uncomfortable talking to people. Seven of the adults completed the Anxiety Disorders Interview Schedule for DSM-IV [ADIS-IV; Brown et al., 1996] as part of the Mervis et al. [2015] study; five (71.4%) met criteria for social phobia. Two reported past medical treatment for depression, and two stated that they had been treated for bipolar disorder. Three adults reported that they had been in trouble with the law.
Endocrinological
Five children were being treated for growth hormone deficiency (9.4%), a problem not previously reported in Dup7. Median height for Group 1 was at the 50th centile. Nine children (17%) had height ≤ 5th centile and six (11%) had height at the 95th centile. One child was being treated for hypothyroidism. One teenage girl was being evaluated for delayed puberty. One girl was treated for salt wasting congenital adrenal hyperplasia. One adult began treatment for hypothyroidism following her research clinic visit. One adult had type 2 diabetes mellitus.
Ophthalmological and Otolaryngological
Esotropia was diagnosed in six children (11.3%); one was treated surgically. Two children had exotropia. Three of the children had hearing loss; two of the three wore hearing aids. Thirteen of the child participants (24.5%) had chronic otitis media in early childhood and eight were treated with ventilating tubes. Three children had frenulectomy for ankyloglossia. Eight children had tonsillectomy and adenoidectomy. One child had obstructive sleep apnea treated with CPAP.
Cardiovascular
Neonatal diagnoses of cardiovascular problems included septal defects in two, subvalvar aortic stenosis in one, and PDA in four. Four additional children were diagnosed with PDA between ages 1 and 8 years, for a rate of PDA of 15.1% for Group 1.
Recently, two groups of investigators reported aortic dilation in individuals with Dup7 [Zarate et al., 2014; Parrott et al., 2015]. At about the same time, we learned that one of the participants in Group 2 (patient #55) had just had repair of an aneurysm of the ascending aorta (diameter = 5.3cm) and of the abdominal aorta at age 66 years. We then suggested echocardiographic evaluation for ascending aorta dilation to the other participants. At the time of this report, 26 had echocardiograms (Table VI) and 12 demonstrated aortic dilation (46.2%). The age range of the affected individuals was 1.18 – 66 years.
Table VI.
Echocardiogram Results
| Number | Age Echo 1 | Echo 1 Result | Age Echo 2 | Echo 2 Resulta |
|---|---|---|---|---|
| 42 | 0.2 | PDA, subvalvar aortic stenosis | 5 | trace aortic insufficiency |
| 53 | 1 | VSD, ASD, mitral and tricuspid regurgitation | 10 | normal |
| 9 | 1 | VSD | 11 | dilated aortic root and ascending aorta (Z = 3.7) |
| 64 | 1.18 | ascending aorta dilation (Z = 5.48) | 1.38 | ascending aorta dilation (Z = 5.55) |
| 7 | 2 | normal | ||
| 8 | 2 | aortic dilation (Z = 3) | 4 | aortic dilation (Z = 2.62) |
| 14 | 2.66 | normal | ||
| 47 | 3.8 | normal | ||
| 6 | 4 | PDA, normal aorta | ||
| 12 | 4 | normal | ||
| 10 | 4.8 | normal | ||
| 11 | 5 | mild aortic dilation | ||
| 41 | 8 | trivial PDA, ascending aorta dilation (Z = 2.2) | ||
| 13 | 8 | aortic dilation | ||
| 52 | 8.5 | normal | ||
| 26 | 9 | normal | ||
| 27 | 9 | normal | ||
| 23 | 11 | normal | ||
| 28 | 11.5 | aortic dilation (Z = 2.09) | ||
| 32 | 12 | mild tricuspid insufficiency | ||
| 35 | 15 | aortic dilation | ||
| 21 | 17 | normal (Z = 1.4) | ||
| 38 | 18 | ascending aorta dilation (Z = 2.1) | ||
| 37 | 20 | normal | 24 | aortic root and ascending aorta dilation |
| 54 | 38 | Ascending aorta dilation (4.3cm) | ||
| 55 | 66 | ascending aorta aneurysm (5.3cm), mitral regurgitation, aortic insufficiency |
Note: Z scores were not available in all reports. Abbreviations: echo = echocardiogram; PDA = patent ductus arteriosus; VSD = ventricular septal defect; ASD = atrial septal defect.
For those participants who had two echocardiograms, the second one was done in 2014 specifically to evaluate for aortic dilation.
Gastrointestinal
Four children had significant problems with feeding requiring feeding via gastrostomy in early childhood; one was still tube fed at age 12 years. Four children had gastroesophageal reflux in infancy. Chronic constipation was a significant problem in 35 of 53 (66.0%) of Group 1. Of those with chronic constipation, 7 (20.0%) had encopresis. Four children required hospital admissions for disimpaction and colon clean out. One child had a motility study demonstrating slow motility and required a colostomy. Manometry in one 9-year-old girl was positive for low anal sphincter tone. Ganglion cells were normal in two children who had bowel biopsy. Constipation was reported to be a problem in 3 of 11 (27.3%) adults in Group 2.
Genitourinary
A renal ultrasound had been performed in 22 individuals in Group 1. Eighteen (81.8%; 8 girls, 10 boys) had normal studies. Of the remaining individuals, two boys had hydronephrosis, one girl had unilateral renal agenesis and Mullerian agenesis, and one girl had unilateral renal agenesis and uterus didelphys. One girl was diagnosed with delayed bladder emptying. Three boys required orchiopexy and two boys had bilateral inguinal herniorraphy. One adult female in Group 2 had unilateral renal agenesis.
Musculoskeletal
Musculoskeletal problems were noted in 15 children (28.3%) including hyperextensible joints (five), pectus excavatum (two), lordosis (two), pes planus (two), and avascular necrosis of the hip (one). The three children with club feet noted at birth were successfully treated with casting.
Integumentary
Of the 47 children < age 14 years at the time of the examination, 21 (44.7%) had cutis marmorata.
DISCUSSION
Dup7 (MIM 609757) is caused by the reciprocal duplication of the 26 genes that are deleted in WS (MIM 194050). As the use of chromosome microarray increases in the evaluation of patients with malformations, developmental delay, and/or behavior problems, more individuals with Dup7 will be identified. The results of this systematic study (the largest case series reported) inform clinicians and families about the variability of the phenotype, common medical problems, and the natural history of the syndrome. In the remainder of the Discussion, we offer recommendations for health care supervision for individuals with Dup7 based on the findings from our case series (Table VII). We also compare and contrast the characteristics associated with Dup7 to those associated with WS and briefly discuss future research.
Table VII.
Suggested Evaluations and Health Surveillance/Treatment for Dup7
| System | Initial Evaluation | Surveillance/Treatment |
|---|---|---|
| Genetic | Evaluate parents for duplication | Provide genetic counseling |
| Neurological | Neurological examination MRI of the brain Physical therapy evaluation Occupational therapy evaluation |
Monitor head circumference Treat seizures Physical therapy for Developmental Coordination Disorder Occupational therapy if needed |
| Psychological/Psychiatric | Developmental evaluation Neuropsychological evaluation Gold-standard autism evaluation |
Treat ADHD, Anxiety, ODD Autism treatment if needed |
| Speech and Language | Speech-language evaluation | Speech therapy for Speech Sound Disorder, language therapy if language disorder is present |
| Endocrinological | If short stature is present, test for growth hormone deficiency | Monitor growth |
| Cardiovascular | Evaluate for PDA Echocardiogram measurement of the aortic root and ascending aorta with calculation of Z scores |
Yearly echocardiogram, but more frequently if aortic dilation is detected |
| Gastrointestinal | Evaluate for constipation | Aggressively treat constipation |
| Genitourinary | Renal ultrasound Evaluate for cryptorchidism |
If renal malformation is present in a female, evaluate Mullerian structures |
Abbreviations: ADHD = attention deficit hyperactivity disorder; ODD = oppositional defiant disorder; PDA = Patent ductus arteriosus
Genetics
While the facial phenotype is recognizable, it is likely that most children with Dup7 will be diagnosed when evaluated for developmental delay. Diagnosis of a child should prompt genetic studies on the parents (FISH, qPCR, MLPA, or chromosome microarray) because 27% of probands will have an affected parent, who may have a milder phenotype. While some parents with Dup7 reported in the literature were said to have a normal phenotype, detailed clinical assessment was not performed. In our experience, all of the parents had sufficient clinical findings (craniofacial, medical, behavioral, speech, and/or developmental) to make a diagnosis of Dup7. WS is also inherited in an autosomal dominant fashion, but few affected individuals reproduce, resulting in rare familial cases. As a comparison, in the most common autosomal deletion (22q11.2 deletion, MIM 192430) and reciprocal duplication (22q11.2 duplication, MIM 608383) syndromes, 10% of individuals with the deletion have an affected parent [McDonald-McGinn et al., 2001] while 70% of individuals with the duplication have a parent with the duplication, who may have a milder phenotype [Wentzel et al., 2008]. Just as in WS, the WS region inversion polymorphism [Hobart et al., 2010] has now been documented in parents of individuals with Dup7. The current study did not have sufficient numbers to determine if the presence of the inversion polymorphism increases the chance to have a child with Dup7, but we have previously shown that individuals with the inversion polymorphism have a fivefold-increased likelihood of having a child with WS [Hobart et al., 2010], and they likely also have an increased chance to have a child with Dup7 [Hobart et al, 2010]. It is important that families of a proband be offered genetic counseling. Parent of origin has been determined for a small number of individuals with de novo Dup7; the numbers of mothers and fathers are near equal, which is expected since WS has an equal proportion of maternal and paternal parent of origin [Hobart et al., 2010].
Neurology, Development, and Behavior
Dup7 has its greatest impact on the neurological system. There are structural and functional abnormalities of the central nervous system demonstrated on MRI neuroimaging and neurological examination in over 80%. The 5.6% prevalence of hydrocephalus requiring shunting as well as the presence of macrocephaly in 50% justifies obtaining MRI of the brain for affected individuals at the time of diagnosis. Head circumference should be routinely measured, especially in young children. In WS, microcephaly is more common, found in one third of affected individuals [Pankau et al., 1994]. The presence of cerebellar vermis hypoplasia in Dup7 is interesting in contradistinction to the increased cerebellar volume in WS relative to total brain volume [Jones et al., 2002; Prabhakaran et al., 2014] and suggests that dosage sensitive gene(s) in the WS region are important in structural development of the central nervous system. In WS, the mismatch between reduced posterior fossa size coupled with increased cerebellar volume may contribute to Chiari 1 malformation found in some affected individuals [Pober & Filiano, 1995; Mercuri et al., 1997]. One brain volumetric study comparing children with WS, children with Dup7, and typically-developing controls showed that relative to total brain volume, the ventricles were larger and the cerebellum was smaller in Dup7 compared to both controls and WS [Prabhakaran et al., 2014]. The clinical MRI findings in the present study are consistent with those observations. Other differences noted in the volumetric study included a larger putamen volume and caudate volume in Dup7 relative to total brain volume compared to both controls and WS, and smaller hippocampus volume and amygdala volume relative to total brain volume in Dup7 compared to WS (but not in comparison to controls) [Prabhakaran et al,. 2014]. Future studies comparing brain structure and function of WS and Dup7 should employ additional methodologies including voxel based morphometry, diffusion tensor imaging, and functional MRI. One functional MRI study in a single individual with Dup7 showed lack of normal activation of the emotion processing areas (amygdala, orbital frontal cortex) on a facial expression task [Prontera et al., 2014]. Taken together, the data suggest that dosage sensitive genes affect brain structure and function as they relate to both social phobia (Dup7) and social disinhibition (WS).
The problems of hypotonia and abnormalities of gait and station combine to produce Developmental Coordination Disorder in three quarters of children with Dup7. Physical therapy assessment is indicated, with therapy provided as necessary. Both Dup7 and WS are associated with difficulties with balance: 52% of children aged 5 years or older in this series compared to 93% of children with WS [Gagliardi et al., 2007]. One key difference in the neurological examination is that 42% of children with WS develop hyperreflexia in the lower extremities over time [Morris et al., 1988; Gagliardi et al., 2007], while only two individuals with Dup7 (3.8%) had this sign. Mild dysmetria is present in both groups: Dup7, 11% in this series; WS, 31% [Gagliardi et al., 2007]. Adults with WS frequently have cerebellar signs including ataxia, dysmetria, and tremor [Pober & Morris 2007]; a systematic study of adults with Dup7 will be required to determine if cerebellar signs persist into adulthood. In most children with Dup7 the first noted delayed developmental milestone is speech; Speech Sound Disorder is present in 80%. Early speech evaluation and therapy is important in Dup7 to improve verbal communication. Because cognitive ability varies widely in Dup7, psychological assessment is indicated, even though the median IQ or DQ is in the low average range [Mervis et al., 2015].
Families struggle with the maladaptive behavior common in Dup7 that includes disorders of attention, anxiety, and opposition. Individuals with Dup 7 should be screened and treated for these disorders. Compared to WS in which social disinhibition is common, individuals with Dup7 are more likely to have separation anxiety or social phobia [Mervis et al., 2012; Mervis et al., 2015], another interesting contrast between the effects of deletion versus duplication on the phenotype. Both syndromes are associated with a high rate of specific phobia: 53.2% for Dup7 [Mervis et al., 2015] and 53.8% for WS [Leyfer et al., 2006]. Medical management of behavior for individuals with comorbid disorders of anxiety and attention may be challenging; pediatric psychiatric assessment and treatment may be beneficial. Autism spectrum disorders are more common in Dup7 than in the general population. In this series, some young children had been diagnosed with autism because they had anxiety and delayed speech, but did not meet the criteria for an autism diagnosis when assessed by gold standard instruments. Other children did meet criteria for ASD based on gold-standard assessment. Careful and systematic differential diagnosis is needed.
Endocrinological
This study is the first to report growth hormone deficiency in a Dup7 cohort. Because the median height in Dup7 is at the 50th centile, a child with Dup7 and short stature should have growth hormone studies. There have been rare case reports of growth hormone deficiency in WS [e.g., Kuijpers et al., 1999; Xekouki et al., 2005], but growth hormone deficiency was not increased in a series of patients with WS [Partsch et al., 1994] even though short stature is a common feature.
Cardiovascular
Congenital heart disease is a relatively low frequency finding in Dup7 with PDA being the most common anomaly. Of much greater concern is the presence of aortic dilation with a prevalence of 46.2% in this series. Individuals with Dup7 should have regular echocardiographic measurement of the aorta (aortic root, sinotubular junction, and ascending aorta) with calculation of Z scores in order to monitor for potential progression of dilation. There have not been sufficient numbers of individuals with Dup7 studied longitudinally to know the optimal frequency for monitoring; a prospective longitudinal multi-center study would be ideal to provide evidence-based recommendations [Parrott et al., 2015]. The most common cardiovascular abnormality in WS is supravalvar aortic stenosis (SVAS) [Morris et al., 1988] which is due to deletion of the elastin gene, located in the WS region [Ewart et al., 1993]. In WS, the SVAS may worsen, especially in the first five years of life, and 20 – 30% require corrective surgery [Collins et al., 2010]. It is likely that duplication of ELN, encoding the important structural protein elastin, predisposes to aortic dilation in Dup7. No histopathologic study of aortic tissue removed during aortic surgery in Dup7 has yet been reported; it would be interesting to compare elastic fiber morphology and lamellar unit structure of the aorta in the two conditions.
Gastrointestinal and Genitourinary
Chronic constipation was a serious problem in two thirds of children with Dup7 and is likely due to a combination of factors such as central hypotonia, slow gut motility, and stool withholding related to anxiety. Early aggressive treatment of constipation is warranted, because development of encopresis (20%) further compounds the difficulty with peer interactions. Chronic constipation is also a problem for ~50% of individuals with WS [Morris et al., 1988], with an increased prevalence of diverticulitis [Stagi et al, 2010].
A renal ultrasound is warranted at the time of diagnosis of Dup7 because kidney problems (unilateral renal agenesis, hydronephrosis) have been described in 18% of those studied. Females with renal malformation or agenesis should have investigation of Mullerian structures. Structural abnormalities of the urinary tract are reported in 35 – 50% of individuals with WS [Pober et al., 1993; Sforzini et al., 2002].
Future Directions
This study documents the clinical features, medical problems, phenotypic variability, and natural history of Dup7. The phenotypic information presented for this relatively large case series allows formulation of initial medical recommendations for evaluation and surveillance with the goal of improving outcome through early intervention and prevention of possible complications. Additional prospective and longitudinal studies of the Dup7 population will be required to refine these recommendations based on evidence. Especially important topics for future study include prospective studies regarding aortic dilation and evaluation of correlations between central nervous system structure and function compared to neurological, developmental, and behavior abnormalities. The present study also serves as a baseline to begin to address genotype phenotype correlations by comparing the classic Dup7 phenotype to the phenotypes associated with specific shorter or longer duplications.
Supplementary Material
Figure 4.

Facial features of Dup7 in mid childhood. Top row left to right: 5.7 years, 5.91 years, 6.78 years, 7.09 years, 7.3 years. Bottom row left to right: 7.99 years, 9.75 years, 10 years (front and profile), 10.3 years.
Figure 5.

Facial features of adolescents with Dup7. Top row left to right: 10.5 years (front and profile), 11 years (front and profile). Middle row left to right: 12.18 years, 13.25 years, 13.67 years, 13.99 years. Bottom row left to right: 13.99 years, 15.28 years (front and profile), 18.9 years.
Acknowledgments
We are grateful to the participants and their families. We thank Cecilia Rios for contributing the FISH photographs for Figure 1. We thank Holly H. Ardinger, Alexander Asamoah, Kimberly A. Farinella, Maricela Gulbronson, Kelly Jackson, Anna Sosa, and Anne Tsai for referring patients to the study.
Grant sponsor: Simons Foundation; Grant number: SFARI # 238896
Grant sponsor: National Institute of Neurological Disorders and Stroke; Grant number: R01 NS35102
Grant sponsor: National Institute of Child Health and Human Development; Grant number: R37 HD29957
References
- Berg JS, Brunetti-Pierri N, Peters SU, Kang S-HL, Fong CT, Salamone J, Freedenberg D, Hannig VL, Prock LA, Miller DT, Raffalli P, Harris DJ, Erickson RP, Cunniff C, Clark GD, Blazo MA, Peiffer DA, Gunderson KL, Sahoo T, Patel A, Lupski JR, Beaudet AL, Cheung SW. Speech delay and autism spectrum behaviors are frequently associated with duplication of the 7q11.23 Williams-Beuren syndrome region. Genet Med. 2007;9:427–441. doi: 10.1097/gim.0b013e3180986192. [DOI] [PubMed] [Google Scholar]
- Brown TA, DiNardo P, Barlow DH. Anxiety Disorder Interview Schedule Adult Version (ADIS-IV) San Antonio, TX: Graywind Publications; 1996. [Google Scholar]
- Collins RT, 2nd, Kaplan P, Somes GW, Rome JJ. Long-term outcomes of patients with cardiovascular abnormalities and Williams syndrome. Am J Cardiol. 2010;105:874–878. doi: 10.1016/j.amjcard.2009.10.069. [DOI] [PubMed] [Google Scholar]
- Değerliyurt A, Ceylaner S, Ozdağ H. A 7q11.23 microduplication patient with cerebral palsy and facial dysmorphism. Genet Couns. 2012;23:263–267. [PubMed] [Google Scholar]
- Depienne C, Heron D, Betancur C, Benyahia B, Trouillard O, Bouteiller D, Verloes A, Leguern E, Leboyer M, Brice A. Autism, language delay and mental retardation in a patient with 7q11 duplication. J Med Genet. 2007;44:452–458. doi: 10.1136/jmg.2006.047092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depienne C, Heron D, Betancur C, Benyahia B, Trouillard O, Bouteiller D, Verloes A, Leguern E, Leboyer M, Brice A. Autism, language delay and mental retardation in a patient with 7q11 duplication. BMJ Case Rep. 2009 doi: 10.1136/bcr.05.2009.1911. adapted with permission from Depienne et al. 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit A, McKee S, Mansour S, Mehta SG, Tanteles GA, Anastasiadou V, Patsalis PC, Martin K, McCullough S, Suri M, Sarkar A. 7q11.23 microduplication: a recognizable phenotype. Clin Genet. 2013;83:155–161. doi: 10.1111/j.1399-0004.2012.01862.x. [DOI] [PubMed] [Google Scholar]
- Elliott CD. Differential Ability Scales. 2nd. San Antonio, TX: Psychological Corporation; 2007. [Google Scholar]
- Ewart AK, Morris CA, Atkinson D, Jin W, Sternes K, Spallone P, Stock AD, Leppert M, Keating MT. Hemizygosity at the elastin locus in a developmental disorder, Williams syndrome. Nat Genet. 1993;5:11–16. doi: 10.1038/ng0993-11. [DOI] [PubMed] [Google Scholar]
- Gagliardi C, Martelli S, Burt MD, Borgatti R. Evolution of neurologic features in Williams syndrome. Pediatr Neurol. 2007;36:301–306. doi: 10.1016/j.pediatrneurol.2007.01.001. [DOI] [PubMed] [Google Scholar]
- Gidley Larson JC, Mostofsky SH, Goldberg MC, Cutting LE, Denckla MB, Mahone EM. Effects of gender and age on motor exam in typically developing children. Dev Neuropsychol. 2007;32:543–562. doi: 10.1080/87565640701361013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobart HH, Morris CA, Mervis CB, Pani AM, Kistler DJ, Rios CM, Kimberley KW, Gregg RG, Bray-Ward P. Inversion of the Williams syndrome region is a common polymorphism found more frequently in parents of children with Williams syndrome. Am J Med Genet C. 2010;154C:220–228. doi: 10.1002/ajmg.c.30258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones W, Hesselink J, Courchesne E, Duncan T, Matsuda K, Bellugi U. Cerebellar abnormalities in infants and toddlers with Williams syndrome. Dev Med Child Neurol. 2002;44:688–694. doi: 10.1017/s0012162201002766. [DOI] [PubMed] [Google Scholar]
- Kirchhoff M, Bisgaard AM, Bryndorf T, Gerdes T. MLPA analysis for a panel of syndromes with mental retardation reveals imbalances in 5.8% of patients with mental retardation and dysmorphic features, including duplications of the Sotos syndrome and Williams-Beuren syndrome regions. Eur J Med Genet. 2007;50:33–42. doi: 10.1016/j.ejmg.2006.10.002. [DOI] [PubMed] [Google Scholar]
- Klein-Tasman BP, Mervis CB. Distinctive personality characteristics of 8-, 9-, and 10-year-old children with Williams syndrome. Dev Neuropsychol. 2003;23:271–292. doi: 10.1080/87565641.2003.9651895. [DOI] [PubMed] [Google Scholar]
- Klein-Tasman BP, Mervis CB, Lord C, Phillips KD. Socio-communicative deficits in young children with Williams syndrome: Performance on the Autism Diagnostic Observation Schedule. Child Neuropsychol. 2007;13:444–467. doi: 10.1080/09297040601033680. [DOI] [PubMed] [Google Scholar]
- Kriek M, White SJ, Szuhai K, Knijnenburg J, van Ommen GJ, den Dunnen JT, Breuning MH. Copy number variation in regions flanked (or unflanked) by duplicons among patients with developmental delay and/or congenital malformations; detection of reciprocal and partial Williams-Beuren duplications. Eur J Hum Genet. 2006;14:180–189. doi: 10.1038/sj.ejhg.5201540. [DOI] [PubMed] [Google Scholar]
- Kuijpers GM, De Vroede M, Knol HE, Jansen M. Growth hormone treatment in a child with Williams-Beuren syndrome: a case report. Eur J Pediatr. 1999;158:451–454. doi: 10.1007/s004310051118. [DOI] [PubMed] [Google Scholar]
- Leyfer OT, Woodruff-Borden J, Klein-Tasman BP, Fricke JS, Mervis CB. Prevalence of psychiatric disorders in 4 to 16-year-olds with Williams syndrome. Am J Med Genet Part B. 2006;141B:615–622. doi: 10.1002/ajmg.b.30344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lincoln AJ, Searcy YM, Jones W, Lord C. Social interaction behaviors discriminate young children with autism and Williams syndrome. J Am Acad Child Adolesc Psychiatry. 2007;46:323–331. doi: 10.1097/chi.0b013e31802b9522. [DOI] [PubMed] [Google Scholar]
- Malenfant P, Liu X, Hudson ML, Qiao Y, Hrynchak M, Riendeau N, Hildebrand MJ, Cohen IL, Chudley AE, Forster-Gibson C, Mickelson EC, Rajcan-Separovic E, Lewis ME, Holden JJ. Association of GTF2i in the Williams-Beuren syndrome critical region with autism spectrum disorders. J Autism Dev Disord. 2012;42:1459–1469. doi: 10.1007/s10803-011-1389-4. [DOI] [PubMed] [Google Scholar]
- McDonald-McGinn DM, Tonnesen MK, Laufer-Cahana A, Finucane B, Driscoll DA, Emanuel BS, Zackai EH. Phenotype of the 22q11.2 deletion in individuals identified through an affected relative: cast a wide FISHing net! Genet Med. 2001;3:23–29. doi: 10.1097/00125817-200101000-00006. [DOI] [PubMed] [Google Scholar]
- McGrew SG, Peters BR, Crittendon JA, Veenstra-Vanderweele J. Diagnostic yield of chromosomal microarray analysis in an autism primary care practice: which guidelines to implement? J Autism Dev Disord. 2012;42:1582–1591. doi: 10.1007/s10803-011-1398-3. [DOI] [PubMed] [Google Scholar]
- Mercuri E, Atkinson J, Braddick O, Rutherford MA, Cowan FM, Counsell SJ, Dubowitz LM, Bydder G. Chiari I malformation in asymptomatic young children with Williams syndrome: clinical and MRI study. Eur J Paediatr Neurol. 1997;1:177–181. doi: 10.1016/s1090-3798(97)80055-4. [DOI] [PubMed] [Google Scholar]
- Merritt JL, Lindor NM. Further clinical description of duplication of Williams-Beuren region presenting with congenital glaucoma and brachycephaly. Am J Med Genet A. 2008;146A:1055–1058. doi: 10.1002/ajmg.a.32235. [DOI] [PubMed] [Google Scholar]
- Mervis CB, Dida J, Lam E, Crawford-Zelli NA, Young EJ, Henderson DR, Onay T, Morris CA, Woodruff-Borden J, Yeomans J, Osborne LR. Duplication of GFT2I results in separation anxiety in mice and humans. Am J Hum Genet. 2013;90:1064–1070. doi: 10.1016/j.ajhg.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mervis CB, Klein-Tasman BP, Huffman MJ, Velleman SL, Pitts CH, Henderson DR, Woodruff-Borden J, Morris CA, Osborne LR. Children with 7q11.23 duplication syndrome: Psychological characteristics. Am J Med Genet Part A. 2015;167A:1436–1450. doi: 10.1002/ajmg.a.37071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris CA, Demsey SA, Leonard CO, Dilts C, Blackburn BL. Natural history of Williams syndrome: Physical characteristics. J Pediatr. 1988;113:328–336. doi: 10.1016/s0022-3476(88)80272-5. [DOI] [PubMed] [Google Scholar]
- Morris CA, Mervis CB, Hobart HH, Gregg RG, Bertrand J, Ensing GJ, Sommer A, Moore CA, Hopkin RJ, Spallone PA, Keating MT, Osborne L. GTF2I hemizygosity implicated in mental retardation in Williams syndrome: genotype-phenotype analysis of five families with deletions in the Williams syndrome region. Am J Med Genet A. 2003;123A:45–59. doi: 10.1002/ajmg.a.20496. [DOI] [PubMed] [Google Scholar]
- Mulle JG, Pulver AE, McGrath JA, Wolyniec PS, Dodd AF, Cutler DJ, Sebat J, Malhotra D, Nestadt G, Conrad DF, Hurles M, Barnes CP, Ikeda M, Iwata N, Levinson DF, Gejman PV, Sanders AR, Duan J, Mitchell AA, Peter I, Sklar P, O’Dushlaine CT, Grozeva D, O’Donovan MC, Owen MJ, Hultman CM, Kähler AK, Sullivan PF, Molecular Genetics of Schizophrenia Consortium. Kirov G, Warren ST. Reciprocal duplication of the Williams-Beuren syndrome deletion on chromosome 7q11.23 is associated with schizophrenia. Biol Psychiatry. 2014;75:371–377. doi: 10.1016/j.biopsych.2013.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orellana C, Bernabeu J, Monfort S, Roselló M, Oltra S, Ferrer I, Quiroga R, Martínez-Garay I, Martínez F. Duplication of the Williams-Beuren critical region: case report and further delineation of the phenotypic spectrum. J Med Genet. 2008;45:187–189. doi: 10.1136/jmg.2007.054064. [DOI] [PubMed] [Google Scholar]
- Pankau R, Partsch CJ, Neblung A, Gosch A, Wessel A. Head circumference of children with Williams-Beuren syndrome. Am J Med Genet. 1994;52:285–290. doi: 10.1002/ajmg.1320520307. [DOI] [PubMed] [Google Scholar]
- Parrott A, James J, Goldenberg P, Hinton RB, Miller E, Shikany A, Aylsworth AS, Kaiser-Rogers K, Ferns SJ, Lalani SR, Ware SM. Aortopathy in the 7q11.23 microduplication syndrome. Am J Med Genet A. 2015;167A:363–370. doi: 10.1002/ajmg.a.36859. [DOI] [PubMed] [Google Scholar]
- Partsch CJ, Pankau R, Blum WF, Gosch A, Wessel A. Hormonal regulation in children and adults with Williams-Beuren syndrome. Am J Med Genet. 1994;51:251–257. doi: 10.1002/ajmg.1320510316. [DOI] [PubMed] [Google Scholar]
- Pober BR, Filiano JJ. Association of Chiari I malformation and Williams syndrome. Pediatr Neurol. 1995;12:84–88. doi: 10.1016/0887-8994(94)00117-k. [DOI] [PubMed] [Google Scholar]
- Pober BR, Lacro RV, Rice C, Mandell V, Teele RL. Renal findings in 40 individuals with Williams syndrome. Am J Med Genet. 1993;46:271–274. doi: 10.1002/ajmg.1320460306. [DOI] [PubMed] [Google Scholar]
- Pober BR, Morris CA. Diagnosis and management of medical problems in adults with Williams-Beuren syndrome. Am J Med Genet C Semin Med Genet. 2007;145C:280–290. doi: 10.1002/ajmg.c.30139. [DOI] [PubMed] [Google Scholar]
- Prabhakaran R, Kippenhan JS, Sottile M, Mervis CB, Roe K, Eisenberg D, Gregory M, Yankowitz L, Nash T, Insel C, Murray S, Mikhaiel JP, Elliot M, Nguyen T-V, Masdeu J, Osborne LR, Kohn P, Jabbi M, Wei S-M, Czarapata J, Berman KF. Structural brain alterations in children with copy number variation of the Williams syndrome genetic region (7q11.23) and normal IQs. Society for Neuroscience; Washington, DC: 2014. [Google Scholar]
- Prontera P, Serino D, Caldini B, Scarponi L, Merla G, Testa G, Muti M, Napolioni V, Mazzotta G, Piccirilli M, Donti E. Brief report: Functional MRI of a patient with 7q11.23 duplication syndrome and autism. J Autism Dev Disord. 2014 doi: 10.1007/s10803-014-2117-7. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Sanders SJ, Ercan-Sencicek AG, Hus V, Luo R, Murtha MT, Moreno-De-Luca D, Chu SH, Moreau MP, Gupta AR, Thomson SA, Mason CE, Bilguvar K, Celestino-Soper PB, Choi M, Crawford EL, Davis L, Wright NR, Dhodapkar RM, DiCola M, DiLullo NM, Fernandez TV, Fielding-Singh V, Fishman DO, Frahm S, Garagaloyan R, Goh GS, Kammela S, Klei L, Lowe JK, Lund SC, McGrew AD, Meyer KA, Moffat WJ, Murdoch JD, O’Roak BJ, Ober GT, Pottenger RS, Raubeson MJ, Song Y, Wang Q, Yaspan BL, Yu TW, Yurkiewicz IR, Beaudet AL, Cantor RM, Curland M, Grice DE, Günel M, Lifton RP, Mane SM, Martin DM, Shaw CA, Sheldon M, Tischfield JA, Walsh CA, Morrow EM, Ledbetter DH, Fombonne E, Lord C, Martin CL, Brooks AI, Sutcliffe JS, Cook EH, Jr, Geschwind D, Roeder K, Devlin B, State MW. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron. 2011;70:863–885. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sforzini C, Milani D, Fossali E, Barbato A, Grumieri G, Bianchetti MG, Selicorni A. Renal tract ultrasonography and calcium homeostasis in Williams-Beuren syndrome. Pediatr Nephrol. 2002;17:899–902. doi: 10.1007/s00467-002-0889-z. [DOI] [PubMed] [Google Scholar]
- Silverman WK, Albano AM. The Anxiety Disorders Interview Schedule for DSM-IV: Parent Interview Schedule. San Antonio, TX: Graywind Publications; 1996. [Google Scholar]
- Somerville MJ, Mervis CB, Young EJ, Seo E-J, del Campo M, Bamforth S, Peregrine E, Loo W, Lilley M, Pérez-Jurado LA, Morris CA, Scherer SW, Osborne LR. Severe expressive-language delay related to duplication of the Williams-Beuren locus. N Engl J Med. 2005;353:1694–1701. doi: 10.1056/NEJMoa051962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stagi S, Lapi E, Chiarelli F, de Martino M. Incidence of diverticular disease and complicated diverticular disease in young patients with Williams syndrome. Pediatr Surg Int. 2010;26:943–944. doi: 10.1007/s00383-010-2666-6. [DOI] [PubMed] [Google Scholar]
- Stock AD, Spallone PA, Dennis TR, Netski D, Morris CA, Mervis CB, Hobart HH. Heat shock protein 27 gene: chromosomal and molecular location and relationship to Williams syndrome. Am J Med Genet A. 2003;120A:320–325. doi: 10.1002/ajmg.a.20055. [DOI] [PubMed] [Google Scholar]
- Tam E, Young EJ, Morris CA, Marshall CR, Loo W, Scherer SW, Mervis CB, Osborne LR. The common inversion of the Williams-Beuren syndrome region at 7q11.23 does not cause clinical symptoms. Am J Med Genet A. 2008;146A:1797–1806. doi: 10.1002/ajmg.a.32360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tordjman S, Anderson GM, Botbol M, Toutain A, Sarda P, Carlier M, Saugier-Veber P, Baumann C, Cohen D, Lagneaux C, Tabet AC, Verloes A. Autistic disorder in patients with Williams-Beuren syndrome: a reconsideration of the Williams-Beuren syndrome phenotype. PLoS One. 2012;27:e30778. doi: 10.1371/journal.pone.0030778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torniero C, dalla Bernardina B, Novara F, Cerini R, Bonaglia C, Pramparo T, Ciccone R, Guerrini R, Zuffardi O. Dysmorphic features, simplified gyral pattern and 7q11.23 duplication reciprocal to the Williams-Beuren deletion. Eur J Hum Genet. 2008;16:880–887. doi: 10.1038/ejhg.2008.42. [DOI] [PubMed] [Google Scholar]
- Torniero C, dalla Bernardina B, Novara F, Vetro A, Ricca I, Darra F, Pramparo T, Guerrini R, Zuffardi O. Cortical dysplasia of the left temporal lobe might explain severe expressive-language delay in patients with duplication of the Williams-Beuren locus. Eur J Hum Genet. 2007;15:62–67. doi: 10.1038/sj.ejhg.5201730. [DOI] [PubMed] [Google Scholar]
- Van der Aa N, Rooms L, Vandeweyer G, van den Ende J, Reyniers E, Fichera M, Romano C, Delle Chiaie B, Mortier G, Menten B, Destrée A, Maystadt I, Männik K, Kurg A, Reimand T, McMullan D, Oley C, Brueton L, Bongers EM, van Bon BW, Pfund R, Jacquemont S, Ferrarini A, Martinet D, Schrander-Stumpel C, Stegmann AP, Frints SG, de Vries BB, Ceulemans B, Kooy RF. Fourteen new cases contribute to the characterization of the 7q11.23 microduplication syndrome. Eur J Med Genet. 2009;52:94–100. doi: 10.1016/j.ejmg.2009.02.006. [DOI] [PubMed] [Google Scholar]
- Velleman SL, Mervis CB. Children with 7q11.23 duplication syndrome: Speech, language, cognitive, and behavioral characteristics and their implications for intervention. Perspect Lang Learn Educ. 2011;18:108–116. doi: 10.1044/lle18.3.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wentzel C, Fernström M, Ohrner Y, Annerén G, Thuresson AC. Clinical variability of the 22q11.2 duplication syndrome. Eur J Med Genet. 2008;51:501–510. doi: 10.1016/j.ejmg.2008.07.005. [DOI] [PubMed] [Google Scholar]
- Xekouki P, Fryssira H, Maniati-Christidi M, Amenta S, Karavitakis EM, Kanaka-Gantenbein C, Dacou-Voutetakis C. Growth hormone deficiency in a child with Williams-Beuren syndrome. The response to growth hormone therapy. J Pediatr Endocrinol Metab. 2005;18:205–207. doi: 10.1515/jpem.2005.18.2.205. [DOI] [PubMed] [Google Scholar]
- Zarate YA, Lepard T, Sellars E, Kaylor JA, Alfaro MP, Sailey C, Schaefer GB, Collins RT., 2nd Cardiovascular and genitourinary anomalies in patients with duplications within the Williams syndrome critical region: phenotypic expansion and review of the literature. Am J Med Genet A. 2014;164A:1998–2002. doi: 10.1002/ajmg.a.36601. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.