

Abstract
There are at present no published studies providing a global overview of changes in bladder metabolism resulting from diabetes. Such studies have the potential to provide mechanistic insight into the development of diabetic bladder disorder (DBD). In the present study, we compared the metabolome of detrusor and urothelial layer in a 1-mo streptozotocin-induced rat model of type 1 diabetes with nondiabetic controls. Our studies revealed that diabetes caused both common and differential changes in the detrusor and urothelial layer's metabolome. Diabetes resulted in similar changes in the levels of previously described diabetic markers in both tissues, such as glucose, lactate, 2-hydroxybutyrate, branched-chain amino acid degradation products, bile acids, and 1,5-anhydroglucitol, as well as markers of oxidative stress. In the detrusor (but not the urothelial layer), diabetes caused activation of the pentose-phosphate and polyol pathways, concomitant with a reduction in the TCA cycle and β-oxidation. Changes in detrusor energy-generating pathways resulted in an accumulation of sorbitol that, through generation of advanced glycation end products, is likely to play a central role in the development of DBD. In the diabetic urothelial layer there was decreased flux of glucose via glycolysis and changes in lipid metabolism, particularly prostaglandin synthesis, which also potentially contributes to detrusor dysfunction.
Keywords: diabetic bladder disorder, metabolomics, urothelial, detrusor, metabolism
diabetic bladder dysfunction (DBD; also known as diabetic cystopathy) refers to a number of bladder symptoms occurring in patients with diabetes mellitus. The clinical symptoms of DBD present as storage problems (overactive bladder and urge incontinence), voiding problems (poor emptying and overflow incontinence), and several other phenotypes (such as decreased sensation and increased capacity) (1, 9, 12, 16, 22–24). The prevalence of DBD among diabetic individuals is between 43 and 87% (8, 15, 17). Although DBD is not life threatening, it is severely detrimental to the quality of life of patients (2) and has a societal economic cost of greater than $50 billion/yr (11, 26).
The pathophysiology of DBD is multifactorial and includes alterations in detrusor, urothelial, and neuronal dysfunctions (9, 37). The roles of these tissues in the overall function of bladder differ; the detrusor determines the tone of the bladder, whereas the urothelial layer serves both as a barrier and in a regulatory role of detrusor tone. The differing roles of these tissues in bladder function are reflected by different pathophysiological responses to diabetes. The involvement of detrusor in DBD has been attributed to several mechanisms that affect regulation and development of smooth muscle tone; these include increased muscarinic receptor density (31, 32), overstretch-induced hypertrophy, increased calcium sensitivity or increased calcium channel activity (34), alteration in intracellular signaling, and other genetic changes (9). Diabetic urothelial dysfunction has been attributed to compromised barrier function, alterations in mechanosensitivity, and impaired mediator release such as endogenous prostaglandin (27, 28).
Diabetes is a disease of metabolism, and overlying the mechanisms of DBD development are elevated glucose levels, which can result in metabolic perturbation. However, few if any studies have determined the effects of diabetes on the metabolism of bladder or whether there are differential effects on the urothelial layer and detrusor. The recently developed field of metabolomics allows the determination of complete metabolite profiles of tissues. The goal of metabolomics is to provide systematic measurement and biological interpretation of small biochemical molecules (MW 50-1,500 Da) within a biological sample such as urine, plasma, or tissue. Metabolomics has been applied to understand the pathology of several diseases, such as cancer, type 2 diabetes, and inherent metabolic diseases, to study effects of external stimuli on diseases and to identify new biomarkers that could be used in diagnosis and prognosis (7, 29). However, although there are a large number of review articles on metabolomics and its application to oncologic and diabetes research (5, 30, 39), its application to studying any benign urological disease has not been reported.
Streptozotocin-treated rats are a commonly used model of type 1 diabetes, and we have published a detailed study on the development and progression of urogenital dysfunction in this animal model (19). Cystometric studies demonstrated that after 1 mo of diabetes, animals exhibited bladder dysfunction physiologically similar to DBD in patients. In the present study, we applied metabolomics to this animal model to compare the metabolome of the urothelial layer and detrusor of nondiabetic (control) and diabetic rats to gain insight into the differential metabolism in urothelial and detrusor tissue, and the metabolic changes in these tissues with the onset of diabetes. The results from our studies have the potential to identify novel strategies that can ameliorate diabetic bladder pathophysiology.
METHODS
Animals and tissues.
All experimental protocols were approved by the Institutional Animal Care and Use Committee of the Albert Einstein College of Medicine. Eight male Fischer 344 rats were made diabetic by an intraperitoneal injection with 35 mg/kg of streptozotocin (STZ) dissolved in citrate buffer (60 ml of 0.1 M citric acid and 40 ml of 0.2 m Na2HPO4, pH 4.6), as described previously (33). Sixteen age-matched nondiabetic controls (controls) received an injection of vehicle (citrate buffer). Rats were housed in individual cages and allowed free access to food and water. Hyperglycemia was confirmed by the presence of blood glucose levels (determined with the OneTouch Ultra 2 Glucometer; Life Scan, Milpitas, CA) of 250 mg/dl for 3 consecutive days. After 1 mo the animals were euthanized, and bladders were removed and immediately placed into cold phosphate-buffered saline (137 mM NaCl, 8 mM Na2HPO4, 2.7 mM KCl, and 1.47 mM KH2PO4, pH 7.4) to manually separate the detrusor and urothelial layers. We refer to the tissue that is peeled away from the luminal surface of the bladder as the “urothelial layer” as it is not “pure” urothelium but contains both urothelium and lamina propria. Buffer was then aspirated via Kimwipes and flash-frozen in liquid nitrogen using custom-made calipers. Tissues were transferred into tubes and stored at minus 80°C prior to shipping for metabolomic profiling (performed by Metabolon, as described below). Average characteristics of the animals are shown in Table 1. A minimum of 50 mg of tissue is required to perform the metabolomic analysis described below. As shown in Table 1, there was insufficient urothelial layer from control animals to perform the analysis, and therefore, it was necessary to pool tissue from two control animals.
Table 1.
Animal characteristics
| Total Animal Weight, g |
||||||
|---|---|---|---|---|---|---|
| Treatment | Initial | 1 Mo of STZ* | Urothelial Layer, mg | Detrusor, mg | Blood Glucose, mg/dl | Urine Glucose, mg/dl |
| Nondiabetic (n = 16) | 273 ± 5 | 293 ± 4# | 24 ± 2# | 99 ± 9# | 83 ± 1# | |
| Diabetes (n = 8) | 248 ± 3 | 199 ± 7# | 54 ± 6# | 217 ± 20# | 414 ± 25# | 500–2,000 |
Values are reported as means ± SE; n = no. of animals. STZ, streptozotocin.
Unpaired 2-sample t-test, P ≤ 0.05.
Metabolomics.
Metabolomic analysis was performed on eight diabetic and nondiabetic detrusor and urothelial tissue samples. Sample preparation was carried out as described previously (25). Briefly, an automated liquid handler (Hamilton LabStar, Salt Lake City, UT) was used to add methanol containing recovery standards to the experimental samples to facilitate protein precipitation. Following centrifugation, the supernatants were split into aliquots for analysis on four platforms: ultra-high-performance liquid chromatography/tandem mass spectrometry (UHPLC/MS/MS) optimized for basic species, UHPLC/MS/MS optimized for acidic species, UHPLC/MS/MS optimized for polar species, and gas chromatography/mass spectrometry (GC/MS). All aliquots were dried under nitrogen and vacuum desiccated. The samples were subsequently reconstituted in 50 μl of 0.1% formic acid in water (acidic conditions) or in 50 μl of 6.5 mM ammonium bicarbonate in water, pH 8 (basic conditions) for the UHPLC/MS/MS analyses or derivatized to a final volume of 50 μl for GC/MS analysis using equal parts bistrimethyl-silyl-trifluoroacetamide and solvent mixture acetonitrile-dichloromethane-cyclohexane (5:4:1) with 5% triethylamine at 60°C for 1 h. In addition, three types of controls were analyzed in concert with the experimental samples; aliquots of a “client matrix” (formed by pooling a small amount of each sample) served as technical replicates throughout the data set, extracted water samples served as process blanks, and a cocktail of standards spiked into every analyzed sample allowed for instrument performance monitoring. Experimental samples and controls were randomized across all platform run days.
For the UHLC/MS/MS analysis, aliquots were separated using a Waters Acquity UPLC (Waters, Millford, MA) and analyzed using a Q-Exactive high-resolution/accurate mass spectrometer (Thermo Fisher Scientific, Waltham, MA) that consisted of an electrospray ionization (ESI) source and Orbitrap mass analyzer. Derivatized samples for GC/MS were separated on a 5% phenyldimethyl silicone column with helium as the carrier gas and a temperature ramp from 60 to 340°C and then analyzed on a Thermo-Finnigan Trace DSQ MS (Thermo Fisher Scientific) operated at unit mass resolving power with electron impact ionization and an atomic mass unit scan range of 50–750.
Metabolites were identified by automated comparison of the ion features in the experimental samples to a reference library of chemical standard entries that included retention time, molecular weight (m/z), preferred adducts, and in-source fragments as well as associated MS spectra and were curated by visual inspection for quality control using software developed at Metabolon (4).
Statistical analysis.
For data display purposes and statistical analysis, each metabolite was rescaled to set the median equal to 1. In addition, any missing values were assumed to be below the limits of detection, and these values were imputed with the compound minimum (minimum value imputation). Following median scaling and imputation of missing values, statistical analysis of log-transformed data was performed using “R” (http://cran.r-project.org/), which is a freely available, open-source software package. Metabolites that differed significantly between the experimental groups were determined using ANOVA contrasts. P values ≤0.05 were considered statistically significant, and P values <0.10 were reported as trends. Multiple comparisons were accounted for by estimating the false discovery rate using q values. The data were rescaled prior to Pearson's correlation analysis. Pearson's correlation was calculated to determine direct from indirect metabolite associations.
RESULTS
The metabolome of bladder.
In total, 484 and 522 identifiable metabolites were detected at significant levels in the detrusor and urothelial layer, respectively. As shown in Table 2, 103 metabolites were significantly (P ≤ 0.05) changed in expression in the urothelial layer of diabetic compared with nondiabetic controls (55% upregulated and 45% downregulated). In the detrusor, 143 metabolites were significantly changed (P ≤ 0.05) in expression in the diabetic compared with nondiabetic controls, (62% upregulated and 38% downregulated). We also considered a trend toward change in expression levels if the P value lay between 0.05 < P < 0.10, which accounted for 35 urothelial and 44 detrusor metabolites (Table 2). In the following analyses, we compared which specific metabolites and their associated metabolic pathways were changed in common between detrusor and urothelial layer in response to diabetes and which changes were tissue specific.
Table 2.
Total no. of metabolites changed in expression in the diabetic and nondiabetic urothelial layer and detrusor
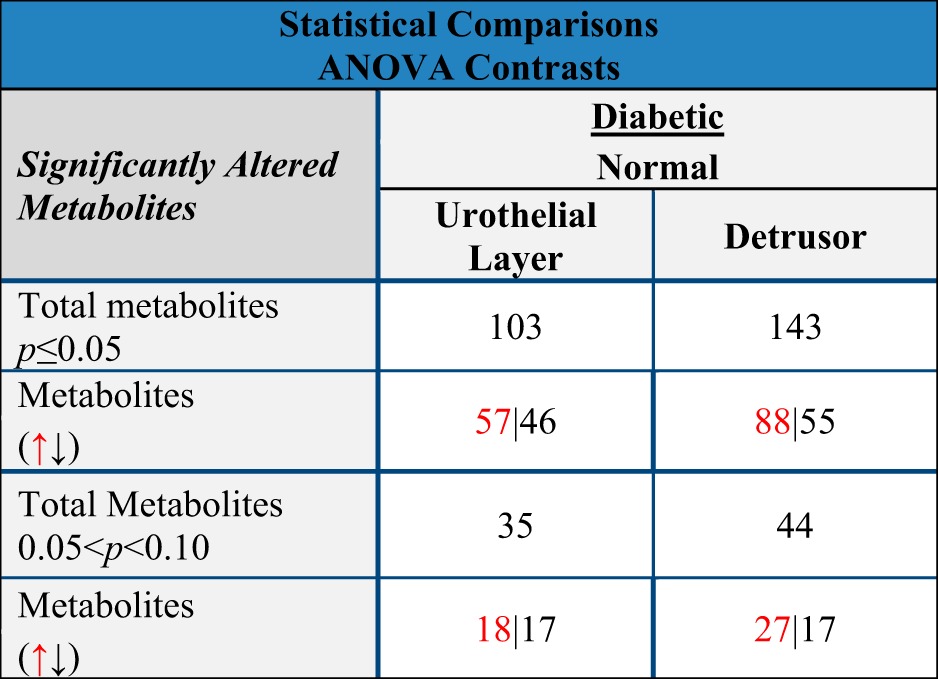
Red upward arrow, increase; black downward arrow, decrease.
Common changes in metabolite levels caused by hyperglycemia in the detrusor and urothelial layer.
In Table 3, we show a complete list of metabolites that have changed levels of expression in both the urothelial layer and detrusor in response to diabetes. In this section, we highlight some of the more significant of these metabolites or families of metabolites. Several previously reported metabolic indicators of hyperglycemia in the blood were reflected by changed expression in bladder tissues with the onset of diabetes. For example, diabetes-induced hyperglycemia in the blood was reflected by significantly increased levels of glucose in the detrusor of apprxoimately fourfold and a trend toward higher glucose levels in the urothelial layer. There were also significant reductions in a biochemical marker of short-term glycemic control, 1,5-anhydroglucitol, in both tissues.
Table 3.
Common diabetes-mediated changes in metabolite levels in detrusor and urothelial layer
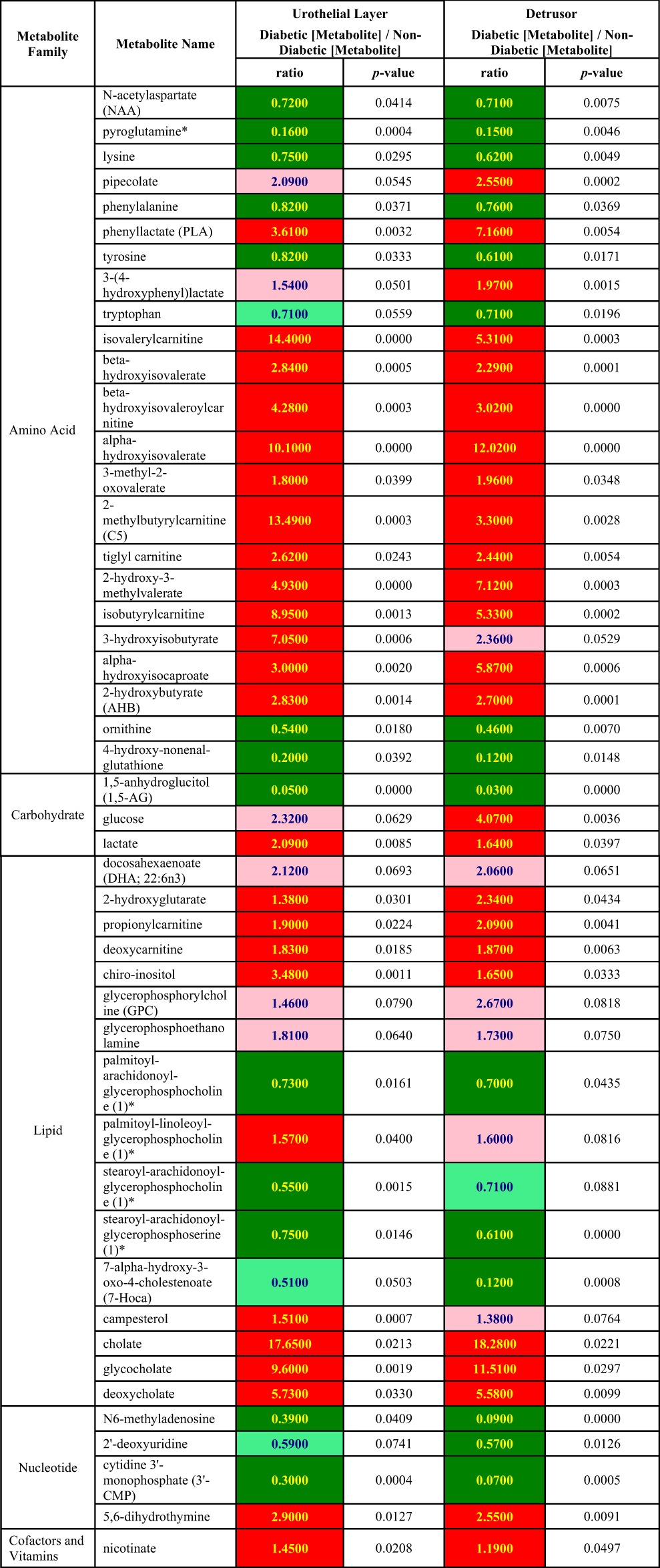
ANOVA contrast analysis was performed for specific comparisons between diabetic groups and control groups. Dark green, significant difference (P ≤ 0.05) between the groups shown, metabolite ratio of <1.00; light green, a trend/narrowly missed statistical cutoff for significance 0.05 < P < 0.10, metabolite ratio of <1.00; red, significant difference.
Numerous studies have indicated that diabetes is associated with an increase in circulating branched chain amino acids (BCAAs). This is reflected by upregulation of numerous catabolites produced from the BCAAs valine, isoleucine, and leucine in both the detrusor and urothelial layer such as 2-methylbutrylcarnitine, 3-hydroxisobutyrate, 3-methyl-2-oxovalerate, and 4-methyl-2-oxopentanoate. Other nonbranched amino acids and their metabolites (such as lysine, phenylalanine, tyrosine, and tryptophan) were modestly (<2-fold) downregulated in both tissues. In addition, the nonproteogenic amino acid ornithine was downregulated approximately twofold.
Several reports suggest that hepatic dysfunction occurs in both diabetic humans and animal models (20). There were elevated levels in the primary bile acids cholate and glycocholate, the secondary bile acid deoxycholate, and campesterol, a factor involved in bile acid reabsorbtion in both the detrusor and urothelial layer from diabetic rats suggesting that this comorbidity occurs in the STZ animal model.
Although intermediates of glycolysis were unchanged with diabetes in both tissues, two indicators of changes in energy-generating pathways, lactate and 2-hydroxybutyrate, were significantly upregulated in both the detrusor and the urothelial layer. However, as discussed below, there is evidence that the reason for upregulation in each tissue is due to different mechanisms.
Differential changes in metabolite levels between the detrusor and urothelial layer caused by hyperglycemia.
Table 4 compares differential changes in metabolite levels that occur in the detrusor and urothelial layer with the onset of hyperglycemia. For example, in the diabetic compared with nondiabetic detrusor there are significantly elevated levels of fructose (2.25-fold), sorbital (5.13-fold), and ribulose/xylulose 5-phosphate (2.7-fold), suggesting activation of the pentose phosphate pathway. In addition, in the detrusor, levels of acetylphosphate increased significantly (1.53-fold, P < 0.05), whereas there was a trend to lowered phosphate, suggesting perturbation of oxidative phosphorylation in the detrusor. There was also a trend for reductions in lysolipids, and free medium-chain fatty acids and elevation in dicarboxlyated fatty acids may reflect decreased synthesis of structural lipids and increased lipid peroxidation. The increased levels of propionlycarnitine, valerylcarnitine, and octanoylcarnitine without ketobody changes indicated that β-oxidations were suppressed as well.
Table 4.
Different diebetes-mediated changes in metbaolite levels in detrusor and urothelial layer
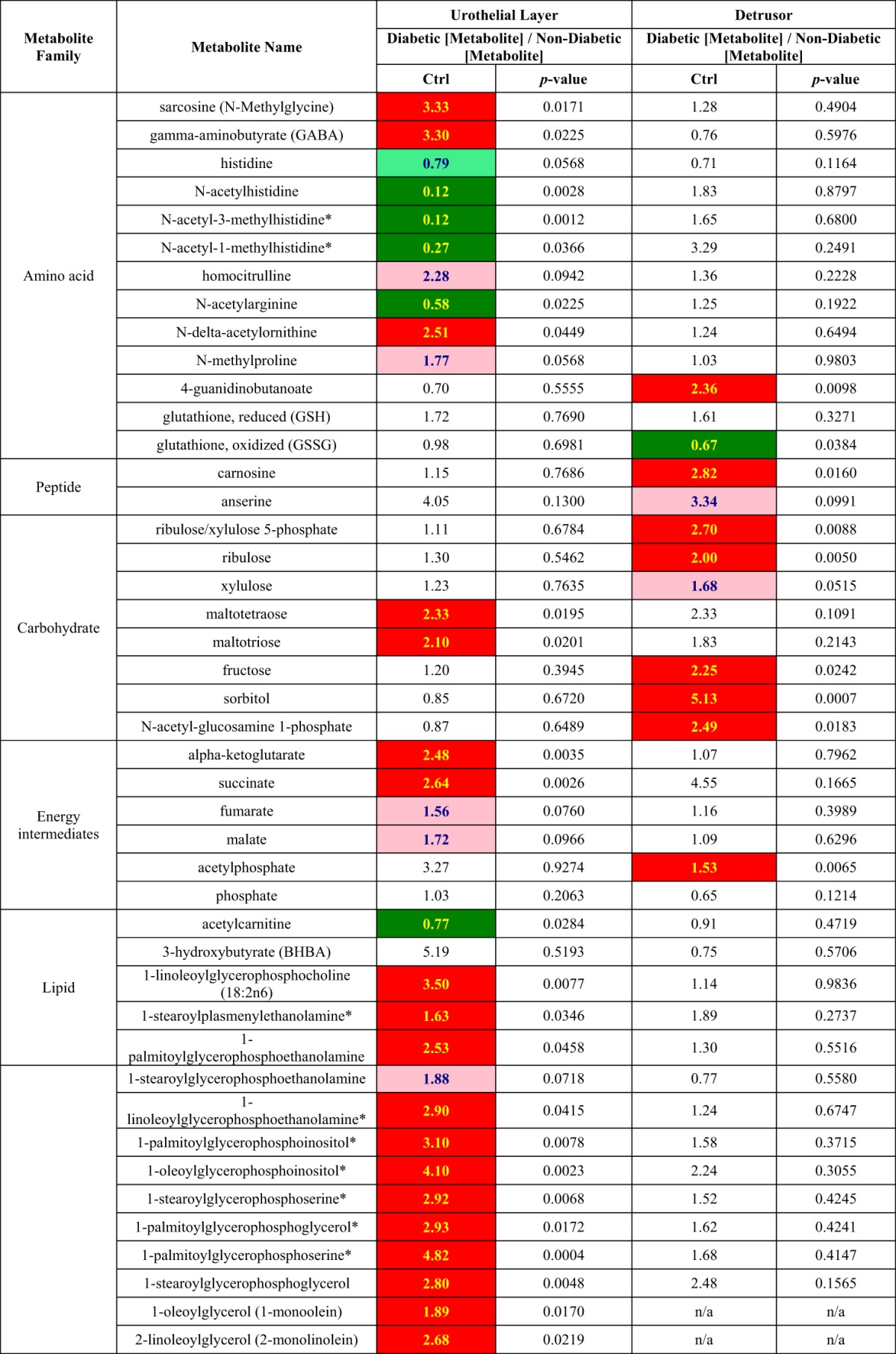
ANOVA contrast analysis was performed for specific comparison between diabetic groups and control groups. Dark green, significant difference (P ≤ 0.05) between the groups shown, metabolite ratio of <1.00; light green, a trend/narrowly missed statistical cutoff for significance 0.05 < P < 0.10, metabolite ratio of <1.00; red, significant difference (P ≤ 0.05) between the groups shown, metabolite ratio of ≥1.00; pink, narrowly missed statistical cutoff for significance 0.05 < P < 0.10, metabolite ratio of ≥1.00.
In contrast to the detrusor, in the diabetic urothelial layer, several components of the TCA cycle were either significantly elevated, such as α-ketoglutarate (2.48-fold) and succinate (2.64-fold), or had a trend for upregulation, such as fumarate (1.56-fold, P < 0.1) and malate (1.72-fold, P < 0.1). In the diabetic urothelial layer there were also elevated levels of several phospholipid/lysolipids, free polyunsaturated fatty acids, and ketobodies, which are components of pathways involved in synthesis of membrane lipids.
DISCUSSION
This is the first report to apply metabolomics to study global changes in metabolism in an animal model of DBD. We also took the novel approach of investigating the effects of diabetes separately on the detrusor and urothelial layer. Through these studies we were able to demonstrate both common and differential effects of hyperglycemia on the metabolism of these different tissues.
The time point used in these studies (1 mo of diabetes caused by streptozotocin) has been reported by our group and others as the earliest at which bladder dysfunction is manifested in this animal model (19, 36). Therefore, we believe that the bladder pathophysiology at this time point is likely a direct consequence of diabetes-induced changes in the metabolism of the detrusor and urothelial layer. One month of hyperglycemia results in a persistent increase in detrusor overactivity with no evidence of detrusor decompensation (19, 36). As described below, longer times of hyperglycemia (from 12 wk onward) result in progression of DBD from a bladder with a compensated overactive phenotype to one exhibiting decompensation, underactivity, and hypertrophy. This later decompensated stage in DBD has been suggested to result from a combination of both polyuria and continued hyperglycemic-induced metabolic stress (19).
Metabolites and pathways affected in both detrusor and urothelial layer by diabetes.
Several previously identified serum or tissue markers of hyperglycemia, such as glucose, 1,5-anhydroglucitol, and BCAAs, were commonly changed in levels in both the urothelial layer and detrusor.
Interestingly, with the exception of an elevation in lactate levels, the intermediates of glycolysis were not changed in either tissue. The conversion of pyruvate to lactate is coupled to oxidation of NADH to NAD. The tissue lactate/pyruvate ratio is considered a reliable indicator of the cytosolic ratio of free NADH/NAD (10), and the 3-hydroxybutyrate/acetoacetate ratio is a good parameter of the mitochondrial NADH/NAD (reduction of 3-hydroxybutyrate to acetoacetate is accompanied with oxidation of NADH to NAD). Previous studies have suggested that hyperglycemia may induce redox imbalance by increasing the ratio of NADH/NAD (35). Consistent with these reports, significant elevation of lactate in our studies suggests that diabetes results in an increased cystolic NADH/NAD ratio in the bladder. Since 3-hydroxybutyrate was not significantly altered in diabetic tissues and acetoacetate was not included in our data set, we were unable to deduce the changes of NADH/NAD in mitochondria in diabetic tissue. However, NAD level was significantly reduced in diabetic detrusor. Thus, redox imbalance already occurs after 1 mo in the streptozotocin-induced diabetic bladder, which may then result in some of the pathophysiology associated with DBD (10).
Previous studies (6) have observed that diabetes is associated with reduced circulating levels of methionine and cysteine, and it has been suggested that this may reflect increased flux into glutathione synthesis. Consistent with these observations, our data showed a trend in diabetic detrusor toward lower levels of methionine and cysteine accompanied by increased levels of reduced glutathione. The ratio of reduced (GSH) to oxidized glutathione (GSSG) was observed to increase with diabetes. Reduced glutathione (GSH) is considered an important scavenger of reactive oxygen species, and the GSH/GSSG ratio is a useful marker of oxidative stress (40). Based on our metabolic profiles, GSSG was significantly decreased in diabetic detrusor (GSH with a trend for increase in diabetic urothelial layer), suggesting that at the 1-mo time point diabetes has resulted in the activation of this mechanism to reduced oxidative stress, but it is not overwhelmed.
Ascorbic acid is suggested to play a protective role in the development of pathophysiology associated with diabetes. Previous studies showed that type 1 diabetic subjects have a reduction of threonic acid in circulation, a breakdown product of the antioxidant ascorbic acid, suggesting impaired ascorbic acid metabolism (6). In this study, a reduction of threonic acid was observed in diabetic detrusor, whereas the dehydroascorbic acid was decreased in the diabetic urothelial layer. The dehydroascorbic acid is an oxidized and trafficking form of ascorbic acid, which helps ascorbic acid relocalize into the endoplasmic reticulum and mitochondria to protect against oxidative stress wherein. The presence of reduced ascorbate metabolites in the bladder may indicate an attenuation of the antioxidant function of ascorbic acid.
There were elevated levels of both primary and secondary bile acids in both the urothelial layer and detrusor, suggesting impaired hepatic function in the STZ-diabetic rat. Bile acids exhibit toxic properties, and ectopic accumulation of these metabolites in the bladder may also be a contributing factor to DBD.
Overall, the commonly changed metabolites in the detrusor and urothelial layer suggest that previously determined changes in levels of metabolic markers of diabetes in the serum and other tissues are reflected by changes in bladder tissue. There is also an increase in the NADH/NAD ratio that in turn could impact several metabolic pathways, metabolites are activated indicative and protective of oxidative stress, and there is an increase in potentially toxic bile acid concentrations.
Differential pathways perturbed by diabetes in detrusor and urothelial layer.
The data in Table 4 suggest that diabetes effects the energy-generating pathways differently in the detrusor and urothelial layer. These changes in the pathways are analyzed in Table 5, where Pearson's correlation is used to compare levels of glucose with metabolite levels in the downstream energy-generating pathways, glycolysis, and the TCA cycle. Normally, tissues maintain intracellular glucose hemostasis by balancing glucose uptake and degradation. Table 5 demonstrates that in the nondiabetic urothelial layer there is a positive correlation of glucose with glycolytic and TCA cycle intermediates, suggesting that glucose homeostasis is being achieved through these pathways that break down glucose and generate energy. However, with diabetes the metabolic flux in the urothelial layer is altered dramatically, such that the increased levels of glucose in the tissue are not reflected by increased glycolysis, resulting in a negative correlation between glucose and the glycolytic intermediates. Since the levels of glucose-6-phosphate (G6P) are also negatively correlated with the increased levels of glucose, this suggests that in the diabetic urothelial layer, the conversion of glucose to G6P by hexokinase is the rate-limiting step. As noted in Table 4, several TCA intermediates displayed significantly increased levels in the diabetic urothelial layer, and the Pearson correlation (Table 5) demonstrated a similar flux as in the nondiabetic urothelial layer, implying that the TCA cycle was activated in the diabetic urothelial layer.
Table 5.
Pearson's correlation of glucose to other metabolites in the nondiabetic and diabetic detrusor and urothelial layer
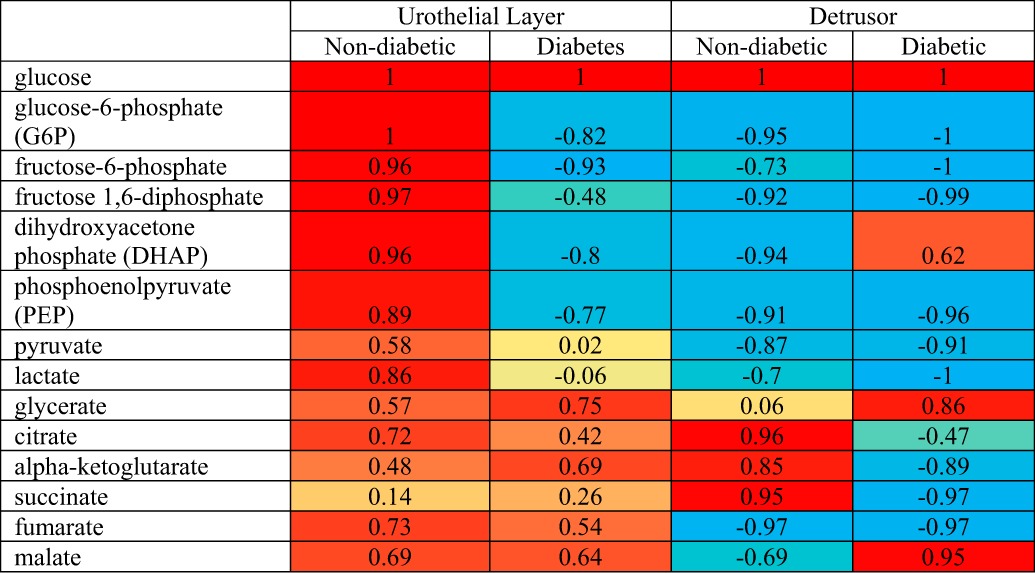
Correlation value ranges from −1 to 1. A negative value indicates a negative relationship and a positive value implies a positive correlation. Red, yellow, and blue color codes represent values statistically not different from +1, 0, and −1, respectively. Values between 0 to +1 and 0 to −1 are shown as a proportionally mixed combination of 2 colors (red and yellow and blue and yellow, respectively).
In contrast to the urothelial layer, in the detrusor the correlation of glucose with the majority of glycolytic intermediates was negative in both the diabetic and nondiabetic animals (Table 5), suggesting that the glycolytic pathway retained the capacity to handle higher glucose levels after 1 mo of diabetes. However, there is evidence that hyperglycemia suppresses or perturbs the TCA cycle in detrusor. In the nondiabetic detrusor, there is a positive correlation between glucose and several of the TCA cycle intermediates (citrate, α-ketoglutarate, and succinate). However, with the onset of diabetes, these same TCA cycle intermediates were negatively correlated with glucose. The disturbance of the TCA in the diabetic detrusor is correlated with increased activity of the other two pathways: the sorbitol and pentose phosphorylation pathways. Previous work in the whole bladder has suggested that in the hyperglycemic state the hexokinase (which converts glucose into G6P) becomes saturated and the affinity of aldose reductase for glucose increases, causing an increased production and accumulation of sorbitol. In addition, Daneshgari et al. (3a) suggested that there is increased aldose reductase expression in human detrusor cells under hyperglycemic conditions. Subsequently, sorbitol dehydrogenase transforms sorbitol to fructose, and our evidence demonstrates that both metabolites are elevated in the diabetic detrusor (Table 4). Sorbitol can potentially glycate nitrogen atoms on proteins (such as collagen) generating advanced glycation end (AGE) products, which would impact normal detrusor function.
In addition, activation of these pathways is accompanied by oxidation (and consumption) of NADPH to NADP and an increase in the NADH/NAD. NADPH and NAD are necessary cofactors in redox reactions, and their intracellular reduction leads to decreased synthesis of glutathione and other putative antioxidants, such as taurine, with an increased production of reactive oxygen species, which would also contribute to the pathophysiology of detrusor tissue. For example, in a rabbit model of alloxan-induced diabetes, decreased contractility of the detrusor has been associated with increased lipid peroxidation products (3) that are generated by reactive oxygen species.
The differential changes in the energy-generating pathways caused by diabetes in the detrusor and urothelial layer are highlighted in Fig. 1. In this figure, it is noted that individual metabolites are compared at absolute levels between the diabetic and nondiabetic tissue, whereas the activity of “pathways” is compared as the flux of glucose through that pathway. For example, in the diabetic compared with nondiabetic detrusor and urothelial layer, glucose is elevated, but the levels of intermediates of glycolysis remain the same, resulting in a net reduction in glucose flux through glycolysis.
Fig. 1.

Comparison of changes in the energy generating pathways between the nondiabetic and diabetic detrusor (A) and urothelial layer (B). If there are changes in absolute level of individual metabolites (boldface lowercase), then this is indicated by colored font (black, no change; ↑red, increased level; ↓green, decreased level). Energy-generating pathways are shown in boldface uppercase. The changes in flux of glucose through these pathways (determined by the Pearson correlation; see Table 5) are indicated by colored font (black, no change; ↑red, increased glucose flux; ↓green, decreased glucose flux). GSSG, glutathione disulfide; GSH, glutathione; SDH, sorbitol dehydrogenase; AR, aldose reductase; LDH, lactatedehydrogenase, GAPDH, glyceraldehyde-3-P dehydrogenase.
Diabetes is also commonly associated with disturbance of lipid metabolism (6, 13, 38). In this study, alterations in lipid metabolism were clearly delineated between the urothelial layer and detrusor. The synthesis of long-chain fatty acids is dependent on the NADPH availability, whereas degradation of fatty acid is affected by an increase in mitochondrial NADH/NAD+. As discussed above, because of the shift in the diabetic detrusor toward glucose oxidation via sorbitol to fructose and pentose phosphorylation, there would be an increase in the NAD(P)H/NAD(P) ratio. The diabetic detrusor displayed significant reductions the long-chain fatty acid [10-heptadecenoate (17:1n7)] and two medium-chain fatty acids [caproate (6:0) and caprylate (8:0)] (Table 3) as well as a concomitant increase in several acyl-carnitines (e.g., valerylcarnitine and octanoylcarnitine), implying that the reductions in free fatty acids represented a shift toward acyl-carnitines, which are the derivatives of fatty acids used to transport them into mitochondria (Table 3). However, since we observed no increase in acetylcarnitine and ketone bodies (the mitochochondrial downstream metabolites of fatty acid catabolism), our data suggest that β-oxidation was inhibited in the diabetic detrusor. Furthermore, our metabolic profile showed significant reductions in several phospholipids (containing arachidonic acid, such as palmitoyl-arachidonoyl-glycerophosphocholine and stearoyl-arachidonoyl-glycerophosphocholine) and increases in sphingolipids (e.g., oleoyl sphingomyelin). Phospho- and sphingolipids are major components of the cell membrane, and the alterations in these structural lipids may both modulate membrane architecture and disturb the functions of various receptors and channels on the membrane (21).
We also noted that diabetes caused changes in the pathways of lipid metabolism in the urothelial layer, although these were distinct, and in some case opposite, to the changes observed in the detrusor (Table 4). For example, there were significant increases in polyunsaturated fatty acids {e.g., stearidonate [18:4n3], eicosapentaenoate [20:5n3], and docosahexaenoate [22:6n3] and lysolipids [e.g., 1-linoleoylglycerophosphocholine (18:2n6), 1-palmitoylglycerophosphoethanolamine, and 1-stearoylglycerophosphoglycerol, etc.]} and dramatic reductions in sphingolipid. These changes could potentially impact urothelial layer membrane fluidity and permeability and thereby attenuate urothelial barrier functions in the diabetic animal. The urothelial layer also synthesizes prostanoids such as prostaglandin E2 (PGE2) and prostaglandin F2α (PGF2α), which act as important mediators of detrusor function. Previous research has demonstrated impaired prostaglandin release from urothelial preparations from type 1 diabetic rats (28). In patients with bladder overactivity, there are conflicting reports concerning urinary levels of PGE2 and PGF2α. They are reported as either significantly increased (14) or not significantly different from those in healthy controls (18). Our metabolomics profile demonstrates that although the prostaglandin precursor arachidonic acid (AA) was not changed in diabetic urothelial layer, prostaglandin PGE2, prostaglandin D2, and PGF2α were significantly decreased. The cause of these decreased levels may result from several mechanisms (such as a reduction in their synthetic pathways from AA or an increase in export and release of prostaglandins from the urothelial layer). However, given that prostaglandins are important regulators of detrusor activity, our results suggest that changes in the dynamics of prostaglandin synthesis are likely to play a key role in the development of DBD.
Chronicity of diabetic bladder disease.
DBD is a progressive disease in both the animal model used in these studies and patients (17, 19, 36), and therefore, an expansion of these studies to consider temporal effects of diabetes on bladder metabolism is warranted. As described above, since 1 mo of streptozotocin-induced diabetes is the earliest time point when bladder pathophysiology manifests, we believe it represents a time point where changes in the metabolism of the detrusor and urothelial layer (resulting from hyperglycemia) have a direct effect on bladder physiology; the changes in the metabolism we document here are likely directly resulting in the compensated overactive bladder phenotype. With longer periods of diabetes-induced hyperglycemia (from 12 wk onward), the bladder phenotype progresses toward decompensation, underactivity, and hypertrophy. This decompensated phase is believed to result primarily from a combination of polyuria and continued hyperglycemic-induced metabolic and oxidative stress (36). Therefore, although progression to the decompensated state is likely to be reflected by differences in metabolism compared with the compensated stage, it will be challenging to ascribe whether these changes in metabolism are a direct cause of or a response to diabetic pathophysiology. For example, changes in metabolism may result in response to the altered physiology/anatomy of the decompensated bladder. Alternatively, accumulation of AGE products and damage caused by oxidative stress might influence transcription and activity of enzymes and thereby metabolism and physiology. Therefore, future experiments delineating the cause and effect of diabetes on the pathophysiological progression of DBD will not only require temporal studies on diabetic animals but also need comparison with nondiabetic animal models of the decompensated state.
Conclusions.
In both the urothelial layer and detrusor, after 1 mo there was strong evidence of oxidative stress through the upregulation of protective metabolites and pathways. There were striking differential changes caused by diabetes on the metabolism of the detrusor and urothelial layer. Changes in the energy-generating pathways of the detrusor resulted in an accumulation of sorbitol, which through generation of AGE products would likely play a key role in the development of DBD. In the urothelial layer, changes in prostaglandin synthesis would also likely impact detrusor function.
GRANTS
Y. Wang is a recipient of the Lilly Innovation Fellowship Award, and K. P. Davies is supported by the National Institute of Diabetes and Digestive and Kidney Diseases (R01-DK-107807).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Y.W. and K.P.D. conception and design of research; Y.W. performed experiments; Y.W. and K.P.D. analyzed data; Y.W. and K.P.D. prepared figures; Y.W. and K.P.D. drafted manuscript; G.G.D. and K.P.D. edited and revised manuscript; K.P.D. interpreted results of experiments; K.P.D. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Metabolon for performing metabolic analysis, in particular Dr. Nicki Bell.
REFERENCES
- 1.Baneerjee MK, Das AK, Basha A, Ganesan TS, Chandrasekhar S. Reversibility of diabetic bladder dysfunction with early and good control. J Assoc Physicians India 33: 337–338, 1985. [PubMed] [Google Scholar]
- 2.Bradway C, Coyne KS, Irwin D, Kopp Z. Lower urinary tract symptoms in women—a common but neglected problem. J Am Acad Nurse Pract 20: 311–318, 2008. [DOI] [PubMed] [Google Scholar]
- 3.Changolkar AK, Hypolite JA, Disanto M, Oates PJ, Wein AJ, Chacko S. Diabetes induced decrease in detrusor smooth muscle force is associated with oxidative stress and overactivity of aldose reductase. J Urol 173: 309–313, 2005. [DOI] [PubMed] [Google Scholar]
- 3a.Daneshgari F, Liu G, Birder L, Hanna-Mitchell AT, Chacko S. Diabetic bladder dysfunction: current translational knowledge. J Urol 182 6Suppl: S18–S26, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dehaven CD, Evans AM, Dai H, Lawton KA. Organization of GC/MS and LC/MS metabolomics data into chemical libraries. J Cheminform 2: 9, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dumas ME, Kinross J, Nicholson JK. Metabolic phenotyping and systems biology approaches to understanding metabolic syndrome and fatty liver disease. Gastroenterology 146: 46–62, 2014. [DOI] [PubMed] [Google Scholar]
- 6.Fahrmann J, Grapov D, Yang J, Hammock B, Fiehn O, Bell GI, Hara M. Systemic alterations in the metabolome of diabetic NOD mice delineate increased oxidative stress accompanied by reduced inflammation and hypertriglyceremia. Am J Physiol Endocrinol Metab 308: E978–E989, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Friedrich N. Metabolomics in diabetes research. J Endocrinol 215: 29–42, 2012. [DOI] [PubMed] [Google Scholar]
- 8.Frimodt-Møller C. Diabetic cystopathy: epidemiology and related disorders. Ann Intern Med 92: 318–321, 1980. [DOI] [PubMed] [Google Scholar]
- 9.Golbidi S, Laher I. Bladder dysfunction in diabetes mellitus. Front Pharmacol 1: 136, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Imai S, Guarente L. NAD+ and sirtuins in aging and disease. Trends Cell Biol 24: 464–471, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Irwin DE, Mungapen L, Milsom I, Kopp Z, Reeves P, Kelleher C. The economic impact of overactive bladder syndrome in six Western countries. BJU Int 103: 202–209, 2009. [DOI] [PubMed] [Google Scholar]
- 12.Kaplan SA, Blaivas JG. Diabetic cystopathy. J Diabetes Complications 2: 133–139, 1988. [DOI] [PubMed] [Google Scholar]
- 13.Kim HJ, Kim JH, Noh S, Hur HJ, Sung MJ, Hwang JT, Park JH, Yang HJ, Kim MS, Kwon DY, Yoon SH. Metabolomic analysis of livers and serum from high-fat diet induced obese mice. J Proteome Res 10: 722–731, 2011. [DOI] [PubMed] [Google Scholar]
- 14.Kim JC, Park EY, Seo SI, Park YH, Hwang TK. Nerve growth factor and prostaglandins in the urine of female patients with overactive bladder. J Urol 175: 1773–1776, 2006. [DOI] [PubMed] [Google Scholar]
- 15.Lee WC, Wu HP, Tai TY, Liu SP, Chen J, Yu HJ. Effects of diabetes on female voiding behavior. J Urol 172: 989–992, 2004. [DOI] [PubMed] [Google Scholar]
- 16.Lee WC, Wu HP, Tai TY, Yu HJ, Chiang PH. Investigation of urodynamic characteristics and bladder sensory function in the early stages of diabetic bladder dysfunction in women with type 2 diabetes. J Urol 181: 198–203, 2009. [DOI] [PubMed] [Google Scholar]
- 17.Liu GM, Daneshgari F. Diabetic bladder dysfunction. Chinese Med J-Peking 127: 1357–1364, 2014. [PMC free article] [PubMed] [Google Scholar]
- 18.Liu HT, Tyagi P, Chancellor MB, Kuo HC. Urinary nerve growth factor but not prostaglandin E2 increases in patients with interstitial cystitis/bladder pain syndrome and detrusor overactivity. BJU Int 106: 1681–1685, 2010. [DOI] [PubMed] [Google Scholar]
- 19.Melman A, Zotova E, Kim M, Arezzo J, Davies K, DiSanto M, Tar M. Longitudinal studies of time-dependent changes in both bladder and erectile function after streptozotocin-induced diabetes in Fischer 344 male rats. BJU Int 104: 1292–1300, 2009. [DOI] [PubMed] [Google Scholar]
- 20.Milliat F, Gripois D, Blouquit ME, Ferezou J, Serougne C, Fidge NH, Lutton C. Short and long-term effects of streptozotocin on dietary cholesterol absorption, plasma lipoproteins and liver lipoprotein receptors in RICO rats. Exp Clin Endocrinol Diabetes 108: 436–446, 2000. [DOI] [PubMed] [Google Scholar]
- 21.Mills JK, Needham D. Lysolipid incorporation in dipalmitoylphosphatidylcholine bilayer membranes enhances the ion permeability and drug release rates at the membrane phase transition. Biochim Biophys Acta 1716: 77–96, 2005. [DOI] [PubMed] [Google Scholar]
- 22.Moller CF. Diabetic cystopathy. III:Urinary bladder dysfunction in relation to bacteriuria. Dan Med Bull 23: 287–291, 1976. [PubMed] [Google Scholar]
- 23.Moller CF. Diabetic cystopathy.I: A clinical study of the frequency of bladder dysfunction in diabetics. Dan Med Bull 23: 267–278, 1976. [PubMed] [Google Scholar]
- 24.Moller CF. Diabetic cystopathy.II: Relationship to some late-diabetic manifestations. Dan Med Bull 23: 279–287, 1976. [PubMed] [Google Scholar]
- 25.Ohta T, Masutomi N, Tsutsui N, Sakairi T, Mitchell M, Milburn MV, Ryals JA, Beebe KD, Guo L. Untargeted metabolomic profiling as an evaluative tool of fenofibrate-induced toxicology in Fischer 344 male rats. Toxicol Pathol 37: 521–535, 2009. [DOI] [PubMed] [Google Scholar]
- 26.Onukwugha E, Zuckerman IH, McNally D, Coyne KS, Vats V, Mullins CD. The total economic burden of overactive bladder in the United States: a disease-specific approach. Am J Manag Care 15: S90–S97, 2009. [PubMed] [Google Scholar]
- 27.Pinna C, Bolego C, Puglisi L. Effect of substance P and capsaicin on urinary bladder of diabetic rats and the role of the epithelium. Eur J Pharmacol 271: 151–158, 1994. [DOI] [PubMed] [Google Scholar]
- 28.Pinna C, Zanardo R, Puglisi L. Prostaglandin-release impairment in the bladder epithelium of streptozotocin-induced diabetic rats. Eur J Pharmacol 388: 267–273, 2000. [DOI] [PubMed] [Google Scholar]
- 29.Spratlin JL, Serkova NJ, Eckhardt SG. Clinical applications of metabolomics in oncology: a review. Clin Cancer Res 15: 431–440, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suhre K. Metabolic profiling in diabetes. J Endocrinol 221: R75–R85, 2014. [DOI] [PubMed] [Google Scholar]
- 31.Tong YC, Cheng JT. Alteration of M-3 subtype muscarinic receptors in the diabetic rat urinary bladder. Pharmacology 64: 148–151, 2002. [DOI] [PubMed] [Google Scholar]
- 32.Tong YC, Chin WT, Cheng JT. Alterations in urinary bladder M-2-muscarinic receptor protein and mRNA in 2-week streptozotocin-induced diabetic rats. Neurosci Lett 277: 173–176, 1999. [DOI] [PubMed] [Google Scholar]
- 33.Wang Y, Tar MT, Fu S, Melman A, Davies KP. Diabetes attenuates urothelial modulation of detrusor contractility and spontaneous activity. Int J Urol 21: 1059–1064, 2014. [DOI] [PubMed] [Google Scholar]
- 34.Waring JV, Wendt IR. Effects of streptozotocin-induced diabetes mellitus on intracellular calcium and contraction of longitudinal smooth muscle from rat urinary bladder. J Urol 163: 323–330, 2000. [PubMed] [Google Scholar]
- 35.Williamson JR, Chang K, Frangos M, Hasan KS, Ido Y, Kawamura T, Nyengaard JR, van den Enden M, Kilo C, Tilton RG. Hyperglycemic pseudohypoxia and diabetic complications. Diabetes 42: 801–813, 1993. [DOI] [PubMed] [Google Scholar]
- 36.Xiao N, Wang Z, Huang Y, Daneshgari F, Liu G. Roles of polyuria and hyperglycemia in bladder dysfunction in diabetes. J Urol 189: 1130–1136, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yoshimura N, Chancellor MB, Andersson KE, Christ GJ. Recent advances in understanding the biology of diabetes-associated bladder complications and novel therapy. BJU Int 95: 733–738, 2005. [DOI] [PubMed] [Google Scholar]
- 38.Zhang S, Nagana Gowda GA, Asiago V, Shanaiah N, Barbas C, Raftery D. Correlative and quantitative 1H NMR-based metabolomics reveals specific metabolic pathway disturbances in diabetic rats. Anal Biochem 383: 76–84, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zimny D, Szatkowska M, Polubok J, Maciaszek J, Machaj M, Barg E. [The use of metabolomics in medicine - some examples of oncological and metabolic diseases]. Pediatr Endocrinol Diabetes Metab 20: 55–62, 2015. [DOI] [PubMed] [Google Scholar]
- 40.Zitka O, Skalickova S, Gumulec J, Masarik M, Adam V, Hubalek J, Trnkova L, Kruseova J, Eckschlager T, Kizek R. Redox status expressed as GSH:GSSG ratio as a marker for oxidative stress in paediatric tumour patients. Oncol Lett 4: 1247–1253, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]