

Abstract
Burn trauma results in prolonged hypermetabolism and skeletal muscle wasting. How hypermetabolism contributes to muscle wasting in burn patients remains unknown. We hypothesized that oxidative stress, cytosolic protein degradation, and mitochondrial stress as a result of hypermetabolism contribute to muscle cachexia postburn. Patients (n = 14) with burns covering >30% of their total body surface area were studied. Controls (n = 13) were young healthy adults. We found that burn patients were profoundly hypermetabolic at both the skeletal muscle and systemic levels, indicating increased oxygen consumption by mitochondria. In skeletal muscle of burn patients, concurrent activation of mTORC1 signaling and elevation in the fractional synthetic rate paralleled increased levels of proteasomes and elevated fractional breakdown rate. Burn patients had greater levels of oxidative stress markers as well as higher expression of mtUPR-related genes and proteins, suggesting that burns increased mitochondrial stress and protein damage. Indeed, upregulation of cytoprotective genes suggests hypermetabolism-induced oxidative stress postburn. In parallel to mtUPR activation postburn, mitochondrial-specific proteases (LONP1 and CLPP) and mitochondrial translocases (TIM23, TIM17B, and TOM40) were upregulated, suggesting increased mitochondrial protein degradation and transport of preprotein, respectively. Our data demonstrate that proteolysis occurs in both the cytosolic and mitochondrial compartments of skeletal muscle in severely burned patients. Increased mitochondrial protein turnover may be associated with increased protein damage due to hypermetabolism-induced oxidative stress and activation of mtUPR. Our results suggest a novel role for the mitochondria in burn-induced cachexia.
Keywords: burn injury, cachexia, hypermetabolism-induced oxidative stress, mitochondrial unfolded protein response, mitochondria proteases
burn trauma results in a profound hypermetabolic and hypercatabolic stress response that persists for several months after injury (12). Several studies have implicated catecholamines as the primary mediators of hypermetabolism (26, 67) in a state in which increased whole body oxygen consumption supports greater ATP turnover and thermogenesis in critically injured burn patients. Skeletal muscle cachexia resulting from a chronic imbalance between protein synthesis and breakdown (12, 18) leads to significant long-term morbidity in burn survivors (67). Whereas the degree of hypermetabolism and skeletal muscle catabolism is correlated in burn survivors (18), whether a common mechanism links burn-induced hypermetabolism and protein wasting remains unclear.
It has been shown previously that skeletal muscle becomes hypermetabolic in response to burns (68). Recent reports from our laboratory demonstrated that skeletal muscle mitochondria contribute to the hypermetabolic response in burn patients (49, 51, 60). Given their role in oxidative phosphorylation, mitochondria are a principal site of cellular reactive oxygen species (ROS) generation (42, 62). Respiratory complexes in the electron transport chain, with the exception of complex IV, may leak electrons to oxygen, forming superoxide anions (O2−) (12, 13, 62). The superoxide anion is a precursor to many other ROS and can activate other oxidative reactions (62, 64). A persistent elevation in mitochondrial respiration and superoxide production in skeletal muscle predisposes mitochondrial proteins to oxidation. Although mitochondria are normally capable of eliminating superoxide, sustained increases in the mitochondrial respiration and thus superoxide production in the skeletal muscle of burn victims may result in the chronic damage to mitochondrial proteins and activation of mitochondrial unfolded protein response (mtUPR) (11, 38, 41, 61).
Hypermetabolism-induced oxidative stress may contribute to mtUPR activation when the proteotoxic stress within the mitochondrion exceeds protein-folding capacity of chaperones (41, 44). When the protein-folding capacity of the chaperones is exceeded, accumulation of misfolded or aggregated protein ensues. To protect the cellular compartments from the deleterious effects of protein misfolding and aggregation (21, 44), stressed cells use two mechanisms. They activate organelle-specific proteases such as LONP-1, CLPP, and YME1L1 to help degrade misfolded and damaged proteins (4, 44). In addition, mtUPR is activated to help increase chaperone capacity and reestablish homeostasis within the mitochondrial protein-folding environment (41, 44). This can be accomplished by transducing the stress response to the nucleus to help induce transcription factors responsible for proteostatic surveillance and synthesis of new peptide (32, 41).
Given their locality to the respiratory complexes, mitochondrial proteins are more susceptible to oxidative damage than cytoplasmic proteins (13, 62, 63). However, since most mitochondrial proteins are encoded in the nucleus, new proteins must be translocated from the cytoplasm via mitochondrial translocase proteins to replace damaged proteins (45, 61, 66). In this study, we hypothesized that persistent hypermetabolism-induced oxidative stress leads to cumulative oxidative damage of mitochondrial proteins and activation of the unfolded protein stress response. We also hypothesized that these result in an increase in mitochondrial protein turnover, which leads to an upregulation of mitochondrial translocase proteins and influx of replacement preproteins from cytoplasm (nucleus) of the skeletal muscle cell. Our results show that protein degradation takes place in both mitochondrial and cytosolic cellular compartments of skeletal muscle in burn patients. Therefore, our data provide a novel mechanistic association between burn-induced hypermetabolism and muscle wasting.
MATERIALS AND METHODS
Subjects
The study protocol was reviewed and approved by the Institutional Review Board at the University of Texas Medical Branch (UTMB), Galveston, TX. We recruited 14 adult burned patients who were admitted to the Blocker Burn Unit at UTMB, and teenage children admitted to Shriners Hospitals for Children (Galveston, TX) with total burn surface area (TBSA) of >30% were recruited. Upon admission, all burn patients received standard burn care, including fluid resuscitation and total wound excision. Sequential wound grafting was performed based on the individual need of the patients until burn wounds were closed. Patients were placed on a standard nutritional protocol and received a low-fat diet (82% carbohydrate, 3% fat, 15% protein) (Vivonex T.E.N.; Nestle Health Science, Minneapolis, MN) based on their burn size (1,500 kcal/m2 body area + 1,500 kcal/m2 body area burned). Nutrition was provided via nasogastric tube until patients were able to tolerate oral feeding. Resting energy expenditure (REE) was determined by indirect calorimetry (65) and compared with the Harris-Benedict-derived REE (56) to determine the degree of systemic hypermetabolism.
We recruited 13 healthy control subjects from the Galveston County, TX, area via local advertisements to serve as unburned controls. These participants were admitted to the Clinical Research Center at the UTMB. Informed and written consent was obtained before participation in the study. After an overnight fast, muscle biopsy was collected from m. vastus lateralis using a suction-adapted Bergstrom needle (6).
High-Resolution Respirometry
Skeletal muscle mitochondrial respiration (metabolism) was determined as described initially (48). Briefly, ∼10 mg of skeletal muscle was permeabilized by agitating myofiber bundles in 2 ml of mitochondrial preservation buffer containing 20 mM saponin at 4°C for ∼30 min. Thereafter, myofiber bundles were washed in 2 ml of mitochondrial respiration buffer containing 0.5 mM EGTA, 3 mM MgCl2, 60 mM K-lactobionate, 20 mM taurine, 10 mM KH2PO4, 20 mM HEPES, 110 mM sucrose, and 1 mg/ml essential fatty acid-free bovine serum albumin (pH 7.1) at 4°C for ∼15 min. Next, ∼2–4 mg of myofiber bundles were blotted onto filter paper and weighed on a precision microbalance (Mettler-Toledo, Zaventem, Belgium) prior to being transferred to an Oxygraph-O2K respirometer chamber (Oroboros Instruments, Innsbruck, Austria), where they were suspended in 2 ml of mitochondrial respiration buffer. Respiration measurements were made on the same day as sample collection, typically within 6 h of harvest. During the experiments temperature was maintained at 37°C, and O2 concentration was within 250–400 nmol/ml. Mitochondrial O2 flux was computed and recorded at 2- to 4-s intervals by DatLab software (Oroboros Instruments). Mitochondrial respiratory capacity and function were assayed in permeabilized skeletal muscle samples by the addition of a saturating concentration of substrates (1.5 mM octanoyl-l-carnitine, 5 mM pyruvate, 2 mM malate, 10 mM glutamate, and 10 mM succinate), followed by the addition of adenosine diphosphate (5 mM), cytochrome c (10 mM), and the mitochondrial uncoupler carbonyl cyanide m-chlorophenyl hydrazin to a final concentration of 5 mM. Resulting values were normalized using citrate synthase protein.
Skeletal Muscle Protein Kinetics Study
The fractional synthetic rate (FSR) and fractional breakdown rate (FBR) of mixed-muscle proteins were determined according to the protocol described previously (12). Briefly, baseline plasma samples were collected to determine background isotopic enrichment prior to bolus injections of l-[ring-13C6]Phe (5.56 mg/kg) and l-[15N]Phe (5.40 mg/kg) administered intravenously at 0 and 30 min, respectively. Blood samples were collected into heparinized sample tubes at various time points (5, 10, 15, 20, 29, 35, 40, 50, and 60 min) after the injection of the initial tracer bolus. Muscle samples were collected from the m. vastus lateralis at 10 and 60 min after the injection of the initial tracer bolus with the aid of suction-adapted Bergstrom needle (6). Muscle samples were rinsed with saline to remove all visible blood and then rapidly frozen in liquid nitrogen.
Plasma and muscle phenylalanine enrichments were determined in tert-butyldimethylsilyl derivatives using an Agilent 6890 gas chromatograph-mass spectrometer. Ions were selectively monitored at mass-to-charge ratios of 336, 337, 340, 342, and 346 for phenylalanine enrichment. Isotopic enrichments were expressed as tracer-to-tracer ratio. Muscle concentrations of free intracellular phenylalanine were calculated from the internal standard (46). The precursor-product method was utilized to calculate muscle FSR (Eq. 1), whereas muscle FBR was calculated from the decay in intracellular and plasma enrichment (Eq. 2) (70):
| (1) |
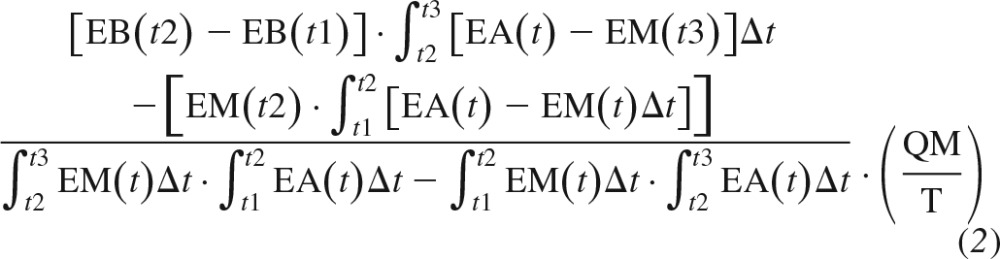 |
FSR and FBR data were obtained in healthy young men in a previous study by our group (16) and are presented here as heathy (normal) values to provide a comparison with data obtained in burn victims.
Gene Expression
RNA was extracted from ∼50 to 100 mg of muscle tissue from both control subjects and burn patients using the Pure Link RNA isolation kit according to the manufacturer's protocol (Thermo Fisher Scientific, Waltham, MA). Isolated RNA was quantitated using a Biotek Take3 microvolume plate reader (Biotek Instruments). cDNA was synthesized using a High-Capacity RNA-to-cDNA Kit according to the manufacturer's protocol (Thermo Fisher Scientific). Gene expression was analyzed in both burn patients and healthy controls using quantitative PCR performed with the StepOne Plus real-time PCR system (Applied Biosytems, Waltham, MA) and using Gene Expression master mix with the following TaqMan gene assays: Hs00998404_m1 [Lon peptidase 1 (LONP1)], Hs00204609_m1 [YME1 like 1 ATPase (YME1L1)], Hs00231457_m1 [nuclear factor, erythroid 2 like 1 (NFE2L1)], Hs00975961_g1 [nuclear factor, erythroid 2 like 2 (NFE2L2)], Hs00202227_m1 [Kelch-like ECH-associated protein 1 (KEAP1)], Hs00997642_m1 [valosin-containing protein (VCP/P97)], Hs00427357_m1 [proteasome subunit-β type-1 (PSMB1)], Hs01002946_m1 [proteasome subunit-β type-2 (PSMB2)], Hs00605652_m1 [proteasome subunit-β type-5 (PSMB5)], Hs01027360_g1 [proteasome subunit-α type-1 (PSMA1)], Hs01113429_m1 [proteasome non-ATPase regulatory subunit 14 (PSMD14)], Hs00160631_m1 [26S proteasome non-ATPase regulatory subunit 1 (PSMD1)], Hs01029472_g1 [26S protease regulatory subunit 8 (PSMC5)], Hs00426616_g1 [aconitase 2 (ACO2)], Hs00158095_m1 (ACO1), Hs00533490_m1 [superoxide dismutase 1 (SOD1)], Hs00167309_m1 (SOD2), Hs00428953_g1 [peroxiredoxin 3 (PRDX3)], Hs00156308_m1 [catalase (CAT)], Hs00357891_S1 [transcription factor jun-B (JUNB)], Hs00232586_m1 [activating transcription factor 6 (ATF6)], Hs01922818_s1 [CCAAT/enhancer-binding protein (CEBPγ)], Hs00270923_s1 (CEBPβ), Hs00358796_g1 [C/EBP homologous protein (CHOP/DDIT3]), Hs00430663_g1 [ubiquitin-like protein 5 (UBL5)], Hs00269972_s1 (CEBPα), Hs00177102_m1 [c-Jun NH2-terminal protein kinase 2 (JNK2/MAPK9)], Hs00195655_m1 (CLPP), Hs01103582_s1 [c-jun transcription factor (c-JUN)], Hs01654720_g1 [heat shock protein (HSP)10], Hs01036753_g1 [60-kDa heat shock protein, mitochondrial (HSP60)], Hs03043881_g1 [heat shock protein 90 AB (HSP90AB)], and Hs02758991_g1 (GAPDH) (Thermo Fisher Scientific). Expression of the target genes was normalized to that of the housekeeping gene GAPDH. Relative gene expression data analyses were performed using DataAssist Software version 3.01 (Life Technologies) to derive ΔΔCT values. Final data are expressed in fold difference of burn patients to control.
Western Blotting Analysis
Antibodies.
Antibodies to LONP1 (no. 15440-1-AP), TIMM17B (no. 11062-1-AP), and TIM23 (no. 11123-1-AP) were from Proteintech (Rosemont, IL); PSMD14 (no. 7662S), SOD1 (no. 2770S), SOD2 (13194S), catalase (14097S), ACO2 (6922S), p-4E-BP1 (eukaryotic initiation factor 4E binding protein 1; Thr37/46) (no. 2855S), phospho-S6 ribosomal protein (Ser240/244; no.5364S), p-mTOR (mammalian target of rapamycin) (Ser2448; no. 5536S), P-p70 S6 kinase (Thr389; no. 9234S), p-eEF2 (eukaryotic elongation factor 2) (Thr56; no.2331S), p-eIF2α (eukaryotic initiation factor 2)(Ser51; no. 9721S), p-eE2K (Ser366; no.3691S), CHOP (no. 2895S), HSP60 (4869S), HSP90 (4877S), and JunB (no. 3753S) were from Cell Signaling Technology (Denver, MA); NFE2L1/NrF1(sc-13031) was from Santa Cruz Biotechnology (Dallas, TX); NFE2L2/NrF2 (ab62352), proteasome 20S α + β (ab22673), proteasome 20S C2 (ab109530), YME1L1 (ab170123), 4-hydroxynonenal (ab48506), nitrotyrotyrosine (ab125106), TOM40 (ab51884), PRDX3 (ab73349), ATF-6 (ab203119), rabbit horseradish-peroxidase-labeled anti-mouse (ab6728), and donkey horseradish peroxidase-labeled anti-rabbit (ab6802) were from Abcam (Cambridge, MA); and GAPDH (no. G9545) was from Sigma-Aldrich (St. Louis, MO).
Gel electrophoresis and protein detection.
Proteins were separated using SDS-polyacrylamide gel electrophoresis (PAGE) and transferred to nitrocellulose membranes. Muscle samples were homogenized and protein was extracted using T-PER tissue extraction reagent (Thermo Fisher Scientific, Rockford, IL) containing protease and phosphatase inhibitors. Twenty micrograms of the skeletal muscle homogenate protein from control and patient muscle samples was suspended in NuPAGE LDS Sample Buffer, heated at 70°C for 10 min, rapidly cooled on ice, and then loaded onto 4–12% Bis-tris gels or 3–8% Tris-acetate gel depending on the molecular weight of the protein of interest (Thermo Fisher Scientific, Waltham, MA). Gels were run for 120 min at 100 V using the X-SureLock Mini-Cell electrophoresis system (Life Technologies). NuPAGE 2-(N-morpholino) ethanesulfonic acid-sodium dodecyl sulfate (MES-SDS), MOPS (3-morpholinopropane-1-sulfonic acid), or Tris-acetate SDS running buffer (Life Technologies) was used, depending on the molecular weight of protein of interest. The protein was then transferred to a nitrocellulose membrane using transfer buffer (Thermo Fisher Scientific, Waltham, MA) containing 20% methanol at 20 V for 2 h. Blots were blocked using 5% (wt/vol) nonfat milk (Bio-Rad, Hercules, CA) or bovine serum albumin (Cell Signaling Tecnbology) in TBS with 0.1% Tween 20 (TBST) for 1 h at room temperature. Membrane blots were then incubated with the antibody of interest in 5% nonfat milk or BSA in TBST at 4°C overnight. Blots were washed three times with TBST for 15 min each before probing with either donkey anti-rabbit or rabbit anti-mouse secondary horseradish peroxidase-conjugated antibodies (Abcam, Cambridge, MA) at a 1:10,000 dilution for 1 h at room temperature. Blots were washed with TBST and visualized by autoradiography after incubation of blots in Pierce ECL Western Blotting Substrate (Thermo Scientific, Rockford, IL) for 1 min at room temperature according to the manufacturer's instructions. Autoradiographs were then scanned and band areas measured using ImageJ 1.48 software (National Institutes of Health, Bethesda, MD). Resulting values were normalized to those obtained for GAPDH.
Statistics
Statistical analysis was performed, and graphs were created using GraphPad Prism version 6 (GraphPad Software, La Jolla, CA). Student's t-tests were used to compare controls and burn patient data, with P < 0.05 being considered significant. Both control and burn patient data are presented as means ± SE, with one exception; data related to subject/patient characteristics are presented as means ± SD.
RESULTS
Subject Characteristics
As shown in Table 1, healthy controls (n = 13) and burn patients (n = 14) did not differ with regard to age, weight, height, or body mass index at the time of their admission to the intensive care unit. Burn patients had large injuries, encompassing 63 ± 22% of their TBSA, with full-thickness burn lesions encompassing 54 ± 23% of the TBSA. Both weight and body mass index of burn victims declined significantly during hospitalization (P < 0.05). On average, burn patients were hospitalized for 58 ± 46 days.
Table 1.
Subject characteristics
| Characteristic | Control Subjects (n = 13) | Burn Patients (n = 14) |
|---|---|---|
| Age, yr | 26.3 ± 4.7 | 33.9 ± 18.1 |
| Height, m | 1.8 ± 0.1 | 1.7 ± 0.1 |
| Weight on admission, kg | 83.1 ± 14.5 | 83.5 ± 23.1 |
| BMI on admission, kg/m2 | 25.4 ± 3.1 | 26.7 ± 5.4 |
| Weight on study date, kg | 74.4 ± 17.5* | |
| Length of stay, days | 58 ± 46 | |
| Days postburn | 18.8 ± 9 | |
| Weight at discharge, kg | 65.6 ± 15.7* | |
| BMI at discharge, kg/m2 | 21.4 ± 3.7* | |
| Burn size, %TBSA | 63 ± 22 | |
| Full-thickness burn, %TBSA | 54 ± 23 |
Values are ± SD. %TBSA, %total body surface area.
P < 0.05 vs. admission measurement.
Hypermetabolic Hypercatabolic Response to Burns
The metabolic rate in burn patients was 43 ± 7% above normal (Fig. 1A). At the level of skeletal muscle, burn patients had greater coupled (P) and leak (L) mitochondrial respiration, indicating that, per mitochondria, O2 consumption was greater in burn patients compared with controls (Fig. 1B). Burn patients lost 18 ± 3% (15 ± 3 kg) of their body mass during hospitalization (P < 0.05 vs. healthy controls; Fig. 1C). Lean body mass (Fig. 1D) and bone mineral content (Fig. 1E) were lower in burn patients at hospital discharge than in healthy controls.
Fig. 1.

Effect of burn trauma on resting energy expenditure (REE; A), mitochondrial leak and coupled respiration (B), body weight (C), lean body mass (D), and bone mineral content (BMC; E) and the fractional synthetic rate (FSR), fractional breakdown rate (FBR), and net balance (NB) of mixed muscle proteins (F). Healthy control FSR, FBR, and NB data were taken from Gundermann et al. (16). Activation of mTORC1 signaling and protein synthesis in human muscle following blood flow restriction exercise is inhibited by rapamycin. Values are presented as means ± SE. *P < 0.05 vs. control; **P < 0.01 vs. control; ***P < 0.001 vs. control.
Compared with a previously published (16) healthy control group, skeletal muscle protein turnover was significantly altered in burn patients. Skeletal muscle FSR was 1.6-fold higher in burn patients (0.086 ± 0.013 vs. 0.06 ± 0.006%/h, P < 0.05; Fig. 1F). Skeletal muscle FBR was 3.5-fold greater in burn patients than in healthy controls (0.354 ± 0.089 vs. 0.100 ± 0.010%/h P < 0.001; Fig. 1F). Net protein balance (i.e., difference between FSR and FBR) was approximately sixfold higher in burn patients (−0.268 ± 0.091%/h) than in healthy controls (−0.045 ± 0.010%/h; Fig. 1F).
mTOR Signaling
Protein synthesis in burn patients and healthy controls was further explored through Western blot analysis of the master growth regulator mTOR and its targets p-70S6K and 4E-BP1 (Fig. 2). Phosphorylation of mTOR at Ser2448 was significantly higher in burn patients compared with healthy controls (P < 0.01; Fig. 2A). In addition, phosphorylation of 4E-BP1 at Thr37/46, S6rp at Ser240/244, and p-eEF2 at Thr56 was greater in burn patients (P < 0.05; Fig. 2, B–D). However, phosphorylation of p70S6K at Thr389, eukaryotic elongation factor factor-2 kinase at (Eef2k) at Ser366, and EeF2 at Ser51 did not differ between the two groups (P > 0.05; Fig 2, E–G).
Fig. 2.

Effects of burn trauma on mammalian target of rapamycin (mTOR) signaling in skeletal muscle. Phosphorylation of mTOR at Ser2448 (A), eukaryotic initiation factor 4E binding protein (4E-BP1) at Thr37/46 (B), S6rp at Ser240/244 (C), and eukaryotic elongation factor 2 (eEF2) at Thr56 (D) was significantly greater in burn patients, whereas phosphorylation of p70S6K at Thr389 (E), eukaryotic elongation factor factor-2 kinase at Ser366 (Eef2k; F), and eEF2 at Ser51 (G) did not differ between groups. Data were normalized to a loading control (GAPDH; n = 4 healthy controls and n = 5 burn patients). Values are presented means ± SE. P < 0.05 vs. control.
Mitochondrial Oxidative Stress
One of the primary objectives of this study was to determine whether hypermetabolism was associated with mitochondrial oxidative stress in skeletal muscle. We demonstrated that burn trauma induced upregulation of SOD1, SOD2, and PRDX3 genes by 2.6-, 16.1-, and 1.5-fold, respectively, compared with healthy controls (P < 0.001, P < 0.05, and P < 0.01, respectively; Fig. 3A). Similarly, catalase gene expression was 5.2-fold greater in burn patients than in controls (P < 0.05; Fig. 3A). SOD1, SOD2, and CAT proteins were all elevated in burn patients (P < 0.001, P < 0.01, and P < 0.05, respectively; Fig. 3, D–F). Conversely, PRDX3 protein did not differ between groups (P > 0.05; Fig. 3E). Nitrosylated tyrosine, a hallmark of amino acid oxidation and cell damage (54), was ∼80% higher in burn patients compared with controls (P < 0.01; Fig. 3B). Levels of 4-hydroxynoneal (HNE), a by-product of lipid peroxidation, were not significantly different between burn patients and healthy controls (Fig. 3C).
Fig. 3.

Effect of burn trauma on skeletal muscle oxidative stress and related factors. A: fold difference in SOD1, SOD2, catalase (CAT), peroxidoxine-3 (PRDX3), aconitase (ACO)1, and ACO2 mRNA levels between healthy controls and burn patients (n = 8/group). B–H: Western blot analysis of nitrotyrosilated protein (B), 4-hydroxynonenal (HNE; C), superoxide dismutase (SOD)1 (D), SOD2 (E), CAT (F), PRDX3 (G), and ACO2 (H). Data were normalized to a loading control (GAPDH) (n = 4 for control and n = 5 for burn group). Values are presented as group means ± SE. *P < 0.05 vs. control; **P < 0.01 vs. control; ***P < 0.001 vs. control.
Aconitase
Mitochondrial aconitase (ACO2) inactivation is a known source of hydroxyl radical formation. Cytosolic aconitase (ACO1) gene expression was 2.5-fold higher in burn patients (P < 0.01; Fig. 3A). ACO2 mRNA levels were comparable between groups (P > 0.05; Fig. 3A), but ACO2 protein levels were higher in burn patients than in controls (P < 0.001; Fig. 3H).
Mitochondrial Unfolded Protein Response
Stress response to oxidative stress or accumulation of misfolded proteins in the mitochondrial matrix and intermembrane space may result in the induction of mtUPR genes to help bolster chaperone capacity (44). Accumulation of unfolded proteins within mitochondrial matrix stimulates JNK2 and c-JUN to upregulate CHOP (41). CHOP subsequently activates chaperones to restore the mitochondrial protein-folding environment. Quantitative PCR analysis revealed that, compared with controls, burn patients had 1.5-fold higher JNK2 expression, 4.4-fold higher c-JUN expression, and 2.6-fold higher CHOP gene expression (P < 0.05; Fig. 4A). Other factors needed for the activation of CHOP were elevated in burn patients. JUNB increased 103.3-fold (P < 0.01 vs. control), CEBPβ 4.0-fold (P < 0.01), CEBPα 8.5-fold (P < 0.001), and CEBPγ threefold (P > 0.05; Fig. 4A). Furthermore, UBL5 was 2.5-fold higher in burn patients than in controls (P < 0.001; Fig. 4A), suggesting that mitochondrial stress was transduced to the cytoplasm. The ultimate goal of the mtUPR is the upregulation of chaperones. Indeed, burn patients had 2.7-fold higher heat shock protein (HSP)90 mRNA levels, 3.7-fold higher HsP60 mRNA levels, and a 2.6-fold higher HSP10 mRNA (P < 0.01 for all; Fig. 4A). Burn patients had not only higher expression of mtUPR genes than controls but also higher HSP90 and CHOP proteins (P < 0.05; Fig. 4, B and C). HSP60 and JUNB were also expressed to a higher degree in burn patients, but this was not significant (P > 0.05; Fig. 4, D and F). ATF6 protein was slightly higher in burn patients (P = 0.596; Fig. 4E).
Fig. 4.

Effect of burn trauma on expression of factors related to the mitochondrial unfolded protein response. Fold difference of mRNA levels of Jun-B, activating transcription factor 6 (ATF-6), CCAAT/enhancer-binding protein-γ (C/EBPγ), C/EBP homologous protein (CHOP)/DDIT3, heat shock protein (HSP)10, HSP60, HSP90, c-JUN, JNK2, ubiquitin-like protein 5 (UBL5), and C/EBPα between controls and burn patients (n = 8/group). Western blot analysis of HSP90 (B), CHOP (C), HSP60 (D), ATF6 (E), and Jun-B (F). Data were normalized to a loading control (GAPDH) (n = 4 for control and n = 5 for burn group). Values are presented as means ± SE. *P < 0.05 vs. control; **P < 0.01 vs. control; ***P < 0.001 vs. control.
Cytoprotective Transcription Factors
In response to oxidative stress, KEAP1/NFE2L2 signaling augments antioxidant capacity (23). NFE2L2 mRNA expression was 4.7-fold higher in burn patients than in controls, whereas KEAP1 was 1.8-fold higher (P < 0.01; Fig. 5A). Surprisingly, NFE2L2 protein levels were lower in burn patients than in controls despite significant transcriptional upregulation (P < 0.05; Fig. 5C). Furthermore, investigation of other genes revealed that NFE2L1 and VCP/P97 were 2.2- and 2.5-fold greater in burn patients than healthy controls, respectively (P < 0.001; Fig. 5A). NFE2L1 protein levels were also lower in burn patients (P < 0.01; Fig. 5D) most likely because both NFE2L1 and NFE2L2 are from the same family of human genes encoding basic leucine zipper (bZIP) transcription factors.
Fig. 5.

Effect of burn trauma on expression of cytoprotective transcription factors and proteosomal subunits. A: fold difference in mRNA levels of Kelch-Like ECH-associated protein 1 (KEAP1), nuclear factor, erythroid 2 like 2 (NFE2L2), nuclear factor, erythroid 2 like 1 (NFE2L1), and valosin-containing protein (VCP/p97) between controls and burn patients (n = 8/group). B: fold difference in the mRNA expression of 20S and 19S in controls and burn patients (n = 8/group). C–G: Western blot analysis of NFE2L2 (NrF2; C), NFE2L1 (NrF1; D), proteasome non-ATPase regulatory subunit 14 (PSMD14; E), proteasome 20S C2 (F), and proteasome 20Sα and -β (G). Data were normalized to a loading control (GAPDH; n = 4 for control and n = 5 for burn group). Values are presented as means ± SE. *P < 0.05 vs. control; **P < 0.01 vs. control; ***P < 0.001 vs. control.
20S and 19S Proteasomes
Ubiquitin-mediated proteolysis is central to muscle atrophy (2). The 26S proteasome plays an important role in the degradation of ubiquitinated proteins and is composed of a 20S core and 19S regulatory subunits (35). 20S and 19S subunits mRNAs were upregulated in burn patients compared with healthy controls (Fig. 5B). Specifically, PSMB1 mRNA was increased 2.6-fold (P < 0.001 vs. control), PSMB2 mRNA 3.6-fold (P < 0.01), PSMB5 mRNA 1.8-fold (P < 0.01), and PSMA1 3.0-fold (P < 0.00; Fig. 5B). In addition, burn patients showed 2.3-fold higher expression of PSMD1 mRNA, 2.8-fold higher expression of PSMD14 mRNA, and 2.8-fold higher expression of PSMC5 mRNA (P < 0.01; Fig. 5B). Interestingly, proteasomal protein expression showed a trend toward being greater in burn patients than in controls (Fig. 5, E–G), although the increase was significant for only the α- and β-subunits of 20S (P < 0.05; Fig. 5G). Furthermore, we found that mitochondrial respiration in skeletal muscle was significantly correlated with proteasome gene expression (r = 0.71, P < 0.01), suggesting a correlation between cellular respiration rates and the induction of proteolytic machinery in the cell cytosol.
Mitochondrial Proteases (LONP1, CLPP, and YME1L1) and Translocase Proteins
LONP1 and CLPP are mitochondrial proteases responsible for degradation of oxidized or unfolded protein within the matrix, whereas YME1L1 is responsible for degrading damaged proteins in the intermembrane space (61). LONP1 and CLPP mRNA levels were 2.4- and 2.2-fold higher, respectively, in burn patients compared with healthy controls (P < 0.001; Fig. 6A). Accordingly, burn patients had ∼60% more LONP1 protein abundance than healthy controls (P < 0.01; Fig. 6B). YME1L1 mRNA was twofold higher in burn patients than in controls (P < 0.01; Fig. 6A). This was accompanied by lower YME1L1 protein expression (P < 0.01; Fig. 6C). Interestingly, we found that mitochondrial respiration in skeletal muscle was significantly correlated with LONP1 gene expression (r = 0.86, P < 0.001), suggesting a strong correlation between cellular respiration rates and induction of mitochondrial proteases.
Fig. 6.

Effect of burn trauma on expression of mitochondrial proteases and translocases. A: fold difference in mRNA expression of lon peptidase 1 (LONP1), YME1 like 1 ATPase (YME1L1), and caseinolytic mitochondrial matrix peptidase proteolytic subunit (CLpP) in controls and burn patients (n = 8/group). B–G: Western blot analysis of LONP1 (B), YME1L1 (C), TIM23 (D), TIM 17A (E), TIM17B (F), and TOM40 (G). Data were normalized to a loading control (GAPDH; n = 4 for control and n = 5 for burn group). Values are presented as means ± SE. *P < 0.05 vs. control; **P < 0.01 vs. control; ***P < 0.001 vs. control.
Mitochondrial membrane translocase proteins were also analyzed, as increased expression of proteins suggests increased mitochondrial protein demand in burn patients' muscle. Expression of the inner membrane translocase protein 23 (TIM23) was ∼50% higher in burn patients than in controls (P < 0.05; Fig. 6D), whereas expression of the inner membrane translocase protein 17B (TIM17B) was ∼55% higher in burn patients (P < 0.05; Fig. 6E). Expression of the outer membrane translocase protein 40 (TOM40), the main constituent of the general import pore, was significantly greater in burn patients than in controls (P < 0.05; Fig. 6F).
DISCUSSION
The present study demonstrates a novel association between hypermetabolism and skeletal muscle protein degradation in burn patients. Elimination of damaged proteins is carried out mainly by proteasomes (28–30), which are primarily found in the nucleus, endoplasmic reticulum (ER), and cytoplasm of the cell but not in the mitochondria (43). In the context of severe burn injury and the profound hypermetabolism that accompanies this unique type of trauma, we hypothesized that mitochondrial oxidative stress and mtUPR also contribute to the marked muscle cachexia seen in burn patients. To address this hypothesis, we studied skeletal muscle protein turnover, markers of oxidative stress, mtUPR, and the proteolytic machinery residing in both the cytoplasmic and mitochondrial compartments of skeletal muscle in healthy individuals and severely burned patients exhibiting hypermetabolism and cachexia.
Although both FSR and FBR of skeletal muscle proteins were elevated in burn patients compared with data published previously by our group from healthy young men (16), massively elevated skeletal muscle FBR was the primary contributor to the protein-wasting phenotype postburn. This finding is in agreement with our previous findings using cross-limb arterial-venous balance methods (7, 19) and tracer bolus injection approaches (12). This previous study showed that skeletal muscle protein metabolism is profoundly altered in burn victims. Not surprisingly, in the ∼8 wk that they were hospitalized after injury, burn patients lost ∼15 kg of body mass (∼18% of their body mass at admission to the UTMB). Indeed, burn patients had significantly less body mass and in particular lower lean mass and bone mineral content than healthy controls.
In agreement with our stable isotope data showing that FSR of skeletal muscle proteins is greater in burn patients than in healthy controls, the molecular machinery involved in protein anabolism is also upregulated in the skeletal muscle of burn patients. Phosphorylation of mTORC1 and its downstream targets 4E-BP1 and S6rp was significantly greater in burn patients. Burn patients exhibit a catabolic endocrine response and undergo prolonged denervation postinjury, making the observation of greater protein synthesis in skeletal muscle somewhat curious. However, we have shown previously that massive skeletal muscle proteolysis results in increased intracellular free amino acid concentrations within muscle cells (19). This chronic elevation in the concentration of intracellular amino acids, including leucine, may explain why anabolic signaling and protein FSR are greater in burn patients (12, 19, 47, 52). Nevertheless, increased skeletal muscle FSR in burn patients did not match the increase in skeletal muscle FBR, resulting in a greater net loss of amino acids than seen in healthy controls. It is important to note that the increased phosphorylation of eEF2 at Thr56 (p-eEF2-Thr56) eEF2 is a GTP-dependent translocase that is responsible for the movement of nascent peptidyl-tRNAs from the A-site to the P-site of the ribosome (22). p-eEF2-Thr56 is involved in the inhibitory regulation of this step during protein translation elongation. Therefore, phosphorylation of eEF2 at Thr56 is inversely related to the rate of protein elongation, thereby contributing to the overall decrease of protein synthesis (24). This process might have been activated postburn to meet the significant amount of energy needed for protein breakdown. Protein degradation in the cytoplasm, nucleus, and mitochondria requires pathways that are highly energy dependent (15). Energy costs for protein turnover have been estimated to account for up to one-third of the total energy production during cell replication (34). This heightened energy demand needed for protein turnover and resulting in reduced peptide elongation might have contributed to the disproportional ratio of FSR to FBR in burn patients.
Burn patients were profoundly hypermetabolic, with resting energy expenditure ∼45% above normal values. Our data show that, in addition to exhibiting systemic hypermetabolism, burn patients experience hypermetabolism at the level of skeletal muscle. This indicates greater oxygen consumption by mitochondria of burn survivors. The increased oxygen consumption may further exacerbate the rate of superoxide production in the mitochondrial matrix, leading to mitochondrial stress and protein damage (40).
Approximately 0.2 to 0.5% of the oxygen consumed by the mitochondrion leaked out as superoxide (3). This implies that postburn hypermetabolism will increase exposure of mitochondria proteins to superoxide and possibly lead to oxidative protein damage in skeletal muscle mitochondria. Meanwhile, oxidative damage increases the likelihood of a protein being sequestered for a degradative fate. Oxidation disrupts protein structure and function, and it can result in the formation of protein aggregates that interfere with cell homeostasis (43, 53, 62). In the current study, we found that skeletal muscle of burn patients had an increased accumulation of nitrotyrosine, an indicator of greater protein oxidation. Within the mitochondrial matrix, SOD2 helps convert O2− to H2O2, which can easily be detoxified by glutathione peroxidase, but in the event of increased production of H2O2, hydroxyl radicals (OH) will be produced (3). The hydroxyl radical can indiscriminately react with nucleic acid, amino acids, and lipids. Tyrosine nitration occurs when tyrosine is exposed to a peroxynitrite anion (ONOO−) and nitrogen dioxide (NO2), both of which are by-products of nitric oxide (NO) metabolism in the presence of superoxide (O2−) and hydrogen peroxide (H2O2) (54). The marked hypermetabolism seen in burn patients likely augments mitochondrial O2− production. Alternatively, burn-induced mitochondrial dysfunction may also increase ROS generation, which in turn may increase uncoupling in the mitochondria as a compensatory mechanism against mitochondrial oxidative stress (50). Increased expression of HNE, a marker of lipid peroxidation, further corroborates oxidative protein damage in burn patients. Accumulating evidence suggests that lipid peroxidation can form protein abducts and exacerbates mitochondria dysfunction (3, 55). Importantly, HNE-induced protein adducts are not reversible, and studies suggest that profound accumulation of HNE-induced adducts can decrease proteasomal activity (23). Our findings are consistent with this idea that burn patients exhibit significantly increased mRNA and protein levels of proteasomes and antioxidants (catalase, SOD1, SOD2, and PRDX3). Upregulation of these proteins may have occurred in response to increased oxidative stress and served as a preventive measure to limit oxidative damage to proteins postburn (31, 43, 53). Unabated ROS generation can result in accumulation of oxidative protein damage and resultant unfolded proteins response stress due to the proteotoxic stress within mitochondria. The pathological effects of accumulation of unfolded proteins are not limited to the subcellular compartment and are dangerous for the entire cell (41, 48). Any possible pathological effects may be prevented through activation of the mtUPR and mitochondria proteases.
The mtUPR is induced when proteotoxic stress exceeds protein folding capacity by chaperones and the proteotoxic stress is sensed and transduced to the nucleus to reestablish homeostasis within the mitochondrial protein-folding environment (32, 41, 44). Accumulation of unfolded proteins within mitochondria results in the upregulation of JNK2 and c-JUN transcription factors, further resultin g in the stimulation of the transcription factors CHOP and C/EBPβ (44). CHOP and C/EBPβ transcription factors bind to specific mtUPR promoter elements and activate target genes such as HSP60 and CLPP (41, 44). Upregulation of JNK2, c-JUN, CHOP, C/EBPβ, C/EBPα, HSP60, CLPP, JUNB, and ATF6 genes suggests that mtUPR is activated in skeletal muscle of burn patients. These data indicate profound proteotoxic stress within the mitochondria environment and, most importantly, suggest that this stress is transduced to the nucleus to help combat the accumulation of unfolded proteins within skeletal muscle mitochondria in burn patients. Upregulation of both HSP60 and HSP10 chaperones further supports our hypothesis that in response to proteotoxic stress new proteins have to be transported into the mitochondria to meet increased protein degradation. Specifically, a prior study by Zhao et al. (71) shows that accumulation of unfolded protein within the mitochondrial matrix results in the upregulation of CHOP, HSP60, HSP10, CLPP, and mtDnaJ. This further confirms the association of mitochondrial stress with burn trauma. Important to note is that activation of UPR may result in profound mitochondrial dysfunction and possibly mitophagy (58). Interestingly, data showing mtUPR in burn patients are scarce. Most burn-related studies have focused on ER stress (14, 25). Our data also suggest activation of ER stress in skeletal muscle of burn patients, as reflected by upregulation of ATF6 and CHOP mRNA and protein. Furthermore, CHOP has been shown to be activated during both mitochondrial proteotoxic stress and extreme ER stress (27, 71), and this suggests the possibility of ER stress induction postburn. Although both CHOP and C/EBPβ contain AP-1 binding sites in their promoters, this is a major requirement for the induction of mtUPR but not for ER stress (27). The upregulation of AP-1 (JUNB) mRNA by 100-fold strongly suggests activation of mtUPR after severe burn trauma. Therefore, our study is the first to show that burn patient skeletal muscle cells experience not only endoplasmic reticular stress but also activation of mtUPR stress. There seems to be a cross-talk between mitochondrial stress and ER stress besides the communication between the mitochondria and the nucleus. Furthermore, UBL-5 protein levels have been reported to be increased in response to acute or chronic stress related to unfolded mitochondrial protein stress to help activate mtUPR (10). In addition to performing this function, it can combine with a yet to be characterized protein (DVE-1) when the stress is transduced to the cytoplasm to help upregulate the HSP60 gene (5, 10, 20, 27). Hence, UBL-5 mRNA upregulation suggests that, after burns, the stress response in skeletal muscle mitochondria is transduced to the cytoplasm to help activate the nuclear steps required for mounting a response to the threat of mitochondrial protein misfolding (5, 20).
For protein turnover within the cytoplasm to proceed during stress, stress-related transcription factors are activated (23). The transcription factors nuclear factor erythroid 2-related factor 2 (Nrf2/NFE2L2) and NFE2L1 regulate adaptive responses to oxidative stress (1). NFE2L2 and NFE2L1 are members of the CNC subfamily of bZIP transcription factors, which regulate the expression of a large number of antioxidants and cytoprotective genes (23, 36, 43). Specifically, NFE2L1 and NFE2L2 have been demonstrated to be important in the induction of proteasomes (23, 36, 37). Both prevent the accumulation of terminally misfolded or oxidatively damaged proteins by upregulating ubiquitin-proteasome system and antioxidant enzymes (23, 36). Whereas NFE2L1 mediates the induction of genes encoding 26S proteasome subunits during proteasome inhibition, NFE2L2 induces 26S subunits only during oxidative stress after oxidative modification of KEAP-1 (53, 59). The 20S proteasome, which is responsible for the degradation of oxidized proteins, is induced specifically by NFE2L2 (17, 23). A study by Lee et. al. (36) showed that deletion of NFE2L1 impairs proteasome activation in the liver. The significant upregulation of PSMB1, PSMB2, PSMB5, and PSMA1 proteasome genes (20S proteasome) found in burn patients suggests that upregulation of NFE2L1 and NFE2L2 was associated with cytoprotective functions. Concurrent upregulation of 19S (PSMD1, PSMD14, and PSMC5) proteasomes, which are responsible for processing ubiquitinated proteins, strongly suggests that protein degradation was increased in burn patients due to protein damage. Our data are consistent with the findings of Majetschak et al. (39), who observed elevated levels of circulating 20S proteasomes. Together, these findings support the notion that proteasomal activity plays a role in muscle wasting after burns. In addition, induction of valosin-containing protein (VCP/p97) is central in the degradation of damaged protein in the ER and helps in the induction of NrF1/NFE2L1 (31, 53, 69, 72). The increased expression of ATF6, a marker of ER stress, suggests that after burns the skeletal muscle ER experiences profound unfolded protein stress. This might have contributed to the greater expression of VCP/p97 genes postburn.
The main responsibility of ubiquitin-proteasomal system is the maintenance of proteins within cytoplasm (23). Proteasomes are not present within the mitochondrial matrix, which is separated from the cytoplasm by a double membrane. Intriguingly, mitochondria are a major source ROS, making mitochondrial proteins susceptible to oxidative damage (42, 62). The untoward effects of persistent ROS generation within mitochondria are counteracted through activation of a mitochondrial quality control system, including the ATPases Associated With Diverse Cellular Activities (AAA+) proteolytic family, to promptly remove oxidized proteins (43, 57). The AAA proteases YME1L1, CLPP, and LONP1 play an essential role in the removal of damaged mitochondrial proteins (38, 43, 63). Lon peptidase 1 (LONP1) degrades oxidized and misfolded proteins within the mitochondrial matrix, whereas CLPP is required for the activation of mtUPR and degradation of misfolded proteins (20). YME1L1 is present within the mitochondrial intermembrane space, where it performs a similar function (4). Our data show that LONP1, CLPP, and YME1L1 mRNA levels were significantly elevated in burn patients. Elevated LONP1 and lower YME1L1 proteins further suggest that both proteins are activated in response to mitochondria protein stress. The higher level of YME1L1 mRNA might be a compensatory mechanism of the decreased YME1L1 protein expression to meet increased demand. These data also suggest that, after burns, increased accumulation of oxidized and misfolded proteins in skeletal muscle mitochondria results in the induction of LONP1, CLPP, and YME1L1. This response likely functions to prevent oxidized protein from creating toxic aggregates that would lead to gross cellular dysfunction (43). Overall, the activation of LONP-1, CLPP, YME1L1, and proteasomes supports our hypothesis of increased protein turnover in skeletal muscle postburn.
In addition to degrading misfolded or unfolded protein, LONP1 is one of the main proteases responsible for the degradation of oxidized protein within mitochondria (9, 63). Mitochondrial proteins with complex organization such as Fe/S cluster proteins are generally highly susceptible to oxidation-dependent degradation (43, 64). LONP1 preferentially degrades oxidatively inactivated mitochondrial aconitase (ACO2) because it contains Fe/S. This likely prevents extensive accumulation of oxidized proteins and aggregates (8). Mitochondrial aconitase (ACO2) inactivation is a major source of mitochondrial OH (11, 64). Mitochondrial aconitase protein levels were higher in burn patients, perhaps suggesting a compensatory response to the degradation of inactivated ACO2. Inactivation of mitochondrial aconitase ([4Fe-4S]2+) by O− promotes the production of Fe2+ and H2O2, both of which can contribute to oxidative stress via the Fenton reaction and reduction mechanisms, respectively (13, 64).
Despite the predisposition of mitochondria to oxidative stress and protein damage, the mitochondrial genome encodes only 1% of the 1,500 proteins needed for normal mitochondrial function (33, 66). The remaining mitochondrial proteins are synthesized in the cytoplasm as precursor proteins and transported into the mitochondrion via translocase protein complexes (66). Some of the translocase proteins that maintain the sorting and specificity of the polypeptide to be imported include TIM23, TIM17, and TOM40 complexes (33, 45). Elevated expression of TIM23, TIM17B, and TOM40 proteins in burn patients suggests that protein demand by skeletal muscle mitochondria is increased after injury, which is likely due to greater mitochondrial proteolysis by LONP1, CLPP, and YME1L1. In addition, mitochondrial stress-mediated export of peptides leads to nuclear translocation of ATFS-1 in complex with transcriptional regulators UBL-5 and DVE-1 as well as upregulation of chaperones, proteases, and mitochondrial import proteins (20). In response to decreased protein-folding load in mitochondria, cytosolic protein translation may be inhibited by activating a signaling cascade involving GCN-2-mediated phosphorylation of elf2α (20). In parallel with these signaling events, the rate of mitochondrial protein import may diminish in response to decreased cytosolic protein translation by upregulation of YME1L1-mediated degradation of TIM17A. Hence, this cascade may have contributed to the overall protein deficit in burn patients. The elevated translocase proteins further support a role for the mitochondrion in muscle protein wasting following burn trauma, where increased transport of cytosolic proteins replaces those damaged and degraded within the mitochondria, ultimately increasing protein turnover in both the cytoplasm and mitochondrion in skeletal muscle after burns.
The current study provides novel data indicating that skeletal muscle protein degradation in burn patients takes place in both the cytosol and mitochondria. Systemic and skeletal muscle hypermetabolism (i.e., increased mitochondrial respiration) after burn was accompanied by increased oxidative stress and an mtUPR, as evidenced by higher levels of nitrotyrosine and SOD2, CHOP, JNK2, and HSP60. Persistent mitochondrial superoxide leak creates a proteotoxic environment within the mitochondrion, leading to increased oxidative stress damage and mtUPR, with subsequent degradation of mitochondrial proteins. Activation of mtUPR stress and UBL5 levels suggest that hypermetabolism-induced stress is not limited to mitochondria but may also be transduced to the cytoplasm, where it contributes to protein degradation. Increased mitochondrial protease levels in the skeletal muscle of burn patients were accompanied by a concurrent increase in the abundance of mitochondrial translocase complexes, suggesting that new proteins were transferred into mitochondria to replace oxidatively damaged or misfolded proteins and subsequently degraded. The increase in translocase proteins, taken with the mtUPR activation, suggests a mechanism to help compensate for insufficient chaperones capacity and reestablish homeostatic protein folding within the mitochondrial compartment.
In summary, we provide evidence that burn-induced hypermetabolism is associated with skeletal muscle wasting. Oxidative stress and activation of mtUPR with subsequent degradation of proteins in both the mitochondria and cytosol of the skeletal muscle cell compartments contribute to muscle wasting postburn. Thus, the mitochondrion is a cellular organelle of interest in terms of future strategies aimed at blunting muscle catabolism in burn patients.
GRANTS
Funding was provided by the National Institutes of Health (NIH; P50-GM-060388, R01-GM-56687, R01-AR-049877, and T32-GM-8256), the Institute for Translational Sciences at UTMB [supported in part by a Clinical and Translational Science Award (UL1TR000071) from the National Center for Advancing Translational Sciences, NIH], Shriners Hospitals for Children (80100, 84080, 84090, and 85310), and the Department of Surgery at UTMB.
DISCLOSURES
The authors have no conflicts of interest, financial or otherwise, to report.
AUTHOR CONTRIBUTIONS
J.O.O., C.P., D.N.H., and L.S.S. conception and design of research; J.O.O., D.R.A., and A.P. performed experiments; J.O.O., C.P., T.C., D.R.A., and A.P. analyzed data; J.O.O., C.P., D.N.H., T.A.Z., B.B.R., and L.S.S. interpreted results of experiments; J.O.O. and C.P. prepared figures; J.O.O., C.P., and L.S.S. drafted manuscript; J.O.O., C.P., D.N.H., T.C., D.R.A., A.P., M.C., T.A.Z., P.T.R., B.B.R., and L.S.S. edited and revised manuscript; J.O.O., C.P., D.N.H., T.C., D.R.A., A.P., M.C., T.A.Z., P.T.R., B.B.R., and L.S.S. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Geping Fang, Gurjit Singh, and Anahi Delgadillo for their technical assistance and Dr. Kasie Cole-Edwards for proofreading the manuscript. In addition, we thank the clinical research coordinators, physicians' assistants, and research nurses at the Blocker Burn Unit and the Institute for Translational Science Clinical Research Center at the University of Texas Medical Branch for assisting consenting patients and participants and for assisting in data collection.
REFERENCES
- 1.Al-Sawaf O, Clarner T, Fragoulis A, Kan YW, Pufe T, Streetz K, Wruck CJ. Nrf2 in health and disease: current and future clinical implications. Clin Sci (Lond) 129: 989–999, 2015. [DOI] [PubMed] [Google Scholar]
- 2.Attaix D, Ventadour S, Codran A, Béchet D, Taillandier D, Combaret L. The ubiquitin-proteasome system and skeletal muscle wasting. Essays Biochem 41: 173–186, 2005. [DOI] [PubMed] [Google Scholar]
- 3.Auger C, Alhasawi A, Contavadoo M, Appanna VD. Dysfunctional mitochondrial bioenergetics and the pathogenesis of hepatic disorders. Front Cell Dev Biol 3: 40, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baker BM, Haynes CM. Mitochondrial protein quality control during biogenesis and aging. Trends Biochem Sci 36: 254–261, 2011. [DOI] [PubMed] [Google Scholar]
- 5.Benedetti C, Haynes CM, Yang Y, Harding HP, Ron D. Ubiquitin-like protein 5 positively regulates chaperone gene expression in the mitochondrial unfolded protein response. Genetics 174: 229–239, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bergström J. Percutaneous needle biopsy of skeletal muscle in physiological and clinical research. Scand J Clin Lab Invest 35: 609–616, 1975. [PubMed] [Google Scholar]
- 7.Biolo G, Fleming RY, Maggi SP, Nguyen TT, Herndon DN, Wolfe RR. Inverse regulation of protein turnover and amino acid transport in skeletal muscle of hypercatabolic patients. J Clin Endocrinol Metab 87: 3378–3384, 2002. [DOI] [PubMed] [Google Scholar]
- 8.Bota DA, Davies KJ. Lon protease preferentially degrades oxidized mitochondrial aconitase by an ATP-stimulated mechanism. Nat Cell Biol 4: 674–680, 2002. [DOI] [PubMed] [Google Scholar]
- 9.Bota DA, Van Remmen H, Davies KJ. Modulation of Lon protease activity and aconitase turnover during aging and oxidative stress. FEBS Lett 532: 103–106, 2002. [DOI] [PubMed] [Google Scholar]
- 10.Broadley SA, Hartl FU. Mitochondrial stress signaling: a pathway unfolds. Trends Cell Biol 18: 1–4, 2008. [DOI] [PubMed] [Google Scholar]
- 11.Cantu D, Schaack J, Patel M. Oxidative inactivation of mitochondrial aconitase results in iron and H2O2-mediated neurotoxicity in rat primary mesencephalic cultures. PLoS One 4: e7095, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chao T, Herndon D, Porter C, Chondronikola M, Chaidemenou A, Abdelrahman D, Bohanon F, Andersen C, Sidossis L. Skeletal Muscle Protein Breakdown Remains Elevated in Pediatric Burn Survivors up to One-Year Post-Injury. Shock 44: 397–401, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen YR, Zweier JL. Cardiac mitochondria and reactive oxygen species generation. Circ Res 114: 524–537, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diao L, Marshall AH, Dai X, Bogdanovic E, Abdullahi A, Amini-Nik S, Jeschke MG. Burn plus lipopolysaccharide augments endoplasmic reticulum stress and NLRP3 inflammasome activation and reduces PGC-1alpha in liver. Shock 41: 138–144, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gottesman S, Maurizi MR, Wickner S. Regulatory subunits of energy-dependent proteases. Cell 91: 435–438, 1997. [DOI] [PubMed] [Google Scholar]
- 16.Gundermann D, Walker D, Reidy P, Borack M, Dickinson J, Volpi E, Rasmussen B. Activation of mTORC1 signaling and protein synthesis in human muscle following blood flow restriction exercise is inhibited by rapamycin. Am J Physiol Endocrinol Metab 306: E1198–E1204, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Han W, Ming M, Zhao R, Pi J, Wu C, He YY. Nrf1 CNC-bZIP protein promotes cell survival and nucleotide excision repair through maintaining glutathione homeostasis. J Biol Chem 287: 18788–18795, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hart DW, Wolf SE, Mlcak R, Chinkes DL, Ramzy PI, Obeng MK, Ferrando AA, Wolfe RR, Herndon DN. Persistence of muscle catabolism after severe burn. Surgery 128: 312–319, 2000. [DOI] [PubMed] [Google Scholar]
- 19.Hart DW, Wolf SE, Mlcak R, Chinkes DL, Ramzy PI, Obeng MK, Ferrando AA, Wolfe RR, Herndon DN. Persistence of muscle catabolism after severe burn. Surgery 128: 312–319, 2000. [DOI] [PubMed] [Google Scholar]
- 20.Held NM, Houtkooper RH. Mitochondrial quality control pathways as determinants of metabolic health. Bioessays 37: 867–876, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hetz C, Chevet E, Harding HP. Targeting the unfolded protein response in disease. Nat Rev Drug Discov 12: 703–719, 2013. [DOI] [PubMed] [Google Scholar]
- 22.Hizli AA, Chi Y, Swanger J, Carter JH, Liao Y, Welcker M, Ryazanov AG, Clurman BE. Phosphorylation of eukaryotic elongation factor 2 (eEF2) by cyclin A-cyclin-dependent kinase 2 regulates its inhibition by eEF2 kinase. Mol Cell Biol 33: 596–604, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hohn TJ, Grune T. The proteasome and the degradation of oxidized proteins: part III-Redox regulation of the proteasomal system. Redox Biol 2: 388–394, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hong-Brown LQ, Brown CR, Huber DS, Lang CH. Lopinavir impairs protein synthesis and induces eEF2 phosphorylation via the activation of AMP-activated protein kinase. J Cell Biochem 105: 814–823, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jeschke MG, Finnerty CC, Herndon DN, Song J, Boehning D, Tompkins RG, Baker HV, Gauglitz GG. Severe injury is associated with insulin resistance, endoplasmic reticulum stress response, and unfolded protein response. Ann Surg 255: 370–378, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jeschke MG, Finnerty CC, Suman OE, Kulp G, Mlcak RP, Herndon DN. The effect of oxandrolone on the endocrinologic, inflammatory, and hypermetabolic responses during the acute phase postburn. Ann Surg 246: 351–360; discussion 360–352, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jovaisaite V, Mouchiroud L, Auwerx J. The mitochondrial unfolded protein response, a conserved stress response pathway with implications in health and disease. J Exp Biol 217: 137–143, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jung T, Grune T. The proteasome and its role in the degradation of oxidized proteins. IUBMB Life 60: 743–752, 2008. [DOI] [PubMed] [Google Scholar]
- 29.Jung T, Grune T. The proteasome and the degradation of oxidized proteins: Part I-structure of proteasomes. Redox Biol 1: 178–182, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jung T, Höhn A, Grune T. The proteasome and the degradation of oxidized proteins: Part II - protein oxidation and proteasomal degradation. Redox Biol 2: 99–104, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kobayashi T, Manno A, Kakizuka A. Involvement of valosin-containing protein (VCP)/p97 in the formation and clearance of abnormal protein aggregates. Genes Cells 12: 889–901, 2007. [DOI] [PubMed] [Google Scholar]
- 32.Kotiadis VN, Duchen MR, Osellame LD. Mitochondrial quality control and communications with the nucleus are important in maintaining mitochondrial function and cell health. Biochim Biophys Acta 1840: 1254–1265, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kutik S, Guiard B, Meyer HE, Wiedemann N, Pfanner N. Cooperation of translocase complexes in mitochondrial protein import. J Cell Biol 179: 585–591, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lahtvee PJ, Seiman A, Arike L, Adamberg K, Vilu R. Protein turnover forms one of the highest maintenance costs in Lactococcus lactis. Microbiology 160: 1501–1512, 2014. [DOI] [PubMed] [Google Scholar]
- 35.Lecker SH, Goldberg AL, Mitch WE. Protein degradation by the ubiquitin-proteasome pathway in normal and disease states. J Am Soc Nephrol 17: 1807–1819, 2006. [DOI] [PubMed] [Google Scholar]
- 36.Lee CS, Ho DV, Chan JY. Nuclear factor-erythroid 2-related factor 1 regulates expression of proteasome genes in hepatocytes and protects against endoplasmic reticulum stress and steatosis in mice. FEBS J 280: 3609–3620, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee CS, Lee C, Hu T, Nguyen JM, Zhang J, Martin MV, Vawter MP, Huang EJ, Chan JY. Loss of nuclear factor E2-related factor 1 in the brain leads to dysregulation of proteasome gene expression and neurodegeneration. Proc Natl Acad Sci USA 108: 8408–8413, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lionaki E, Tavernarakis N. Oxidative stress and mitochondrial protein quality control in aging. J Proteomics 92: 181–194, 2013. [DOI] [PubMed] [Google Scholar]
- 39.Majetschak M, Zedler S, Romero J, Albright JM, Kraft R, Kovacs EJ, Faist E, Gamelli RL. Circulating proteasomes after burn injury. J Burn Care Res 31: 243–250, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Manoli I, Alesci S, Blackman MR, Su YA, Rennert OM, Chrousos GP. Mitochondria as key components of the stress response. Trends Endocrinol Metab 18: 190–198, 2007. [DOI] [PubMed] [Google Scholar]
- 41.Mottis A, Jovaisaite V, Auwerx J. The mitochondrial unfolded protein response in mammalian physiology. Mamm Genome 25: 424–433, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J 417: 1–13, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ngo JK, Pomatto LC, Davies KJ. Upregulation of the mitochondrial Lon Protease allows adaptation to acute oxidative stress but dysregulation is associated with chronic stress, disease, and aging. Redox Biol 1: 258–264, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pellegrino MW, Nargund AM, Haynes CM. Signaling the mitochondrial unfolded protein response. Biochim Biophys Acta 1833: 410–416, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pfanner N, Geissler A. Versatility of the mitochondrial protein import machinery. Nat Rev Mol Cell Biol 2: 339–349, 2001. [DOI] [PubMed] [Google Scholar]
- 46.Phillips SM, Tipton KD, Aarsland A, Wolf SE, Wolfe RR. Mixed muscle protein synthesis and breakdown after resistance exercise in humans. Am J Physiol Endocrinol Metab 273: E99–E107, 1997. [DOI] [PubMed] [Google Scholar]
- 47.Porter C, Cotter M, Diaz E, Jennings K, Herndon D, Børsheim E. Amino acid infusion fails to stimulate skeletal muscle protein synthesis up to 1 year after injury in children with severe burns. J Trauma Acute Care Surg 74: 1480–1485, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Porter C, Herndon DN, Bhattarai N, Ogunbileje JO, Szczesny B, Szabo C, Toliver-Kinsky T, Sidossis LS. Differential acute and chronic effects of burn trauma on murine skeletal muscle bioenergetics. Burns 42: 112–122, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Porter C, Herndon DN, Bhattarai N, Ogunbileje JO, Szczesny B, Szabo C, Toliver-Kinsky T, Sidossis LS. Severe Burn Injury Induces Thermogenically Functional Mitochondria in Murine White Adipose Tissue. Shock 44: 258–264, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Porter C, Herndon DN, Borsheim E, Bhattarai N, Chao T, Reidy PT, Rasmussen BB, Andersen CR, Suman OE, Sidossis LS. Long-Term Skeletal Muscle Mitochondrial Dysfunction is Associated with Hypermetabolism in Severely Burned Children. J Burn Care Res 37: 53–63, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Porter C, Herndon DN, Borsheim E, Chao T, Reidy PT, Borack MS, Rasmussen BB, Chondronikola M, Saraf MK, Sidossis LS. Uncoupled skeletal muscle mitochondria contribute to hypermetabolism in severely burned adults. Am J Physiol Endocrinol Metab 307: E462–E467, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Porter C, Hurren N, Herndon D, Børsheim E. Whole body and skeletal muscle protein turnover in recovery from burns. Int J Burns Trauma 3: 9–17, 2013. [PMC free article] [PubMed] [Google Scholar]
- 53.Radhakrishnan SK, den Besten W, Deshaies RJ. p97-dependent retrotranslocation and proteolytic processing govern formation of active Nrf1 upon proteasome inhibition. Elife 3: e01856, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Radi R. Nitric oxide, oxidants, and protein tyrosine nitration. Proc Natl Acad Sci USA 101: 4003–4008, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Roede JR, Jones DP. Reactive species and mitochondrial dysfunction: mechanistic significance of 4-hydroxynonenal. Environ Mol Mutagen 51: 380–390, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Roza A, Shizgal H. The Harris Benedict equation reevaluated: resting energy requirements and the body cell mass. Am J Clin Nutr 40: 168–182, 1984. [DOI] [PubMed] [Google Scholar]
- 57.Sauer RT, Baker TA. AAA+ proteases: ATP-fueled machines of protein destruction. Annu Rev Biochem 80: 587–612, 2011. [DOI] [PubMed] [Google Scholar]
- 58.Senft D, Ronai ZA. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem Sci 40: 141–148, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sha Z, Goldberg AL. Proteasome-mediated processing of Nrf1 is essential for coordinate induction of all proteasome subunits and p97. Curr Biol 24: 1573–1583, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sidossis LS, Porter C, Saraf MK, Borsheim E, Radhakrishnan RS, Chao T, Ali A, Chondronikola M, Mlcak R, Finnerty CC, Hawkins HK, Toliver-Kinsky T, Herndon DN. Browning of Subcutaneous White Adipose Tissue in Humans after Severe Adrenergic Stress. Cell Metab 22: 219–227, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Taylor EB, Rutter J. Mitochondrial quality control by the ubiquitin-proteasome system. Biochem Soc Trans 39: 1509–1513, 2011. [DOI] [PubMed] [Google Scholar]
- 62.Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol 552: 335–344, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ugarte N, Petropoulos I, Friguet B. Oxidized mitochondrial protein degradation and repair in aging and oxidative stress. Antioxid Redox Signal 13: 539–549, 2010. [DOI] [PubMed] [Google Scholar]
- 64.Vasquez-Vivar J, Kalyanaraman B, Kennedy MC. Mitochondrial aconitase is a source of hydroxyl radical. An electron spin resonance investigation. J Biol Chem 275: 14064–14069, 2000. [DOI] [PubMed] [Google Scholar]
- 65.Weir J. New methods for calculating metabolic rate with special reference to protein metabolism. J Physiol 109: 1–9, 1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wiedemann N, Frazier AE, Pfanner N. The protein import machinery of mitochondria. J Biol Chem 279: 14473–14476, 2004. [DOI] [PubMed] [Google Scholar]
- 67.Williams FN, Herndon DN, Jeschke MG. The hypermetabolic response to burn injury and interventions to modify this response. Clin Plast Surg 36: 583–596, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wilmore D, Aulick L. Systemic responses to injury and the healing wound. JPEN J Parenter Enteral Nutr 4: 147–151, 1980. [DOI] [PubMed] [Google Scholar]
- 69.Yamanaka K, Sasagawa Y, Ogura T. Recent advances in p97/VCP/Cdc48 cellular functions. Biochim Biophys Acta 1823: 130–137, 2012. [DOI] [PubMed] [Google Scholar]
- 70.Zhang XJ, Chinkes DL, Wolfe RR. Measurement of muscle protein fractional synthesis and breakdown rates from a pulse tracer injection. Am J Physiol Endocrinol Metab 283: E753–E764, 2002. [DOI] [PubMed] [Google Scholar]
- 71.Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, Hoogenraad NJ. A mitochondrial specific stress response in mammalian cells. EMBO J 21: 4411–4419, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhong X, Shen Y, Ballar P, Apostolou A, Agami R, Fang S. AAA ATPase p97/valosin-containing protein interacts with gp78, a ubiquitin ligase for endoplasmic reticulum-associated degradation. J Biol Chem 279: 45676–45684, 2004. [DOI] [PubMed] [Google Scholar]