

Abstract
The loss of strength in combination with constant fatigue is a burden on cancer patients undergoing chemotherapy. Doxorubicin, a standard chemotherapy drug used in the clinic, causes skeletal muscle dysfunction and increases mitochondrial H2O2. We hypothesized that the combined effect of cancer and chemotherapy in an immunocompetent breast cancer mouse model (E0771) would compromise skeletal muscle mitochondrial respiratory function, leading to an increase in H2O2-emitting potential and impaired muscle function. Here, we demonstrate that cancer chemotherapy decreases mitochondrial respiratory capacity supported with complex I (pyruvate/glutamate/malate) and complex II (succinate) substrates. Mitochondrial H2O2-emitting potential was altered in skeletal muscle, and global protein oxidation was elevated with cancer chemotherapy. Muscle contractile function was impaired following exposure to cancer chemotherapy. Genetically engineering the overexpression of catalase in mitochondria of muscle attenuated mitochondrial H2O2 emission and protein oxidation, preserving mitochondrial and whole muscle function despite cancer chemotherapy. These findings suggest mitochondrial oxidants as a mediator of cancer chemotherapy-induced skeletal muscle dysfunction.
Keywords: cancer, chemotherapy, mitochondria, skeletal muscle, reactive oxygen species
the american cancer society predicts that approximately 230,000 new cases of invasive breast cancer will be diagnosed this year in the US (1). These new cases are in addition to the 2.8 million women with confirmed breast cancer diagnoses. Doxorubicin is a chemotherapeutic anthracycline commonly prescribed to treat numerous human malignancies, including breast cancer in postmenopausal women (6, 33). The combined effect of both cancer and chemotherapy leads to disabling muscle weakness and fatigue for patients (18, 35). The loss of strength in combination with constant fatigue is a burden for patients not only during therapy but up to 10 yr following the cessation of therapy (5).
Changes that occur within skeletal muscle can play a significant role in the decline of physical function for cancer patients. These changes are manifested in the clinic as a sense of tiredness or perceived weakness. The loss of muscle mass, reduced cardiorespiratory fitness, or peripheral muscle fatigue can all contribute to functional impairment. Prior research suggests that deteriorating physical function in cancer patients may be due to an effect specifically on skeletal muscle, resulting in peripheral muscle weakness and fatigue. Using a healthy rodent model, the chemotherapy drug doxorubicin has been shown to decrease both hindlimb and respiratory muscle strength, along with accelerating fatigue (11, 13, 14). Chemotherapy alone has a detrimental effect on muscle function. However, little is known how functional capacity is affected by chemotherapy in the context of cancer, more closely resembling what happens to patients in the clinic.
Potential mediators of chemotherapy-induced muscle weakness and fatigue are reactive oxygen species (ROS). Exposure to the chemotherapeutic agent doxorubicin leads to an increase in ROS in skeletal muscle, as evidenced by elevated cytosolic oxidant activity (13) and markers of protein oxidation (13, 36). Mitochondrial H2O2-emitting potential is elevated in both skeletal and cardiac muscle following doxorubicin administration (12, 26), and treatment of cultured myotubes or rats with a cell-permeable mitochondrial-targeting peptide (MTP-131) that reduces ROS production also attenuates doxorubicin-induced oxidant production and accumulation of oxidation markers (15, 26). Collectively, these findings suggest that mitochondria are the major source of ROS production in skeletal muscle in response to doxorubicin treatment.
The objective of this study was to determine the exact nature and extent to which mitochondrial function is impacted by cancer chemotherapy, specifically in skeletal muscle. It was hypothesized that chemotherapy in the context of cancer would inhibit skeletal muscle mitochondrial respiration, leading to an increase in H2O2-emitting potential. To determine whether increased mitochondrial H2O2 production is the primary factor leading to compromised muscle function in response to cancer chemotherapy, a genetic mouse model (MCAT) in which catalase is targeted to the mitochondria was employed. Mitochondrial function was evaluated in permeabilized fiber bundles (PmFBs) from soleus muscle of wild-type mice in an immunocompetent cancer rodent model exposed to breast tumor cells (E0771) and the chemotherapeutic agent doxorubicin. The results indicate that cancer chemotherapy impairs mitochondrial respiration in hindlimb skeletal muscle. Interestingly, exposure to tumor cells and chemotherapy individually increases mitochondrial H2O2-emitting potential, whereas the combination of cancer chemotherapy diminishes the effects of either treatment alone. Scavenging of mitochondrial oxidants abolishes the mitochondrial dysfunction caused by cancer chemotherapy and protects against contractile dysfunction.
METHODS
Overview of experimental design.
Figure 1A illustrates the experimental design. Female wild-type and heterozygous mice expressing the human catalase gene targeted specifically to the mitochondrial matrix in striated muscle (MCAT) (34) on a C57BL/6N background were ovariectomized at 12 wk of age to simulate menopause. Following recovery from the surgery, mice assigned to the tumor-bearing (TB) groups were inoculated subcutaneously on the right pad of the fourth mammary gland with mouse breast cancer cells (E0771) suspended in phosphate-buffered saline (PBS). When the tumor reached a mean size of 100–150 mm2 (Fig. 1B), mice assigned to the doxorubicin (DOX) + TB groups received a single intraperitoneal injection of doxorubicin (DOX; 20 mg/kg in PBS), as described previously (12, 13). Control animals received the same volume of vehicle (PBS). Mice were monitored and weighed daily. Functional measurements and PmFBs were prepared from soleus muscles 72 h postinjection. Tibialis anterior (TA) muscles were frozen in liquid nitrogen for Western blot analysis. Experimental groups included WT control (CTRL; n = 10), WT tumor bearing (TB; n = 10), WT doxorubicin (DOX, n = 10), WT TB + DOX (n = 9), MCAT CTRL (n = 12), MCAT TB (n = 10), MCAT DOX (n = 11), and MCAT TB + DOX (n = 10).
Fig. 1.

Mammary tumor growth in wild-type (WT) and mitochondrial catalase-expressing mice. A: schematic diagram illustrating the experimental protocol. Mice assigned to the tumor-bearing (TB) groups were injected subcutaneously in the mammary fat pad with E0771 cells and euthanized once tumor size reached 100–150 mm2. Mice assigned to the doxorubicin (DOX) + TB groups received a single intraperitoneal injection of DOX (20 mg/kg) once tumor size reached 100–150 mm2 and were euthanized 72 h postinjection. B: growth of tumor size in female C57BL/6N WT and heterozygous mice expressing catalase in the mitochondrial matrix (MCAT). Data are means ± SE. WT TB, n = 10; WT TB + DOX, n = 9; MCAT TB, n = 10; MCAT TB + DOX, n = 10.
Reagents and cell lines.
Doxorubicin was purchased from Bedford Laboratories (Bedford, OH). Protease and phosphatase inhibitors were purchased from Roche Diagnostics (Indianapolis, IN). RIPA Lysis and Extraction Buffer was purchased from ThermoFisher Scientific (Waltham, MA). The Oxidized Protein Western blot detection kit was purchased from Abcam (Cambridge, UK). All other chemicals were purchased from Sigma-Aldrich (St. Louis, MO). The mouse breast cancer cells (E0771) were originally developed at Wake Forest University Health Sciences and provided by Dr. Lee Jones at Duke University. E0771 cells were maintained as monolayer cultures in RPMI Medium 1640 (Gibco) supplemented with 10% fetal bovine serum and 1% antibiotic antimycotic solution and incubated at 37°C in a humidified 5% CO2/air-injected atmosphere.
Animal care.
All mice were housed in the Department of Comparative Medicine at East Carolina University in a temperature- and light-controlled room and given free access to food and water. All procedures were approved by the university's Institutional Animal Care and Use Committee. For the ovariectomy procedure, mice were anesthetized and incision sites shaved and cleaned with iodine solution. Standard aseptic procedures were observed. Dorsal incisions were made in the lumbar region to reveal the dorsal fat pads covering the ovaries. Ovaries were removed through cauterization. After ovariectomy, muscle incisions were sutured and the skin incisions closed with sterile suture wound clips. Mice were given meloxicam (5 mg/kg orally) prior to surgery and 24 h postsurgery. Wound clips were removed 7 days following surgery.
Following recovery from the ovariectomy procedure, mice were inoculated subcutaneously on the right pad of the fourth mammary gland with 100 μl of 5 × 105 E0771 cells suspended in PBS using a 22-gauge needle. Tumor growth was monitored every other day in two perpendicular dimensions parallel with the surface of the mice using a slide caliper. Skeletal muscle was obtained from anesthetized mice by intraperitoneal injection with ketamine-xylazine (90 and 10 mg/kg). Following surgery, mice were euthanized by cervical dislocation under anesthesia.
Determination of body composition.
Measurements of fat and lean body mass were determined using the EchoMRI-500 (Houston, TX) in accordance with the manufacturer's instructions.
Permeabilized fiber bundle preparation.
Procedures were performed as described previously (2, 12, 30). In brief, fiber bundles from the soleus muscle were separated with fine forceps in ice-cold buffer X (in mM: 50 K-MES, 35 KCl, 7.23 K2EGTA, 2.77 CaK2EGTA, 20 imidazole, 20 taurine, 5.7 ATP, 14.3 PCr, and 6.56 MgCl2-6H2O, pH 7.1). Once separated, fiber bundles were permeabilized in buffer X with 30 μg/ml saponin for 30 min and then washed in ice-cold buffer Z (in mM: 105 K-MES, 30 KCl, 1 EGTA, 10 KH2PO4, 5 MgCl2-6H2O, and 0.5 mg/ml BSA, pH 7.1) until analysis.
Mitochondrial respiration.
High-resolution O2 consumption measurements were conducted using the OROBOROS Oxygraph-2k (Oroboros Instruments, Innsbruck, Austria) at 37°C with an initial chamber concentration of 300–350 μM oxygen. All experiments were run in buffer Z containing 20 mM creatine monohydrate and 25 μM blebbistatin to inhibit contraction, as described previously (30). Concentrations of individual substrates for respiration protocols were palmitoyl-CoA (50 μM)/carnitine (5 mM), malate (2 mM), ADP (4 mM), pyruvate (1 mM), glutamate (10 mM), and succinate (10 mM). At the end of each protocol, cytochrome c (10 μM) was added to test for mitochondrial membrane integrity, and any PmFBs that generated a >10% increase in respiration following the addition were not included in data analysis. At the conclusion of each experiment, PmFBs were washed in dH2O and dried via freeze-drying (Labconco, Kansas City, MO). Polarographic oxygen measurements are expressed as picomoles per second per milligram dry weight.
Mitochondrial H2O2 emission.
H2O2 emission was measured fluorometrically at 37°C via Amplex Ultra Red [10 μM/horseradish peroxidase (1 U/ml) detection system (Eex/Eem = 565/600)]. Fluorescence was monitored by a fluorolog-3 spectrofluorometer (Horiba Jobin Yvon, Edison, NJ) with temperature control and magnetic stirring. For each protocol, a background fluorescence rate was established in the presence of a PmFB following the addition of subsequent substrates. After correcting for the rate of change in background fluorescence, the concentration of H2O2 (pmol) was calculated from previously established resorufin fluorescence intensity standard curves with known concentrations of H2O2. Following each experiment, PmFBs were dried and weighed as described above. Concentrations of individual substrates/inhibitors for H2O2 emission protocols were succinate (10 mM), pyruvate (1 mM), carnitine (5 mM), auranofin (AF; 1 μM), and bis-chloroethylnitrosourea (BCNU; 100 μM).
Contractile function.
Measurements of muscle contractile function were performed as described previously (11, 13). In brief, upon removal, one end of the soleus muscle was tied to a dual-mode muscle lever system (Aurora Scientific) and the other end secured to a glass rod using a 4.0 silk suture. Following 15 min of thermoequilibration (32°C), the muscles were plated at optimal length, and force-frequencey measurements began. The soleus was stimulated with a supramaximal current (600 mA) with pulse and train durations of 0.3 and 500 ms. The force-frequency relationship was determined using contractions evoked at 2-min intervals using stimulus frequencies of 1, 15, 30, 50, 80, 120, 150, 200, and 250 (Hz). All data were recorded and analyzed using commercial software (Dynamic Muscle Control and Analysis Software; Aurora Scientific).
Oxidized protein Western blot.
To detect changes in oxidative modification of proteins, TA muscles were homogenized in ice-cold RIPA lysis and extraction buffer containing protease and phosphatase inhibitors per the manufacturer's directions. Protein carbonyl groups were measured using the Oxidized Protein Western Blot Detection kit according to the manufacturer's instructions.
Statistics.
Results are reported as means ± SE. Statistical analyses were performed using commercial software (Prism GraphPad, La Jolla, CA). Results were analyzed using an ANOVA test for the comparison of multiple means. When an overall difference was detected by an ANOVA, a post hoc Bonferroni's test was performed. Statistical significance was accepted at P < 0.05.
RESULTS
In agreement with our previous work (11–13), wild-type (WT) mice exposed to the chemotherapeutic agent doxorubicin display a significant decline in body weight (%change of preexposure weight: WT DOX −12.3 ± 2.9, n = 10; WT TB + DOX −9.0 ± 1.1, n = 9; P < 0.01). Compared with their preexposure mass, whole body fat (%change: DOX −22.6 ± 6.2, TB + DOX −27.2 ± 5.9, P < 0.01) and lean mass (%change: WT DOX −11.1 ± 2.6, WT TB + DOX −5.5 ± 1.3, P < 0.01) were decreased following doxorubicin exposure. WT control and tumor-bearing mice displayed a normal increase in whole body weight, fat, and lean mass over the experimental period (data not shown).
MCAT mice displayed a similar decrease in body weight compared with WT mice (%change of preexposure weight: MCAT DOX −14.3 ± 3.1, n = 11; MCAT TB + DOX −6.3 ± 2.7, n = 10; P < 0.01). Whole body fat (%change: MCAT DOX −28.9 ± 5.6, MCAT TB + DOX −27.1 ± 6.7, P < 0.05) and lean mass (%change: MCAT DOX −11.9 ± 1.8, MCAT TB + DOX −12.3 ± 1.5, P < 0.05) were decreased in MCAT mice compared with their preexposure mass following doxorubicin administration. MCAT control and tumor-bearing mice exhibited a normal increase in whole body weight, fat, and lean mass over the experimental period (data not shown).
Skeletal muscle mitochondrial respiration following cancer chemotherapy.
Our previous work has shown that the chemotherapeutic agent doxorubicin alone depresses mitochondrial respiration in skeletal muscle of healthy rodents (12), affecting both complex I- and complex II-supported respiration. To determine the combined effect of tumor cells and doxorubicin on skeletal muscle mitochondrial function, PmFBs were isolated from the soleus muscle following exposure. Mitochondrial respiration of various complexes in the electron transport system (ETS) was measured sequentially.
Mitochondrial citrate synthase activity was similar among groups (data not shown), suggesting that cancer/chemotherapy did not influence muscle mitochondrial density. Tumor-bearing WT mice showed a significant decrease in maximal ADP-stimulated respiration supported by complex I (pyruvate/glutamate) and complex II (succinate) substrates (Fig. 2A). Doxorubicin administration alone also reduced both complex I- and complex II-supported respiration. No further reduction in respiration was observed in tumor-bearing mice treated with doxorubicin. Interestingly, during respiration supported by fatty acid oxidation, maximal ADP-stimulated respiration was reduced only in the cancer plus chemotherapy treatment group.
Fig. 2.

Cancer chemotherapy impairs mitochondrial respiratory capacity in skeletal muscle. Permeabilized myofibers were prepared following exposure to tumor cell (TB) and doxorubicin (DOX) administration in both wild-type mice [control (CTRL), n = 9; TB, n = 9; DOX, n = 10, TB + DOX, n = 9; A] and mice expressing catalase in the mitochondrial matrix (MCAT; CTRL, n = 11; MCAT TB, n = 10; MCAT DOX, n = 11; MCAT TB + DOX, n = 10; B). Mitochondrial respiration was measured sequentially in the presence of maximal palmitoyl-CoA + carnitine (P/Car), malate (Mal), ADP (D), pyruvate (Pyr), glutamate (Glut), and succinate (Succ). Data are means ± SE. *P < 0.05 vs. CTRL
MCAT mice exposed to tumor cells alone were protected from the decrease in maximal ADP-stimulated oxygen consumption during respiration supported by complex I (pyruvate/glutamate) and complex II (succinate) substrates (Fig. 2B). However, doxorubicin treatment induced a decrease in maximal respiration in MCAT mice (Fig. 2B) similar to that in WT mice (Fig. 2A). A similar trend was evident for the group exposed to both tumor cells and doxorubicin, although none reached statistical significance (P = 0.1 vs. CTRL; Fig. 2B), suggesting that the presence of catalase did provide at least some protection.
Cancer chemotherapy alters mitochondrial H2O2-emitting potential.
The chemotherapeutic agent doxorubicin is known to increase oxidant activity in skeletal muscle (13), specifically altering mitochondrial H2O2-emitting potential (12). Few studies have determined whether skeletal muscle from tumor-bearing animals has altered mitochondrial H2O2-emitting potential, which could lead to elevated oxidant activity and ultimately dysfunction. We evaluated the H2O2-emitting potential of mitochondria in PmFBs following exposure to both tumor cells and doxorubicin.
WT tumor-bearing mice display a significant increase (∼22%) in mitochondrial H2O2-emitting potential in PmFBs during respiration supported by the complex II substrate succinate (Fig. 3A), which elicits superoxide production at complex I (due to reverse electron flow) and potentially at complex II (28, 31). Doxorubicin alone increased the rate of mitochondrial H2O2-emitting potential by ∼94%. Surprisingly, cancer combined with chemotherapy completely blunted the effects of either treatment alone, generating an H2O2-emitting potential no different from controls. H2O2 production by the pyruvate dehydrogenase complex (10) was not different between groups (data not shown).
Fig. 3.

Mitochondrial H2O2 emission in wild-type and mitochondrial catalase-expressing mice following exposure to cancer chemotherapy. Mitochondrial H2O2-emitting potential of permeabilized myofibers from wild-type (A) and mitochondrial catalase-expressing mice (MCAT; B) exposed to tumor cells (TB) and chemotherapy (DOX). Substrate conditions were in the presence of maximal succinate (Succ) and inhibitors of both Thioredoxin reductase and glutathione reductase [auranofin/bis-chloroethylnitrosourea (+AF/BCNU)]. Scavening index (SI) indicates calculated index of maximal scavenging. Data are means ± SE. Wild-type CTRL, n = 9; TB, n = 9; DOX, n = 10; TB + DOX, n = 8; MCAT CTRL, n = 8; MCAT TB, n = 8; MCAT DOX, n = 10; MCAT TB + DOX, n = 10. *P < 0.05 vs. CTRL.
To determine the potential effect of cancer chemotherapy on the mitochondrial redox-buffering system in skeletal muscle, succinate-mediated H2O2 production was studied in the presence of inhibitors of both glutathione reductase (BCNU) and thioredoxin reductase (AF), as described previously (10). In the presence of both reductase inhibitors, matrix H2O2 scavenging is inhibited, and thus H2O2 emission reflects total H2O2 production (i.e., emission = production − scavenging). The scavenging index was calculated by subtracting the emission rate (+Succ rate) from the total H2O2 production (+AF/BCNU rate). Compared with controls, the scavenging index was decreased for all treated groups (Fig. 3A), suggesting that the ability to scavenge oxidants is decreased with cancer chemotherapy.
Heterozygous mice expressing skeletal muscle mitochondrial catalase in the matrix were completely protected from the increase in mitochondria H2O2 production caused by cancer chemotherapy (Fig. 3B). As expected, removal of the mitochondrial redox-buffering system by inhibitors (AF/BCNU) in MCAT mice had minimal effect on H2O2 emission (Fig. 3B).
Cancer chemotherapy promotes oxidative modifications of muscle proteins.
Findings from our previous work have shown the chemotherapeutic agent doxorubicin alone increases oxidative modifications of myofibrillar proteins in skeletal muscle (13). Elevated cellular oxidants can result in posttranslational modifications of proteins that can affect whole muscle function. We evaluated the levels of reactive carbonyl derivatives in tibialis anterior muscle following exposure to both tumor cells and doxorubicin.
Tumor-bearing WT mice showed a significant increase in hindlimb muscle global protein carbonylation (Fig. 4A). Administration of the chemotherapeutic agent doxorubicin alone or in combination with tumor cells induced a similar increase in levels of reactive protein carbonyls. Heterozygous mice expressing skeletal muscle mitochondrial catalase in the matrix were completely protected from the increase in global protein carbonylation caused by cancer chemotherapy (Fig. 4B). These findings suggest that the presence of mitochondrial catalase in skeletal muscle is protective of cancer chemotherapy-induced oxidative damage.
Fig. 4.

Detection of global protein oxidation in wild-type and mitochondrial catalase-expressing mice (MCAT) following exposure to cancer chemotherapy. Protein carbonyls in tibialis anterior muscles from wild-type (A) and MCAT (B) exposed to tumor cells (TB) and chemotherapy (DOX). Representative Western blots are shown to the right of quantified data. Data are means ± SE. *P < 0.05 vs. CTRL; n = 6/group.
Function of soleus following cancer chemotherapy.
To determine the possible effect of cancer chemotherapy on whole muscle function, measurements of contractile properties were performed on the contralateral soleus muscle. Soleus weight was decreased significantly in WT mice exposed to doxorubicin, along with a decrease in calculated cross-sectional area (Table 1). A similar trend was evident for the tumor-bearing WT mice, although the group did not reach statistical significance (P = 0.1 vs. CTRL; Table 1).
Table 1.
Soleus muscle characteristics of female C57BL6/N wild-type following exposure to tumor cells and DOX
| CTRL (n = 10) | TB (n = 10) | DOX (n = 10) | TB + DOX (n = 9) | |
|---|---|---|---|---|
| Length, (L0) cm | 1.23 ± 0.008 | 1.24 ± 0.03 | 1.22 ± 0.015 | 1.24 ± 0.014 |
| Weight, g | 0.0079 ± 0.0002 | 0.0072 ± 0.0002† | 0.0064 ± 0.0002* | 0.0077 ± 0.0003 |
| Cross-sectional area, cm2 | 0.0060 ± 0.0001 | 0.0055 ± 0.0001† | 0.0049 ± 0.0002* | 0.0059 ± 0.0002 |
Data are means ± SE. TB, tumor bearing; DOX, doxorubicin; CTRL, control.
P < 0.05 vs. CTRL; †P = 0.1 vs. CTRL.
The absolute force-frequency relationship of the soleus muscle is displayed in Fig. 5A. In WT mice, maximal isometric tetanic force was decreased in all experimental groups by 12–14% compared with controls (Fig. 5B). When normalized for cross-sectional area, specific force was not different between groups (Fig. 5, C and D).
Fig. 5.
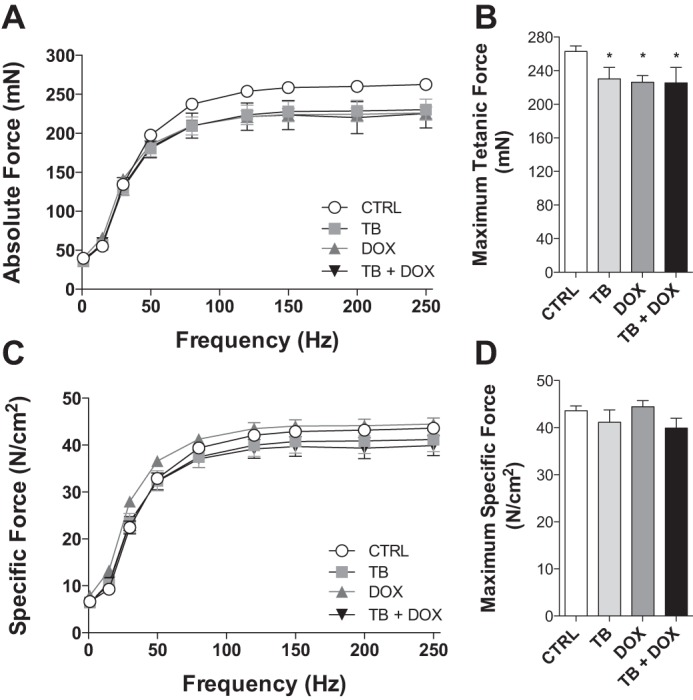
Cancer chemotherapy alters contractile function of hindlimb muscle from wild-type mice. Soleus contractile properties in wild-type mice exposed to tumor cells (TB) and chemotherapy (DOX). Absolute force-frequency relationship (A), maximal tetanic force (B), specific force-frequency relationship (C), and maximum specific force (D). Data are means ± SE. *P < 0.05 vs. CTRL.
MCAT mice were not protected against the loss of whole muscle weight following cancer chemotherapy (Table 2). Exposure to tumor cells or doxorubicin alone caused a ∼13–16% decrease in soleus weight. The combined effect of cancer chemotherapy produced a similar decrease in soleus weight and cross-sectional area in MCAT mice (Table 2).
Table 2.
Soleus muscle characteristics of female heterozygous MCAT following exposure to tumor cells and DOX
| MCAT CTRL (n = 11) | MCAT TB (n = 10) | MCAT DOX (n = 11) | MCAT TB + DOX (n = 9) | |
|---|---|---|---|---|
| Length (L0), cm | 1.29 ± 0.016 | 1.28 ± 0.018 | 1.26 ± 0.009 | 1.31 ± 0.011 |
| Weight, g | 0.0084 ± 0.0001 | 0.0073 ± 0.0002* | 0.0070 ± 0.0002* | 0.0074 ± 0.0002* |
| Cross-sectional area, cm2 | 0.0062 ± 0.0001 | 0.0054 ± 0.0001* | 0.0053 ± 0.0001* | 0.0053 ± 0.0003* |
Data are means ± SE. MCAT, mice expressing catalase in the mitochondrial matrix; TB, tumor bearing; DOX, doxorubicin.
P < 0.05 vs. MCAT CTRL.
Interestingly, MCAT mice at baseline were characterized by lower maximal isometric tetanic force compared with wild-type mice at baseline (compare Fig. 6B, CTRL, with Fig. 5B, CTRL). In contrast with WT mice, cancer chemotherapy did not cause a decrease in maximal isometric tetanic force in MCAT mice, nor did exposure to doxorubicin alone or in combination with tumor cells (Fig. 6, A and B), suggesting that the presence of catalase in the mitochondria did provide some protection against the loss of absolute muscle force. Specific force normalized for cross-sectional area was not different between groups (Fig. 6, C and D).
Fig. 6.

Overexpression of skeletal muscle mitochondrial catalase protects against cancer chemotherapy contractile dysfunction. Soleus contractile properties in mice expressing catalase in the mitochondrial matrix (MCAT) exposed to tumor cells (TB) and chemotherapy (DOX). Absolute force-frequency relationship (A), maximal tetanic force (B), specific force-frequency relationship (C), and maximum specific force (D). Data are means ± SE.
DISCUSSION
The present experiments provide novel insight into how the combined insults of cancer and chemotherapy affect mitochondria of skeletal muscle. Tumor bearing, the chemotherapeutic agent doxorubicin, and the combination of tumor cells and doxorubicin diminish both complex I- and complex II-supported respiration in skeletal muscle of wild-type mice. Furthermore, mitochondrial H2O2-emitting potential is elevated in skeletal muscle of both tumor-bearing and doxorubicin-exposed mice in conjunction with a decline in scavenging capacity. Surprisingly, the increase in mitochondrial H2O2-emitting potential is diminished when tumor-bearing mice receive doxorubicin, although global protein oxidation in skeletal muscle remains elevated. Scavenging H2O2 in the mitochondrial matrix partially protects against impaired skeletal muscle mitochondrial function and contractile dysfunction caused by cancer and chemotherapy. These findings provide evidence that elevated mitochondrial H2O2 emission may be a contributing underlying mechanism by which cancer chemotherapy causes debilitating muscle weakness and fatigue.
Cancer alone can negatively affect skeletal muscle mitochondrial respiration. Both complex I- and complex II-supported respiration was impaired in permeabilized myofibers from soleus hindlimb muscle of tumor-bearing mice. Few studies have assessed skeletal muscle mitochondrial respiratory capacity in the immunocompetent mouse model of breast cancer utilizing E0771 cells. However, similar findings have been established in other rodent cancer models using isolated mitochondria from hindlimb muscles. Tumor-bearing female rodents exhibit significant decreases in activities of complex I, complex II, and complex IV of the ETS from hindlimb muscle of lung (9) and intestinal cancer models (7).
Exposing healthy wild-type mice to chemotherapy alone decreases mitochondrial respiration. Following doxorubicin administration, soleus mitochondrial NADH- and FADH2-supported respiratory capacity is diminished. These findings are consistent with our previous work and other groups demonstrating that doxorubicin causes skeletal muscle mitochondrial dysfunction in a time-dependent manner (12, 17, 26).
Although doxorubicin alone can decrease the tumor burden on the rodent breast cancer model by reducing tumor volume (25), the combined effect of both the tumor and chemotherapeutic drug is not without negative effects. Cancer chemotherapy significantly decreased skeletal muscle mitochondrial respiratory capacity in wild-type mice. Interestingly, the effects were not additive, as respiratory capacity was depressed in the cancer chemotherapy experimental group to approximately the same degree as cancer and chemotherapy alone (∼20%). Very few studies have assessed mitochondrial function in non-tumor-bearing tissue in a preclinical rodent model of combined cancer chemotherapy. The majority of studies indirectly assess mitochondria by focusing on the apoptotic effects of anticancer drugs, particularly through the mitochondrial permeability transition pathway (20). In cardiac muscle, alterations in mitochondrial apopotic gene expression (e.g., Bbc3, Bad, and Casp7) and elevated caspase-3 activity are indicators of mitochondrial dysfunction in an immunocompetent female rat breast cancer model (8, 16). These studies imply that mitochondria in noncancerous cells are negatively affected by cancer chemotherapy. Our present work provides new data on how combined cancer chemotherapy affects mitochondria of skeletal muscle, suggesting a critical role in the impaired physical function experienced by patients.
Mitochondrial H2O2 emission and protein carbonylation, a global marker of protein oxidation, were increased in skeletal muscle from wild-type mice following exposure to both tumor cells and doxorubicin alone. Evidence of elevated oxidants in other rodent cancer models are indicated by increased markers of protein oxidation in hindlimb muscle of tumor-bearing mice (4), suggesting a disruption in the redox balance of the cell. Although cancer alone can jeopardize the redox buffering of a cell, the majority of studies point to chemotherapy as the main culprit. Min et al. (26) reported elevated H2O2 emission in both PmFBs from cardiac and skeletal muscle following doxorubicin administration, along with elevated markers of protein oxidation. Elevated H2O2-emitting potential in skeletal muscle induced by doxorubicin is likely due to redox modifications within the matrix (e.g., the ETS, redox-buffering system) (12). If persistent, elevated H2O2 emission can shift the redox environment of the cell to a more oxidized state. The findings of the present study also suggest that oxidant scavenging is depressed within the mitochondria. Consistent with these findings, prolonged exposure to the burden of tumor-bearing or chemotherapy has previously been shown to decrease superoxide dismutase (23) and glutathione (24, 27) content. Collectively, these findings suggest that mitochondrial redox buffering circuits are negatively affected by cancer and/or chemotherapy, compromising the ability to maintain the proper redox state of key structural and functional proteins in skeletal muscle.
Surprisingly, the combined effect of cancer with chemotherapy blunted the increase in mitochondrial H2O2 emission in skeletal muscle caused by the two insults individually despite a significant decrease in mitochondrial respiratory capacity. The reduced H2O2-emitting potential was not accounted for by an increase in mitochondrial scavenging capacity, suggesting that the combined effect of cancer chemotherapy decreased H2O2 production. Tumor size was not different with doxorubicin treatment, suggesting that cancer burden was similar. State 4 respiration was not increased in the cancer chemotherapy condition, at least not under the substrate conditions tested (Fig. 2), indicating that elevated proton conductance due to uncoupling likely did not account for the decrease in H2O2 production. Another possibility is that the drop in H2O2 production reflected a precipitous decline in mitochondrial function in response to the combination of cancer plus chemotherapy. In a previous study in rats, we also found that mitochondrial H2O2 emission was lower 3 days after doxorubicin injection alone but elevated at earlier time points (12). The lower H2O2 production coincided with lower membrane potential and greater susceptibility to Ca2+-induced opening of the permeability transition pore, both of which are indicative of compromised mitochondrial function. In the present study, global protein oxidation remained elevated with combined cancer chemotherapy, consistent with the notion that the lower H2O2 production did not reflect an improvement in mitochondrial function. The collective implications of these findings are of course constrained by the limited time course of such studies. Determining exactly how the combination of cancer and chemotherapy specifically modulates superoxide/H2O2 production and overall mitochondrial function in skeletal muscle mitochondria over time will require further experiments.
The chemotherapeutic agent doxorubicin alone caused the mice to lose weight following exposure, as shown previously (11–13, 32). Whole body fat and lean mass were significantly decreased, a phenomenon that occurs in the clinic. Catabolism and muscle wasting are significant contributors to the decline in physical function in cancer patients (38). Decrements in femoral quadriceps muscle thickness are detected by ultrasound within the first 4 wk of chemotherapy (21). The loss of muscle mass contributes to the muscle weakness caused by cancer chemotherapy and plays a role in the observed drop in absolute force of wild-type mice exposed to tumor cells and doxorubicin. In the present study, there was no change in specific force, which is the absolute force normalized for muscle cross-section. Our findings suggest that cancer chemotherapy affects skeletal muscle function via two pathways: 1) the loss of muscle mass and 2) a decline in contractile function, both of which can directly compromise the physical performance of patients.
To determine whether the elevated H2O2-emitting potential in skeletal muscle may mediate the mitochondrial dysfunction caused by cancer chemotherapy, mice that express mitochondrial-targeted catalase in skeletal muscle (MCAT) were exposed to tumor cells and doxorubicin. The increase in mitochondrial H2O2-emitting potential following tumor cell inoculation or doxorubicin administration alone was completely blunted in MCAT mice. Elevated protein oxidation in skeletal muscle caused by cancer chemotherapy was ablated in MCAT mice. In addition, the cancer chemotherapy-induced decline in mitochondrial respiration was restored in MCAT mice, suggesting that mitochondrial H2O2 may be a mechanism contributing to the mitochondrial dysfunction induced by cancer chemotherapy. Not only were MCAT mice protected on a cellular level from the negative effects of cancer chemotherapy, but the cancer chemotherapy-induced decrease in skeletal muscle isometric tetanic force was blunted in MCAT mice. The protection against the decline in contractile function occurred despite the MCAT mice exhibiting cancer chemotherapy-induced loss of muscle mass. It is important to note that although overexpression of catalase did appear to protect against the loss of maximal isometric tetanic force induced by cancer chemotherapy, maximal force was initially lower in MCAT controls relative to wild-type controls. Previous reports have demonstrated that a more oxidized cellular redox state increases maximal force in single fibers, an effect that is prevented by inclusion of an oxidant-buffering agent (3, 22). Therefore, the presence of catalase would be predicted to reset the maximal force to a lower value, consistent with the observed findings. The implication is that optimal force depends to some degree on thiol oxidation within proteins of the contractile machinery and that the added oxidant-buffering capacity provided by catalase held this new redox set point despite elevated H2O2 production induced by the cancer chemotherapy.
Mitochondrial-targeted antioxidants are a viable intervention for protecting against the negative effects of cancer chemotherapy. Administration of the mitochondrial-targeted peptide MTP-131 also protects against the loss of skeletal muscle function caused by doxorubicin (26). MTP-131 is thought to exert its cytoprotective effects at least in part by reducing H2O2 emission (37). Nonmitochondrial targeted approaches appear to be less effective, as several studies have utilized the antioxidant NAC (N-acetylcysteine) to replenish the loss of glutathione caused by cancer chemotherapy and found little to no protection against mitochondrial dysfunction in tumor-bearing mice (9) or following doxorubicin administration (29, 39).
In summary, the findings of the present study suggest that increased mitochondrial ROS production is a significant contributor to the development of cancer chemotherapy-induced mitochondrial and skeletal muscle dysfunction. Our current findings and the work of others (26) indicate that mitochondrial-targeted antioxidants may be an effective adjunct therapeutic approach for cancer patients undergoing chemotherapy to help prevent muscle weakness and loss. The reduction in muscle mass and function is both an acute and long-term problem for cancer patients (17, 19), and thus limiting these distressing side effects could lead to improved therapy retention rates and enhanced quality of life for patients long term.
GRANTS
This project was supported by National Institutes of Health Grants R01-DK-073488 (P. D. Neufer) and F32-AR-061946 (L. A. Gilliam).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
L.A.G., D.S.L., L.R.R., and P.D.N. conception and design of research; L.A.G., D.S.L., L.R.R., M.J.T., T.E.R., C.-T.L., and B.L.C. performed experiments; L.A.G. analyzed data; L.A.G. interpreted results of experiments; L.A.G. prepared figures; L.A.G. drafted manuscript; L.A.G., D.S.L., L.R.R., M.J.T., T.E.R., C.-T.L., B.L.C., and P.D.N. edited and revised manuscript; L.A.G. and P.D.N. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Matthew Rosenbaum for instructing and assisting in the ovariectomy surgeries.
REFERENCES
- 1.American Cancer Society. Cancer Facts & Figures 2015. Atlanta, GA: American Cancer Society, 2015. [Google Scholar]
- 2.Anderson EJ, Lustig ME, Boyle KE, Woodlief TL, Kane DA, Lin CT, Price JW 3rd, Kang L, Rabinovitch PS, Szeto HH, Houmard JA, Cortright RN, Wasserman DH, Neufer PD. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest 119: 573–581, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andrade FH, Reid MB, Allen DG, Westerblad H. Effect of hydrogen peroxide and dithiothreitol on contractile function of single skeletal muscle fibres from the mouse. J Physiol 509: 565–575, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barreiro E, de la Puente B, Busquets S, Lopez-Soriano FJ, Gea J, Argiles JM. Both oxidative and nitrosative stress are associated with muscle wasting in tumour-bearing rats. FEBS Lett 579: 1646–1652, 2005. [DOI] [PubMed] [Google Scholar]
- 5.Bower JE, Ganz PA, Desmond KA, Bernaards C, Rowland JH, Meyerowitz BE, Belin TR. Fatigue in long-term breast carcinoma survivors: a longitudinal investigation. Cancer 106: 751–758, 2006. [DOI] [PubMed] [Google Scholar]
- 6.Chabner BA, Ryan DP, Paz-Ares L, Garcia-Carbonero R, Calabresi P, Hardman JG, Limbird LE, Gilman AG. Goodman & Gilman's The Pharmacologic Basis of Therapeutics. New York: McGraw-Hill, Medical Publishing Division, 2001, p. 1389–1459. [Google Scholar]
- 7.Cummings J, Willmott N, Calman KC. Effect of a subcutaneously growing Walker 256 carcinosarcoma on host tissue mitochondrial function and magnesium content. Cancer Res 44: 1333–1336, 1984. [PubMed] [Google Scholar]
- 8.Dickey JS, Gonzalez Y, Aryal B, Mog S, Nakamura AJ, Redon CE, Baxa U, Rosen E, Cheng G, Zielonka J, Parekh P, Mason KP, Joseph J, Kalyanaraman B, Bonner W, Herman E, Shacter E, Rao VA. Mito-tempol and dexrazoxane exhibit cardioprotective and chemotherapeutic effects through specific protein oxidation and autophagy in a syngeneic breast tumor preclinical model. PLoS One 8: e70575, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fermoselle C, García-Arumí E, Puig-Vilanova E, Andreu AL, Urtreger AJ, de Kier Joffé ED, Tejedor A, Puente-Maestu L, Barreiro E. Mitochondrial dysfunction and therapeutic approaches in respiratory and limb muscles of cancer cachectic mice. Exp Physiol 98: 1349–1365, 2013. [DOI] [PubMed] [Google Scholar]
- 10.Fisher-Wellman KH, Lin CT, Ryan TE, Reese LR, Gilliam LA, Cathey BL, Lark DS, Smith CD, Muoio DM, Neufer PD. Pyruvate dehydrogenase complex and nicotinamide nucleotide transhydrogenase constitute an energy-consuming redox circuit. Biochem J 467: 271–280, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gilliam LA, Ferreira LF, Bruton JD, Moylan JS, Westerblad H, St Clair DK, Reid MB. Doxorubicin acts through tumor necrosis factor receptor subtype 1 to cause dysfunction of murine skeletal muscle. J Appl Physiol 107: 1935–1942, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gilliam LA, Fisher-Wellman KH, Lin CT, Maples JM, Cathey BL, Neufer PD. The anticancer agent doxorubicin disrupts mitochondrial energy metabolism and redox balance in skeletal muscle. Free Radic Biol Med 65: 988–996, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gilliam LA, Moylan JS, Callahan LA, Sumandea MP, Reid MB. Doxorubicin causes diaphragm weakness in murine models of cancer chemotherapy. Muscle Nerve 43: 94–102, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gilliam LA, Moylan JS, Ferreira LF, Reid MB. TNF/TNFR1 signaling mediates doxorubicin-induced diaphragm weakness. Am J Physiol Lung Cell Mol Physiol 300: L225–L231, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gilliam LA, Moylan JS, Patterson EW, Smith JD, Wilson AS, Rabbani Z, Reid MB. Doxorubicin acts via mitochondrial ROS to stimulate catabolism in C2C12 myotubes. Am J Physiol Cell Physiol 302: C195–C202, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gonzalez Y, Pokrzywinski KL, Rosen ET, Mog S, Aryal B, Chehab LM, Vijay V, Moland CL, Desai VG, Dickey JS, Rao VA. Reproductive hormone levels and differential mitochondria-related oxidative gene expression as potential mechanisms for gender differences in cardiosensitivity to Doxorubicin in tumor-bearing spontaneously hypertensive rats. Cancer Chemother Pharmacol 76: 447–459, 2015. [DOI] [PubMed] [Google Scholar]
- 17.Gouspillou G, Scheede-Bergdahl C, Spendiff S, Vuda M, Meehan B, Mlynarski H, Archer-Lahlou E, Sgarioto N, Purves-Smith FM, Konokhova Y, Rak J, Chevalier S, Taivassalo T, Hepple RT, Jagoe RT. Anthracycline-containing chemotherapy causes long-term impairment of mitochondrial respiration and increased reactive oxygen species release in skeletal muscle. Sci Rep 5: 8717, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jacobsen PB, Hann DM, Azzarello LM, Horton J, Balducci L, Lyman GH. Fatigue in women receiving adjuvant chemotherapy for breast cancer: characteristics, course, and correlates. J Pain Symptom Manage 18: 233–242, 1999. [DOI] [PubMed] [Google Scholar]
- 19.Jones LW, Watson D, Herndon JE 2nd, Eves ND, Haithcock BE, Loewen G, Kohman L. Peak oxygen consumption and long-term all-cause mortality in nonsmall cell lung cancer. Cancer 116: 4825–4832, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kluza J, Marchetti P, Gallego MA, Lancel S, Fournier C, Loyens A, Beauvillain JC, Bailly C. Mitochondrial proliferation during apoptosis induced by anticancer agents: effects of doxorubicin and mitoxantrone on cancer and cardiac cells. Oncogene 23: 7018–7030, 2004. [DOI] [PubMed] [Google Scholar]
- 21.Koskelo EK, Saarinen UM, Siimes MA. Skeletal muscle wasting and protein-energy malnutrition in children with a newly diagnosed acute leukemia. Cancer 66: 373–376, 1990. [DOI] [PubMed] [Google Scholar]
- 22.Lamb GD, Posterino GS. Effects of oxidation and reduction on contractile function in skeletal muscle fibres of the rat. J Physiol 546: 149–163, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leuthauser SW, Oberley LW, Oberley TD, Loven DP. Lowered superoxide dismutase activity in distant organs of tumor-bearing mice. J Natl Cancer Inst 72: 1065–1074, 1984. [PubMed] [Google Scholar]
- 24.Li T, Danelisen I, Singal PK. Early changes in myocardial antioxidant enzymes in rats treated with adriamycin. Mol Cell Biochem 232: 19–26, 2002. [DOI] [PubMed] [Google Scholar]
- 25.Mattarollo SR, Loi S, Duret H, Ma Y, Zitvogel L, Smyth MJ. Pivotal role of innate and adaptive immunity in anthracycline chemotherapy of established tumors. Cancer Res 71: 4809–4820, 2011. [DOI] [PubMed] [Google Scholar]
- 26.Min K, Kwon OS, Smuder AJ, Wiggs MP, Sollanek KJ, Christou DD, Yoo JK, Hwang MH, Szeto HH, Kavazis AN, Powers SK. Increased mitochondrial emission of reactive oxygen species and calpain activation are required for doxorubicin-induced cardiac and skeletal muscle myopathy. J Physiol 593: 2017–2036, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mohamed HE, Asker ME, Ali SI, el-Fattah TM. Protection against doxorubicin cardiomyopathy in rats: role of phosphodiesterase inhibitors type 4. J Pharm Pharmacol 56: 757–768, 2004. [DOI] [PubMed] [Google Scholar]
- 28.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J 417: 1–13, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Myers C, Bonow R, Palmeri S, Jenkins J, Corden B, Locker G, Doroshow J, Epstein S. A randomized controlled trial assessing the prevention of doxorubicin cardiomyopathy by N-acetylcysteine. Semin Oncol 10: 53–55, 1983. [PubMed] [Google Scholar]
- 30.Perry CG, Kane DA, Lin CT, Kozy R, Cathey BL, Lark DS, Kane CL, Brophy PM, Gavin TP, Anderson EJ, Neufer PD. Inhibiting myosin-ATPase reveals a dynamic range of mitochondrial respiratory control in skeletal muscle. Biochem J 437: 215–222, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Quinlan CL, Orr AL, Perevoshchikova IV, Treberg JR, Ackrell BA, Brand MD. Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J Biol Chem 287: 27255–27264, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rephaeli A, Waks-Yona S, Nudelman A, Tarasenko I, Tarasenko N, Phillips DR, Cutts SM, Kessler-Icekson G. Anticancer prodrugs of butyric acid and formaldehyde protect against doxorubicin-induced cardiotoxicity. Br J Cancer 96: 1667–1674, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saeed MF, Premecz S, Goyal V, Singal PK, Jassal DS. Catching broken hearts: pre-clinical detection of doxorubicin and trastuzumab mediated cardiac dysfunction in the breast cancer setting. Can J Physiol Pharmacol 92: 546–550, 2014. [DOI] [PubMed] [Google Scholar]
- 34.Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, Coskun PE, Ladiges W, Wolf N, Van Remmen H, Wallace DC, Rabinovitch PS. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 308: 1909–1911, 2005. [DOI] [PubMed] [Google Scholar]
- 35.Schwartz AL. Daily fatigue patterns and effect of exercise in women with breast cancer. Cancer Pract 8: 16–24, 2000. [DOI] [PubMed] [Google Scholar]
- 36.Smuder AJ, Kavazis AN, Min K, Powers SK. Exercise protects against doxorubicin-induced oxidative stress and proteolysis in skeletal muscle. J Appl Physiol 110: 935–942, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Szeto HH. Development of mitochondria-targeted aromatic-cationic peptides for neurodegenerative diseases. Ann NY Acad Sci 1147: 112–121, 2008. [DOI] [PubMed] [Google Scholar]
- 38.Tozer RG, Tai P, Falconer W, Ducruet T, Karabadjian A, Bounous G, Molson JH, Droge W. Cysteine-rich protein reverses weight loss in lung cancer patients receiving chemotherapy or radiotherapy. Antioxid Redox Signal 10: 395–402, 2008. [DOI] [PubMed] [Google Scholar]
- 39.Unverferth DV, Jagadeesh JM, Unverferth BJ, Magorien RD, Leier CV, Balcerzak SP. Attempt to prevent doxorubicin-induced acute human myocardial morphologic damage with acetylcysteine. J Natl Cancer Inst 71: 917–920, 1983. [PubMed] [Google Scholar]