

Abstract
Frontal cortical dysfunction is thought to contribute to cognitive and behavioral features of autism spectrum disorders; however, underlying mechanisms are poorly understood. The present study sought to define how loss of Mecp2, the gene mutated in Rett syndrome (RTT), disrupts function in the murine medial prefrontal cortex (mPFC) using acute brain slices and behavioral testing. Compared with wildtype, pyramidal neurons in the Mecp2 null mPFC exhibit significant reductions in excitatory postsynaptic currents, the duration of excitatory UP-states, evoked population activity, and the ratio of NMDA:AMPA currents, as well as an increase in the relative fraction of NR2B currents. These functional changes are associated with reductions in the density of excitatory dendritic spines, the ratio of vesicular glutamate to GABA transporters and GluN1 expression. In contrast to recent reports on circuit defects in other brain regions, we observed no effect of Mecp2 loss on inhibitory synaptic currents or expression of the inhibitory marker parvalbumin. Consistent with mPFC hypofunction, Mecp2 nulls exhibit respiratory dysregulation in response to behavioral arousal. Our data highlight functional hypoconnectivity in the mPFC as a potential substrate for behavioral disruption in RTT and other disorders associated with reduced expression of Mecp2 in frontal cortical regions.
Keywords: AMPA, E/I balance, EPSC, Fluo-4 AM, GABA, IPSC, MeCP2, medial prefrontal cortex, NMDA, Rett syndrome, synaptic
Introduction
Dysfunction of the prefrontal cortex is thought to contribute to cognitive and behavioral deficits in diverse neurodevelopmental disorders, including autism spectrum disorder (ASD), Rett syndrome (RTT), Angelman syndrome, Prader–Willi syndrome, Down syndrome, and attention deficit hyperactivity disorder (ADHD) (Akbarian et al. 2001; Nagarajan et al. 2006; Emond et al. 2009; Yizhar et al. 2011; Katz et al. 2012; Zikopoulos and Barbas 2013; Li et al. 2014). However, mechanisms that link the molecular etiologies of any of these disorders to altered prefrontal cortical function are poorly understood. To approach this issue, the present study sought to define how loss of Mecp2, the gene mutated in RTT (Amir et al. 1999), alters circuit function in the medial prefrontal cortex (mPFC). RTT is a complex disorder characterized by neurological regression during postnatal development and by severe impairments in cognition, motor function and respiratory and autonomic control. In addition, RTT patients exhibit autistic behaviors and an increased risk of seizures. Moreover, there is evidence that dysregulation of Mecp2 expression in frontal cortical regions also contributes to other neurodevelopmental syndromes, including Angelman, Prader–Willi, and Down's (Samaco et al. 2004; Nagarajan et al. 2006). Therefore, understanding the role of Mecp2 in the regulation of synaptic connectivity in the mPFC may shed light on the pathogenesis of overlapping features of these disorders, including autistic behaviors. In general, cortical dysfunction in Mecp2 mutants is thought to result from disruptions in structural and functional connectivity at the microcircuit level rather than gross anatomical changes (Armstrong 2005; Wood et al. 2009; Wood and Shepherd 2010; Shepherd and Katz 2011; Kron et al. 2012); however, the effects of Mecp2 mutations on circuit function in the mPFC are unknown.
Neocortical neurons in Mecp2 mutant mouse models of RTT display apparently normal intrinsic properties and long-term potentiation; however, many neocortical circuits appear to be hypofunctional due to a shift in excitatory/inhibitory (E/I) synaptic balance (Dani et al. 2005; Moretti et al. 2006; Dani and Nelson 2009; Gogolla et al. 2009; Wood et al. 2009; Blackman et al. 2012; Kron et al. 2012; Na et al. 2013). It has been suggested that E/I imbalance stems from reduced glutamatergic excitatory drive, increased inhibitory connectivity, or a combination thereof (Dani et al. 2005; Wood et al. 2009; Durand et al. 2012). In fact, there is growing evidence that mechanisms of E/I imbalance in Mecp2 null mice differ markedly among cortical regions. For example, although inhibitory hyperconnectivity has been reported in visual cortex (Durand et al. 2012), reduced excitatory drive without significant alterations in inhibitory input has been reported in frontal-motor cortex (Wood et al. 2009). Moreover, in contrast to cortex, subcortical regions including hindbrain and hippocampus exhibit hyperexcitability (Calfa et al. 2011; Shepherd and Katz 2011; Kron et al. 2012) and the specific inputs to subcortical structures display phenotypes that are dependent on their anatomical origin (Shahbazian et al. 2002; Gambino et al. 2010; Wither et al. 2013).
We previously found that loss of Mecp2 results in reduced expression of the immediate early gene product Fos, a surrogate marker of neuronal activity, in midline limbic structures, including the mPFC (Kron et al. 2012). Although these data suggest that mPFC neurons are less active in Mecp2 mutants compared with wild type (Wt) controls, underlying mechanisms have not been defined. Therefore, we examined circuit dysfunction in the mPFC in Mecp2 knockout mice using a range of techniques to quantify E/I synaptic balance, NMDA and AMPA responses, neuronal population excitability, synaptic protein expression, and dendritic spine density. In addition, we analyzed the respiratory component of the orienting reflex, a behavioral response that is dysregulated following mPFC lesions in normal animals (Frysztak and Neafsey 1991). Our findings suggest that loss of Mecp2 results in reduced excitatory synaptic connectivity in the mPFC and that mechanisms of synaptic and circuit dysfunction differ in important ways from hypoconnectivity phenotypes described in other cortical regions in Mecp2 mutants. Moreover, we find that behavioral disruption of breathing in Mecp2 mutants is consistent with mPFC hypofunction, highlighting the mPFC as a potential substrate of behavioral dysregulation in RTT.
Materials and Methods
All procedures and protocols used in this study adhere to published guidelines of the National Institutes of Health and were approved by the Institutional Animal Care and Use Committee at Case Western Reserve University.
All experiments were performed on Wt and Mecp2tm1.1Jae null mice from our local mouse colony (Chen et al. 2001). Every animal was independently genotyped twice to confirm its genetic identity. Across all the experiments performed animals were used within the range of 32–42 days old. Only Mecp2 null animals with a Bird score between 6 and 10 were included in our analysis (Guy et al. 2007). Mecp2tm1.1Jae null mice typically display disease phenotypes at the ages used in this study and disease onset is somewhat earlier than in other strains (Katz et al. 2012).
Electrophysiology
Mouse brain slices were cut from the mPFC of young adult (32–42 days postnatal) Wt and Mecp2 null mice deeply anesthetized with isoflurane (4%). The brain was dissected and temporarily (<30 s) placed in ice-cold (1–4°C) oxygenated (95% O2, 5% CO2) artificial cerebral spinal fluid (ACSF), composed of the following (in mM): 126 NaCl, 1 NaH2PO4, 25 NaHCO3, 25 dextrose, 3 KCl, 2 MgSO4, and 2 CaCl2. Brain slices were cut in oxygenated ice-cold ASCF into 350 µm slices and placed in a holding chamber with 30–35°C ACSF that gradually (<30 min) equilibrated to room temperature (25°C).
Brain slices were transferred from the holding chamber after 1–2 h and placed in a submersion-recording chamber with a heated water jacket (Warner Instruments, Hamden, CT). Slices were perfused with oxygenated 30–35°C gravity-fed ACSF (2–4 mL/min). ASCF temperature was maintained with a ThermoClamp-1 (Automate Scientific, Berkeley, CA) inline heater and temperature controller. Neurons were visualized with an Olympus BX51WI microscope (Olympus America, Central Valley, PA) equipped with a water immersion objective (20× with 2× post magnification) with near-infrared wavelength illumination, differential interference contrast optics (DIC), and a CCD camera (OLY-150, Olympus America, Central Valley, PA) with contrast enhancement (Sceniak and Maciver 2006; Sceniak and Sabo 2010).
Membrane potentials and currents were collected using a Multi-clamp 700B patch-clamp amplifier (Axon Instruments, Foster City, CA) and digitized with a Digidata 1440A analog to digital converter which was controlled by the pClamp 10.2 software package (Axon Instruments). In order to measure intrinsic membrane properties and spiking activity, recording electrodes were filled with a K-gluconate-based internal solution composed of the following (in mM): 100 K-gluconate, 20 KCl, 10 phosphocreatine, 5 MgCl2, 10 HEPES, 4 Na-ATP, 0.3 Na-GTP (pH 7.3 and 290–300 mOsm). Spontaneous excitatory and inhibitory synaptic currents were recorded using the following Cs-methylsulfonate internal electrode solution (in mM): 120 Cs-methylsulfonate, 12 CsCl, 0.1 EGTA, 2 MgCl2, 10 HEPES, 2 Na-ATP, 0.25 Na-GTP, 10 phosphocreatine, and 5 QX314 (pH 7.3 and 290–300 mOsm). Electrode resistance ranged from 4–8 MΩ and whole-cell patch recordings were made with seal resistances of 1–3 GΩ. Successful whole-cell recordings had resting membrane potentials of −57 mV or more negative. Average electrode series resistance ranged from 10–30 MΩ after whole-cell patching. Recordings with seals <1 GΩ or resting potentials greater than −57 mV were not included in the analysis. Electrical current pulses were delivered to the tissue using high impedance bipolar tungsten metal electrodes (FHC, Bowdoin ME) and a constant current source. Current pulses were optimized according to the recorded current response output to achieve specific failure rates, as with UP-state measures (<90%), or particular response amplitudes, as with AMPA recordings (roughly 100 pA or twice the minimal stimulation amplitude).
Time-locked current responses were analyzed offline to determine the amplitude and integrated current charge. Intrinsic membrane properties were estimated through pClamp and through post processing in Matlab (The Mathworks, Natick, MA). All synaptic current data were analyzed and quantified using custom written analysis routines in Matlab.
Recordings were made from excitatory pyramidal neurons located within layer 5 of the mPFC. Neurons were selected to establish the most homogeneous population possible based on spiking and intrinsic electrical membrane properties and confirmed through histological reconstructions using biocytin fills (Sceniak and Maciver 2006; Sceniak and Sabo 2010). Based on their morphologies and intrinsic properties, the cells we recorded from were most likely all type A pyramidal neurons (Hattox and Nelson 2007).
Miniature excitatory postsynaptic currents (mEPSCs) were recorded (Vhold = −70 mV) using Cs-methylsulfonate internal electrode solution as described above with tetrodotoxin (TTX) (1 µM) and picrotoxin (100 µM) added to the ACSF bath perfusion. Miniature inhibitory postsynaptic currents (mIPSCs) were recorded (Vhold = −70 mV) using a KCl-based internal electrode solution in order to amplify individual events with the following (in mM): 100 KCl, 0.2 EGTA, 5 MgCl2, 40 HEPES, 2 Na-ATP, 0.3 Na-GTP, 10 phosphocreatine and 5 QX314 (pH 7.3 and 290–300 mOsm). The mIPSCs were pharmacologically isolated through bath perfusion with TTX (1 µM), CNQX (10 µM), and APV (100 µM). All mEPSC and mIPSC recordings were conducted at room temperature (27°C) in order to provide recordings with identifiable unitary events. Post hoc analysis of mEPSC and mIPSC recordings was performed using Clampex (Axon Instruments) in order to identify the individual mEPSCs or mIPSCs after baseline subtraction. The amplitude and time occurrence of individual events was collected in order to determine the amplitude and instantaneous frequency (inter-event frequency or 1/inter-event period). In a subset of experiments in which pharmacological isolation of mEPSCs and mIPSCs was verified by blocking events with either CNQX or picrotoxin respectively, no remaining events were observed in the recordings.
Spontaneous Excitatory and Inhibitory Synaptic Currents
To estimate the excitatory to inhibitory synaptic balance within the same cell, spontaneous synaptic currents were measured using whole-cell patch-clamp recording while the brain slice was perfused with a high-conductance ACSF containing the following (in mM concentrations): 126 NaCl, 1 NaH2PO4, 25 NaHCO3, 25 dextrose, 3.5 KCl, 0.5 MgCl2, and 1 CaCl2. The ionic balance of the high-conductance ACSF was set to push the resting membrane potential closer to firing threshold and therefore increase excitability (Haider et al. 2006). Excitatory and inhibitory synaptic currents were estimated electrically by holding the membrane potential under voltage-clamp at the reversal potential of synaptic inhibition (approximately −50 mV) or synaptic excitation (approximately +10 mV) respectively (Dani et al. 2005; Dani and Nelson 2009).
NMDA to AMPA Synaptic Current Ratio
AMPA and NMDA currents were measured within the same cell using pharmacological isolation combined with electrical isolation (Ye et al. 2005). The identical electrical stimulus amplitude (optimized for each cell to produce AMPA responses 100% of the time with roughly 2× the amplitude for minimal stimulus responses) was used to measure AMPA, NMDA, and NR2B components within a given cell, in order to make quantitative comparisons of the NMDA-to-AMPA ratio. Initial estimates of AMPA currents were made from recordings using whole-cell voltage-clamp techniques (Vhold = −70 mV) in normal ACSF in the presence of the GABAergic blocker picrotoxin (100 μm). Synaptic responses were evoked through a bipolar stimulating electrode placed in the white matter and calculated as the time-locked average of 10 repeats of the electrical stimulus. Evoked NMDA synaptic currents were measured within the same cell in the presence of 10 μM CNQX (Vhold = +45 mV). The NR2B insensitive component of the NMDA current was estimated as the average response to electrical stimulation as in the presence 3 μM ifenprodil. The NR2B component was mathematically estimated as the difference between the total NMDA current and the NR2B insensitive component.
Fluo-4 AM Calcium Imaging
Brain slices were prepared as described above and placed in a holding chamber for 1–2 h. The slices were then incubated in Fluo-4 AM (Molecular Probes, Cat# F14201) solution (2.5 μM Fluo-4 AM 0.5 μL pluronic per 1 mL ACSF) at 34°C for 30 min in the dark, then placed in normal room temperature ACSF for 1 h to recover and reduce background fluorescence (Cossart et al. 2005; MacLean and Yuste 2009).
Slices were imaged at 0.2 s exposures using fluorescence light imaging timed with an automatic shutter to avoid photo-bleaching. Imaging sequences (50 frames) were timed to electrical stimulation evoked through a bipolar stimulating electrode placed in the white matter. The change in fluorescence was calculated as deviations in fluorescence (ΔF) normalized to background (F0) or ΔF/F0. The mean background fluorescence, F0, was estimated from the first 10 frames prior to electrical stimulation within the region of interest. The change in fluorescence, ΔF, was calculated as the difference in fluorescence intensity from the initial baseline and estimated across all 50 frames. Plots indicate the maximum ΔF/F0 evoked from electrical stimulation. The stimulus intensity was adjusted using a constant current source and stimulating electrode resistance was constant across all experiments.
The brain slices were imaged on an upright fluorescence microscope (BX51WI, Olympus) using a low power objective (4×/0.13 FN26.5, UPAN-FLN, Olympus) and a CCD camera (Retiga EXi, Qimaging). Image sequences were collected using maximum gain (30×) and an exposure frame of 0.2 s. The fluorescence light source (X-cite series 120, EXFO) was used with a GFP filter (Olympus) and a shutter controller (Lambda SC). Image sequences were collected using the Matlab Image acquisition toolbox (Mathworks, in conjunction with QCam drivers).
Histology
Animals were deeply anesthetized by inhalation of isoflurane and perfused transcardially with PBS followed by 4% paraformaldehyde and 4% sucrose in PBS at 37°C. Brains were postfixed in 4% paraformaldehyde for 10 min and then stored in PBS at 4°C. Brain slices were prepared from either frozen brains using a cryostat (40 μm) or at room temperature using a vibratome (Leica VT1000S; 350 μm) for immunohistochemical or diolistic analysis, respectively (Sceniak and Maciver 2006; Kron et al. 2012).
Immunohistochemistry
Fixed brain slices were exposed to antibodies for VGLUT1, VGAT, or parvalbumin (Synaptic Systems; catalog numbers #135304, #131003, and #195011, respectively). Floating coronal brain slices (40 μm) were incubated in primary antibodies overnight and then in fluorescent conjugated secondary antibodies (Life Technologies; part numbers A11034, A11035, A11073, and A11074). Mounted brain sections were imaged on a confocal fluorescence microscope (20×, Nikon) and images were obtained across all cortical layers. Images were acquired from 3 brain sections from each of 5 Wt and 5 Mecp2 null animals. To generate low magnification images of the horizontal mPFC brain slice, as in Fig. 8, individual images of 40 µm DAPI-stained sections were collected using a light microscope (Zeiss Axiophot) and a CCD digital camera (Q-Imaging) and combined into a photo montage.
Figure 8.
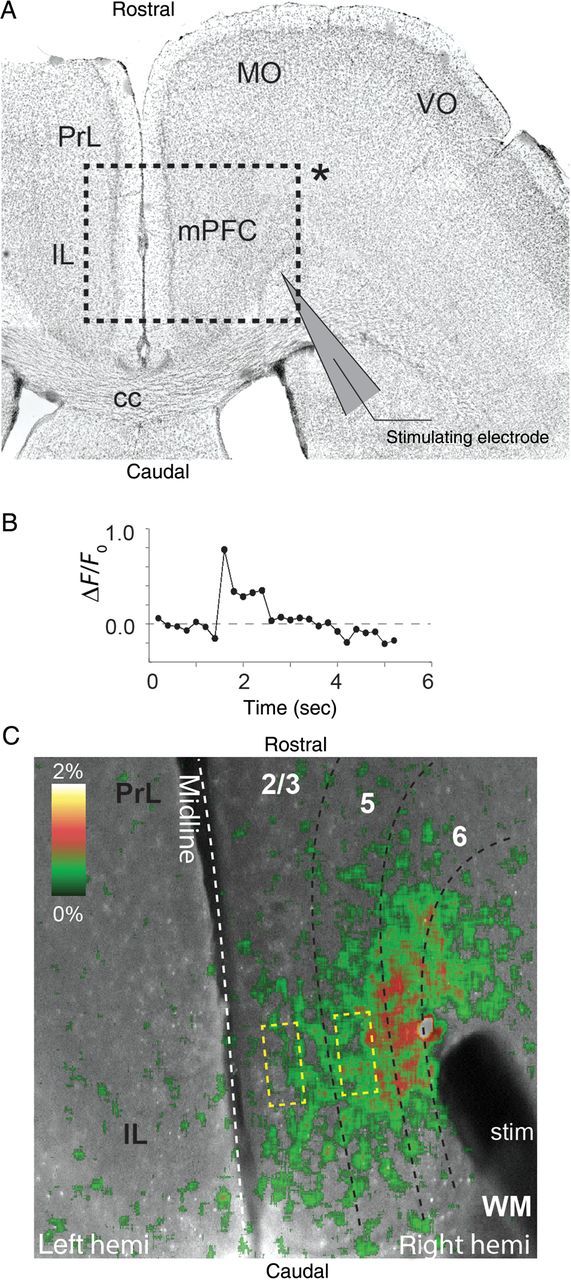
Calcium imaging analysis of electrically evoked signal propagation in mPFC. (A) Representative DAPI stained horizontally sectioned brain slice of mPFC, showing the (inset indicated by *) region of interest studied with Fluo-4AM calcium imaging. The stimulating electrode was positioned within the white matter that projects into the mPFC. (B) Calcium signal responses were calculated normalized to baseline level (ΔF/F0) in response to electrical stimulation. Image sequences were sampled (100 frames or 20 s) to capture calcium measures before and after electrical stimulation. (C) Representative image of Fluo-4AM imaging of mPFC. Electrical stimulation through bipolar stimulating electrode placed in the white matter evoked propagation of current through cortical layers. The pseudo-colored image illustrates the intensity of the current spread through the cortex.
DiOlistic Labeling and Dendritic Spine Analyses
DiOlistic neuronal labeling was performed as described by others (Gan et al. 2000; Staffend and Meisel 2011). Briefly, 5 mg of the carbocyanine fluorescent dye, DiI (Molecular Probes), was dissolved in 100 µL methylene chloride and applied to 15 mg of 1.7 µm tungsten microcarrier particles (Bio-Rad). Fixed horizontal brain slices were shot with DiI-coated tungsten bullets at 200 psi using a Helios gene gun (Bio-Rad) through a 3.0 µm pore size, polycarbonate membrane filter (EMD Millipore). DiI-labeled brain slices were mounted in ProLong Gold AntiFade Reagent with DAPI (Molecular Probes).
Z-stack images of DiI-labeled layer 5 pyramidal neurons in the mPFC of Mecp2 null and Wt mice were obtained using a Zeiss LSM 510 META confocal microscope. DiI-labeled pyramidal neurons were traced in confocal image stacks using Neurolucida (MBF Bioscience, Inc.) and dendritic spines on the apical oblique dendrites were mapped and used to define dendritic spine density (per µm) and length (in µm) using Neurolucida Explorer (MBF Bioscience, Inc.). Spine counts were obtained from 20 µm dendritic segments. Dendritic spine length was measured in 10 µm segments and divided by the total number of spines in each segment to calculate average spine length on the apical oblique dendrites. All spine analysis was performed blind and decoded by a third party.
Western Blots
Mice were euthanized with an overdose of inhaled isoflurane. Brain tissue was rapidly dissected over ice, frozen in dry ice, and stored at −80°C until use. Tissue was homogenized in RIPA buffer with HALT protease inhibitors (20 mM Tris, pH 8.0, 137 mM NaCl, 1% Igepal CA-630, 10% glycerol, 1 mM PMSF, 10 μg/mL aprotinin, 1 μg/mL leupeptin, 500 μM orthovanadate; Thermo Scientific). The homogenates were centrifuged at 14 000 × g for 10 min and the supernatant was collected. Protein concentrations of the samples were determined using the BCA Protein Assay Kit (Pierce). Rabbit polyclonal anti-GluA1 (1:1000, Synaptic Systems, Cat#182 003), anti-GluA1 (1:1000, BD Pharmingen, Cat# 556308), anti-NR2A (1:500, Life Technologies, Cat#A6473), and anti-NR2B (1:500, Life Technologies, Cat#A6474), with goat anti-rabbit peroxidase conjugated secondary antibody (1:500, Jackson Laboratories, Cat#111035003) as well as mouse monoclonal anti-β-actin-peroxidase antibody (1:40 000, Sigma-Aldrich, Cat#A3854) were used for western blots and detected using the ECL Chemiluminescence System (GE Healthcare). Protein levels were determined through densitometric analysis using Quantity One software (Bio-Rad). Analysis was performed blind by a third party.
Respiratory Measurements
Breathing was recorded in unrestrained Wt and null mice using a whole-body plethysmograph (Buxco II; Buxco Research Systems, Wilmington, NC) in which a constant bias flow supply connected to the animal recording chamber ensured continuous inflow of fresh air (1 L/min). Only episodes of quiet breathing, totaling at least 10 min, were used for analysis. Quiet breathing was defined as periods when the animal was not moving and had all 4 paws on the chamber platform for a minimum of 3 min. Ambient temperature was maintained between 23 and 25°C. Plethysmographic recordings were obtained before, during, and after exposure to a single 50 ms broad-spectrum auditory pulse (80 dB white noise). The acoustic stimulus was presented through 2 speakers (Advent) placed outside the plethysmographic chamber and the intensity level was calibrated using a sound meter (Med Associates, ANL-929A-PC; Vermont, US) placed inside the chamber. Breathing traces were analyzed using Biosystem XA software (Buxco Research Systems, Wilmington, NC).
Data Analysis
All data analyses were performed using custom-written functions in Matlab and functions written in ImageJ (NIH software). Confocal images were analyzed using either ImageJ (NIH software) or Neurolucida (MBF Bioscience, Inc.) for immunohistochemical staining and DiOlistic labeling, respectively. All data are expressed as the mean ± SEM unless otherwise stated. Statistical significance was determined using the two-tailed Student's t-test, the Mann–Whitney U-test for non-parametric data or the analysis of variance (ANOVA) where appropriate. The nested ANOVA (Matlab) was used to determine significance for spine density analysis where dendritic segments were grouped according to cell origin and genotype.
Results
E/I Balance is Disrupted in the mPFC in Mecp2 Null Mice
To define effects of Mecp2 loss on synaptic drive in the mPFC, spontaneous synaptic currents were measured using whole-cell voltage-clamp in high-conductance ACSF (see Materials and Methods) to evoke maximal synaptic activity. Excitatory and inhibitory currents were isolated within the same cell by adjusting the holding potential to either the reversal potential for inhibition (−50 mV; Fig. 1A) or excitation (+10 mV; Fig. 1A) and optimizing for each individual cell (Dani et al. 2005). Several 30 s long current recordings were collected for excitatory and inhibitory (n = 7 repeats) synaptic spontaneous inputs for each cell (14 Wt and 12 null neurons; Fig. 1). Therefore, we were able collect consecutive estimates for excitatory glutamatergic and inhibitory GABAergic currents within the same cell, allowing us to estimate the E/I balance on a cell-by-cell basis.
Figure 1.
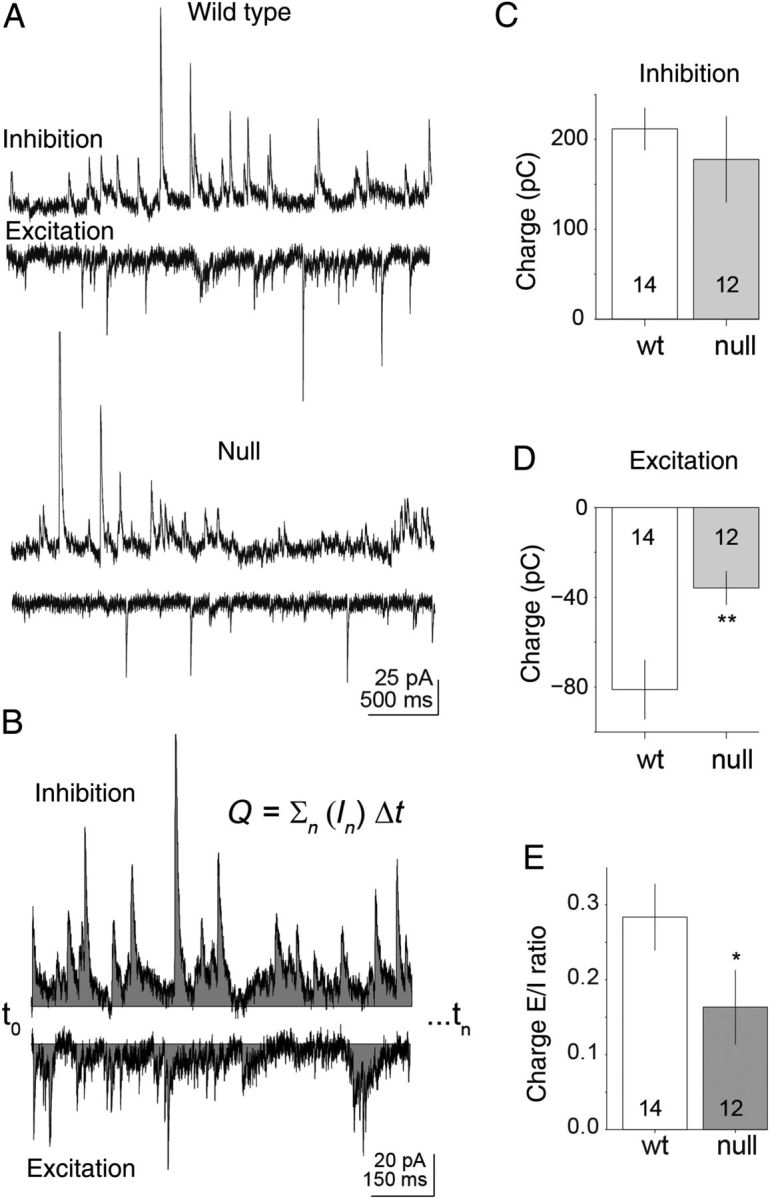
E/I balance is shifted toward hypofunction in Mecp2 null layer 5 pyramidal neurons of the mPFC. Inhibitory and excitatory synaptic currents were recorded within the same cell by holding the membrane potential under voltage-clamp recording mode fixed at either the reversal potential for excitatory (Vhold≈ +10 mV) or inhibitory (Vhold ≈ −50 mV) synaptic potentials. (A) Representative spontaneous synaptic current recordings are shown for inhibitory and excitatory synaptic responses in Wt and Mecp2 null pyramidal neurons. (B) Representation of the charge calculation used for excitation and inhibition. Charge was calculated as the summed current (over 60 s) of either excitatory or inhibitory recording. (C–E) The mean inhibitory and excitatory synaptic charge is shown for Wt (n = 14) versus Mecp2 null mice (n = 12) excitatory layer 5 pyramidal neurons of the mPFC. There is a significant (P = 0.012; Mann–Whitney U-test) decrease in the net charge of excitatory synaptic currents in Mecp2 null excitatory pyramidal layer 5 neurons compared with Wt. The net inhibitory synaptic charge was not significantly different between Wt and null. The E/I ratio for Mecp2 null excitatory pyramidal layer 5 neurons shows a significant decrease compared with Wt (P = 0.02; Mann–Whitney U-test), reflecting hypofunction in Mecp2 null mPFC excitatory pyramidal layer 5 neurons as a result of reduced excitatory synaptic input.
The synaptic charge or integrated current was calculated across all repeats (calculated as the charge over 60 s; Fig. 1B) for the electrically isolated excitatory and inhibitory currents in Wt and Mecp2 null neurons (Fig. 1D–F). On average, there was a reduction in the mean excitatory current charge in Mecp2 null neurons compared with Wt (median = −40 and −81 pC, respectively; n = 12 and 14, P = 0.012, Mann–Whitney U-test; Fig. 1E). Within the same cells, there was no significant difference in the inhibitory current charge between Mecp2 null and Wt neurons (median = 178 and 211, respectively; Fig. 1D). The E/I balance was significantly reduced in the Mecp2 null neurons compared with Wt (median = 0.16 and 0.28, respectively; P = 0.023 Mann–Whitney U-test), but the shift results from a reduction in excitation rather than an increase in inhibition (Fig. 1F).
Although there was a shift in the E/I balance of synaptic activity toward reduced excitatory drive during high levels of synaptic activity, there was no significant difference (Mann Whitney U-test) in the amplitude of miniature excitatory or inhibitory postsynaptic currents as measured through pharmacologically isolated synaptic recordings (Figs 2,3). Pharmacologically isolated mEPSCs were recorded in the presence of TTX and picrotoxin and were blocked completely with CNQX (Fig. 2A). Across the population of recorded cells (n = 7 Wt and 10 null), all individually isolated events (n = 895 Wt and 1282 null) were analyzed for event frequency and amplitude after baseline subtraction (Fig. 2B–E). The median amplitude of mEPSCs between Wt and Mecp2 null neurons was virtually identical (median = −12.5 and −12.4 pA, respectively; Mann Whitney-U test). The median instantaneous frequency of excitatory events was significantly reduced by only 10 Hz (median = 75 Hz and 64 Hz Wt and null respectively; P = 9.7 × 10−4, Mann Whitney-U test). Similarly, mIPSCs displayed no significant difference in amplitude for Wt and Mecp2 null neurons (median = −23.4 and −23.0 pA, respectively; Fig. 3A–C). However, the mIPSC frequency was statistically lower (median = 50.9 and 42.2 Hz; Fig. 3B–E; P = 3.9 × 10−5, Mann Whitney U-test). The cumulative distributions for both amplitude and frequency were statistically different for mEPSCs because of differences in distribution shape and large sample size (P = 0.002 and 0.0049 amplitude and frequency respectively; Kolmogorov–Smirnov test). However, only the mIPSC event frequency cumulative distributions were statistically different (P = 5.7 × 10−5; Kolmogorov–Smirnov test).
Figure 2.

Excitatory miniature postsynaptic currents (mEPSC) amplitude showed no difference between Wt and Mecp2 null neurons. (A) Representative current recordings of mEPSCs recorded from excitatory pyramidal neurons in layer 5 of mPFC in the presence on picrotoxin and tetrodotoxin (TTX) for Wt and Mecp2 null brain slices. (B,C) Across the population (n = 7 Wt neurons and 10 Mecp2 null neurons) of mEPSC events (n = 895 Wt and 1281 null events), there was no significant difference between the median amplitudes (median = −12.5 and −12.4 pA, respectively), of Wt and Mecp2 null events (Mann Whitney U-test). However, there were subtle differences in the cumulative distributions of grouped events (P = 0.002, Kolmogorov–Smirnov test). (D,E) There was a small but significant reduction in median mEPSC frequency between Wt and Mecp2 null neurons (75 and 64 Hz, respectively; P = 9.7 × 10–4, Mann Whitney U-test). The cumulative distributions were also significantly different in shape (P = 0.005, Kolmogorov–Smirnov test).
Figure 3.

Inhibitory miniature postsynaptic current (mIPSC) amplitudes showed no significant difference between Wt and Mecp2 null neurons. (A) Representative current recordings of mIPSCs recorded from excitatory pyramidal neurons in layer 5 of mPFC in the presence of TTX, CNQX, and APV and with the chloride gradient reversed through a KCl-based internal recording solution. (B,C) Across the population (n = 13 Wt neurons and 11 Mecp2 null neurons; 1286 and 899 events, respectively) of mIPSCs there was no significant difference in the median amplitude between Wt and Mecp2 null events (median = −23.4 and −23 pA respectively; Mann Whitney U-test) or cumulative distribution shape (Kolmogorov–Smirnov test). (D,E) The median frequency of mIPSCs for Wt compared with Mecp2 null neurons was reduced by a small but significant amount (50.9 and 42.4 Hz respectively; Mann Whitney U-test), and the cumulative distributions of mIPSC frequency were also significantly different (P = 5.7 × 10−5; Kolmogorov–Smirnov test).
These results indicate that the amplitudes of individual miniature events are preserved across Wt and Mecp2 null neurons. Therefore, postsynaptic function does not appear to be significantly impaired, but the overall number of connections is likely altered with fewer excitatory connections. This reduced excitatory synaptic connectivity of mPFC excitatory pyramidal layer 5 neurons is not accompanied by increases in inhibitory connectivity. In fact, mIPSC frequency is reduced in Mecp2 null mPFC layer 5 excitatory pyramidal neurons (Figs 2,3) which may be consistent with reduced output, or reduced numbers of synapses from inhibitory neurons.
Compared with Wt, excitatory pyramidal layer 5 neurons in Mecp2 null animals did not exhibit alterations in intrinsic membrane properties, including input resistance (null <Rin> = 105 ± 10 MΩ and Wt <Rin> = 98 ± 7.8 MΩ), membrane capacitance (null <Cm> = 127 ± 4.2 pF and Wt <Cm> = 142 ± 6.5 pF) or resting membrane potential (null <Vm> = −63 ± 1.2 mV and Wt <Vm> = −65 ± 0.8 mV). In addition, there was no significant difference in action potential half-height duration between Wt and Mecp2 null neurons (Fig. 4A,B). There was a significant, but small increase in the mean action potential amplitude of Wt versus Mecp2 null neurons (Fig. 4C).
Figure 4.
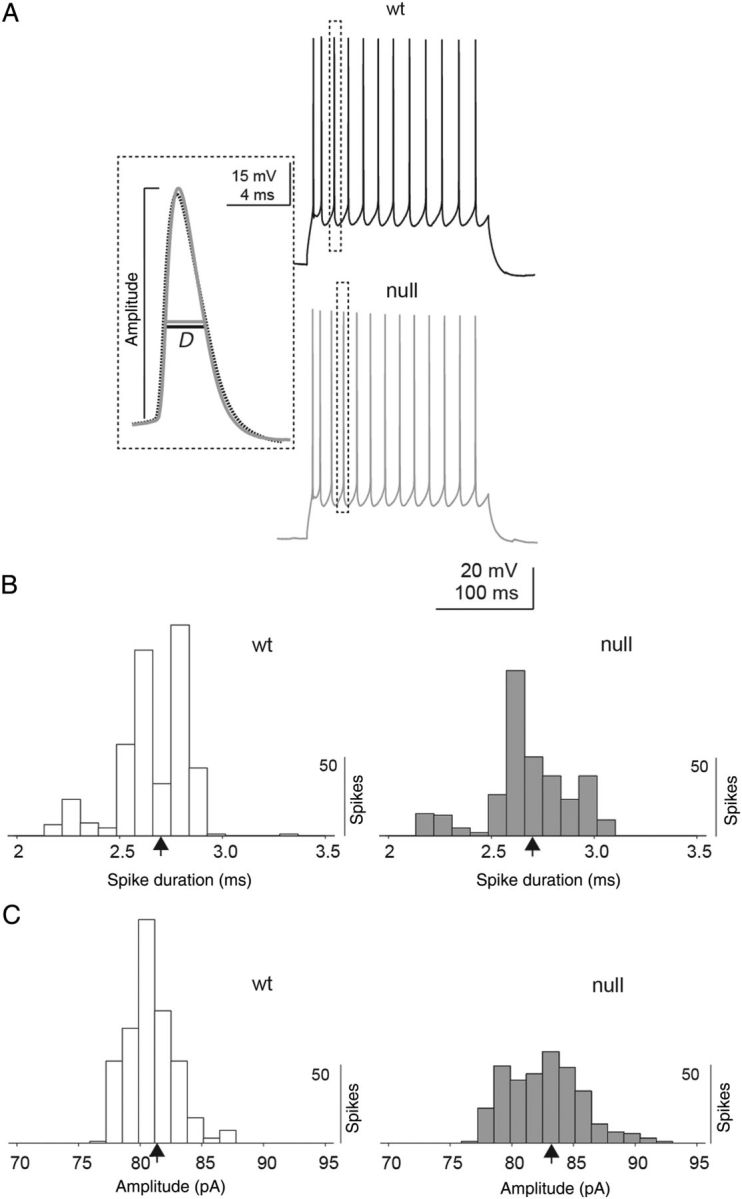
Intrinsic action potential properties between Wt and Mecp2 null neurons. (A) Representative voltage traces from Wt and Mecp2 null excitatory pyramidal layer 5 mPFC neurons in response to current injections. Individual action potential properties were analyzed (see dashed box and inset) to determine the amplitude and half-height spike duration (D) between Wt and Mecp2 null neurons. (B) Across the population of neurons examined (n = 5 neurons with 7 repeats per neuron for both Wt and Mecp2 null animals), spike duration was not significantly different between Wt and Mecp2 null neurons (mean duration = 2.70 and 2.73 ms, respectively). (C) There was a small but significant increase in the mean spike amplitude between Wt and Mecp2 null neurons (81.5 and 83.3 mV, respectively; P = 3e−17, Mann Whitney U-test).
Mecp2 Null Neurons Exhibit a Shift in the Balance of NMDA-to-AMPA Currents
Excitatory synaptic responses in the mPFC are composed of AMPA and NMDA components (Hirsch and Crepel 1990). Although a shift in the NMDA-to-AMPA ratio has been reported in Mecp2 null subcortical circuits (Gambino et al. 2010) and whole-brain lysates (Maliszewska-Cyna et al. 2010), the NMDA/AMPA ratio has not previously been examined in prefrontal cortical circuits.
The NMDA and AMPA current components evoked by electrical stimulation were recorded within the same layer 5 mPFC neurons in order to determine the NMDA-to-AMPA ratio, using pharmacological manipulation of the bath perfusion to isolate the components (Fig 5; see Ye et al. 2005). AMPA currents were recorded in the presence of 100 μm picrotoxin with the holding potential hyperpolarized (Vhold = −70 mV). NMDA currents were recorded in the same cells in the presence of picrotoxin and CNQX with the membrane potential depolarized (Vhold = + 45 mV). The NR2B components were estimated within these same cells by recording the electrically evoked synaptic current (using the identical stimulation pulse amplitude to estimate the AMPA and NMDA components) in the presence of ifenprodil (3 μm) and subtracting this current from the NMDA current. Therefore, we were able to estimate the ratio of NMDA/AMPA as well as the fraction of (NR2B)/(presumptive NR2A) within the same cells. Neocortical neurons contain NMDA currents that are predominantly NR2A and NR2B, but with a small fraction of NR2C or NR2D (Sanz-Clemente et al. 2013). Therefore the NR2A current contains a small fraction of other currents. However, there are no selective blockers that allow one to isolate NR2A for mouse neocortical neurons.
Figure 5.
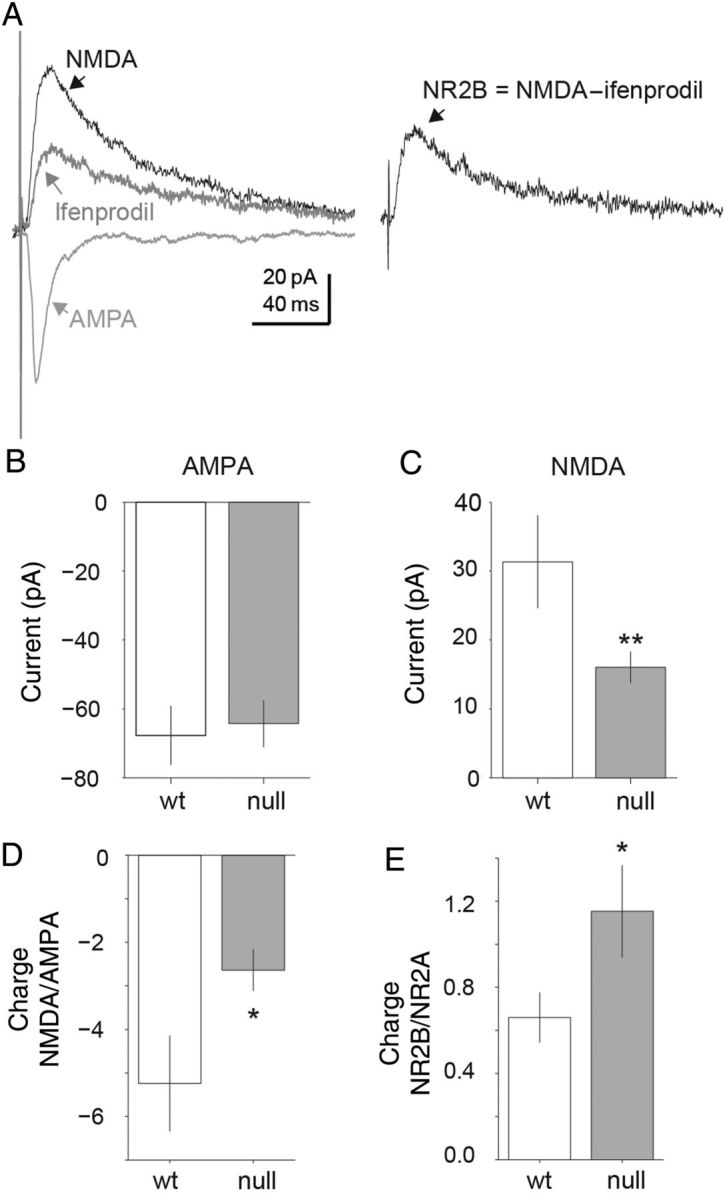
Representative traces of electrically evoked AMPA and NMDA currents measured within the same cell. (A) Evoked AMPA currents measured in the presence of picrotoxin (Vhold = −70 mV). Evoked NMDA current measured in the presence of picrotoxin and CNQX (Vhold = +40 mV). Evoked NMDA NR2B component isolated by comparing the evoked currents in the absence and presence of ifenprodil. NR2B component calculated as the difference between the NMDA current and the ifenprodil insensitive component (NR2B = NMDA −;ifenprodil). (B–E) Mecp2 null excitatory pyramidal layer 5 mPFC neurons displayed a reduced NMDA-to-AMPA ratio. (B,C) For evoked AMPA currents matched in amplitude (Wt n = 7; Mecp2 null n = 12) there was a significantly lower NMDA component for evoked currents in Mecp2 null neurons compared with Wt (mean = 16 and 31 pA, respectively; P = 0.03; two-tailed Student's t-test). (D) Population averages of Mecp2 null compared with Wt excitatory pyramidal neurons displayed significant decreases in the NMDA/AMPA ratio based on integrated current charge (mean = −2.6 and −5.2, respectively; P = 0.026; two-tailed Student's t-test). (E) There is also a significant increase in the NR2B/NR2A current ratio in Mecp2 null (n = 10) neurons compared with Wt (n = 5) neurons (mean = 0.66 and 1.2, respectively; P = 0.02, two-tailed Student's t-test). Therefore Mecp2 null excitatory pyramidal layer 5 neurons displayed increased NR2B components of the total NMDA current compared with Wt neurons.
For electrical stimulation pulses that elicited AMPA responses that were statistically identical in amplitude in Mecp2 null (n = 7) and Wt (n = 10) neurons (mean = −64 and −67 pA, respectively; two-tailed Student's t-test; Fig. 5B), the NMDA current responses were significantly lower for the Mecp2 neurons compared with Wt (median = 16 and 32 pA, respectively; P = 0.033, two-tailed Student's t-test; Fig. 5C). Across the population of recorded cells, there was a significant shift toward reduced NMDA to AMPA ratio in Mecp2 null neurons compared with Wt (median ratio = −2.6 and −5.2 respectively; P = 0.026, two-tailed Student's t-test; Fig. 5D). In addition, the fraction of NR2B current within the NMDA responses was significantly higher for the Mecp2 null neurons compared with Wt (median fraction = 1.15 and 0.66, respectively; P = 0.018, two-tailed Student's t-test; Fig. 5E).
To determine whether or not the abnormal ratio of NMDA:AMPA currents in Mecp2 mutants was caused by genotype effects on expression of glutamate receptor subunits, we compared expression of GluN1 and GluA1 proteins in mPFC homogenates from Wt and null animals by western blot (Fig. 6; null (n = 7); Wt (n = 8)). The level of GluN1 expression normalized to Wt levels was significantly reduced in Mecp2 null samples compared with Wt (mean % Wt = 79.88 ± 4.32%; P = 0.017, two-tailed Student's t-test; Fig. 6A,B). There was also a significant reduction in the GluN1/GluA1 ratio in null compared with Wt mPFC brain homogenates (mean % Wt = 83.58 ± 3.09% normalized to Wt; P = 0.007, two-tailed Student's t-test; Fig. 6D). However, the expression level of GluA1 normalized to Wt was not significantly different in Mecp2 null samples compared with Wt (mean = 95.81 ± 2.75%; Fig. 6C). To determine if the increase in NR2B current component in Mecp2 mutants was caused by a genotype effect on the expression of NMDA receptor subunits, we compared the expression of NR2A and NR2B in mPFC homogenates from Wt and null animals by western blot (Supplementary Fig. 1; null (n = 8); Wt (n = 8)). In contrast to our electrophysiological finding of increased NR2B current in mutant pyramidal neurons, these experiments revealed no difference in the expression of NR2A (mean = 95.75 ± 9.45% of Wt) and NR2B (mean = 96.46 ± 8.24% of Wt) between mPFC lysates from Wt and null animals. This apparent discrepancy may indicate that the effects of MeCP2 loss on NR2B current expression are restricted to a subset of mPFC neurons, for example, layer 5 pyramidal cells and therefore not detectable in whole mPFC lysates. Alternatively, loss of MeCP2 may affect functional properties of NR2B receptors rather than levels of protein expression.
Figure 6.

GluN1 expression levels are significantly reduced in Mecp2 null mPFC compared with Wt. (A) Isolated mPFC was biochemically analyzed for NMDA and AMPA protein composition with GluN1 and GluA1 antibodies using western blots. (B) The GluN1 levels were significantly reduced (normalized to Wt levels) across the samples tested (n = 8; mean = 79.88 ± 4.32%; P = 0.017, two-tailed Student's t-test). GluA1 levels were not significantly different (mean = 95.81 ± 2.75% of Wt level). The GluN1/GluA1 ratio was also significantly reduced (mean = 83.58 ± 3.09% of Wt level; P = 0.007, two-tailed Student's t-test).
Mecp2 Null Neurons Display a Shift in the Dynamics of E/I UP-state Activity
UP-state activity results from a synchronized barrage of synaptic input and has been reported both in vitro and in vivo (Haider et al. 2006). The properties of the synaptic barrages are a reflection of the network connectivity when measured in acute brain slices (Destexhe et al. 2001; Fellous et al. 2003; Rudolph et al. 2004; Sceniak and Sabo 2010). We used measures of excitatory and inhibitory UP-state activity in acute brain slices in order to evaluate synaptic drive and connectivity in Mecp2 null versus Wt mPFC (Gibson et al. 2008).
In the presence of high-conductance ACSF (see Materials and Methods), electrical stimulation elicited brief strong UP-state responses that were reliably evoked in excitatory layer 5 pyramidal neurons in the mPFC. Similar to the spontaneous currents described above, the membrane holding potential was adjusted on a cell-by-cell basis in order to electrically isolate either excitatory glutamatergic or inhibitory GABAergic currents within the same cell. The excitatory and inhibitory components of the UP-states were quantitatively analyzed for event amplitude, duration, and integrated charge (Fig. 7A–H). As has been previously reported, the inhibitory conductance is roughly 2–4 times that of excitation (Destexhe et al. 2001; Fellous et al. 2003; Rudolph et al. 2004; Sceniak and Sabo 2010). Across the population of responses measured (n = 12), there was a significant decrease in the duration of excitatory UP-state components in Mecp2 null neurons compared with Wt (mean = 329 and 527 ms, respectively; P = 0.035, two-tailed Student's t-test; Fig. 7H). However, the inhibitory duration was not significantly different (mean = 360 and 515 ms). In addition, the excitatory and inhibitory amplitude and charge were not significantly different between Wt and Mecp2 null neurons (Fig. 7F,G). These results point to a synaptic barrage within the Mecp2 null mPFC that is more synchronized or transient in temporal signature.
Figure 7.

Excitatory UP-state activity was altered in Mecp2 null excitatory pyramidal layer 5 mPFC neurons. (A–D) Representative excitatory (A,B) and inhibitory (C,D) currents during electrically evoked UP-states measured within excitatory pyramidal layer 5 mPFC neurons of both Wt and Mecp2 null brain slices. (E) As in Figure 1, inhibitory and excitatory currents were electrically isolated by holding the membrane potential at either the reversal potential for excitatory (Vhold ≈ +10 mV) or inhibitory (Vhold ≈ −50 mV) synaptic currents in voltage-clamp mode. Electrical pulses delivered to the white matter elicited UP-states and either the excitatory or inhibitory components were recorded depending on the holding potential of the cell. UP-states were isolated from background based on 2-SD from baseline activity. (F–H) Summary of excitatory and inhibitory current high-conductance UP-state components of Mecp2 null (n = 14) versus Wt (n = 12) excitatory pyramidal layer 5 mPFC neurons. Across the population, the temporal dynamics of the excitatory synaptic charge was distinct in Mecp2 null neurons. (H) Overall there was a significant (P = 0.035, two-tailed Student's t-test) decrease in the mean duration of the excitatory component of the evoked UP-states in Mecp2 null neurons (mean Enull = 329 ms) compared with Wt (mean EWt = 527 ms). On average, UP-states in Mecp2 null neurons displayed more transient excitatory response dynamics compared with Wt neurons.
Calcium Imaging Reveals Reduced Signal Propagation Through the Mutant mPFC
In addition to UP-state activity, we examined the degree of population activity within the mPFC circuits using calcium imaging (Fig. 8). Horizontally sectioned brain slices (Fig. 8A) were incubated in Fluo-4 AM calcium indicator dye to visually quantify the population activity across cortical layers of mPFC in response to white matter electrical current pulse stimulation. Calcium responses were estimated as the normalized change in fluorescent intensity (ΔF0/F0). Time-lapse images (100 frames, 0.2 s exposure per frame) of the calcium dynamics were collected and processed to determine the ΔF0/F0 calculated for each frame on a pixel-by-pixel basis. The ΔF0/F0 displayed a stereotypical peak after electrical stimulation that lasted 1.4–6 s (Fig. 8A). Increases in calcium signals above background revealed spatial structure as well (Fig. 8B). The pattern of activity typically emanated from the stimulating electrode and spread toward the pia mater. Across all brain slices recorded (n = 5 each, Wt and null), the pattern of activity was greatest in deeper layers and gradually decreased in upper layers with a spatially restricted pattern in the direction normal to the surface.
To compare responsiveness to electrical stimulation in Wt versus Mecp2 null mPFC, the ΔF0/F0 was calculated as the average (n = 3 repeats) peak response (over 100 frames or 20 s) to electrical stimuli from 0.25 to 3 mA (Fig. 9). The ΔF0/F0 measure was estimated as the average peak to a given electrical stimulus measured within a defined region of interest in layer 2/3 and layer 5 within the cloud of increased activity (Fig. 8B). Mecp2 null mPFC brain slices displayed reduced responsiveness compared with Wt for the identical stimulus intensity especially at near threshold stimuli (see vertical arrows; Fig. 9A–D). Across the population (n = 5), there was a significant decrease in the ΔF0/F0 for Mecp2 null slices compared with Wt for layer 2/3 (mean = 0.17% and 0.48%, respectively; P = 0.02, two-tailed Student's t-test) and layer 5 (mean = 0.80% and 0.29%, respectively; P = 0.041, two-tailed Student's t-test). Therefore, the Mecp2 null brain slices show a significantly weaker responsiveness to electrical stimulation than Wt, consistent with the decrease in excitatory synaptic connectivity (Fig. 1).
Figure 9.

Mecp2 null mPFC brain slices display reduced calcium responses to evoked stimulation compared with Wt. (A–D) Calcium responses are shown as a function of stimulus current pulse intensity (mA) for the population of brain slices examined for Mecp2 null (black lines, n = 5) and Wt (gray lines, n = 5) animals. Vertical arrows indicate the threshold level stimulus for a significant change in response level. (E) Summary of responses (ΔF/F0) measured in layer 2/3 using Fluo-4AM calcium imaging of mPFC in Mecp2 null versus Wt mice. There is a significant reduction in the threshold level responses in Mecp2 null versus Wt. brain slices (mean, null = 0.18% and Wt = 0.48%; P = 0.02, two-tailed Student's t-test). (F) Layer 5 estimates of calcium response to electrical stimulation are also reduced in Mecp2 null slices compared with Wt (mean null = 0.29% and Wt = 0.80%; P = 0.04, two-tailed Student's t-test).
Mecp2 Genotype Affects the Balance of Excitatory and Inhibitory Synaptic Protein Expression in mPFC
Our electrophysiological data suggest a shift towards reduced excitatory connectivity in the Mecp2 null mPFC compared with Wt. To define potential underlying molecular mechanisms we used quantitative immunohistochemical analysis (see Materials and Methods) to compare expression of the vesicular glutamate transporter 1 (VGLUT1), a presynaptic marker for glutamatergic function, the vesicular GABA transporter (VGAT), a marker of GABAergic function, as well as expression of parvalbumin (PV), a marker of inhibitory interneurons that has previously been shown to be increased in visual cortex of Mecp2 mutants (Durand et al. 2012), in Wt and Mecp2 null mPFC (Fig. 10). Confocal images were used to quantify the density of fluorescent signals for VGLUT1, VGAT, or PV for regions of interest positioned within layers 2/3 or 5 (Fig. 10A). Low magnification (20×) confocal images were collected with microscope settings and gain fixed across each color channel for all images collected (set independently for the red and green channels) for Mecp2 null and Wt brain slices, in order to make quantitative comparisons.
Figure 10.
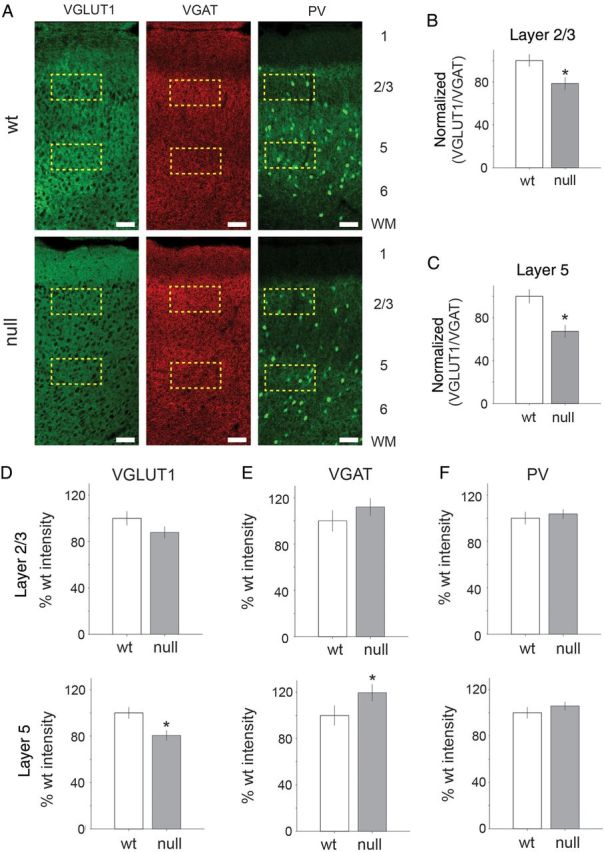
Excitatory and inhibitory presynaptic protein distributions are significantly altered in Mecp2 null brain slices. (A) Representative confocal images of signals from VGLUT1, VGAT, and PV antibody labeling in Wt and Mecp2 null mPFC brain slices. Scale bar indicates 100 µm. Dashed yellow rectangles represent regions of interest defined for sampled layers 2/3 and 5 of mPFC. Mean intensity was estimated in the region of interest across all brain sections (n = 3) for Mecp2 null (n = 5) and Wt (n = 5) animals. (B,C) Across the population of sampled brain slices, there was a significant reduction in the normalized (with respect to the Wt level) VGLUT1 to VGAT ratio in layer 2/3 (mean = 78%; P = 6.5e−5, two-tailed Student's t-test) and layer 5 (mean = 67%; P = 1.0e−7, two-tailed Student's t-test). Across the brain slice sections sampled (Mecp2 null n = 15, Wt = 15), the laminar distribution of presynaptic GABA and glutamate markers is shown for VGLUT1, VGAT, and PV. (D) There was a significant decrease in VGLUT1 expression level in layer 5 for Mecp2 null compared with Wt (mean = 80%; P = 0.0072, two-tailed Student's t-test). (E) VGAT expression levels were elevated in layer 5 in Mecp2 null tissue (mean 120% of Wt level; P = 0.024, two-tailed Student's t-test). (F) The parvalbumin expression levels were not significantly increased in any layer.
Across the population of sampled brain slices (3 brain section images per animal), we compared the normalized VGLUT1/VGAT ratio for Mecp2 null and Wt animals (5 Wt and 5 Mecp2 null; Fig. 10B,C). The VGLUT1/VGAT ratio was calculated from the mean intensity for each marker measured within individual brain slices colabeled with VGLUT1 and VGAT antibodies imaged with either red (546 nm) or green (488 nm) secondary antibodies. The VGLUT1/VGAT ratio (normalized to Wt level) was significantly reduced in Mecp2 null tissue for layer 2/3 (mean % Wt = 78.5%; P = 6.5e−5; two-tailed Student's t-test; Fig. 10B) and layer 5 (mean % Wt = 67.3%; P = 1e−7, two-tailed Student's t-test; Fig. 10C).
The decrease in VGLUT1/VGAT ratio appears to have resulted from a combination of a trend toward a decrease in VGLUT1 and an increase in VGAT (Fig. 10D–F). However, only layer 5 showed significant individual changes (normalized to Wt level) in both VGLUT1 (mean = 81%; P = 0.007, two-tailed Student's t-test) and VGAT (mean = 120%; P = 0.02, two-tailed Student's t-test; Fig. 10D–E). Changes in average VGAT or VGLUT1 levels were not significantly different in layer 2/3. In addition, parvalbumin levels were not significantly different between Mecp2 null and Wt tissue in layers 2/3 or 5 (Fig. 10F). Therefore, the balance of glutamate and GABA appears to be shifted toward a lower VGLUT1/VGAT ratio with the largest change occurring within layer 5. The increase in VGAT expression was not associated with a change in parvalbumin expression in any cortical layer.
VGLUT1 and VGAT are both synaptic proteins that are found predominantly at synapses after early development; however, parvalbumin is expressed throughout the soma and processes of FS (fast spiking) inhibitory interneurons and is not specifically localized at the synapse. Therefore, we quantified the distribution of parvalbumin-positive cell bodies and synaptic puncta in Wt and Mecp2 null tissue to identify potential effects of Mecp2 loss on the parvalbumin-positive population in more detail (Fig. 11). We found no significant difference in the number of parvalbumin-positive somata between Wt and Mecp2 null mPFC (Fig. 11A–D). Using high magnification (63×) confocal imaging, we analyzed the density of PV positive synaptic puncta (Fig. 11E) (shown as white circular outlines in the inset image in Fig. 11E). Across the population of sampled brain slices (5 Wt and 5 Mecp2 null neurons, 3 brain slices per animal), there was no significant difference in the density of parvalbumin-positive puncta between Wt and Mecp2 null samples within either layers 2/3 or 5 of mPFC (Fig. 11F). In light of a previous report demonstrating that loss of MeCP2 results in increased expression of PV in visual cortex (Durand et al. 2012), we also examined PV expression in the visual cortex in the present study. As in the mPFC, we found no significant differences in PV expression (measured as mean intensity) in any layer of visual cortex. However, we did observe a small, but significant increase in the number of PV-positive somata within layer 2/3 of visual cortex in Mecp2 null animals compared with Wt (see Supplementary Fig. 2).
Figure 11.

Parvalbumin expression levels were similar in Wt and Mecp2 null mPFC. (A) Representative confocal images (20× magnification) of Wt and Mecp2 null mPFC brain slices stained with parvalbumin antibodies. Scale bar indicates 100 µm. Dashed yellow rectangles represent regions of interest defined for sampled layers 2/3 and 5 of mPFC. (B) Inset images (indicated by *) below each image in (A) represent digitally magnified (scale bar is 100 µm) regions of layer 2/3 defined by the dashed yellow boxes above. White arrows indicate identified cell bodies. (C) Mean fluorescence intensity was not different for Wt versus Mecp2 null mPFC layers 2/3 or 5. (D) The mean cell body count was also not significantly different between Wt and Mecp2 null mPFC layers 2/3 or 5. (E) Representative confocal image of mPFC layer 2/3 under high magnification (63×). The dashed yellow rectangle represents the region of interest that is digitally zoomed in the overlapping inset (indicated by *). White dashed circular outlines in the inset indicate identified paravalbumin positive synaptic puncta. (F) Across the population of brain slices analyzed (5 Wt and 5 Mecp2 null animals with 3 brain slices each) there was no significant difference in the puncta density for Wt versus Mecp2 null mPFC layers 2/3 or 5.
Reduced Dendritic Spine Density in the mPFC of Mecp2 Null Mice
We next sought to determine whether or not changes in E/I function are associated with structural synaptic changes that might also alter excitatory connectivity. Cortical pyramidal neurons in Mecp2 null mice have previously been shown to exhibit reduced dendritic length and/or branching as well as reduced dendritic spine density (Jugloff et al. 2005; Belichenko et al. 2008; Belichenko, Belichenko et al. 2009; Stuss et al. 2012). However, the effects of Mecp2 loss on dendritic morphology in mPFC have not previously been defined. Therefore, to determine whether decreased excitatory functional connectivity is associated with dendritic structural deficits we used DiOlistic neuronal labeling and confocal microscopy to compare the morphology of the apical dendrites of layer 5 pyramidal neurons in Wt and Mecp2 null mice (Fig. 12), including the length of individual dendritic spines and spine density per micrometer. All analyses of spine morphology were conducted blind to genotype (Wt or Mecp2 null). Across the population of sampled neurons (n = 5 Mecp2 null and 9 Wt cells) spine density was significantly lower in Mecp2 null (mean = 0.84 ± 0.12 spines/µm) compared with Wt (mean = 1.02 ± 0.10 spines/µm) neurons (P = 0.01; nested ANOVA; Fig. 12B). In contrast, there was no significant difference in the length of dendritic spines between Mecp2 null (mean = 1.8 ± 0.07 µm) and Wt (mean = 1.67 ± 0.02 µm) neurons (Fig. 12B).
Figure 12.

Reduced dendritic spine density in Mecp2 null excitatory pyramidal layer 5 neurons compared with Wt. (A) Representative confocal images of dendrite samples used to quantify spine density and length estimates in Mecp2 null and Wt neurons. (B) Across the population of cells studied, there was a significantly lower estimate of spine density (spines/µm) in dendritic branches from Mecp2 null neurons compared with Wt (mean = 0.84 and 1.02 spines/µm, respectively; P = 0.01, nested ANOVA). Dendritic spine length was not significantly different in Mecp2 null versus Wt neurons (1.66 and 1.76 µm, respectively).
Mecp2 Null Mice Exhibit Behavioral Disruption of Breathing Consistent with mPFC Hypofunction
The mPFC has previously been shown to play a critical role in modulating the influence of behavioral state on homeostatic functions, including respiration and autonomic control. For example, lesions of the mPFC result in exaggerated and prolonged increases in breathing frequency in response to auditory fear conditioning (Frysztak and Neafsey 1991). Given that RTT patients exhibit behavioral disruption of breathing (Ramirez et al. 2013), and our findings of hypofunction in the mPFC of Mecp2 null mice, we hypothesized that these animals would exhibit abnormalities in respiratory control similar to those seen after mPFC lesions. To approach this issue we compared the respiratory component of the orienting reflex (OR) in 6–8-week-old Wt and Mecp2 null mice. The OR is evoked by stimuli below the intensity threshold for acoustic startle and is normally accompanied by a transient increase in respiratory frequency that returns to baseline within a few seconds (Nalivaiko 2011). Acoustic startle testing revealed that the threshold for startle is between 90 and 100 dB in Wt mice, and 100 and 110 dB in nulls (Fig. 13A) and, on that basis, we chose 80 dB for OR testing. Animals were exposed to a 50-msec pulse of white noise at 80 dB during plethysmographic recording of respiration. Video monitoring revealed that, in animals of both genotypes, this stimulus was sufficient to elicit an orienting response (ears pricked up) without evoking a startle response (Supplementary Videos 1,2). Both Wt and Mecp2 null mice responded to the auditory pulse with a transient increase in respiratory frequency that returned to baseline within a few seconds after the stimulus (Fig. 13B). However, the magnitude of the frequency increase was significantly larger in mutants compared with Wt (Fig. 13D). In addition, following this transient response, respiratory frequency increased further in mutants, but not in Wt, and remained elevated for at least 1 min (Fig. 13C,E). These data demonstrate that the amplitude and duration of the breathing component of the OR is exaggerated in Mecp2 null animals compared with Wt, consistent with hypofunction of the mPFC.
Figure 13.

Mecp2 null mice exhibit abnormal respiratory responses to an auditory arousal stimulus. (A) Acoustic startle testing revealed that the startle threshold in 6–8 week-old null and Wt mice is 110 dB (#) and 100 dB *, respectively (repeated-measures ANOVA). Mecp2 nulls also exhibit reduced startle responses compared with Wt at 110 and 120 dB (*P < 0.05, ANOVA). (B) Representative pairs of plethysmographic traces and instantaneous frequency plots recorded from Wt (top) and Mecp2 null (bottom) mice during the 10 s before and the 10 s after presentation of an auditory arousal stimulus that was below the threshold for evoking a startle response (50msec, 80 dB; stim). (C) Summary data showing mean respiratory frequency recorded from 6–8 week-old Wt and Mecp2 null mice before and after presentation of the auditory arousal stimulus (stim); time is represented on the x-axis, divided into 10 s bins. (D,E) Summary data showing (D) the maximum change in respiratory frequency recorded within the first 10 s following the auditory stimulus and (E) the average change in respiratory frequency recorded during the first minute following the auditory stimulus. The acute response to auditory stimulation (D) was significantly higher in Mecp2 nulls than in Wt littermates (P < 0.05, t-test) and null animals exhibited a sustained increase in respiratory frequency following the auditory stimulus (E) that was not seen in Wt animals (P < 0.05, t-test).
Discussion
The mPFC is important for high-level decision making and contextual or adaptive learning, particularly for emotional responses (Euston et al. 2012), and prefrontal cortical dysfunction is thought to contribute to the pathophysiology of diverse neurodevelopmental disorders, including ASD, RTT, Angelman syndrome, Prader–Willi syndrome, Down syndrome, and ADHD (Akbarian et al. 2001; Nagarajan et al. 2006; Emond et al. 2009; Yizhar et al. 2011; Katz et al. 2012; Zikopoulos and Barbas 2013; Li et al. 2014). However, relatively little is known about how genetic mutations associated with neurodevelopmental disorders alter mPFC function. Therefore, the present study was designed to provide a detailed analysis of synaptic and circuit function associated with loss of Mecp2, the gene mutated in RTT and an apparent target of misregulation in other neurodevelopmental disorders as well (Samaco et al. 2004; Nagarajan et al. 2006).
Our results indicate that genetic loss of MeCP2 results in hypofunction of the mPFC as evidenced by reduced excitatory postsynaptic currents in layer 5 pyramidal neurons and reduced population activity in response to afferent electrical stimulation. This overall reduction in excitatory synaptic activity was accompanied by decreases in the density of postsynaptic dendritic spines and in the ratio of excitatory to inhibitory presynaptic markers. Excitatory synapses exhibited a reduced ratio of NMDA/AMPA currents and reduced NMDA receptor expression levels. These synaptic deficits were associated with altered network activity including reductions in the duration of excitatory UP-states and evoked population activity in layers 2/3 and 5. Together, these results support the view that reduced excitatory synaptic drive and network activity underlie hypoactivity in the mPFC in Mecp2 mutants (Katz et al. 2012).
Previous studies indicated that synaptic hypofunction in primary sensory neocortex of Mecp2 mutants is due to a shift in excitatory/inhibitory (E/I) synaptic balance resulting from decreased glutamatergic excitation and increased GABAergic inhibition (Dani et al. 2005; Moretti et al. 2006; Dani and Nelson 2009; Gogolla et al. 2009; Wood et al. 2009; Blackman et al. 2012; Kron et al. 2012; Na et al. 2013). However, others have shown that, in frontal-motor cortex of Mecp2 null mice, the spatial spread of excitatory synaptic inputs is reduced without a change in the spatial profile of inhibition (Wood et al. 2009; Wood and Shepherd 2010). Similarly, our results indicate that in the mPFC, excitatory glutamatergic synaptic drive onto mPFC layer 5 pyramidal neurons is reduced in nulls without a change in inhibitory synaptic activity. Together, these data support the view that the effects of Mecp2 loss of function on E/I balance vary among brain regions (Shepherd and Katz 2011) and demonstrate that glutamatergic hypofunction is a dominant feature of synaptic pathophysiology in the Mecp2 null mPFC.
In addition to E/I imbalance, we also observed a reduction in the ratio of NMDA to AMPA currents within excitatory synaptic inputs onto layer 5 pyramidal cells in the mutant mPFC. This shift can likely be explained by our observation that GluN1 protein expression and the ratio of GluN1/GluA1 expression are also reduced in nulls compared with Wt. Reduced GluN1 expression has previously been reported in other brain regions in Mecp2 mice at ages similar to those used in the present study (Metcalf et al. 2006; Maliszewska-Cyna et al. 2010; Blue et al. 2011). However, an increase in NMDA/AMPA currents at cortico-amygdala synapses has also been reported (Gambino et al. 2010), further highlighting regional differences in the effects of Mecp2 loss of function on glutamatergic signaling.
NMDA synaptic currents contribute to overall excitatory synaptic drive and play a crucial role in establishing cortical oscillations (Flint and Connors 1996; Anver et al. 2011). A reduction in frontal cortical NMDA balance has been linked to disease states associated with disruption of action potential timing, synchrony, and potentiation (Jackson et al. 2004; Molina et al. 2014). Therefore, in addition to hypofunction per se, reduced NMDA currents in the mPFC may also disrupt information processing through changes in spike timing or cortical oscillations (Jackson et al. 2004; Anver et al. 2011; Molina et al. 2014).
Although layer 5 cells in the mutant mPFC exhibit a reduction in NMDA current compared with Wt, the NR2B (ifenprodil-sensitive) component is significantly increased. NR2B subunits confer slower NMDA receptor kinetics than NR2A and are typically extrasynaptic in the mature visual cortex (Williams et al. 1993; Philpot et al. 2001). However, our results indicate that a significant NR2B component remains in mature mPFC synaptic responses, consistent with the findings of Wang et al. (2008), and that this component is affected by loss of MeCP2. Additional experiments would be necessary to determine whether extrasynaptic responses are also affected. In addition, during development, there is a switch between the NR2B and NR2A subunit composition of synaptic NMDA receptors, with NR2A becoming dominant after the critical period for synaptic plasticity (Williams et al. 1993; Monyer et al. 1994; Philpot et al. 2001). Thus, our results suggest a relative immaturity in NMDA receptor subunit composition and function in Mecp2 mutants compared with Wt in mPFC (Lee et al. 2008). Our results contrast with the finding of Durand et al. (2012) that loss of MeCP2 in visual cortex results in a relative decrease in NR2B subunit expression compared with NR2A. This difference suggests that the regulation of NMDA receptor subunit expression by MeCP2 is different in the mPFC and visual cortex. Understanding these regional differences is critical, as potential therapeutic strategies aimed at redressing NR2A/NR2B imbalance will need to take such heterogeneity into account. In addition, we did not observe significant differences in NR2A and NR2B expression levels in whole mPFC homogenates by western blot. This may indicate that loss of MeCP2 affects NR2B expression and function within the mPFC in a cell type specific manner that cannot be detected in whole tissue lysates.
Consistent with our analysis of synaptic currents, the E/I balance of presynaptic protein expression was also reduced in the mPFC of Mecp2 null mice. Specifically we found a decrease in VGLUT1 and an increase in VGAT, especially in layer 5. Since synaptic current measures did not indicate an increase in inhibitory input to layer 5 excitatory pyramidal neurons, increased VGAT might reflect changes in inhibitory synapses that either target neurons other than layer 5 pyramidal cells or that have relatively weak effects, for example, due to their spatial distribution within layer 5. In addition, we saw no change in PV expression, which marks the somata and presynaptic terminals of the majority of GABAergic interneurons that target layer 5 pyramidal neurons (Ekstrand et al. 2001). The observation that PV expression is unchanged in the mPFC of Mecp2 null mice contrasts with the finding of Durand and colleagues (Durand et al. 2012) that PV expression, and connectivity of PV interneurons is significantly increased in the thalamic input layer (layer 4) of visual cortex of Mecp2 null mice. This difference between mPFC and visual cortex might result from the fact that rodent mPFC does not contain a thalamic input layer that is homologous to layer 4 of visual cortex (van Aerde and Feldmeyer 2015). Therefore increased PV expression may be a unique feature of visual cortex (or primary sensory cortices in general) in Mecp2 mutants.
Broadly speaking, reduced VGLUT1 protein levels in the mPFC, as in other regions of the Mecp2 null brain (Chao et al. 2007; Blackman et al. 2012) could result from decreased synapse density and/or reduced synaptic VGLUT1 expression. Previous reports indicate that spine density on excitatory neurons is reduced in Mecp2 nulls and varies among dendritic subdomains depending on brain region (Jung et al. 2003; Jugloff et al. 2005; Chao et al. 2007; Belichenko et al. 2008; Belichenko, Belichenko et al. 2009; Belichenko, Wright et al. 2009; Kishi and Macklis 2010; Stuss et al. 2012; Jiang et al. 2013). The fact that spine density was reduced in the apical oblique dendrites of layer 5 pyramidal neurons in the mPFC suggests that at least a portion of the decrease in VGLUT1 that we observed was due to a decrease in the number of synaptic contacts. In addition, VGLUT1 levels may be depressed as a consequence of reduced NMDA currents, as NMDA receptor activation has previously been shown to regulate VGLUT1 expression at synapses (Sceniak et al. 2012). However, given recent evidence that loss of MeCP2 is associated with a global decline in mRNA levels (Li et al. 2013), reduced VGLUT1 expression may result from an overall reduction in transcription, mRNA stability, and/or translation. Indeed, Nguyen et al. (2012) demonstrated that VGLUT1 is under strong posttranscriptional regulation by MeCP2. In this respect it is worth noting that Brain Derived Neurotrophic Factor (BDNF), which is down-regulated in Mecp2 mutants (Chang et al. 2006; Katz 2014; Li and Pozzo-Miller 2014), promotes expression of VGLUT1 mRNA and protein (Chang et al. 2006; Li et al. 2013; Melo et al. 2013; Katz 2014; Li and Pozzo-Miller 2014). These data raise the possibility that therapeutic strategies aimed at increasing BDNF levels or activating the BDNF receptor, TrkB, may be useful for restoring E/I synaptic balance in the context of MeCP2 deficiency (Katz 2014).
Our data indicate that synaptic E/I imbalance in the mPFC of Mecp2 null mice contributes to a disruption of normal cortical network dynamics. Excitatory neurons in null mice exhibited shorter duration evoked excitatory UP-state components. UP-state magnitude and duration are influenced by connectivity of the cortical network rather than intrinsic conductances (Wilson and Kawaguchi 1996; Haider et al. 2006; Gibson et al. 2008). Therefore, a reduction in UP-state duration in Mecp2 null mPFC is unlikely to have resulted from changes in intrinsic membrane properties. The reduction in E/I balance of synaptic currents in Mecp2 null neurons could directly influence cortical UP-state dynamics, because UP-state termination is controlled by the strength of inhibition relative to excitation (Fanselow and Connors 2010). Excitatory UP-states could also be influenced directly by changes in NMDA currents (Milojkovic et al. 2005; Favero and Castro-Alamancos 2013) as well as the spatial organization of synapses on cortical dendrites, which can influence the duration of UP-state synaptic activity by controlling the relative timing or synchrony of synaptic inputs (Schierwagen et al. 2007; Sceniak and Sabo 2010; Wang et al. 2010; Plotkin et al. 2011).
Our analysis of the respiratory component of the orienting reflex to auditory stimulation revealed breathing phenotypes not previously described in Mecp2 mutant mice. As previously shown in rats (Nalivaiko et al. 2012), Wt mice exhibit a transient increase in respiratory frequency that returns to baseline within a few seconds after the auditory pulse. This transient increase is a well-known feature of the OR, which elicits behavioral arousal to novel sensory stimuli that are below the intensity threshold for triggering a startle response (Sokolov 1963). In Mecp2 mutants, however, the amplitude of the initial increase is significantly larger than in Wt and, in addition, is followed by a sustained elevation in respiratory frequency lasting at least 1 min beyond the stimulus. Our finding that exposure to a novel auditory stimulus evokes far greater disruption of the resting breathing pattern in Mecp2 mutants than in Wt is reminiscent of behavioral state-dependent changes in breathing dysfunction reported in RTT patients (Ramirez et al. 2013). Moreover, this phenotype, that is, increased magnitude and duration of respiratory activity in response to auditory arousal, is similar to that observed in rats following bilateral lesions of the mPFC (Frysztak and Neafsey 1991). We hypothesize, therefore, that functional hypoconnectivity in the mPFC underlies or contributes to the exaggerated arousal breathing phenotype in Mecp2 nulls, although further experiments are required to definitively establish such a link. This hypothesis is consistent with the fact that mPFC neurons project to brainstem structures critical for behavioral state-dependent regulation of respiratory motor output, including the periaqueductal gray (Brandão et al. 1993; Keay et al. 1988; Nalivaiko 2011). Increased auditory sensitivity per se seems an unlikely explanation for our results, given our finding that mutants exhibit an increased startle threshold and lower startle amplitude compared with wildtypes.
Functional hypoconnectivity in the mPFC may well contribute to other impairments linked to behavioral state regulation in Mecp2 mutants, including exaggerated pre-pulse inhibition of acoustic startle (Kron et al. 2012) and abnormal fear responses (Gemelli et al. 2006; Stearns et al. 2007; Adachi et al. 2009). Disruption of E/I balance in the mPFC has also been linked to impairments in social behavior (Yizhar et al. 2011), similar to those described in some Mecp2 mutants (Gemelli et al. 2006). Although further work is required to definitively link mPFC hypofunction to any specific behavioral deficits in Mecp2 mutants, our data suggest that the mPFC is one locus at which restoration of E/I synaptic balance may improve behavioral dysregulation in RTT.
Supplementary Material
Supplementary material can be found at: http://www.cercor.oxfordjournals.org/.
Notes
We thank Dr Shasta Sabo for input on the manuscript and technical assistance and Ian Adams and Erica Kimmick for providing significant technical assistance related to the experiments and mouse colony genotyping. Conflict of Interest: None declared.
References
- Adachi M, Autry AE, Covington HE, Monteggia LM. 2009. MeCP2-mediated transcription repression in the basolateral amygdala may underlie heightened anxiety in a mouse model of Rett syndrome. J Neurosci. 29:4218–4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akbarian S, Chen RZ, Gribnau J, Rasmussen TP, Fong H, Jaenisch R, Jones EG. 2001. Expression pattern of the Rett syndrome gene MeCP2 in primate prefrontal cortex. Neurobiol Dis. 8:784–791. [DOI] [PubMed] [Google Scholar]
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. 1999. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 23:185–188. [DOI] [PubMed] [Google Scholar]
- Anver H, Ward PD, Magony A, Vreugdenhil M. 2011. NMDA receptor hypofunction phase couples independent γ-oscillations in the rat visual cortex. Neuropsychopharmacology. 36:519–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong DD. 2005. Neuropathology of Rett syndrome. J Child Neurol. 20:747–753. [DOI] [PubMed] [Google Scholar]
- Belichenko NP, Belichenko PV, Li HH, Mobley WC, Francke U. 2008. Comparative study of brain morphology in Mecp2 mutant mouse models of Rett syndrome. J Comp Neurol. 508:184–195. [DOI] [PubMed] [Google Scholar]
- Belichenko NP, Belichenko PV, Mobley WC. 2009. Evidence for both neuronal cell autonomous and nonautonomous effects of methyl-CpG-binding protein 2 in the cerebral cortex of female mice with Mecp2 mutation. Neurobiol Dis. 34:71–77. [DOI] [PubMed] [Google Scholar]
- Belichenko PV, Wright EE, Belichenko NP, Masliah E, Li HH, Mobley WC, Francke U. 2009. Widespread changes in dendritic and axonal morphology in Mecp2-mutant mouse models of Rett syndrome: evidence for disruption of neuronal networks. J Comp Neurol. 514:240–258. [DOI] [PubMed] [Google Scholar]
- Blackman MP, Djukic B, Nelson SB, Turrigiano GG. 2012. A critical and cell-autonomous role for MeCP2 in synaptic scaling up. J Neurosci. 32:13529–13536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blue ME, Kaufmann WE, Bressler J, Eyring C, O'driscoll C, Naidu S, Johnston MV. 2011. Temporal and regional alterations in NMDA receptor expression in Mecp2-null mice. Anat Rec (Hoboken). 294:1624–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandão ML, Melo LL, Cardoso SH. 1993. Mechanisms of defense in the inferior colliculus. Behav Brain Res. 58:49–55. [DOI] [PubMed] [Google Scholar]
- Calfa G, Hablitz JJ, Pozzo-Miller L. 2011. Network hyperexcitability in hippocampal slices from Mecp2 mutant mice revealed by voltage-sensitive dye imaging. J Neurophysiol. 105:1768–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Q, Khare G, Dani V, Nelson S, Jaenisch R. 2006. The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron. 49:341–348. [DOI] [PubMed] [Google Scholar]
- Chao H-T, Zoghbi HY, Rosenmund C. 2007. MeCP2 controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron. 56:58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen RZ, Akbarian S, Tudor M, Jaenisch R. 2001. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. 27:327–331. [DOI] [PubMed] [Google Scholar]
- Cossart R, Ikegaya Y, Yuste R. 2005. Calcium imaging of cortical networks dynamics. Cell Calcium. 37:451–457. [DOI] [PubMed] [Google Scholar]
- Dani VS, Chang Q, Maffei A, Turrigiano GG, Jaenisch R, Nelson SB. 2005. Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc Natl Acad Sci U S A. 102:12560–12565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dani VS, Nelson SB. 2009. Intact long-term potentiation but reduced connectivity between neocortical layer 5 pyramidal neurons in a mouse model of Rett syndrome. J Neurosci. 29:11263–11270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Destexhe A, Rudolph M, Fellous JM, Sejnowski TJ. 2001. Fluctuating synaptic conductances recreate in vivo-like activity in neocortical neurons. Neuroscience. 107:13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand S, Patrizi A, Quast KB, Hachigian L, Pavlyuk R, Saxena A, Carninci P, Hensch TK, Fagiolini M. 2012. NMDA receptor regulation prevents regression of visual cortical function in the absence of Mecp2. Neuron. 76:1078–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekstrand JJ, Domroese ME, Feig SL, Illig KR, Haberly LB. 2001. Immunocytochemical analysis of basket cells in rat piriform cortex. J Comp Neurol. 434:308–328. [PubMed] [Google Scholar]
- Emond V, Joyal C, Poissant H. 2009. [Structural and functional neuroanatomy of attention-deficit hyperactivity disorder (ADHD)]. L'Encéphale. 35:107–114. [DOI] [PubMed] [Google Scholar]
- Euston DR, Gruber AJ, McNaughton BL. 2012. The role of medial prefrontal cortex in memory and decision making. Neuron. 76:1057–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanselow EE, Connors BW. 2010. The roles of somatostatin-expressing (GIN) and fast-spiking inhibitory interneurons in UP-DOWN states of mouse neocortex. J Neurophysiol. 104:596–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favero M, Castro-Alamancos MA. 2013. Synaptic cooperativity regulates persistent network activity in neocortex. J Neurosci. 33:3151–3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellous J-M, Rudolph M, Destexhe A, Sejnowski TJ. 2003. Synaptic background noise controls the input/output characteristics of single cells in an in vitro model of in vivo activity. Neuroscience. 122:811–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint AC, Connors BW. 1996. Two types of network oscillations in neocortex mediated by distinct glutamate receptor subtypes and neuronal populations. J Neurophysiol. 75:951–957. [DOI] [PubMed] [Google Scholar]
- Frysztak RJ, Neafsey EJ. 1991. The effect of medial frontal cortex lesions on respiration, “freezing”, and ultrasonic vocalizations during conditioned emotional responses in rats. Cereb Cortex. 1:418–425. [DOI] [PubMed] [Google Scholar]
- Gambino F, Khelfaoui M, Poulain B, Bienvenu T, Chelly J, Humeau Y. 2010. Synaptic maturation at cortical projections to the lateral amygdala in a mouse model of Rett syndrome. PLoS One. 5:e11399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan WB, Grutzendler J, Wong WT, Wong RO, Lichtman JW. 2000. Multicolor “DiOlistic” labeling of the nervous system using lipophilic dye combinations. Neuron. 27:219–225. [DOI] [PubMed] [Google Scholar]
- Gemelli T, Berton O, Nelson ED, Perrotti LI, Jaenisch R, Monteggia LM. 2006. Postnatal loss of methyl-CpG binding protein 2 in the forebrain is sufficient to mediate behavioral aspects of Rett syndrome in mice. Biol Psychiatry. 59:468–476. [DOI] [PubMed] [Google Scholar]
- Gibson JR, Bartley AF, Hays SA, Huber KM. 2008. Imbalance of neocortical excitation and inhibition and altered UP states reflect network hyperexcitability in the mouse model of fragile X syndrome. J Neurophysiol. 100:2615–2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogolla N, Leblanc JJ, Quast KB, Südhof TC, Fagiolini M, Hensch TK. 2009. Common circuit defect of excitatory–inhibitory balance in mouse models of autism. J Neurodev Disord. 1:172–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy J, Gan J, Selfridge J, Cobb S, Bird A. 2007. Reversal of neurological defects in a mouse model of Rett syndrome. Science. 315:1143–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haider B, Duque A, Hasenstaub AR, McCormick DA. 2006. Neocortical network activity in vivo is generated through a dynamic balance of excitation and inhibition. J Neurosci. 26:4535–4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattox AM, Nelson SB. 2007. Layer V neurons in mouse cortex projecting to different targets have distinct physiological properties. J Neurophysiol. 98:3330–3340. [DOI] [PubMed] [Google Scholar]
- Hirsch JC, Crepel F. 1990. Use-dependent changes in synaptic efficacy in rat prefrontal neurons in vitro. J Physiol. 427:31–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson ME, Homayoun H, Moghaddam B. 2004. NMDA receptor hypofunction produces concomitant firing rate potentiation and burst activity reduction in the prefrontal cortex. Proc Natl Acad Sci U S A. 101:8467–8472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Ash RT, Baker SA, Suter B, Ferguson A, Park J, Rudy J, Torsky SP, Chao H-T, Zoghbi HY et al. . 2013. Dendritic arborization and spine dynamics are abnormal in the mouse model of MECP2 duplication syndrome. J Neurosci. 33:19518–19533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jugloff DGM, Jung BP, Purushotham D, Logan R, Eubanks JH. 2005. Increased dendritic complexity and axonal length in cultured mouse cortical neurons overexpressing methyl-CpG-binding protein MeCP2. Neurobiol Dis. 19:18–27. [DOI] [PubMed] [Google Scholar]
- Jung BP, Jugloff DGM, Zhang G, Logan R, Brown S, Eubanks JH. 2003. The expression of methyl CpG binding factor MeCP2 correlates with cellular differentiation in the developing rat brain and in cultured cells. J Neurobiol. 55:86–96. [DOI] [PubMed] [Google Scholar]
- Katz DM. 2014. Brain-derived neurotrophic factor and Rett syndrome. Handb Exp Pharmacol. 220:481–495. [DOI] [PubMed] [Google Scholar]
- Katz DM, Berger-Sweeney JE, Eubanks JH, Justice MJ, Neul JL, Pozzo-Miller L, Blue ME, Christian D, Crawley JN, Giustetto M et al. . 2012. Preclinical research in Rett syndrome: setting the foundation for translational success. Dis Models Mech. 5:733–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keay KA, Redgrave P, Dean P. 1988. Cardiovascular and respiratory changes elicited by stimulation of rat superior colliculus. Brain Res Bull. 20:13–26. [DOI] [PubMed] [Google Scholar]
- Kishi N, Macklis JD. 2010. MeCP2 functions largely cell-autonomously, but also non-cell-autonomously, in neuronal maturation and dendritic arborization of cortical pyramidal neurons. Exp Neurol. 222:51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kron M, Howell CJ, Adams IT, Ransbottom M, Christian D, Ogier M, Katz DM. 2012. Brain activity mapping in Mecp2 mutant mice reveals functional deficits in forebrain circuits, including key nodes in the default mode network, that are reversed with ketamine treatment. J Neurosci. 32:13860–13872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Kim W, Ham B-J, Chen W, Bear MF, Yoon B-J. 2008. Activity-dependent NR2B expression is mediated by MeCP2-dependent epigenetic regulation. Biochem Biophys Res Commun. 377:930–934. [DOI] [PubMed] [Google Scholar]
- Li W, Mai X, Liu C. 2014. The default mode network and social understanding of others: what do brain connectivity studies tell us. Front Hum Neurosci. 8:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Pozzo-Miller L. 2014. BDNF deregulation in Rett syndrome. Neuropharmacology. 76(Pt C):737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Wang H, Muffat J, Cheng AW, Orlando DA, Lovén J, Kwok S-M, Feldman DA, Bateup HS, Gao Q et al. . 2013. Global transcriptional and translational repression in human-embryonic-stem-cell-derived Rett syndrome neurons. Cell Stem Cell. 13:446–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean JN, Yuste R. 2009. Imaging action potentials with calcium indicators. Cold Spring Harb Protoc. 2009:1–4. [DOI] [PubMed] [Google Scholar]
- Maliszewska-Cyna E, Bawa D, Eubanks JH. 2010. Diminished prevalence but preserved synaptic distribution of N-methyl-D-aspartate receptor subunits in the methyl CpG binding protein 2(MeCP2)-null mouse brain. Neuroscience. 168:624–632. [DOI] [PubMed] [Google Scholar]
- Melo CV, Mele M, Curcio M, Comprido D, Silva CG, Duarte CB. 2013. BDNF regulates the expression and distribution of vesicular glutamate transporters in cultured hippocampal neurons. PLoS One. 8:e53793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalf BM, Mullaney BC, Johnston MV, Blue ME. 2006. Temporal shift in methyl-CpG binding protein 2 expression in a mouse model of Rett syndrome. Neuroscience. 139:1449–1460. [DOI] [PubMed] [Google Scholar]
- Molina LA, Skelin I, Gruber AJ. 2014. Acute NMDA receptor antagonism disrupts synchronization of action potential firing in rat prefrontal cortex. PLoS One. 9:e85842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. 1994. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 12:529–540. [DOI] [PubMed] [Google Scholar]
- Moretti P, Levenson JM, Battaglia F, Atkinson R, Teague R, Antalffy B, Armstrong D, Arancio O, Sweatt JD, Zoghbi HY. 2006. Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J Neurosci. 26:319–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milojkovic BA, Radojicic MS, Antic SD. 2005. A strict correlation between dendritic and somatic plateau depolarizations in the rat prefrontal cortex pyramidal neurons. J Neurosci. 25:3940–3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Na ES, Nelson ED, Kavalali ET, Monteggia LM. 2013. The impact of MeCP2 loss- or gain-of-function on synaptic plasticity. Neuropsychopharmacology. 38:212–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan RP, Hogart AR, Gwye Y, Martin MR, LaSalle JM. 2006. Reduced MeCP2 expression is frequent in autism frontal cortex and correlates with aberrant MECP2 promoter methylation. Epigenetics. 1:e1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalivaiko E. 2011. Animal models of psychogenic cardiovascular disorders: what we can learn from them and what we cannot. Clin Exp Pharmacol Physiol. 38:115–125. [DOI] [PubMed] [Google Scholar]
- Nalivaiko E, Bondarenko E, Lidström A, Barry RJ. 2012. Respiratory component of the orienting reflex: a novel sensitive index of sensory-induced arousal in rats. Front Physiol. 2:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen MVC, Du F, Felice CA, Shan X, Nigam A, Mandel G, Robinson JK, Ballas N. 2012. MeCP2 is critical for maintaining mature neuronal networks and global brain anatomy during late stages of postnatal brain development and in the mature adult brain. J Neurosci. 32:10021–10034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philpot BD, Weisberg MP, Ramos MS, Sawtell NB, Tang YP, Tsien JZ, Bear MF. 2001. Effect of transgenic overexpression of NR2B on NMDA receptor function and synaptic plasticity in visual cortex. Neuropharmacology. 41:762–770. [DOI] [PubMed] [Google Scholar]
- Plotkin JL, Day M, Surmeier DJ. 2011. Synaptically driven state transitions in distal dendrites of striatal spiny neurons. Nat Neurosci. 14:881–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez J-M, Ward CS, Neul JL. 2013. Breathing challenges in Rett Syndrome: lessons learned from humans and animal models. Respir Physiol Neurobiol. 189:280–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph M, Piwkowska Z, Badoual M, Bal T, Destexhe A. 2004. A method to estimate synaptic conductances from membrane potential fluctuations. J Neurophysiol. 91:2884–2896. [DOI] [PubMed] [Google Scholar]
- Samaco RC, Nagarajan RP, Braunschweig D, LaSalle JM. 2004. Multiple pathways regulate MeCP2 expression in normal brain development and exhibit defects in autism-spectrum disorders. Hum Mol Genet. 13:629–639. [DOI] [PubMed] [Google Scholar]
- Sanz-Clemente A, Nicoll RA, Roche KW. 2013. Diversity in NMDA receptor composition: many regulators, many consequences. Neuroscientist. 19:62–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sceniak MP, Berry CT, Sabo SL. 2012. Facilitation of neocortical presynaptic terminal development by NMDA receptor activation. Neural Dev. 7:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sceniak MP, Maciver MB. 2006. Cellular actions of urethane on rat visual cortical neurons in vitro. J Neurophysiol. 95:3865–3874. [DOI] [PubMed] [Google Scholar]
- Sceniak MP, Sabo SL. 2010. Modulation of firing rate by background synaptic noise statistics in rat visual cortical neurons. J Neurophysiol. 104:2792–2805. [DOI] [PubMed] [Google Scholar]
- Schierwagen A, Alpár A, Gärtner U. 2007. Scaling properties of pyramidal neurons in mice neocortex. Math Biosci. 207:352–364. [DOI] [PubMed] [Google Scholar]
- Shahbazian MD, Antalffy B, Armstrong DL, Zoghbi HY. 2002. Insight into Rett syndrome: MeCP2 levels display tissue- and cell-specific differences and correlate with neuronal maturation. Hum Mol Genet. 11:115–124. [DOI] [PubMed] [Google Scholar]
- Shepherd GMG, Katz DM. 2011. Synaptic microcircuit dysfunction in genetic models of neurodevelopmental disorders: focus on Mecp2 and Met. Curr Opin Neurobiol. 21:827–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolov EN. 1963. [The orienting reflex as a cybernetic system]. Zh Vyssh Nerv Deiat Im I P Pavlova. 13:816–830. [PubMed] [Google Scholar]
- Staffend NA, Meisel RL. 2011. DiOlistic labeling of neurons in tissue slices: a qualitative and quantitative analysis of methodological variations. Front Neuroanat. 5:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stearns NA, Schaevitz LR, Bowling H, Nag N, Berger UV, Berger-Sweeney J. 2007. Behavioral and anatomical abnormalities in Mecp2 mutant mice: a model for Rett syndrome. Neuroscience. 146:907–921. [DOI] [PubMed] [Google Scholar]
- Stuss DP, Boyd JD, Levin DB, Delaney KR. 2012. MeCP2 mutation results in compartment-specific reductions in dendritic branching and spine density in layer 5 motor cortical neurons of YFP-H mice. PLoS One. 7:e31896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Aerde KI, Feldmeyer D. 2015. Morphological and physiological characterization of pyramidal neuron subtypes in rat medial prefrontal cortex. Cereb Cortex. 25:788–805. [DOI] [PubMed] [Google Scholar]
- Wang H, Stradtman GG, Wang X-J, Gao W-J. 2008. A specialized NMDA receptor function in layer 5 recurrent microcircuitry of the adult rat prefrontal cortex. Proc Natl Acad Sci U S A. 105:16791–16796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H-P, Spencer D, Fellous J-M, Sejnowski TJ. 2010. Synchrony of thalamocortical inputs maximizes cortical reliability. Science. 328:106–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams K, Russell SL, Shen YM, Molinoff PB. 1993. Developmental switch in the expression of NMDA receptors occurs in vivo and in vitro. Neuron. 10:267–278. [DOI] [PubMed] [Google Scholar]
- Wilson C, Kawaguchi Y. 1996. The origins of two-state spontaneous membrane potential fluctuations of neostriatal spiny neurons. J Neurosci. 16:2397–2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wither RG, Lang M, Zhang L, Eubanks JH. 2013. Regional MeCP2 expression levels in the female MeCP2-deficient mouse brain correlate with specific behavioral impairments. Exp Neurol. 239:49–59. [DOI] [PubMed] [Google Scholar]
- Wood L, Gray NW, Zhou Z, Greenberg ME, Shepherd GMG. 2009. Synaptic circuit abnormalities of motor-frontal layer 2/3 pyramidal neurons in an RNA interference model of methyl-CpG-binding protein 2 deficiency. J Neurosci. 29:12440–12448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood L, Shepherd GMG. 2010. Synaptic circuit abnormalities of motor-frontal layer 2/3 pyramidal neurons in a mutant mouse model of Rett syndrome. Neurobiol Dis. 38:281–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye G-L, Yi S, Gamkrelidze G, Pasternak JF, Trommer BL. 2005. AMPA and NMDA receptor-mediated currents in developing dentate gyrus granule cells. Brain Res Dev Brain Res. 155:26–32. [DOI] [PubMed] [Google Scholar]
- Yizhar O, Fenno LE, Prigge M, Schneider F, Davidson TJ, O'Shea DJ, Sohal VS, Goshen I, Finkelstein J, Paz JT et al. . 2011. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature. 477:171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zikopoulos B, Barbas H. 2013. Altered neural connectivity in excitatory and inhibitory cortical circuits in autism. Front Hum Neurosci. 7:609. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.