

Abstract
Polyplex particles formed with plasmid DNA (pDNA) and Pluronic P85-block-poly{N-[N-(2-aminoethyl)-2-aminoethyl]aspartamide} (P85-b-P[Asp(DET)]) demonstrated highly effective transfection ability compared to PEG-based block cationomer, PEG-b-P[Asp(DET)]. Ternary polyplexes comprising PEG-b-P[Asp(DET)], poly(ethylene oxide)-b-poly(propylene oxide)-b-poly(ethylene oxide)-b-P[Asp(DET)] (P(EPE)-b-P[Asp(DET)]) used as an analog of P85-b-P[Asp(DET)], and pDNA were prepared in this work aiming at maintaining adequate transfection efficiency while solving the stability issues of the P85-b-P[Asp(DET)] polyplexes. Furthermore, a bioreducible P(EPE)-SS-P[Asp(DET)] possessing a redox potential-sensitive disulfide linkage between the P(EPE) polymer and the cationic block was used as a substitute for P(EPE)-b-P[Asp(DET)] during ternary complex formation to investigate whether the trans-fection ability of the ternary polyplex system could be enhanced by triggered release of P(EPE) polymers from the polyplexes. The ternary complexes showed significant improvement in terms of stability against salt-induced aggregation compared to binary complexes, although the gene delivery ability dropped with the amount of PEG-b-P[Asp(DET)] used for complexation. By manipulating the difference in redox potential between the extracellular and intracellular environments, the reducible ternary complexes achieved higher transfection compared to the non-reducible polyplexes; moreover, the reducible poly-plexes exhibited comparable stability to the non-reducible ones. These results suggest that reducible ternary complexes could provide satisfactory transfection efficiency without comprising the colloidal stability of the particles.
Keywords: pDNA, Non-viral gene delivery, Polyplex, Ternary complex
1. Introduction
Development of non-viral plasmid DNA (pDNA) delivery vehicles, for example, polyplexes [1–3], polymersomes [4], and liposomes [5,6], has been an attractive area of research, aiming for alternatives to virus-based gene therapy. Non-viral gene carriers are usually regarded as a safer gene delivery tool for therapeutic purposes compared to viruses [7]; however, the low level of target gene expression provided by non-viral vectors still prohibits them from practical use [8]. A major cause for this unsatisfactory transfection ability is the colloidal instability of the gene delivering particles at physiological conditions [9].
One of the promising methods for producing stable particles with pDNA is using cationic polymers covalently linked with hydrophilic poly(ethylene glycol) (PEG) [10–12]. Upon electrostatic interaction between anionic pDNA and cationic polymer molecules, a PEG layer will be formed around the complexed pDNA, and then polyplex particles with highly hydrated PEG surface will be result. Among the cationic polymer candidates for pDNA complexation, PEG-based diblock copolymer, poly(ethylene glycol)-block-poly{N-[N-(2-aminoethyl)-2-aminoethyl]aspartamide} (PEG-b-P[Asp(DET)]) has achieved high transfection efficiency in both in vitro and in vivo studies [13–15]. Nonetheless, high N/P ratios are usually crucial to obtain adequate transfection level, and implying that a large excess amount of free polymers should be present with polyplexes. This imposes a barrier for this system to be used in systemic administration as free polymers may not be able to concentrate at the target sites along with the pDNA polyplex particles.
Recently we reported that amphiphilic Pluronic could be used as a substitute for hydrophilic PEG for decorating P[Asp(DET)] polyplex particles to improve transfection ability especially at low N/P ratios. However, it was also demonstrated that the Pluronic P85-based nanoparticles suffered from salt-induced aggregation at physiological salt concentration [16]. Kabanov and coworkers improved the colloidal stability of the Pluronic-based polyplex particles by including free Pluronic polymers during the polyplex formation. The utilization of free Pluronic in those studies also improved the overall transfection efficiency of their systems [17–19]. Unfortunately, preliminary results revealed that free Pluronic could not improve neither the stability nor the transfection level of the P[Asp(DET)]-based polyplexes. Therefore, in order to balance the stability and transfection efficiency of the polyplexes, ternary polyplexes consisting of PEG-b-P[Asp(DET)], poly(ethylene oxide)-b-poly(propylene oxide)-b-poly(ethylene oxide)-b-P[Asp(DET)] (P(EPE)-b-P[Asp(DET)]), and pDNA were prepared and investigated whether the colloidal stability of the ternary complexes could be improved by incorporating PEG-based polymers for complexation while maintaining satisfactory level of transfection.
Another aim of this study was to explore if the transfection ability of the present ternary complex system could be further improved by replacing the P(EPE)-b-P[Asp(DET)] used in ternary complexation with a bioreducible P(EPE)-SS-P[Asp(DET)] polymer, which has a redox potential-responsive disulfide linkage located between the P(EPE) polymer and the cationic block. It has been demonstrated that the reduction of disulfide bond could occur intracellularly in the reduced glutathione-rich cytoplasm [20] or extracellularly due to the presence of free thiols secreted by cells [21] or protein disulfide-isomerase on cellular membrane [22,23]. Moreover, several reviews suggested that amphiphilic polymers and polyelectrolytes (cationic/anionic polymers) interact with lipid bilayer using different mechanisms [24,25] and thus the co-presence of those two species might lead to a more effective membrane perturbation. If the cleavage of disulfide linkage occurs at proximity of cell membrane surface or along the endocytotic pathway after particle internalization, the detachment of P(EPE) polymers from the polyplex particles might allow higher cellular uptake or more efficient endo/lysosomal escape of the particles through the combined membrane-disturbing effects of the amphiphilic P(EPE) and cationic polymers.
Here we described the syntheses of two P(EPE)-based block copolymers, P(EPE)-b-P[Asp(DET)] and bioreducible P(EPE)-SS-P[Asp(DET)], and showed that both of them could form ternary polyplexes with additions of PEG-b-P[Asp(DET)] and pDNA. Characterizations on particle stability at physiological conditions, sensitivity to reducing environment, and transfection ability of the ternary complexes were performed. The ternary complexes revealed higher stability against salt-induced aggregation compared to the P(EPE)-based binary polyplex particles. Moreover, the ternary polyplexes with P(EPE)-detachable property demonstrated improved transfection efficiency compared to the non-bioreducible counterparts.
2. Materials and methods
2.1. Materials
α-Methoxy-ω-amino poly(ethylene glycol) (PEG-NH2, Mn = 12,000 g/mol, Mw/Mn = 1.03) was obtained from Nippon Oil and Fats Co., Ltd. (Tokyo, Japan). Amphiphilic triblock copolymers, α-methoxy-ω-amino poly(ethylene oxide)27-block-poly(propylene oxide)40-block-poly(ethylene oxide)27 (P(EPE)-NH2, Mn = 4800 g/mol, Mw/Mn = 1.10) and α-methoxy-ω-hydroxy poly(ethylene oxide)27-block-poly(propylene oxide)40-block-poly(ethylene oxide)27 (P(EPE)-OH, Mn = 4800 g/mol, Mw/Mn = 1.07), were synthesized by Advanced Polymer Materials Inc. (Montreal, Canada). L-Aspartic acid β-benzyl ester, triphosgene, 4-nitrophenyl chloroformate (p-NPC), branched polyethylenimine (bPEI, Mw = 25,000 g/mol), cysteamine, diethylenetriamine (DET), benzene, N,N-dimethylformamide (DMF), dichloromethane (DCM), hexane, N-methyl-2-pyrrolidone (NMP), and tetrahydrofuran (THF) were purchased from Sigma-Aldrich (St. Louis, MO). DET was distilled by conventional method before use. DCM, DMF, hexane, NMP, and THF were purchased as anhydrous grade and used without further purification. Monomer β-benzyl-L -aspartate N-carboxyanhydride (BLA-NCA) was synthesized from L-aspartic acid β-benzyl ester by the Fuchs-Farthing method using triphosgene and purified through repetitive crystallizations [26]. Dialysis tubings (MWCO’s 1000 and 6–8000) were purchased from Spectra/Por (Rancho Dominguez, CA). The pDNA coding for luciferase in pGL4 vector with a CMV promoter (Promega, Madison, WI) was amplified in DH5α Escherichia coli cells and purified using PureLinkTM HiPure Plasmid Maxiprep Kit (Invitrogen, Carlsbad, CA). Luciferase Assay System Kit, and CellTiter-Blue® Cell Viability Assay were purchased from Promega (Madison, WI). RC DC Protein Assay Kit was purchased from Bio-Rad (Hercules, CA). MDA-MB-231 human breast cancer cell line and A549 human lung cancer cell line were obtained from ATCC.
2.2. 1H NMR and gel permeation chromatography (GPC) analyses
The 1H NMR spectrum of each polymer was obtained with Varian Unity-Inova 400 MHz NMR spectrometer (Palo Alto, CA) with temperature regulated at designated temperature. Chemical shift were reported in ppm relative to the residual protonated solvent resonance. Polymer molecular weight distributions were monitored using Agilent 1100 series equipped with TOSOH TSK-gel G3000PWXL and G4000PWXL columns with temperature regulated at 40 °C and an internal refractive index (RI) detector. DMF with 10 mM LiCl was used as the eluent at a flow rate of 1 ml/min. PEG standards were used for calibration.
2.3. Synthesis of P(EPE)-SS-NH2
Disulfide containing P(EPE) monoamine(P(EPE)-SS-NH2) was prepared from P(EPE)-OH as previously described with modifications [27]. Fifty equivalent of p-NPC (660 mg, 3.14 mmol) was dissolved in benzene and then added dropwise to P(EPE)-OH (301 mg, 0.0627 mmol) dissolved in benzene. The reaction proceeded for 24 h at room temperature under an argon atmosphere. Benzene was then evaporated under reduced pressure and the crude product was dissolved in methanol, precipitated in dry ice-chilled diethylether, and then subjected to centrifugation. The precipitation and centrifugation procedure was repeated 3 times to ensure the complete removal of unreacted p-NPC. The white solid obtained was dissolved in benzene and lyophilized. The degree of activation was determined to be about 100% from the peak intensity ratio of the aryl protons of the nitrophenyl groups (NO2–C6H4–, δ= 7.5 and 8.25 ppm) to the methyl protons of P(EPE) (–CH3, δ= 1.0 ppm) in 1H NMR spectrum taken in DMSO-d6 at 25 °C. The activated P(EPE) was subsequently reacted with cysteamine to obtain P(EPE)-SS-NH2. P(EPE)-p-NPC (0.17 g, 0.0342 mmol) and excess amount of cysteamine (0.54 g, 7 mmol) were dissolved in methanol and mixed. The reaction was allowed to proceed for 48 h at room temperature. GPC result showed that there was no dimer present after 48 h reaction and the reaction mixture was transferred to a dialysis tubing (MWCO:1000) and dialyzed extensively against methanol. The product was then lyophilized from benzene. The degree of amine functionality was confirmed to be about 80% from the peak intensity ratio of the methylene protons of the cysteamine moieties (–CH2–, δ= 2.7–2.9 ppm) to the methyl protons of P(EPE) (–CH3, δ= 1.0 ppm) in 1H NMR spectrum obtained in DMSO-d6 at 80 °C. GPC was used again to monitor the molecular weight distribution after dialysis and it showed that P(EPE)-SS-P(EPE) dimers were formed during dialysis; however, the dimeric molecules were not separated from P(EPE)-SS-NH2 at this stage as they did not affect subsequent polymerization reaction. The dimers would be removed by selective precipitation after the polymerization of PBLA described in the following section.
2.4. Syntheses of PEG-b-/P(EPE)-b-/P(EPE)-SS-PBLA
Block copolymer of PEG-b-poly(β-benzyl-L-aspartate) (PEG-b-PBLA) was synthesized as previously described [28] by ring-opening polymerization using β-benzyl-L -aspartate N-carboxyanhydride (BLA-NCA) and β-methoxy-L-amino poly(ethylene glycol)as the monomer and initiator, respectively. Briefly, vacuum-dried BLA-NCA (1.22 g, 4.90 mmol) dissolved in 3 mL DMF and 20 mL DCM was quickly added in a stream of argon to lyophilized PEG–NH2 (502 mg, 0.0409 mmol) dissolved in 10 mL DCM. The polymerization was allowed to proceed for 96 h at 35 °C under an argon atmosphere. P(EPE)-b-poly(β-benzyl-L -aspartate) (P(EPE)-b-PBLA) and P(EPE)-SS-poly(β-benzyl-L -aspartate) (P(EPE)-SS-PBLA) were synthesized with the same procedure while P(EPE)-NH2 and P(EPE)-SS-NH2 were used as the initiator of the polymerization, respectively. The product was precipitated by adding the reaction mixture into diethylether and then subjected to centrifugation. The precipitation procedure was repeated 3 times to purify the polymer. The polymer obtained was dissolved in benzene and then freeze-dried. The degree of polymerization of the PBLA block of PEG-b-PBLA was determined to be 101 from the peak intensity ratio of the aryl protons of the benzyl groups of the PBLA block (C6H5–,δ= 7.2–7.3 ppm) to the methylene protons of PEG (–OCH2CH2–, δ= 3.5 ppm) in 1H NMR spectrum taken in DMSO-d6 at 25 °C. The degrees of polymerization of the PBLA blocks of P(EPE)-b-PBLA and P(EPE)-SS-PBLA were estimated to be 100 and 104, respectively using the methyl protons of P(EPE) (–CH3, δ= 1.0 ppm) and the aryl protons of the benzyl groups (C6H5–, δ= 7.2–7.3 ppm) of the PBLA block for peak integration.
2.5. Syntheses of PEG-b-/P(EPE)-b-/P(EPE)-SS-P[Asp(DET)]
The side chains of PBLA block of the synthesized PEG-b-PBLA in the previous section were then substituted with DET through aminolysis reaction to obtain PEG-b-P[Asp(DET)] [29]. Freeze-dried PEG-b-PBLA (512 mg, 0.0155 mmol) was dissolved in 12 mL NMP and added dropwise to 8.5 mL (78.3 mmol) distilled DET (50 equiv to benzyl group of PBLA block) dissolved in 8 mL NMP. The reaction mixture was stirred for 1 h at 15 °C under an argon atmosphere. The polymer was then precipitated in diethylether and subsequently dried and dissolved in cold 0.01 N HCl and dialyzed against 0.01 N HCl three times (2 h each) in a dialysis tubing (MWCO: 6–8000) at 4 °C. The polymer solution was then dialyzed against deionized water overnight at 4 °C, lyophilized, and the polymer was collected as the hydrochloride salt form. P(EPE)-b-P[Asp(DET)] and P(EPE)-SS-P[Asp(DET)] were obtained with the same method. Quantitative introduction of DET was also confirmed by the peak intensity ratio of the methylene protons of PEG (–OCH2CH2–, δ= 3.5 ppm) or the methyl protons of P(EPE) (–CH3, δ= 1.0 ppm) to the methylene protons of the DET side chains (–CH2CH2NHCH2CH2–, δ= 2.6–3.6 ppm) in the 1H NMR spectrum taken in D2O at 10 °C.
2.6. Preparation of polyplex
The stock solutions were prepared by dissolving pDNA and synthesized polymers in 10 mM Tris-HCl buffer (pH 7.5) at concentrations of 50 μg/mL and 10 mg/mL, respectively. Binary polyplexes at various N/P ratios were formed by adding polymer solution with different concentrations (1/3 total volume) to pDNA solution (2/3 total volume), vortexing, and then incubating at room temperature for at least 30 min before experiments. The final concentration of pDNA in all samples was kept at 33 μg/mL. Ternary polyplexes were formed using the same procedure except two different polymer solutions were thoroughly mixed before adding to the pDNA solution. The percentage of individual polymer used in forming ternary complexes was calculated based on the amino group percentage of the corresponding polymer at N/P ratio of 4, which was fixed for all ternary polyplexes.
2.7. Dynamic light scattering (DLS) and ζ-potential measurements
The DLS and ζ-potential measurements were done at 25 °C or 37 °C using a Zetasizer Nano-ZS (Malvern Instruments, UK). Polyplex samples formed at 33 μg pDNA/mL with different N/P ratios were adjusted to 10 μg pDNA/mL using 10 mM Tris–HCl buffer (pH 7.5). The polyplex samples (50 μL) were transferred to micro cuvettes and then followed by the measurement. The cumulant diameters of samples were calculated by the Stokes–Einstein equation and the sizes were reported as Z-average diameters. The ζ-potential of polyplexes was estimated using a Dip cell (Malvern Instruments, UK) and the electrophoretic mobility was measured. The ζ-potential was then calculated by the Smoluchowski equation.
2.8. In vitro transfection
MDA-MB-231 human breast cancer cell line and A549 human lung cancer cell line were cultured in DMEM and RPMI 1640 supplemented with 10% FBS and 1% penicillin/streptomycin, respectively. MDA-MB-231 was seeded at 50,000 cells/well and A549 was seeded at 30,000 cells/well on 24-well plates and incubated for 24 h. After the cells reached to about 60% confluence, polyplex solutions (30 μL) were prepared with different polymers synthesized at varying N/P ratios, diluted with 270 μL of fresh culture medium containing 10% FBS, and then added to the cells. The pDNA amount for each well was kept at 1 μg. The polyplexes were allowed to incubate with the cells for 4 h. The polyplex-containing medium was then removed and 1 mL of fresh DMEM with 10% FBS was added to each well. The cells were then maintained at 37 °C for another 44 h before luciferase assay. For luciferase assay, the cells were washed with 1x PBS (1 mL) after medium removal. 140 μL of lysis reagent was then added to each well and the cells were incubated at room temperature for 15 min. 100 μL of luciferase substrate was added to 20 μL of the lysate and the luminescence intensity was measured by an Orion microplate luminometer (Berthold Detection Systems, Oak Ridge, TN). The obtained luciferase expression was then normalized with the amount of total proteins present in the lysates determined by the RC DC Protein Assay Kit.
2.9. In vitro cytotoxicity
MDA-MB-231 cells and A549 cells were seeded at 5000 cells/well and 3000 cells/well, respectively, on 96-well plates in corresponding medium containing 10% FBS and incubated for 24 h. Polyplex solutions (10 μL) prepared at different N/P ratios were diluted with fresh medium (90 μL) and then added to the cells. The cells were incubated with polyplexes for 4 h before exhanging medium. The cells were incubated for another 44 h and the assay was carried out according to the manufacturer’s protocol (CellTiter-Blue® Cell Viability Assay). The fluorescence signals were measured 4 h after adding the dye. The results were represented as percentages of cell viability determined using untreated cells.
2.10. Flow cytometry measurements
pDNA was labeled with Label IT® TrackerTM Intracellular Nucleic Acid Localization Kit, CyTM5 (Mirus, Madison, WI) according to the manufacturer’s protocol. MDA-MB-231 cells were seeded at 100,000 cells/well on 6-well plates in DMEM containing 10% FBS and incubated for 24 h. Polyplex solution prepared with Cy5-labeled pDNA for each polymer at N/P ratio of 4 was added to the cells after dilution with fresh DMEM medium containing 10% FBS and incubated for 4 h. The concentration of pDNA was kept at 2 μg/well. After removing the polyplex-containing medium, the cells were rinsed with PBS 3 times. The cells were trypsinated with 500 μL of trypsin-EDTA and collected by centrifugation at 300 × g for 2 min. The cell pellets were resuspended in 500 μL PBS. The Cy5 fluorescence intensity of each sample was then measured using BD LSR II cytometer (BDBio-sciences, Franklin Lakes, NJ) equipped with a 633 nm laser for excitation.
3. Results
3.1. Syntheses of PEG-b-/P(EPE)-b-/P(EPE)-SS-P[Asp(DET)]
The syntheses of the cationic block copolymers were described in Scheme 1 and the physical characterizations of each polymer were summarized in Table 1. Disulfide containing amphiphilic P(EPE) monoamine was synthesized from activation of the terminal hydroxyl groups of P(EPE)-OH with p-NPC and subsequent reaction between the activated P(EPE) with cysteamine. During this subsequent reaction, the amino group of cysteamine first reacted with the activated P(EPE) to form a carbamate ester; the free thiol group at the other end of cysteamine then reacted with another cysteamine molecule to form a disulfide linkage. Excess amount of cysteamine was employed to prevent the formation of dimeric P(EPE)-SS-P(EPE) while favor the formation of P(EPE)-SS-NH2, which was then used to initiate the polymerization of BLA-NCA. 1H NMR showed that the degree of polymerization of the PBLA block of each polymer was approximately 100 and GPC data confirmed that the molecular weight distribution of each polymer was unimodal with Mw/Mn equal or below 1.13. The obtained copolymers were then subjected to aminolysis reaction with DET, followed by purification, protonation of the DET side chains, and lyophilization. 1H NMR revealed that the benzyl ester protecting groups were completely removed from the obtained cationic copolymers and the substitution rates for DET were close to 100%.
Scheme 1.

Syntheic routes of P(EPE)-SS-NH2 (A) and PEG-b-/P(EPE)-b-/P(EPE)-SS-P[Asp(DET)] block cationomers (B).
Table 1.
Characteristics of the polymers synthesized in this study.
| Polymer | Initiator
|
DP of P[Asp] blockb | Mw/Mnc | Total MW (g/mol)d | ||
|---|---|---|---|---|---|---|
| MW (g/mol)a | Mw/Mna | −NH2% | ||||
| P(EPE)-b-P[Asp(DET)] | 4800 | 1.10 | 70a | 100 | 1.13 | 28500 |
| P(EPE)-SS-P[Asp(DET)] | 4800 | 1.07 | 80b | 104 | 1.13 | 29600 |
| PEG-b-P[Asp(DET)] | 12000 | 1.03 | 97.8a | 101 | 1.09 | 36200 |
Information provided by manufacturers.
Determined by 1H NMR.
Determined by GPC.
Calculated based on the HCl salt form of the polymers.
3.2. Formation and stability of binary polyplexes
Polyplexes at different N/P ratios were formed by mixing pDNA solution with the polymer solution containing either PEG-b-P[Asp(DET)], P(EPE)-b-P[Asp(DET)], or P(EPE)-SS-P[Asp(DET)] directly. The diameters of the polyplexes formed were measured by dynamic light scattering and the cumulant diameters are shown in Fig. 1A. Within the N/P ratio range tested, all the polyplexes possess sizes below 110 nm. The sizes of polyplexes with N/P > 4 maintained at around 70–80 nm for all polyplexes. Larger particle size but no aggregation was observed for the polyplexes formed at N/P 2, which has been determined to be the stoichiometric mixing ratio as approximately 50% of the amino groups of the DET side chains were protonated at pH 7.4 [13], which implies that both the PEG and P(EPE) chains on the polyplex surface could help prohibit the interactions among neutrally charged polyplexes under the conditions tested.
Fig. 1.

Particle sizes (A) and ζ-potential values (B) of polyplexes formed at various N/P ratios in 10 mM Tris-HCl buffer (pH 7.5) at 25 °C.
The ζ-potential of the polyplexes was evaluated, and the results are shown in Fig. 1B. All polyplexes displayed the same trend of ζ-potential change with N/P ratio. The ζ-potential increased rapidly in the range of N/P 1 to 4 and was almost neutral at stoichiometric N/P ratio of 2. The increase in ζ-potential value subsided above N/P 4 and the values stayed at about the same up to N/P 32. Moreover, Fig. 1B demonstrates that in general the ζ-potential values of the P(EPE)-b-P[Asp(DET)] and P(EPE)-SS-P[Asp(DET)] polyplexes were higher than those of PEG-b-P[Asp(DET)] polyplexes. The overall smaller ζ-potential values of PEG-b-P[Asp(DET)] polyplexes revealed that the PEG chains provide a more efficient charge shielding effect. All the P(EPE)-b-P[Asp(DET)] polyplex characterization results obtained in this study were similar to the previously published results obtained using polyplexes formed with P85-b-P[Asp(DET)] [16].
Polyplex colloidal stability was investigated based on the salt-induced particle aggregation at 37 °C using dynamic light scattering as stability is always a major obstacle for nanoparticle development in vivo. Polyplex particles (N/P 4) formed with PEG-b-P[Asp(DET)] showed minimal increase in size during the 1 h incubation. As shown in Fig. 2, the cumulant diameter was maintained below 100 nm. However, the stability of the polyplexes formed with P(EPE)-b-P[Asp(DET)] or P(EPE)-SS-P[Asp(DET)] was inferior to that of PEG-b-P[Asp(DET)] polyplexes. Rapid particle aggregation was observed and the cumulant diameters increased to about 800–900 nm in 1 h.
Fig. 2.
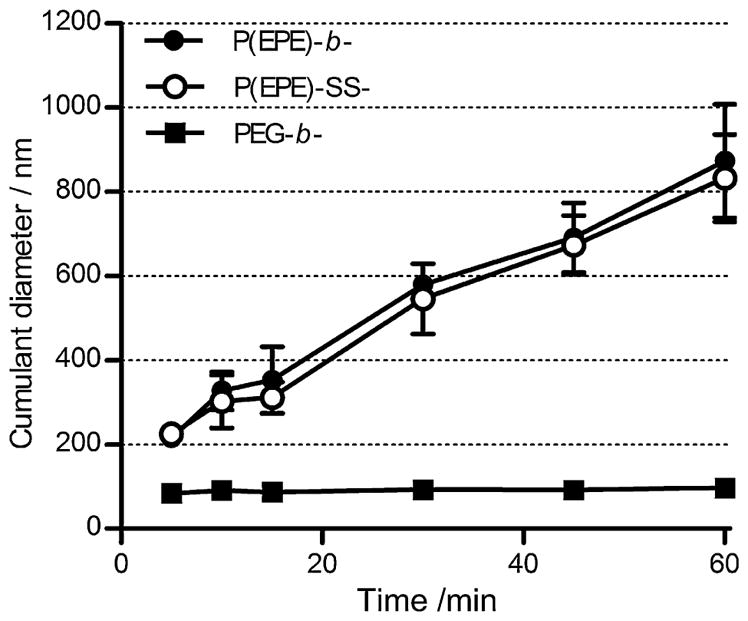
Time-dependent size change of polyplexes formed at N/P 4 in 10 mM Tris-HCl buffer (pH 7.5) containing 150 mM NaCl at 37 °C.
3.3. Reducing environment-sensitivity of P(EPE)-SS-P[Asp(DET)] polyplexes
In order to investigate whether the P(EPE) block copolymers could be released under reducing environment specifically due to the cleavage of the disulfide bond on the P(EPE)-SS-P[Asp(DET)] polymers, the changes in particle size and ζ-potential of poly-plexes were monitored in non-reducing (0 mM DTT) and reducing (10 mM DTT) environments at 25 °C. 10 mM DTT is commonly used to mimic the intracellular reducing environment in the cytoplasm. To look at the size change after the release of the P(EPE) shell, neutral polyplexes were prepared at stoichiometric N/P ratio of 2. Significant increase in particle size for P(EPE)-SS-P[Asp(DET)] polyplexes in reducing milieu was shown in Fig. 3A. In contrast, no such aggregation was observed for the same polyplexes under non-reducing environment which implies that the P(EPE) chains were predominantly stayed on the particle surface. As control poly-plexes, PEG-b-P[Asp(DET)] and P(EPE)-b-P[Asp(DET)] polyplexes (N/P 2) were incubated with 10 mM DTT and there was no change in particle size observed.
Fig. 3.

Time-dependent change of cumulant diameters of polyplexes formed at N/P 2 (A) and ζ-potential values of polyplexes formed at N/P 4 (B) in 10 mM Tris-HCl buffer (pH 7.5) with the presence or absence of 10 mM DTT at 25 °C.
ζ-Potential of the bioreducible polyplexes prepared at N/P 4 in the absence or presence of 10 mM DTT was also measured to further confirm the detachment of P(EPE) block copolymers from the particles under reducing environment. Instantaneous increase in ζ-potential (from +11.5 mV to +23.3 mV) was observed for P(EPE)-SS-P[Asp(DET)] polyplexes within 5 min after 10 mM DTT treatment and the ζ-potential values stayed about the same up to 30 min (Fig. 3B). On the other hand, no such rapid change in ζ-potential was observed for P(EPE)-SS-P[Asp(DET)] polyplexes in the absence of DTT or PEG-b-P[Asp(DET)] and P(EPE)-b-P[Asp(DET)] polyplexes after addition of DTT. These results correlate with the redox potential-dependent size change of the polyplexes and reveal that the release of P(EPE) chains from the P(EPE)-SS-P[Asp(DET)] polyplexes was specific to reducing environment and occurred spontaneously.
3.4. Formation and physical characterizations of ternary polyplexes
As it has been shown in previous section and previous study that the P(EPE)-based polyplexes possess lower colloidal stability compared to the PEG-based counterpart especially at elevated temperature, ternary polyplexes comprised various ratios of PEG-b-P[Asp(DET)] and P(EPE)-b-P[Asp(DET)], and pDNA were prepared aiming at improving the stability of particles and maintaining a satisfactory level of transfection. Furthermore, P(EPE)-SS-P[Asp(DET)] was used instead of P(EPE)-b-P[Asp(DET)] for ternary complex formation to examine whether the site-specific release of P(EPE) chains from the polyplexes could further boost the transfection efficiency based on the possibility of higher cellular uptake and cellular membrane disruption contributed by free P(EPE) amphiphilic copolymers.
Ternary polyplexes were formed at N/P ratio of 4 for all experiments as it has been shown that free polymers exist at N/P > 4 and a low N/P ratio may be more beneficial to in vivo experiments in terms of both transfection efficiency and cytotoxicity [30]. The physical characteristics of the ternary complexes are summarized in Table 2. The sizes of all particles were around 70 nm with low PDI (< 0.14). The ζ-potential measurements of the complexes reveal that the addition of PEG-b-P[Asp(DET)] could reduce the ζ-potential of the polyplexes. The reduction of ζ-potential values increased with the amount of PEG-b-P[Asp(DET)] polymers used. At 75% PEG-b-P[Asp(DET)] composition in the ternary complexes, the ζ-potential was +2.6 mV and +3.6 mV for the PEG-b-/P(EPE)-b-P[Asp(DET)] and PEG-b-/P(EPE)-SS-P[Asp(DET)] ternary polyplexes , respectively. These values are very comparable to the +2.0 mV ζ-potential of PEG-b-P[Asp(DET)] binary polyplex at N/P 4. Moreover, both the particle size and ζ-potential distributions were unimodal for all ternary polyplexes (data not shown).
Table 2.
Physical characteristics of ternary polyplexes.
| Polymer fraction
|
Size (nm)± S.D. | PDI ± S.D. | ζ-potential (mV) ± S.D. | |
|---|---|---|---|---|
| PEG-b- | P(EPE)-b-/P(EPE)-SS- | |||
| 25% | 75% (-b-) | 66.5 ± 0.1 | 0.105 ± 0.003 | 4.2 ± 1.4 |
| 75% (-SS-) | 70.9 ± 3.6 | 0.106 ± 0.036 | 7.0 ± 1.2 | |
| 50% | 50% (-b-) | 70.0 ± 1.6 | 0.078 ± 0.017 | 4.1 ± 1.0 |
| 50% (-SS-) | 70.6 ± 1.0 | 0.093 ± 0.020 | 4.5 ± 0.6 | |
| 75% | 25% (-b-) | 73.1 ± 3.1 | 0.139 ± 0.052 | 2.6 ± 0.3 |
| 25% (-SS-) | 71.4 ± 0.7 | 0.101 ± 0.008 | 3.6 ± 0.4 | |
The redox potential-sensitivity of the PEG-b-/P(EPE)-SS-P[Asp(DET)] ternary polyplexes at various composition ratios was examined by looking at the ζ-potential change 30 min after addition of 10 mM DTT. The change in ζ-potential of the ternary complexes is shown in Fig. 4. Increase of ζ-potential due to the release of P(EPE) chains from polyplexes in reducing condition was observed for ternary complexes prepared at 25% PEG-b-P[Asp(DET)] composition only, but not at 50 and 75%. One possible reason could be the PEG chains shielded the increase of charges after P(EPE) detachment for ternary polyplexes at higher PEG percentage compositions.
Fig. 4.
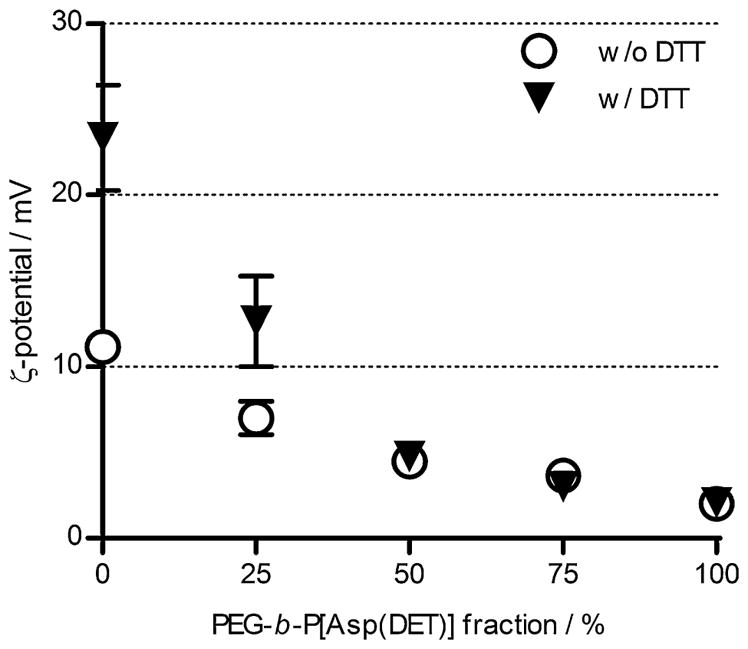
Effect of reducing agent on ζ-potential change of ternary polyplexes. ζ-Potential values of ternary polyplexes prepared at N/P 4 with different ratios of P(EPE)-SS-P[Asp(DET)] and PEG-b-P[Asp(DET)] in 10 mM Tris-HCl buffer (pH 7.5) after 30 min incubation with the presence or absence of 10 mM DTT at 25 °C.
3.5. Stability of ternary polyplexes
The stability of the ternary complexes was assessed by monitoring the particle size change at 37 °C in physiological salt condition of 150 mM NaCl for a 12 h period and the results are shown in Fig. 5. As a control, PEG-b-P[Asp(DET)] binary polyplex prepared at N/P 4 showed a minimal increase in size. The cumulant diameter increased slightly from 90 nm to around 100 nm. The PEG-b-/P(EPE)-b-P[Asp(DET)] ternary complexes showed increase in size ranging from 15 to 50 nm during the 12 h period depending on the amount of PEG-b-P[Asp(DET)] used for complexation. The change in size of particles decreased with the percentage of PEG-b-P[Asp(DET)] employed for ternary complex preparation. As seen in Fig. 5B, the changes in size of the PEG-b-/P(EPE)-SS-P[Asp(DET)] ternary complexes at different mixing ratios showed similar tendency as those of the non-reducible ternary complexes.
Fig. 5.

Time-dependent size change of ternary polyplexes prepared at N/P 4 with various compositions of PEG-b-P[Asp(DET)] and P(EPE)-b-P[Asp(DET)] (A) or P(EPE)-SS-P[Asp(DET)] (B) in 10 mM Tris-HCl buffer (pH 7.5) containing 150 mM NaCl at 37 °C. Percentage values represent the fraction of PEG-b-P[Asp(DET)] used based on charge percentages. Results are expressed as means of triplicate experiments. (Note: error bars omitted for clarity of the graphs.)
3.6. In vitro transfection efficiency and cytotoxicity
It has been previously reported that substituting PEG with Pluronic P85 on the surface of polyplex particles could enhance the transfection efficiency [16,31]. P(EPE)-based polyplexes were used as an analog of P85-based polyplexes for examining their transfection ability in this study. The transfection efficiency of the binary polyplexes was first evaluated in MDA-MB-231 human breast cancer cell line and A549 human lung cancer cell line. As shown in Fig. 6A and B, both the P(EPE)-b-P[Asp(DET)] and P(EPE)-SS-P[Asp(DET)] polyplexes achieved much higher luciferase expression compared to the PEG-based polyplexes in both tested cell lines. The transfection efficiency of the P(EPE)-based polyplexes did not show a N/P ratio-dependent increase and high transfection level was achieved at N/P ratio as low as 4. In contrast, high N/P ratio was necessary for PEG-based polyplexes to obtain a comparable transfection efficiency to the P(EPE)-based polyplexes. Similar trend was observed in our previous study comparing P85-based and PEG-based polyplexes [16]. It was hypothesized that the introduction of disulfide linkage between the P(EPE) polymer and the cationic P[Asp(DET)] block could enhance transfection efficiency of the polyplexes. Performance difference between the P(EPE)-b-P[Asp(DET)] and P(EPE)-SS-P[Asp(DET)] polyplexes in terms of gene delivery ability is shown to be various in the 2 tested cell lines. Overall a 3–9-fold higher transfection was observed with P(EPE)-SS-P[Asp(DET)] polyplexes in MDA-MB-231 cell line, and approximately a 2-fold improvement was achieved in A549 cell line. Cytotoxicity caused by the binary polyplexes was also evaluated based on cellular metabolism in MDA-MB-231 (Fig. 7A) and A549 (Fig. 7B) cell lines. In general, PEG-b-P[Asp(DET)] and P(EPE)-b-P[Asp(DET)] polyplexes showed more than 85% cell viability at all tested N/P ratios in the tested cells. The toxicity level produced by P(EPE)-SS-P[Asp(DET)] polyplexes laid between PEG-b-P[Asp(DET)] or P(EPE)-b-P[Asp(DET)] polyplexes and bPEI polyplexes. In detail, at low N/P ratios, the toxicity of P(EPE)-SS-P[Asp(DET)] polyplexes was negligible and comparable to that of the other two P[Asp(DET)]-based polyplexes. At N/P ratio 32, higher toxicity was observed and the cell viability of the P(EPE)-SS-P[Asp(DET)] polyplex treated cells decreased to 77% and 44% in MDA-MB-231 and A549 cells, respectively; however, the toxicity caused was still lower than that produced by bPEI, which showed around 22% cell viability in both cell lines.
Fig. 6.

Transfection efficiency of binary polyplexes prepared at various N/P ratios in MDA-MB-231 (A) and A549 (B) cell lines. Results are expressed as means ± SD (n = 3). *P < 0.05.
Fig. 7.

Cytotoxicity of binary polyplexes prepared at various N/P ratios in MDA-MB-231 (A) and A549 (B) cell lines.
Transfection efficiency of ternary polyplexes (N/P 4) was then analyzed in MDA-MB-231 (Fig. 8A) and A549 (Fig. 8B) cells. Both of the PEG-b-/P(EPE)-b-P[Asp(DET)] and PEG-b-/P(EPE)-SS-P[Asp(DET)] ternary complexes showed similar trend of transfection ability which decreased with the percentage of PEG-b-P[Asp(DET)] composition used. The P(EPE)-detachable ternary polyplexes demonstrated 2-fold and 1–2 order of magnitude increase in transfection compared to the non-bioreducible ternary complexes in A549 and MDA-MD-231 cells, respectively. Toxicity of the ternary polyplexes was also examined in the same cell lines. Due to the low N/P ratio (N/P 4) used for forming ternary complexes, no significant toxicity was observed for all tested ternary polyplexes. The cells could maintain more than 90% cell viability after ternary complex treatments (data not shown).
Fig. 8.

Transfection efficiency of ternary polyplexes prepared at N/P 4 with different mixing ratio of PEG- and P(EPE)-based polymers in MDA-MB-231 (A) and A549 (B) cell lines. Results are expressed as means ± SD (n = 3). *P < 0.05.
3.7. Cellular uptake study
Cellular internalization of different polyplexes was evaluated by measuring Cy5 fluorescence intensity from MDA-MB-231 cells using flow cytometry after 4 h treatments of polyplexes prepared with Cy5-labeled pDNA at N/P 4 with different polymers. The Cy5 fluorescence intensity decreased with the amount of PEG-b-P[Asp(DET)] polymers used for ternary complex preparation while the PEG-b-P[Asp(DET)] binary polyplexes showed lowest cellular uptake. Furthermore, both the binary and ternary complexes formed with P(EPE)-SS-P[Asp(DET)] demonstrated a higher cellular uptake ranging from 1.4- to 2.4-fold difference compared to the polyplexes formed with P(EPE)-b-P[Asp(DET)] (Fig. 9). The bioreducible nature might allow the P(EPE)-SS-P[Asp(DET)] polyplexes to enter the cells more effectively by releasing P(EPE) polymers from the polyplexes.
Fig. 9.
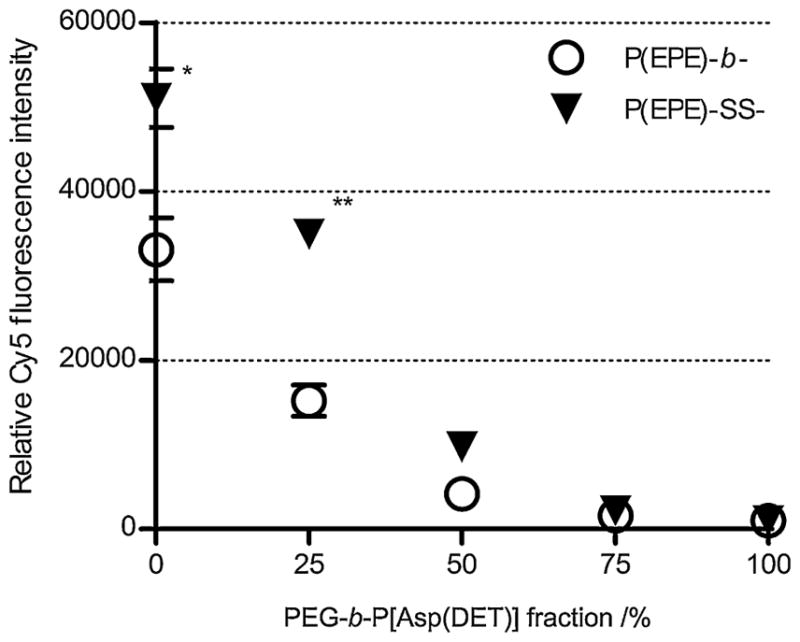
Cellular internalization of Cy5-labeled pDNA polyplexes prepared at N/P 4 with different mixing ratio of PEG- and P(EPE)-based polymers in MDA-MB-231 cells. Results are expressed as means ± SD (n = 3). *P < 0.005. **P < 0.001.
4. Discussion
In this study, we prepared ternary polyplexes surrounded by various ratios of hydrophilic PEG and amphiphilic P(EPE) polymers using different block cationomers based on a poly{N-[N-(2-aminoethyl)-2-aminoethyl]aspartamide} backbone (PEG-b-P[Asp(DET)] and P(EPE)-b-P[Asp(DET)]) aiming at balancing the stability and transfection efficiency of the polyplex particles. Moreover, bioreducible P(EPE)-SS-P[Asp(DET)]) polymer was employed for polyplex formation to assess whether the transfection ability of the current system could be further enhanced by the redox potential-dependent release of free P(EPE) polymers from the polyplexes.
P[Asp(DET)]-based cationic block copolymers were synthesized by the ring-opening polymerization of BLA-NCA using different initiators and the subsequent aminolysis reaction with DET to introduce diaminoethane cationic moieties (Scheme 1). Amphiphilic PEO-b-PPO-b-PEO polymers (P(EPE)) with structure similar to Pluronic P85 having one hydroxyl terminal protected with methoxy group were used instead of modified Pluronic P85 for polymer-ization to ensure the structure of the synthesized polymers by restricting the initiation of polymerization from only one end of the amphiphilic P(EPE) polymers. All the synthesized cationic polymers were able to form polyplexes with pDNA with size less than 110 nm as shown in Fig. 1A. The sizes were reduced to 70–80 nm in the N/P ratio range of 4–32. The maximum ζ-potential measured for the P(EPE)-based polyplexes (with or without disulfide linkage) at the tested N/P ratios was higher than that of PEG-based particles (Fig. 1B), which implies that the PEG layer provides a better charge shielding effect than the P(EPE) layer does and this is consistent with the previous results comparing PEG- and P85-based polyplexes [16]. The better charge shielding effect may attribute to the higher molecular weight of the PEG used; moreover, the amphiphilic nature of P(EPE) may contribute to interactions among the polyplexes and free polymers due to hydrophobic interaction and which can eventually lead to higher ζ-potential values observed for the P(EPE)-based polyplexes.
Intracellular release of free P(EPE) polymer molecules from polyplex particles may further improve the transfection ability of the present system. In order to examine whether the P(EPE)-SS-P[Asp(DET)]) polyplexes possess the specificity in releasing the P(EPE) polymers from the particles, the changes in size and ζ-potential in non-reducing and reducing conditions were investigated. Substantial increase in size was observed for P(EPE)-SS-P[Asp(DET)]) polyplexes at N/P 2 in reducing milieu (Fig. 3A). The observed aggregation demonstrated that the P(EPE) was successfully released from the particles in reducing environment as the detachment of P(EPE) caused the loss of steric hindrance provided by the P(EPE) palisade and at the same time the electrostatic repulsive force was minimal among particles at N/P 2. Such an increase in size was not observed in non-reducing environment. The detachment of P(EPE) from the polyplexes was also confirmed with the prompt increase in ζ-potential in reducing environment but not in non-reducing condition (Fig. 3B). The changes observed imply that the disulfide linkage is stable in the absence of reducing agent but the release of P(EPE) is almost instantaneous once the particles encounter a reducing environment.
The micellization process of Pluronic polymers has been suggested to be temperature-sensitive. The temperature-dependency is due to the rapid dehydration of the poly(propylene oxide) block and which becomes more hydrophobic with increasing temperature [32,33]. This dehydrating property of poly(propylene oxide) may lead to polyplex instability at temperature above room temperature (e.g. 37 °C) when the polyplex surface is covered with the amphiphilic P(EPE) polymers. To investigate the stability of polyplexes at elevated temperature, 37 °C was chosen as this temperature is more relevant to the physiological and in vitro testing conditions. It can be observed in Fig. 2 that the stability of the P(EPE)-based polyplexes against salt-induced aggregation was much lower than that of the PEG-based polyplexes. The increase in size was confirmed to be much more rapid at the physiological temperature of 37 °C than at 25 °C (data not shown). The instability at elevated temperature means that improvements on colloidal stability of the P(EPE)-based polyplexes may be necessary for in vivo applications. Although a lower stability was observed for the P(EPE)-based particles, significantly higher transfection efficiency could be achieved compared to the PEG-based polyplexes especially at low N/P ratios (Fig. 6A, and B). It can also be seen that there is almost no N/P ratio dependency for P(EPE)-based polyplexes in terms of transfection efficiency which demonstrates a clear advantage over the PEG-based counterpart as no excess amount of free polymers are necessary for high level of transfection. Smaller amount of polymers used can also contribute to lower toxicity caused to the target cells. Higher transfection level obtained with the P(EPE)-SS-P[Asp(DET)] polyplexes compared to the P(EPE)-b-P[Asp(DET)] polyplexes suggests that there is improvement in transfection through the redox potential-triggered release of free P(EPE) polymers for the binary polyplex system; however, the degree of enhancement is cell line-dependent.
In order to benefit from both PEG- and P(EPE)-based polymers, ternary polyplexes containing PEG-based, P(EPE)-based cationic polymers, and pDNA were prepared at N/P 4 and tested. The ternary polyplexes showed a size of around 70 nm and the ζ-potential decreased with the proportion of PEG-b-P[Asp(DET)] polymers used (Table 2). All size and ζ-potential distributions of the ternary complexes were unimodal which imply that the population of the polyplex particles consisted of single species instead of mixtures containing individual PEG-based and P(EPE)-based polyplexes. Higher stability was hypothesized as one of the benefits of ternary polyplexes. The dramatic aggregation observed with P(EPE)-based binary polyplexes at 37 °C in physiological salt concentration was successfully prohibited by adding PEG-b-P[Asp(DET)] polymers during ternary complex formation. Change in size was still detectable but significantly reduced to a 15–50 nm increase over a 12 h period depending on the amount of PEG-b-P[Asp(DET)] polymers used (Fig. 5). Similar change in particle size was observed for both ternary complexes formed with P(EPE)-b-P[Asp(DET)] or P(EPE)-SS-P[Asp(DET)] polymers. This reveals that majority of the P(EPE) chains stayed on the particle surface during the 12 h incubation time under the tested conditions.
The transfection ability of the ternary complexes gradually decreased with the amount of PEG-b-P[Asp(DET)] polymers employed for complex formation (Fig. 8). The decrease of trans-fection is probably due to the lower cellular uptake of the particles (Fig. 8) when PEG polymers became more dominant on the surface of the ternary polyplexes. Comparing the reducible and non-reducible ternary polyplexes, the reducible ones demonstrated a 2-fold and 1–2 orders of magnitude higher transfection ability in A549 (Fig. 8A) and MDA-MB-231 cells (Fig. 8B), respectively. The better transfection efficiency of the bioreducible ternary complexes is likely to be related to the release of free P(EPE) polymers under reducing environment. As it has been shown that the reduction of disulfide linkage could occur extracellularly in cell-containing systems by thiol-containing cell surface proteins [22,23] or cell-secreted free thiols and which might affect the cellular uptake of disulfide-possessing nanoparticles [21], flow cytometry was used to monitor the cellular internalization of different poly-plexes into MDA-MB-231 cells in order to predict the possible sites of disulfide reduction and to further elucidate the cause of the higher transfection shown by the bioreducible polyplexes in this study. As seen in Fig. 9, all particles prepared with the reducible P(EPE)-SS-P[Asp(DET)] internalized more into the cells than the non-reducible ones did. These results suggest that at least partial cleavage of the disulfide bonds occurred outside the cells in the 4 h incubation period and the release of P(EPE) polymers subsequently promoted the internalization of the particles. Therefore, the increase in cellular uptake might be one of the reasons for the higher transfection efficiency observed with the bioreducible binary and ternary polyplexes. Although the difference in particle internalization level between the reducible and non-reducible ternary complexes became more marginal when PEG-b-P[Asp(DET)] fraction in the complexes reached 50 and 75%, higher transfection efficiency was still observed with the reducible ternary complexes in MDA-MD-231 cells which implying that other mechanisms might be involved for the improved transfection and the alteration of intracellular trafficking of the particles by free P(EPE) polymers could not be ruled out.
5. Conclusions
Utilizing two different block cationomers, we have prepared ternary pDNA polyplexes with surface covered by hydrophilic PEG polymers and amphiphilic P(EPE) polymers at various ratios for balancing particle stability and transfection efficiency. The incorporation of PEG-b-P[Asp(DET)] to ternary complex formation substantially enhanced the stability of the particles; however, decreased transfection ability of the polyplexes was observed at the same time. To further improve the gene delivery efficiency of the present ternary polyplex system, ternary complexes possessing regulated P(EPE) detachment ability was developed by employing bioreducible P(EPE)-SS-P[Asp(DET)] cationic polymer. The redox potential-sensitive ternary complexes not only maintained similar colloidal stability as the ternary complexes without disulfide linkages, but also demonstrated a 2-fold to 2 orders of magnitude improvement in transfection efficiency.
References
- 1.Osada K, Christie RJ, Kataoka K. J R Soc Interface. 2009;6(Suppl 3):S325. doi: 10.1098/rsif.2008.0547.focus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roesler S, Koch FP, Schmehl T, Weissmann N, Seeger W, Gessler T, Kissel T. J Gene Med. 2011;13:123. doi: 10.1002/jgm.1538. [DOI] [PubMed] [Google Scholar]
- 3.Wagner E, Culmsee C, Boeckle S. Adv Genet. 2005;53:333. [PubMed] [Google Scholar]
- 4.Lomas H, Canton I, MacNeil S, Du J, Armes SP, Ryan AJ, Lewis AL, Battaglia G. Adv Mater. 2007;19:4238. [Google Scholar]
- 5.Kogure K, Akita H, Yamada Y, Harashima H. Adv Drug Deliv Rev. 2008;60:559. doi: 10.1016/j.addr.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 6.Zhang JS, Li S, Huang L. Methods Enzymol. 2003;373:332. doi: 10.1016/S0076-6879(03)73021-3. [DOI] [PubMed] [Google Scholar]
- 7.Glover DJ, Lipps HJ, Jans DA. Nat Rev Genet. 2005;6:299. doi: 10.1038/nrg1577. [DOI] [PubMed] [Google Scholar]
- 8.Pack DW, Hoffman AS, Pun S, Stayton PS. Nat Rev Drug Discov. 2005;4:581. doi: 10.1038/nrd1775. [DOI] [PubMed] [Google Scholar]
- 9.Dash PR, Read ML, Barrett LB, Wolfert MA, Seymour LW. Gene Ther. 1999;6:643. doi: 10.1038/sj.gt.3300843. [DOI] [PubMed] [Google Scholar]
- 10.Merdan T, Kunath K, Petersen H, Bakowsky U, Voigt KH, Kopecek J, Kissel T. Bioconjug Chem. 2005;16:785. doi: 10.1021/bc049743q. [DOI] [PubMed] [Google Scholar]
- 11.Nomoto T, Matsumoto Y, Miyata K, Oba M, Fukushima S, Nishiyama N, Yamasoba T, Kataoka K. J Control Release. 2011;151:104. doi: 10.1016/j.jconrel.2011.02.011. [DOI] [PubMed] [Google Scholar]
- 12.Ogris M, Brunner S, Schuller S, Kircheis R, Wagner E. Gene Ther. 1999;6:595. doi: 10.1038/sj.gt.3300900. [DOI] [PubMed] [Google Scholar]
- 13.Han M, Bae Y, Nishiyama N, Miyata K, Oba M, Kataoka K. J Control Release. 2007;121:38. doi: 10.1016/j.jconrel.2007.05.012. [DOI] [PubMed] [Google Scholar]
- 14.Harada-Shiba M, Takamisawa I, Miyata K, Ishii T, Nishiyama N, Itaka K, Kangawa K, Yoshihara F, Asada Y, Hatakeyama K, Nagaya N, Kataoka K. Mol Ther. 2009;17:1180. doi: 10.1038/mt.2009.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Itaka K, Ohba S, Miyata K, Kawaguchi H, Nakamura K, Takato T, Chung UI, Kataoka K. Mol Ther. 2007;15:1655. doi: 10.1038/sj.mt.6300218. [DOI] [PubMed] [Google Scholar]
- 16.Lai TC, Kataoka K, Kwon GS. Biomaterials. 2011;32:4594. doi: 10.1016/j.biomaterials.2011.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gebhart CL, Sriadibhatla S, Vinogradov S, Lemieux P, Alakhov V, Kabanov AV. Bioconjug Chem. 2002;13:937. doi: 10.1021/bc025504w. [DOI] [PubMed] [Google Scholar]
- 18.Nguyen HK, Lemieux P, Vinogradov SV, Gebhart CL, Guerin N, Paradis G, Bronich TK, Alakhov VY, Kabanov AV. Gene Ther. 2000;7:126. doi: 10.1038/sj.gt.3301052. [DOI] [PubMed] [Google Scholar]
- 19.Yang Z, Sahay G, Sriadibhatla S, Kabanov AV. Bioconjug Chem. 2008;19:1987. doi: 10.1021/bc800144a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meister A, Anderson ME. Annu Rev Biochem. 1983;52:711. doi: 10.1146/annurev.bi.52.070183.003431. [DOI] [PubMed] [Google Scholar]
- 21.Sun WC, Davis PB. J Control Release. 2010;146:118. doi: 10.1016/j.jconrel.2010.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feener EP, Shen WC, Ryser HJP. J Biol Chem. 1990;265:18780. [PubMed] [Google Scholar]
- 23.Mandel R, Ryser HJP, Ghani F, Wu M, Peak D. Proc Natl Acad Sci USA. 1993;90:4112. doi: 10.1073/pnas.90.9.4112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Binder WH. Angew Chem Int Ed. 2008;47:3092. doi: 10.1002/anie.200800269. [DOI] [PubMed] [Google Scholar]
- 25.Tribet C, Vial F. Soft Matter. 2008;4:68. doi: 10.1039/b708431p. [DOI] [PubMed] [Google Scholar]
- 26.Darly WH, Poche D. Tetrahedron Lett. 1988;29:5859. [Google Scholar]
- 27.Li JT, Carlsson J, Lin JN, Caldwell KD. Bioconjug Chem. 1996;7:592. doi: 10.1021/bc960048v. [DOI] [PubMed] [Google Scholar]
- 28.Harada A, Kataoka K. Macromolecules. 1995;28:5294. [Google Scholar]
- 29.Kanayama N, Fukushima S, Nishiyama N, Itaka K, Jang WD, Miyata K, Yamasaki Y, Chung UI, Kataoka K. Chem Med Chem. 2006;1:439. doi: 10.1002/cmdc.200600008. [DOI] [PubMed] [Google Scholar]
- 30.Oba M, Fukushima S, Kanayama N, Aoyagi K, Nishiyama N, Koyama H, Kataoka K. Bioconjug Chem. 2007;18:1415. doi: 10.1021/bc0700133. [DOI] [PubMed] [Google Scholar]
- 31.Bromberg L, Deshmukh S, Temchenko M, Iourtchenko L, Alakhov V, Alvarez-Lorenzo C, Barreiro-Iglesias R, Concheiro A, Hatton TA. Bioconjug Chem. 2005;16:626. doi: 10.1021/bc049749f. [DOI] [PubMed] [Google Scholar]
- 32.Liang X, Guo C, Ma J, Wang J, Chen S, Liu H. J Phys Chem B. 2007;111:13217. doi: 10.1021/jp074990n. [DOI] [PubMed] [Google Scholar]
- 33.Guo C, Wang J, Liu H, Chen J. Langmuir. 1999;15:2703. [Google Scholar]