

Summary
The mTORC1 pathway is involved in the progenitor cell response during liver injury. Here, we show that short-term rapamycin treatment has a persistent effect on the development and progression of focal lesions in an experimental model of progenitor HCC.
Abstract
Hepatocellular carcinoma (HCC) is a heterogeneous disease in which tumor subtypes can be identified based on the presence of adult liver progenitor cells. Having previously identified the mTOR pathway as critical to progenitor cell proliferation in a model of liver injury, we investigated the temporal activation of mTOR signaling in a rat model of hepatic carcinogenesis. The model employed chemical carcinogens and partial hepatectomy to induce progenitor marker-positive HCC. Immunohistochemical staining for phosphorylated ribosomal protein S6 indicated robust mTOR complex 1 (mTORC1) activity in early preneoplastic lesions that peaked during the first week and waned over the subsequent 10 days. Continuous administration of rapamycin by subcutaneous pellet for 70 days markedly reduced the development of focal lesions, but resulted in activation of the PI3K signaling pathway. To test the hypothesis that early mTORC1 activation was critical to the development and progression of preneoplastic foci, we limited rapamycin administration to the 3-week period at the start of the protocol. Focal lesion burden was reduced to a degree indistinguishable from that seen with continuous administration. Short-term rapamycin did not result in the activation of PI3K or mTORC2 pathways. Microarray analysis revealed a persistent effect of short-term mTORC1 inhibition on gene expression that resulted in a genetic signature reminiscent of normal liver. We conclude that mTORC1 activation during the early stages of hepatic carcinogenesis may be critical due to the development of preneoplastic focal lesions in progenitor marker-positive HCC. mTORC1 inhibition may represent an effective chemopreventive strategy for this form of liver cancer.
Introduction
Hepatocellular carcinoma (HCC) is the fifth-leading cause of cancer deaths worldwide (1). At present, potentially curative therapies, including surgical resection, liver transplantation or local ablation, are only available to a small fraction of HCC patients owing to the advanced stage of the disease at diagnosis (2). Like many other cancers, environmental factors, including hepatitis infection, aflatoxin exposure, alcohol abuse and obesity, are risk factors for the development of HCC (2). The course of HCC development is a multistep process involving liver injury and inflammation. As the severity of liver damage increases, the adult hepatocytes undergo senescence and a stem cell compartment is activated, resulting in the proliferation of progenitor cells within the liver (3). This eventually leads to a heterogeneous population of patients, 30–50% of whom develop tumors that express progenitor cell markers (4). These tumors are associated with a unique genetic signature, a higher rate of recurrence and decreased survival compared to progenitor marker-negative HCC (5–7). Better clinical treatments and chemoprevention strategies are clearly needed for HCC patients with advanced disease and for those at risk for the development of liver cancer. Developing such strategies will require a mechanistic understanding of the temporal role of the signaling pathways involved in the pathogenesis of HCC.
The mechanistic target of rapamycin (mTOR) is a nutrient sensing, serine-threonine protein kinase with a key regulatory role in protein translation, autophagy, metabolism, angiogenesis, cellular proliferation and growth. mTOR exists in two distinct multiprotein complexes, mTORC1 and mTORC2 (8). mTORC1, the canonical target of rapamycin, is regulated by growth factors, nutrient availability, cellular energy status and oxygen levels. Its established targets include ribosomal protein S6 kinase (S6K) and the eukaryotic initiation factor 4E (eIF4E)-binding protein-1, 4EBP-1 (9,10). These target proteins are involved in the control of translation initiation, protein synthesis and growth. mTORC1 is also involved in the regulation of RNA polymerase I, gene expression, metabolism and angiogenesis, although the molecular mechanisms have not been fully elucidated (11). mTORC2 has been assigned roles in cellular functions such as protein synthesis, metabolism and control of the actin cytoskeleton (12–15). Insulin stimulation promotes the phosphorylation of Akt by mTORC2, indicating that growth factors regulate this complex. However, the signaling pathway upstream of mTORC2 remains unknown (16). Although long considered to be rapamycin-insensitive, recent data indicate that prolonged exposure to rapamycin leads to dissociation and loss of function of the mTORC2 complex (14,17).
mTORC1 is constitutively activated in a variety of cancers, including advanced HCC (18,19). The aberrant activation of mTORC1 observed in advanced HCC is believed to reflect its role in tumor proliferation, growth and metabolism. However, the temporal role of this pathway in HCC development and progression has not been extensively explored. Previous studies in our laboratory indicate that mTORC1 is involved in the activation and expansion of liver progenitor cells, termed oval cells, in the rat (20). This observation led us to hypothesize that mTORC1 signaling is an early event in the development of progenitor-marker positive HCC. To test this hypothesis, we utilized the well characterized two-step experimental carcinogenesis model established by Solt and Farber (21). In this model, tumors are initiated by administration of diethylnitrosamine (DENA) and promoted by exposure to 2-acetylaminofluorene (2-AAF) combined with partial hepatectomy (PHx). The evolution of HCC in this model is similar to that observed in humans as preneoplastic lesions progress to persistent focal lesions, adenomas and HCC. Studies by Andersen and colleagues (22) concluded that all the carcinomas produced by this protocol express cytokeratin 19, a progenitor cell marker, and that the genetic signature of these tumors could be used to stratify human HCC patients according to clinical outcome.
Materials and methods
Animals
Male Fischer F344 rats (aged 5–6 weeks) were obtained from Charles River Laboratories (Wilmington, MA). Rats were placed on the 2-AAF/PHx protocol to induce oval cells as described previously (23). Where indicated, EGF (0.5mg/g) and insulin (2.5mg/g), or saline vehicle were administered via i.p. injection 15min prior to euthanasia. All animals were housed under standard conditions with access to food and water ad libitum. Rodents were euthanized by exsanguination under isoflurane anesthesia. All animal studies were performed in accordance within the guidelines of the National Institutes of Health and the Rhode Island Hospital Institutional Animal Care and Use Committee.
Hepatic carcinogenesis was induced according to the protocol described by Solt and Farber (21). Briefly, F344 rats were administered an i.p. injection of diethylnitrosamine (DENA, Sigma, St. Louis, MO) at a dose of 200mg/kg, implanted with a time-release 2-acetoaminofluorene pellet (Innovative Research of America, Sarasota, FL) and subjected to 2/3 PHx as described previously (23). Animals were euthanized at time points ranging from 24 to 70 days post-DENA injection. Rats were administered EdU (Invitrogen, Grand Island, NY) 24h prior to euthanasia as described previously (20).
Rats were randomly assigned to treatment groups at the time of PHx. Animals were continuously administered rapamycin by a 40.5-mg, 90-day, slow release pellet (Innovative Research), resulting in a dose of 2.5mg/kg/day based on the average rat weight of 150g (Supplementary Figure 1, available at Carcinogenesis Online). In a second experiment, animals were administered a short course of rapamycin via a 9.45-mg, 21-day, slow release pellet (Innovative Research), also resulting in a dose of 2.5mg/kg/day based on the average rat weight (Supplementary Figure 1, available at Carcinogenesis Online). Control animals received a placebo pellet equivalent to their respective rapamycin group. All pellets were implanted subcutaneously at the time of PHx.
Immunostaining and microscopy
Liver tissue was fixed in 10% neutral buffered formalin, paraffin-embedded and sectioned for histological and immunohistological analysis. In addition to hematoxylin and eosin (H&E) staining, immunohistochemistry was performed using antibodies directed toward ribosomal protein S6, phospho-S6Ser235/236 and phospho-S6Ser240/244, (Cell Signaling Technology, Danvers, MA), placental glutathione S-transferase (MBL International, Woburn, MA), cleaved caspase-3 (Cell Signaling Technology) and phospho-Erk1/2 (Cell Signaling Technology). Dual immunofluorescent staining for GST-P and EdU was carried out as described previously (20). TUNEL staining was performed on cryosections using the TACS XL kit according to the manufacturer’s protocol (Trevigen, Gaithersburg, MD). TUNEL+ cells were quantified from three 20× fields per animal. Dual immunohistochemical staining for GST-P and CK19 (Leica Biosystems, New Castle Upon Tyne, UK) was performed on acetone fixed cryosections. Immunohistochemically stained slides were scanned using the Aperio ScanScope system (Aperio Technologies, Vista, CA) and quantitative analysis performed on fit images using NIH ImageJ software. RGB images were channel split and the blue channel was used to threshold positive staining. Total area of the liver and the area occupied by GST-P+ focal lesions were determined. All sections were analyzed in a blinded fashion.
Western blotting
Frozen liver was homogenized as described previously (24). The homogenates were centrifuged at 1000× g for 15min and the resulting supernatant centrifuged at 9770× g for 20min. A BCA protein assay (Thermo Fisher Scientific, Waltham, MA) was performed. Proteins were separated by SDS-PAGE and transferred to Sequi-Blot PVDF Membrane (Bio-Rad, Hercules, CA). Equal amounts of protein were loaded in each lane based on protein concentration. Membranes were subjected to immunoblot analysis with the following primary antibodies obtained from Cell Signaling Technology: phospho-S6Ser235/236, ribosomal protein S6, Phospho-Akt Ser473, Phospho-AktThr308, Akt and Phospho-Erk1/2 Thr202/Tyr204. Antibodies against Erk1/2 (Millipore, Billerica, MA) and 4E-BP1 (Santa Cruz Biotechnology, Dallas, TX) were also used (24,25). Imaging and densitometry were performed using Labworks 4.5 software (UVP, Upland, CA).
Microarray analysis
Regions of interest were microdissected from FFPE slides using the Arcturus XT™ LCM System (Life Technologies, Grand Island, NY). RNA was extracted using the FFPE RNeasy Kit (Qiagen, Redwood City, CA) and analyzed using Affymetrix Rat Gene ST 1.0 arrays (Affymetrix, Santa Clara, CA). Microarray data are accessible through GEO Series accession number GSE73039 (non-public reviewer link: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=ybwzqqqsnxatdqp&acc=GSE73039) Heat maps and dendrograms were generated using Gene Pattern (26). Venn diagrams were prepared using freely available software from the Whitehead Institute at MIT (http://jura.wi.mit.edu/bio). Gene set enrichment analysis (GSEA) on the full spectrum of genes detected in the array was performed to identify functional gene sets that were enriched in specific experimental groups (27). Inflection points were calculated to determine a fold-change threshold (28). Identification of differentially expressed genes using a false discovery rate (FDR) of <0.05 (29) was performed with Partek Genomic Suite version 6.6 (Partek, St. Louis, MO). Ingenuity pathway analysis (IPA; Qiagen, ww.qiagen.com/ingenuity) was performed as described (30). Input for IPA was significant genes (FDR < 0.05) with a fold-change beyond the inflection point. IPA categories were considered significant when the P value was below the mean minus two standard deviations for the P values obtained from five control gene sets.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 2.01 (San Diego, CA). Results were analyzed using an unpaired t-test or, for multiple comparisons, one-way ANOVA with a post hoc Tukey test.
Results
Continuous rapamycin treatment significantly reduces persistent focal lesion burden
Our previous identification of mTOR as a key regulator of the oval cell response in rodents led us to hypothesize that mTOR signaling would play a critical role in the development and progression of progenitor marker-positive HCC. Rapamycin or placebo control pellets were implanted in DENA-initiated rats at the time of PHx. Rapamycin was administered throughout the duration of the experiment (70 days). The gross appearance of the liver was itself informative, with the placebo group exhibiting nodules on the liver surface (Figure 1A). In contrast, none of the six rats receiving rapamycin exhibited macroscopic nodules. The presence of these large nodules accounted for a significant increase in the liver:carcass weight ratio in the placebo compared to the rapamycin group and to age-matched control rats (Figure 1A). Immunohistochemical staining for GST-P, a marker of neoplastic cells (31), revealed a significant reduction in the total area of GST-P-positive lesions in rapamycin-treated animals (Figure 1B). Previous studies had shown that the early focal lesions in this model are heterogeneous for progenitor cell markers (22,23,32). In order to determine the effect of rapamycin on the phenotype of the cells within the lesions, dual staining for GST-P and CK19 was performed (Figure 1C). Our results were similar to that of Andersen et al. (22), with focal lesions displaying heterogeneous staining for CK19. This ranged from lesions where the majority of cells were CK19+ to lesions that were CK19-. Histopathological examination (Figure 1D) of the liver from placebo-treated rats showed a nodular appearance to the liver parenchyma. This was a result of prominent oval cell hyperplasia that produced bridging between portal areas, along with large numbers of preneoplastic foci. A proportion of the focal lesions showed cellular and nuclear atypia characteristic of early HCC. Scattered apoptotic hepatocytes were present throughout the liver. There was no effect of rapamycin on the percentage of cells undergoing apoptosis (0.6±0.3% in rapamycin versus 1.0±0.6% for placebo) as assessed by TUNEL labeling. Similar results were observed with cleaved caspase-3 staining. The liver histology of the rapamycin-treated rats was relatively normal in appearance with only small preneoplastic focal lesions.
Figure 1.
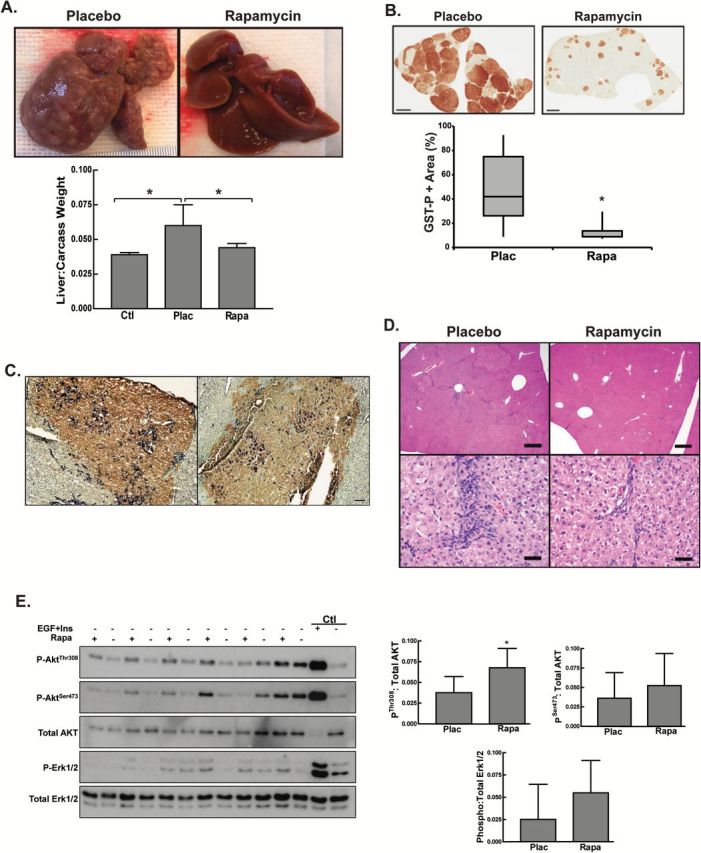
The effect of chronic mTOR inhibition on focal lesions. Rats were placed on the Solt-Farber protocol, administered rapamycin via a 90-day slow release pellet as described in the Materials and Methods section, and euthanized 70 days post-DENA administration. (A) Representative gross images of liver showing numerous lesions in the placebo animals while liver from the rapamycin-treated animals was nearly normal in appearance. Liver weight to carcass weight ratios are shown as the mean + 1 SD for age-matched control (Ctl) and Solt-Farber rats that received placebo (Plac) or rapamycin (Rapa). n ≥ 3 per group. *P < 0.05. (B) Immunohistochemistry for the placental form of glutathione S-transferase (GST-P). Representative Aperio Scans were acquired at 0.8×. Scale bar: 2.5mm. The graph shows quantification of GST-P-positive focal lesions relative to total area as a box and whiskers plot. n ≥ 4 animals per group. *P < 0.05. (C) Dual immunohistochemistry for GST-P (brown) and CK19 (purple). Cryosections were counterstained with methyl green and representative 10× images acquired. Scale bar: 100 µm. (D) H&E staining acquired at 2× (top panel) and 40× (bottom panel). Top panel, Scale bar: 600 µm. Bottom panel, Scale bar: 25 µm. (E) Liver homogenates were prepared from Solt-Farber animals and control adult rats (Ctl) injected with either EGF plus insulin or vehicle 15min prior to euthanasia. Samples were analyzed by immunoblotting with antibodies against phospho-Akt(Thr308), phospho-Akt(Ser473), total Akt, phospho-Erk1/2(Thr202/Tyr204) and total Erk1/2. Densitometric analysis of the basal activity are shown in the graphs as mean + 1SD for the ratio of phospho:total for each protein. n = 6 animals per group. Densitometry was analyzed using an unpaired t-test. *P < 0.05 versus the placebo group.
Prior studies have shown that prolonged rapamycin treatment can result in activation of phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) and mTORC2 signaling by suppressing an S6K1-mediated feedback loop, purportedly leading to cell survival, tumor growth and ‘rapamycin resistance’ (33,34). However, other studies have shown that prolonged rapamycin treatment results in disruption of the mTORC2 complex and decreased Akt signaling (14,17). In order to interrogate the effect of continuous rapamycin treatment on PI3K and mTORC2 signaling in our model, we performed Western blotting of homogenates derived from 70-day livers (Figure 1E). Rapamycin administration resulted in a significant increase in PI3K signaling as assessed by the induction of phospho-AktThr308. We did not observe a significant effect of rapamycin on the mTORC2 site on AktSer473. The absence of a significant effect may have been a consequence of the high degree of variance that, in turn, may be related to the low level of basal activity and heterogeneity of the tissue being analyzed. Rapamycin has also been shown to induce Erk activation in some cancers (34,35). Western blotting for phosphorylated and total Erk1/2 did not demonstrate a significant effect of rapamycin (Figure 1E). Furthermore, immunohistochemistry revealed that P-Erk1/2 was predominantly localized to the oval cells and was absent from the focal lesions (Supplementary Figure 2, available at Carcinogenesis Online).
The mTOR pathway is activated during the promotion of preneoplastic foci
Given the profound growth-inhibiting effect of mTOR inhibition on focal lesions, we investigated the temporal activation of this pathway during the early stages of carcinogenesis (Figure 2A). There was a transient peak of S6 phosphorylation from 3 to 7 days posthepatectomy (days 24–28 of the protocol). S6 phosphorylation waned over the next week, returning to near basal levels by day 35 and remaining at that low level on day 70. Small preneoplastic foci were present as early as day 24 of the protocol. Immunohistochemistry for GST-P clearly distinguished these foci from the surrounding parenchyma (Figure 2B). There was a rapid increase in the size of the lesions from days 24 to 31. The early lesions were heterogeneous for the progenitor cell marker CK19 (Figure 2B). As the lesions developed in the week following PHx, a greater proportion were CK19+ and the staining within the lesions was more homogeneous on days 27 and 31 compared to day 24. Immunohistochemistry for phospho-S6Ser235/236 was consistent with mTOR activation in the preneoplastic foci relative to the surrounding normal cells during the formation and early development of the lesions (Figure 2C). In agreement with the immunoblot, the level of phospho-S6Ser235/236 in the foci was comparable to the surrounding normal cells by day 31. Specificity of staining was confirmed by incubating the slides in blocking peptide and pretreatment with lambda protein phosphatase (Supplementary Figure 3, available at Carcinogenesis Online).
Figure 2.
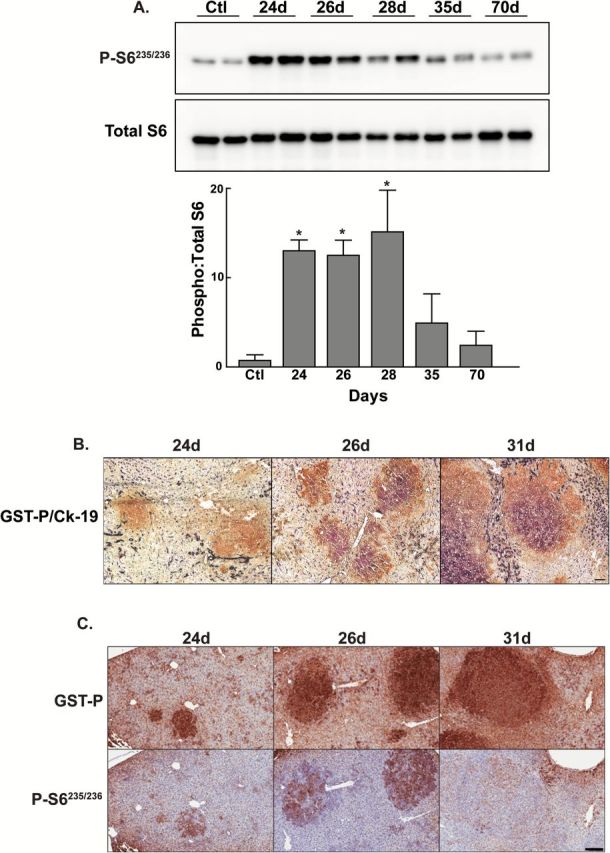
mTORC1 signaling during the initiation and early progression of focal lesions. (A) Representative Western blots for phosphoribosomal protein S6Ser235/236 and total S6 performed on total liver homogenates derived from control (Ctl) or rats placed on the Solt-Farber protocol and euthanized at various days post-DENA administration. The graph shows densitometric data as the mean + 1 SD for the ratio of phospho-S6 to total S6. n = 3 per group. *P < 0.05 versus the control group. (B) Dual immunohistochemistry for GST-P (brown) and CK19 (purple). Cryosections were counterstained with methyl green and representative 10× images acquired. Scale bar: 100 µm. (C) Immunohistochemical staining for GST-P and phospho-S6Ser235/236 was performed on consecutive sections from Solt-Farber animals. Sections were counterstained with hematoxylin. Shown are representative Aperio Scans acquired at 10×. Scale bar: 150 µm.
Short-term rapamycin treatment significantly reduced persistent focal lesion burden
The effectiveness of chronic rapamycin treatment on focal lesion burden and the narrow early window of mTOR activation during the promotion of preneoplastic foci led us to hypothesize that mTOR activity during this early phase was critical to the development and progression of preneoplastic focal lesions. We further hypothesized that limited inhibition during this period of activation would have a persistent effect beyond the period of rapamycin administration. Similar to what was observed in rats treated continuously with rapamycin, there was a significant decrease in focal lesion burden in the short-term rapamycin-treated rats (Figure 3A). The placebo group displayed a significant increase in liver weight to carcass weight ratio compared to age-matched control rats. This increase was prevented by short-term rapamycin administration (Figure 3A).
Figure 3.

The effect of short-term rapamycin administration during the early period of mTOR activation on the subsequent development of focal lesions. Rats were implanted with 21-day slow release rapamycin or placebo pellets on day 21 of the Solt-Farber protocol and euthanized 70 days post-DENA administration. (A) Immunohistochemistry for GST-P. Shown are representative images of Aperio scans acquired at 0.7×. Scale bar: 2.5mm. The left graph illustrates the quantification of lesion burden represented as mean + 1 SD. n ≥ 6 per group. *P < 0.05. The right graph shows the ratio of liver to carcass weight for age-matched control rats (Ctl), and placebo (Plac) or rapamycin-treated (Rapa) Solt-Farber rats. n ≥ 3 per group. *P < 0.05. (B) Cryosections were stained with an antibody against GST-P (red) and a marker of DNA synthesis EdU (green). Slides were counterstained with DAPI (blue) and images acquired at 20×. Scale bar: 100 µm. (C) Cryosections were stained with an antibody against GST-P (brown) and CK19 (purple). Slides were counterstained with methyl green and images acquired at 20×. Scale bar: 100 µm. (D) H&E staining acquired at 2× (top panel) and 40× (bottom panel). Top panel, Scale bar: 600 µm. Bottom panel, Scale bar: 25 µm.
The proliferative activity of the focal lesions and surrounding areas was assessed by dual staining for GST-P and EdU (Figure 3B). The proliferation index in the focal lesions and surrounding areas was low with no difference between the placebo and rapamycin-treated groups (1.77±0.24% in placebo versus 1.36±0.38% in rapamycin). Similar to the chronic treatment group, the focal lesions displayed marked heterogeneity in CK19 staining with no apparent difference between placebo and rapamycin-treated animals (Figure 3C). Histopathological examination revealed similar results to those observed with chronic rapamycin treatment (Figure 3D). The placebo group had a nodular appearance to the liver parenchyma, with oval cell hyperplasia that resulted in bridging between portal areas. Compared with rats in the placebo group, the nodular appearance in the livers of the rapamycin-treated group was less prominent, with decreased oval cell hyperplasia. Similar to the continuously treated group, short-term rapamycin administration did not result in increased hepatocyte apoptosis as assessed by TUNEL labeling (1.2±0.4% in the rapamycin group versus 0.8±0.7% for placebo).
Short-term mTORC1 inhibition and modulation of signaling pathways involved in cell proliferation and growth
To assess whether short-term rapamycin treatment resulted in persistent changes in the activity of mTOR, PI3K and ERK signaling, we performed immunohistochemistry and Western blotting for downstream targets of these signaling pathways (Figure 4A–C). Samples were obtained from animals killed on day 70 of the protocol; i.e. 4 weeks after the end of the period of rapamycin administration. Staining of consecutive sections for GST-P and phospho-S6 revealed minimal heterogeneous phospho-S6 staining in the focal lesions and surrounding liver in placebo-treated animals, while short-term treatment with rapamycin led to a reduction in phospho-S6 staining in the focal lesions and surrounding parenchyma (Figure 4A). Western blotting for phospho-S6 revealed a trend toward a reduction in phospho-S6 in the rapamycin-treated animals (Figure 4C). The basal level of 4E-BP1 phosphorylation in placebo-treated animals was extremely low; we were unable to detect the fully hyperphosphorylated form. Rapamycin treatment did not result in a change in the phosphorylation status of 4EBP-1 (Figure 4B).
Figure 4.
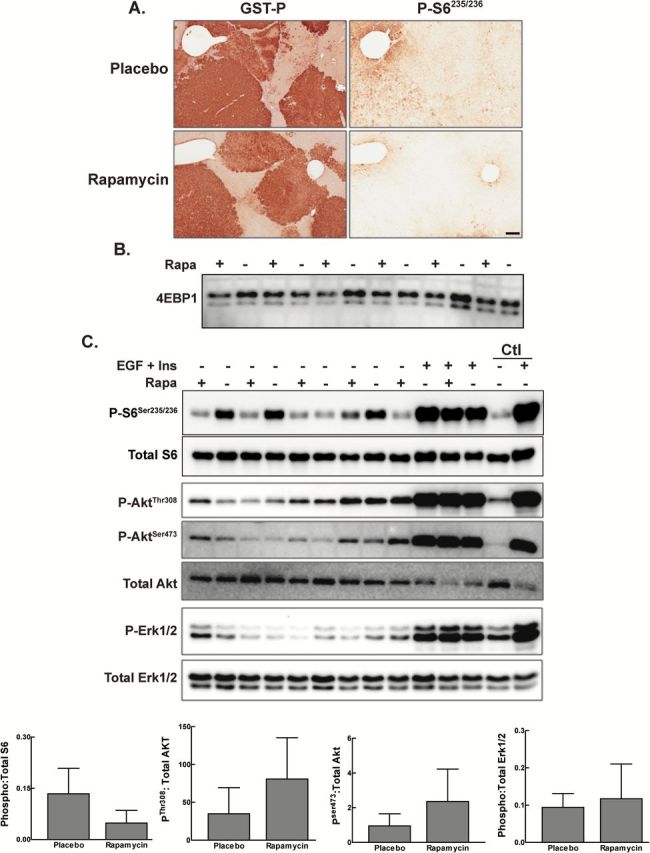
Effect of early mTOR inhibition on PI3K/mTOR and ERK signaling pathways. Rats were implanted with 21-day slow release rapamycin or placebo pellet on day 21 of the Solt-Farber protocol and euthanized 70 days post-DENA administration. (A) Immunohistochemistry for GST-P and phospho- S6Ser235/236 was performed on consecutive sections. Shown are representative Aperio scans acquired at 5×. Scale bar: 200 µm. (B) Total liver homogenates were prepared from rats placed on the Solt-Farber protocol, administered rapamycin or placebo via a 21-day slow release pellet and euthanasized on day 70 post-DENA administration. Homogenates were analyzed by immunoblotting with antibodies against total 4E-BP1. (C) Total liver homogenates were prepared from control adult rats (Ctl) and from rats placed on the Solt-Farber protocol and administered rapamycin via a 21-day slow release pellet. All rats were injected with either EGF plus insulin or vehicle (placebo control) 15min prior to euthanasia on day 70 post-DENA administration. Homogenates were analyzed by immunoblotting with antibodies against phospho-S6(Ser235/236), total S6, phospho-Akt(Thr308), phospho-Akt(Ser473), total Akt, phospho-Erk1/2(Thr202/Tyr204) and total Erk1/2. Densitometric analysis of the basal activity is shown in the graphs as mean + 1SD for the ratio of phospho- to total for each protein. n ≥ 4 animals per group. There were no significant differences identified using an unpaired t-test.
Western blotting for phospho-AktThr308 and phospho-AktSer473 revealed low levels of phosphorylation of these sites that was comparable to that seen in age-matched control rats (Figure 4C). Rapamycin did not have a significant effect on the basal level of phosphorylation at either of these sites. However, animal-to-animal variation was again substantial, and trends were toward greater activity in the rapamycin group. Nonetheless, basal activities remained low, and our data were consistent with the conclusion that short-term rapamycin treatment does not result in substantial activation of the feedback loop to PI3K or changes in mTORC2 signaling. The level of phospho-Erk1/2 in the Solt–Farber animals was similar to control rats, and short-term rapamycin treatment was not associated with changes in Erk1/2 phosphorylation (Figure 4C and Supplementary Figure 2, available at Carcinogenesis Online).
Given the low basal level of both p-Akt and p-Erk1/2, we administered EGF plus insulin to determine whether short-term treatment with rapamycin had led to changes in upstream signaling that would dampen the ability of these pathways to respond to mitogenic stimulation (Figure 4C). We found that there was no difference in the response to EGF and insulin stimulation in the placebo versus rapamycin-treated animals, leading us to conclude that there is not a lasting impact of short-term mTORC1 inhibition on PI3K/AKT, mTORC2 or Erk1/2 signaling.
Short-term mTORC1 inhibition leads to persistent changes in gene expression
Persistent focal lesions uniformly positive for GST-P were microdissected from placebo and short-term rapamycin-treated liver and analyzed by microarray. Samples isolated from normal adult liver and animals placed on a protocol to induce oval cells were also analyzed. Unsupervised hierarchical clustering using the 5% of the genes with the highest coefficient of variation across all samples (Supplementary Figure 4A, available at Carcinogenesis Online) revealed segregation of experimental models; however, the rapamycin and placebo animals did not segregate from one another. This analysis revealed an outlier in the rapamycin-treated group. Additional microarray analyses performed on lesions from that animal demonstrated similar results (Supplementary Figure 4B, available at Carcinogenesis Online). This animal was not an outlier based on any of the other biochemical or physiologic parameters that were measured, leading us to conclude that the array results reflected the heterogeneity of the genetic signature of the focal lesions and response to rapamycin. The raw expression values from the three arrays on this one animal were averaged for subsequent analyses.
GSEA identified gene sets that differed in the focal lesions from rapamycin and placebo animals. This method of analysis is effective at identifying functionally significant changes in gene expression that are associated with relatively small changes in the expression level of numerous genes in a particular experimental condition. A number of KEGG gene sets including Ribosome, Lysosome, Ubiquitin Mediated Proteolysis, Glutathione Metabolism, Renal Cell Carcinoma, and Cell Cycle were enriched in the placebo group (Figure 5A). These sets were accounted for by changes in genes encoding ribosomal proteins, ubiquitin conjugating enzymes, metabolic enzymes and oncogenes. Conversely, the majority of KEGG gene sets enriched in the rapamycin group related to the regulation of fatty acid metabolism and were accounted for by changes in key regulatory enzymes and phospholipases. Changes in hormone receptors, neuropeptides and interleukins accounted for the other gene sets enriched in the rapamycin group.
Figure 5.
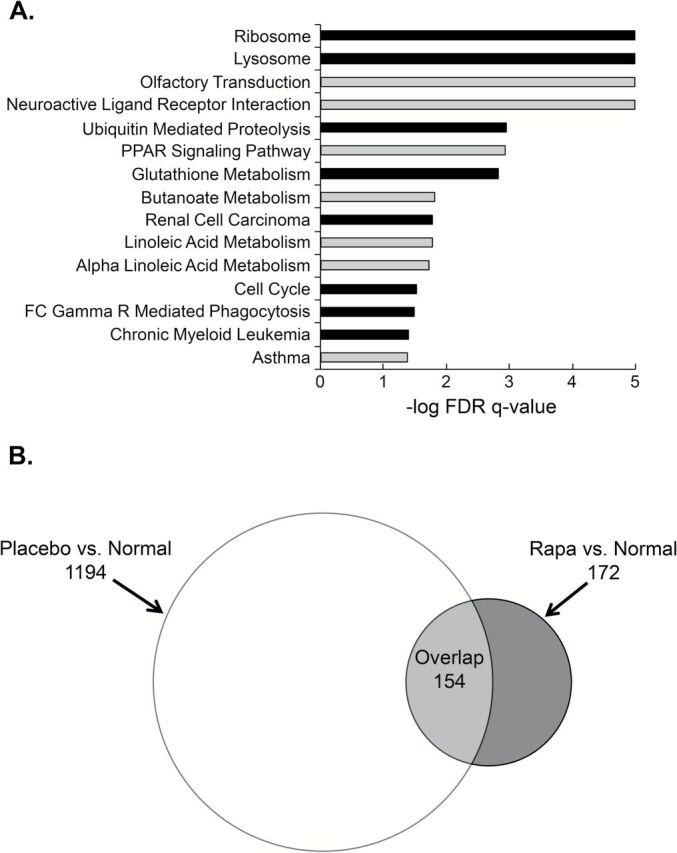
Transient mTORC1 inhibition results in persistent effects on gene expression. (A) GSEA results for KEGG comparing placebo versus rapamycin shown as FDR q-value for gene sets enriched in placebo (black bars) and rapamycin (gray bars) treated focal lesions. (B) Venn diagram of differentially expressed genes (fold change ≥ inflection point) from two comparisons: placebo versus (P/N) normal and rapamycin versus normal (R/N).
We also compared the focal lesions in the Solt-Farber groups to normal liver. We identified the genes whose expression was significantly different in two comparisons, placebo versus normal (P/N) and rapamycin versus normal (R/N), based on a fold-change beyond the inflection point (1.8 for P/N and 2.6 for R/N comparison). This resulted in the inclusion of 1348 genes in the P/N and 326 genes in the R/N comparison (Figure 5B). Of the 326 genes identified in the R/N comparison, 172 were unique to that dataset; 154 of the 326 overlapped with the genes identified in the P/N comparison. Excluding the outlier in the R/N comparison had little effect on the Venn diagram (Supplementary Figure 5, available at Carcinogenesis Online). We interpreted these results as indicating that the focal lesions in the rapamycin-treated animals more closely resembled normal liver than those in the placebo animals.
Genes were selected for IPA based on a FDR < 0.05 and a fold-change beyond the inflection point (Supplementary Table 1A, available at Carcinogenesis Online). No genes met these stringent criteria in the placebo/rapamycin comparison. These criteria resulted in identification of 533 genes for P/N and 69 genes for R/N (Supplementary Table 1B and C). Many of the significant canonical pathways (Figure 6A) identified in the placebo group related to bile acid synthesis and drug metabolism and were accounted for by cytochrome P450 enzymes and solute carriers (Supplementary Table 2A and B, available at Carcinogenesis Online). These pathways were either suppressed or not significant in the rapamycin versus normal comparison. There were many predicted upstream regulators identified in the P/N comparison (Figure 6B). Among these were ligand-dependent nuclear receptors (PPARα, Nr1I2, Nr1I3, RORA, RORC, Nr1H4), enzymes (ACOX1, FECH, GPD1), transcription factors (HNF1A, MAFG, MAFK) and cytokines (IL1B, TNF). The list of genes accounting for these factors included a broad spectrum with cytochrome p450 enzymes and genes involved in lipid metabolism overrepresented (Supplementary Table 2C and D, available at Carcinogenesis Online). PPARα, HNF1α, NR1l3 and HNF4α were predicted to be inhibited while MAFF, MAFG, MAFK, TNF and NFE2L1 were predicted to be activated in the placebo compared to normal liver.
Figure 6.
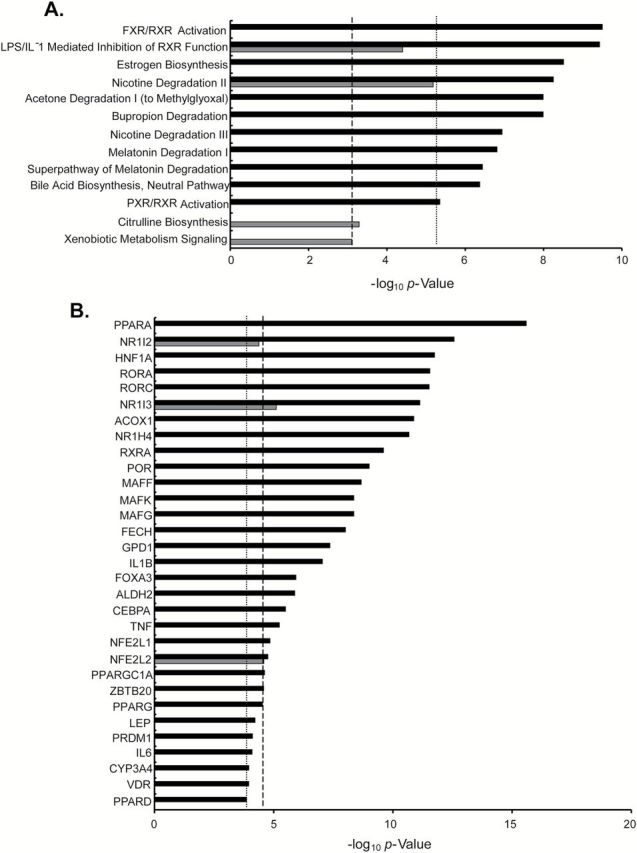
IPA of placebo/normal and rapamycin/normal. Differentially expressed genes (FDR < 0.05 and fold change ≥ inflection point) were included in the analysis of (A) canonical pathways and (B) upstream regulators. Data are shown as unadjusted P values for placebo/normal (black bars) and rapamycin/normal (grays bars). Thresholds for significance based on analysis of control data sets are shown for placebo (dotted line) and rapamycin (dashed line).
As with canonical pathways, the list of regulators identified in the R/N comparison were significantly fewer in number (3 in rapamycin versus normal compared to 31 in placebo versus normal). Three upstream regulators, NR1I2, NR1I3 and NFE2L2, were also identified in the P/N comparison, but were of lower significance in the R/N comparison secondary to there being fewer contributing genes in the latter.
Discussion
We have shown previously that mTORC1 signaling is critical for the oval cell response in two models of liver injury in the rat (20). Analogous progenitor cells in humans are found in individuals with severe liver disease caused by a variety of etiologies, and their abundance correlates with disease severity (36). Studies on the pathogenesis of liver cancer have demonstrated that 55% of small-cell dysplastic foci, the earliest premalignant lesions in humans, consist of progenitor cells and that 30–50% of HCC exhibit progenitor cell markers (4,37,38). Based on our observation that rapamycin attenuates the oval cell response to liver injury (20), we extended our studies to investigate the role of mTOR signaling in the development and expansion of preneoplastic lesions in a rodent model of progenitor-marker positive HCC. mTORC1 was robustly activated in the developing preneoplastic foci during the week immediately following PHx. The activity of this pathway returned to the basal levels seen in age-matched control rats during the second week following PHx. Given the role of mTORC1 in the proliferation of oval cells and the temporal activation of this pathway during the development and early expansion of preneoplastic lesions, we hypothesized that this early activation of mTORC1 was critical to the growth and progression of these lesions.
In a direct comparison of continuous versus early administration of rapamycin, we found that both protocols resulted in the inhibition of macroscopic foci and a significant decrease in microscopic focal lesion burden. In both cases, livers of animals that received rapamycin appeared relatively normal compared to the placebo group, which displayed a nodular appearance owing to the proliferation of oval cells. These results lead us to conclude that mTORC1 activation is a critical early event that leads to the development and growth of preneoplastic foci. Other investigators have similarly observed a salutary effect of rapamycin when given early in carcinogenesis protocols distinct from the one we employed. In experiments carried out by Stelzer and colleagues, HPV transgenic mice were treated topically with the tumor promoter, dimethylbenz[a]CGantracene, to induce squamous cell carcinoma. Administration of rapamycin (5mg/kg/day) prior to the appearance of overt tumors resulted in delayed tumor development and significant reduction in tumor size (35). In another series of studies on a murine model of hereditary tyrosinemia, the rapamycin analogue, RAD001, suppressed the proliferation of hepatocytes with DNA damage resulting in delayed tumor development (39). Mice with liver-specific gain of function of mTORC1 induced by ablation of the negative upstream regulator, Tsc1, develop spontaneous HCC (40). In this model, the chronic activation of mTORC1 led to ER stress and changes in autophagy resulting in liver damage, regeneration and the development of liver tumors of hepatocyte origin. Administration of rapamycin prevented liver damage and the development of hepatoma and HCC. However, rapamycin treatment of older mice with established tumors did not affect tumor burden, number or grade (40).
Similar to other studies, we found that long-term treatment with rapamycin resulted in the activation of PI3K signaling. We did not obtain evidence for a significant effect on TORC2 signaling to Akt. Furthermore, rapamycin treatment did not result in activation of Erk1/2 signaling as has been described for other tumors (34,35). Short-term rapamycin treatment was not associated with a significant increase in phosphorylation of the Akt site associated with PI3K activation. However, there was considerable animal-to-animal variability in all of these analyses. We attribute this to two factors. The first is that basal activity of these pathways is low relative to the stimulated activity seen with administration of EGF plus insulin. Second, our immunostaining results using phospho-specific antibodies all demonstrated marked heterogeneity in the apparent activity of these pathways across the liver tissue. We conclude that secondary activation of PI3K and mTORC2 signaling is unlikely to be occurring in response to short term rapamycin administration. We did not find evidence that Erk1/2 was activated during preneoplastic foci development, suggesting that this pathway does not play a role in the early stages of carcinogenesis in the Solt-Farber model. Similarly, we did not observe changes in 4E-BP1 phosphorylation. The latter is consistent with studies conducted on regenerating rat liver that showed 4E-BP1 phosphorylation was rapamycin insensitive (41). Studies from our lab have shown that the phosphorylation status of 4E-BP1 does not correlate with hepatic cap-dependent translation initiation in the rat (25,42).
The persistence of focal lesions in the short-term rapamycin treatment group led us to question whether these ‘rapamycin resistant’ foci had a more aggressive phenotype or were simply smaller versions of the foci in the placebo group. Microarray analysis revealed that short-term mTORC1 inhibition resulted in persistent effects on gene expression. GSEA showed enrichment of genes encoding ribosomal proteins, ubiquitin-mediated proteolysis and pathways related to cellular proliferation and growth in the foci from placebo animals relative to rapamycin-treated animals. These results parallel the phenotypic differences in the focal lesions, suggesting that mTOR plays a key role in regulating the growth and metabolism of the altered cells in the foci. The gene sets showing the greatest enrichment in the rapamycin foci contained olfactory receptors, which are generally associated with neuronal tissues. However, these receptors are also expressed in the normal adult liver, although their functional significance is unknown (43,44). Further analysis of the changes in gene expression in the focal lesions from our two experimental groups compared to normal liver revealed many fewer changes in the rapamycin treatment group. IPA identified numerous pathways and upstream regulators involved in the regulation of bile acid synthesis, lipid metabolism and hepatocyte growth and differentiation in the placebo focal lesions. The predicted activation state of the core regulatory factors, PPARα, HNF1α, TNF and HNF4α, in the placebo compared to adult liver was consistent with their reported role in liver injury and carcinogenesis (45–47). Previous studies have also identified a link between changes in bile acid synthesis and hepatic carcinogenesis (48). These pathways were either not identified as significant or their significance was suppressed in the rapamycin to normal comparison.
The most direct interpretation of our data is that short-term mTORC1 inhibition results in lesions that more closely resemble normal liver. We speculate that the persistent effect of short-term mTORC1 inhibition on gene expression may result from enhanced cellular differentiation or alterations in the cellular origin of the focal lesions. Our previous studies showed that rapamycin severely dampens the oval cell response (20). It is possible that short-term mTORC1 inhibition in the early phase of carcinogenesis in the Solt-Farber model results in lesions that arise from more well-differentiated hepatic cells. Given the broad but subtle effect of mTORC1 inhibition on gene expression, it is also possible that TORC1 is involved in epigenetic regulation of gene expression.
Our results along with those of Menon et al., suggest that mTOR is critical in the initiation and early progression of HCC, but is not a critical pathway for the growth of established tumors. This suggests that mTOR inhibitors may be effective chemopreventive agents for progenitor marker-positive HCC. The SiLVER study assessing the use of the rapalogue, sirolimus, versus non-mTOR inhibitor immunosuppression in patients undergoing liver transplantation found that there was no significant difference in recurrence-free survival or overall survival (OS) beyond 5 years (49). However, there was a significant benefit of sirolimus-based immunosuppression in recurrence-free survival and OS in younger patients within Milan criteria in the first 3–5 years. There was no difference in adverse events reported between the two groups. Although our histological and gene expression studies did not reveal any adverse effects of rapamycin on liver function, additional experiments designed to specifically interrogate this outcome are warranted as the use of mTOR inhibitors have been associated with increased levels of transaminase, hyperlipidemia, hyperglycemia and hypophosphatemia (50).
In conclusion, our findings reveal a direct role for mTOR in the development and expansion of preneoplastic foci in a rat model of progenitor-marker positive HCC. The persistent effect on focal lesion burden without compensatory changes in the PI3K, Erk1/2 or mTORC2 signaling pathways after a brief period of exposure to rapamycin raises the possibility that mTOR could be targeted as a chemopreventive strategy for HCC and other cancers.
Supplementary material
Supplementary Tables 1 and 2 and Figures 1–7 can be found at http://carcin.oxfordjournals.org/
Funding
National Institutes of Health grants P20RR017695 (J.A.S), R01HD024455, R01DK100301 (P.A.G and J.A.S.) and by the Rhode Island Hospital Department of Pediatrics. Microarray studies performed at Brown University’s Center for Genomics and Proteomics were support by National Institutes of Health grant P30GM103410. A.O.A. was supported by a National Institute of Health Predoctoral Training Grant T32ES007272.
Supplementary Material
Acknowledgements
We thank Joan Boylan for assistance with GSEA, IPA, helpful discussions and critical reading of the manuscript. We thank the COBRE Center for Cancer Research Development Molecular Pathology Core for assistance with the Aperio Scans and laser capture microdissection. We thank Virginia Hovanesian for assistance with image analysis. We also thank Dr Christoph Schorl of Brown University’s Center for Genomics and Proteomics for his advice and performance of the microarrays. J.A.S. and P.A.G. conceived the study. H.F.V., K.E.B. and J.A.S. performed the animal studies. H.F.V. performed the immunostaining and Western blot analyses. A.O.A. performed the gene expression studies analysis of microarray data. N.M.A.P. performed histopathological analyses. H.F.V., A.O.A. and J.A.S. drafted the manuscript. All authors contributed to revisions of the manuscript and have read and approved the final version.
Conflict of Interest Statement: None declared.
Glossary
Abbreviations
- Akt
protein kinase B
- DENA
diethylnitrosamine
- Erk
extracellular signal-related kinase
- GSEA
gene set enrichment analysis
- GST-P
placental glutathione S-transferase
- hpfs
high-powered fields
- HCC
hepatocellular carcinoma
- IPA
ingenuity pathway analysis
- LCM
laser capture microdissection
- mTORC1
mammalian target of rapamycin complex 1
- mTORC2
mammalian target of rapamycin complex 2
- 4EBP-1
eukaryotic initiation factor 4E binding protein-1
- 2-AAF
2-acetylaminofluorene
- PI3K
phosphatidylinositol-4,5-bisphosphate 3-kinase
- PHx
partial hepatectomy
References
- 1. World Cancer Report 2014 (2014) International Agency for Research on Cancer, Lyon. [Google Scholar]
- 2. Sauerland C., et al. (2009) Cancers of the pancreas and hepatobiliary system. Semin. Oncol. Nurs., 25, 76–92. [DOI] [PubMed] [Google Scholar]
- 3. Mangnall D., et al. (2003) The molecular physiology of liver regeneration following partial hepatectomy. Liver Int., 23, 124–138. [DOI] [PubMed] [Google Scholar]
- 4. Libbrecht L., et al. (2000) The immunohistochemical phenotype of dysplastic foci in human liver: correlation with putative progenitor cells. J. Hepatol., 33, 76–84. [DOI] [PubMed] [Google Scholar]
- 5. Yang X.R., et al. (2010) High expression levels of putative hepatic stem/progenitor cell biomarkers related to tumour angiogenesis and poor prognosis of hepatocellular carcinoma. Gut, 59, 953–962. [DOI] [PubMed] [Google Scholar]
- 6. Yamashita T., et al. (2008) EpCAM and alpha-fetoprotein expression defines novel prognostic subtypes of hepatocellular carcinoma. Cancer Res., 68, 1451–1461. [DOI] [PubMed] [Google Scholar]
- 7. Lee J.S., et al. (2006) A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nat. Med., 12, 410–416. [DOI] [PubMed] [Google Scholar]
- 8. Abraham R.T. (2002) Identification of TOR signaling complexes: more TORC for the cell growth engine. Cell, 111, 9–12. [DOI] [PubMed] [Google Scholar]
- 9. Fingar D.C., et al. (2004) Target of rapamycin (TOR): an integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene, 23, 3151–3171. [DOI] [PubMed] [Google Scholar]
- 10. Hay N., et al. (2004) Upstream and downstream of mTOR. Genes Dev., 18, 1926–1945. [DOI] [PubMed] [Google Scholar]
- 11. Wang X., et al. (2011) mTORC1 signaling: what we still don’t know. J. Mol. Cell Biol., 3, 206–220. [DOI] [PubMed] [Google Scholar]
- 12. Jacinto E., et al. (2004) Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol., 6, 1122–1128. [DOI] [PubMed] [Google Scholar]
- 13. Hagiwara A., et al. (2012) Hepatic mTORC2 activates glycolysis and lipogenesis through Akt, glucokinase, and SREBP1c. Cell Metab., 15, 725–738. [DOI] [PubMed] [Google Scholar]
- 14. Lamming D.W., et al. (2012) Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science, 335, 1638–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lamming D.W., et al. (2014) Hepatic signaling by the mechanistic target of rapamycin complex 2 (mTORC2). FASEB J., 28, 300–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zoncu R., et al. (2011) mTOR: from growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol., 12, 21–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sarbassov D.D., et al. (2006) Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell, 22, 159–168. [DOI] [PubMed] [Google Scholar]
- 18. Shaw R.J., et al. (2006) Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature, 441, 424–430. [DOI] [PubMed] [Google Scholar]
- 19. Villanueva A., et al. (2008) Pivotal role of mTOR signaling in hepatocellular carcinoma. Gastroenterology, 135, 1972–83, 1983.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sanders J.A., et al. (2012) The inhibitory effect of rapamycin on the oval cell response and development of preneoplastic foci in the rat. Exp. Mol. Pathol., 93, 40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Farber E., et al. (1977) Newer insights into the pathogenesis of liver cancer. Am. J. Pathol., 89, 477–482. [PMC free article] [PubMed] [Google Scholar]
- 22. Andersen J.B., et al. (2010) Progenitor-derived hepatocellular carcinoma model in the rat. Hepatology, 51, 1401–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Faris R.A., et al. (1991) Antigenic relationship between oval cells and a subpopulation of hepatic foci, nodules, and carcinomas induced by the “resistant hepatocyte” model system. Cancer Res., 51, 1308–1317. [PubMed] [Google Scholar]
- 24. Sanders J.A., et al. (2008) The effect of rapamycin on DNA synthesis in multiple tissues from late gestation fetal and postnatal rats. Am. J. Physiol. Cell Physiol., 295, C406–C413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Anand P., et al. (2006) Rapamycin inhibits liver growth during refeeding in rats via control of ribosomal protein translation but not cap-dependent translation initiation. J. Nutr., 136, 27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Reich M., et al. (2006) GenePattern 2.0. Nat. Genet., 38, 500–501. [DOI] [PubMed] [Google Scholar]
- 27. Subramanian A., et al. (2007) GSEA-P: a desktop application for Gene Set Enrichment Analysis. Bioinformatics, 23, 3251–3253. [DOI] [PubMed] [Google Scholar]
- 28. Chen R.Q., et al. (2009) CDC25B mediates rapamycin-induced oncogenic responses in cancer cells. Cancer Res., 69, 2663–2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Storey J.D., et al. (2003) Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA, 100, 9440–9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Boylan J.M., et al. (2015) Profiling of the fetal and adult rat liver transcriptome and translatome reveals discordant regulation by the mechanistic target of rapamycin (mTOR). Am. J. Physiol. Regul. Integr. Comp. Physiol., 309, R22–R35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Satoh K., et al. (1991) Biochemical characteristics of a preneoplastic marker enzyme glutathione S-transferase P-form(7-7). Arch. Biochem. Biophys., 285, 312–316. [DOI] [PubMed] [Google Scholar]
- 32. Hixson D.C., et al. (1997) Antigenic phenotypes common to rat oval cells, primary hepatocellular carcinomas and developing bile ducts. Carcinogenesis, 18, 1169–1175. [DOI] [PubMed] [Google Scholar]
- 33. O’Reilly K.E., et al. (2006) mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res., 66, 1500–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Carracedo A., et al. (2008) Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J. Clin. Invest., 118, 3065–3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Stelzer M.K., et al. (2010) Rapamycin inhibits anal carcinogenesis in two preclinical animal models. Cancer Prev. Res. (Phila)., 3, 1542–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lowes K.N., et al. (1999) Oval cell numbers in human chronic liver diseases are directly related to disease severity. Am. J. Pathol., 154, 537–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hsia C.C., et al. (1992) Occurrence of oval-type cells in hepatitis B virus-associated human hepatocarcinogenesis. Hepatology, 16, 1327–1333. [DOI] [PubMed] [Google Scholar]
- 38. Van Eyken P., et al. (1988) Cytokeratin expression in hepatocellular carcinoma: an immunohistochemical study. Hum. Pathol., 19, 562–568. [DOI] [PubMed] [Google Scholar]
- 39. Buitrago-Molina L.E., et al. (2009) Rapamycin delays tumor development in murine livers by inhibiting proliferation of hepatocytes with DNA damage. Hepatology, 50, 500–509. [DOI] [PubMed] [Google Scholar]
- 40. Menon S.C., et al. (2012) Effect of ventricular size and function on exercise performance and the electrocardiogram in repaired tetralogy of Fallot with pure pulmonary regurgitation. Ann. Pediatr. Cardiol., 5, 151–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jiang Y.P., et al. (2001) Rapamycin-insensitive regulation of 4e-BP1 in regenerating rat liver. J. Biol. Chem., 276, 10943–10951. [DOI] [PubMed] [Google Scholar]
- 42. Gruppuso P.A., et al. (2008) Hepatic translation control in the late-gestation fetal rat. Am. J. Physiol. Regul. Integr. Comp. Physiol., 295, R558–R567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kang N., et al. (2015) Olfactory marker protein expression is an indicator of olfactory receptor-associated events in non-olfactory tissues. PLoS One, 10, e0116097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Flegel C. Manteniotis S. Osthold S. Hatt H. and Gisselmann G (2013) Expression profile of ectopic olfactory receptors determined by deep sequencing. PLoS One, 8, e55368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang N., et al. (2014) Peroxisome proliferator activated receptor alpha inhibits hepatocarcinogenesis through mediating NF-κB signaling pathway. Oncotarget, 5, 8330–8340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lazarevich N.L., et al. (2004) Progression of HCC in mice is associated with a downregulation in the expression of hepatocyte nuclear factors. Hepatology, 39, 1038–1047. [DOI] [PubMed] [Google Scholar]
- 47. Nakagawa H., et al. (2014) ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell, 26, 331–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li T., et al. (2015) Bile Acid Metabolism and Signaling in Cholestasis, Inflammation, and Cancer. Adv. Pharmacol., 74, 263–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Geissler E.K., et al. (2016) Sirolimus Use in Liver Transplant Recipients With Hepatocellular Carcinoma: A Randomized, Multicenter, Open-Label Phase 3 Trial. Transplantation, 100, 116–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bhat M., et al. (2013) The mTOR pathway in hepatic malignancies. Hepatology, 58, 810–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.