

Do corticosteroids compromise survival in cases of glioblastoma? In a retrospective analysis of clinical cohorts, Pitter et al. identify dexamethasone (DEX) use during radiotherapy as an independent indicator of shorter survival. Moreover, DEX pretreatment reduces survival in irradiated glioblastoma-bearing mice. Anti-VEGFA antibodies may counter oedema without compromising radiation efficacy.
Keywords: astrocytoma, CNS tumour: surgical treatment, glioma, genetics, neurooncology

Do corticosteroids compromise survival in cases of glioblastoma? In a retrospective analysis of clinical cohorts, Pitter et al. identify dexamethasone (DEX) use during radiotherapy as an independent indicator of shorter survival. Moreover, DEX pretreatment reduces survival in irradiated glioblastoma-bearing mice. Anti-VEGFA antibodies may counter oedema without compromising radiation efficacy.
Abstract
Glioblastoma is the most common and most aggressive primary brain tumour. Standard of care consists of surgical resection followed by radiotherapy and concomitant and maintenance temozolomide (temozolomide/radiotherapy→temozolomide). Corticosteroids are commonly used perioperatively to control cerebral oedema and are frequently continued throughout subsequent treatment, notably radiotherapy, for amelioration of side effects. The effects of corticosteroids such as dexamethasone on cell growth in glioma models and on patient survival have remained controversial. We performed a retrospective analysis of glioblastoma patient cohorts to determine the prognostic role of steroid administration. A disease-relevant mouse model of glioblastoma was used to characterize the effects of dexamethasone on tumour cell proliferation and death, and to identify gene signatures associated with these effects. A murine anti-VEGFA antibody was used in parallel as an alternative for oedema control. We applied the dexamethasone-induced gene signature to The Cancer Genome Atlas glioblastoma dataset to explore the association of dexamethasone exposure with outcome. Mouse experiments were used to validate the effects of dexamethasone on survival in vivo. Retrospective clinical analyses identified corticosteroid use during radiotherapy as an independent indicator of shorter survival in three independent patient cohorts. A dexamethasone-associated gene expression signature correlated with shorter survival in The Cancer Genome Atlas patient dataset. In glioma-bearing mice, dexamethasone pretreatment decreased tumour cell proliferation without affecting tumour cell viability, but reduced survival when combined with radiotherapy. Conversely, anti-VEGFA antibody decreased proliferation and increased tumour cell death, but did not affect survival when combined with radiotherapy. Clinical and mouse experimental data suggest that corticosteroids may decrease the effectiveness of treatment and shorten survival in glioblastoma. Dexamethasone-induced anti-proliferative effects may confer protection from radiotherapy- and chemotherapy-induced genotoxic stress. This study highlights the importance of identifying alternative agents such as vascular endothelial growth factor antagonists for managing oedema in glioblastoma patients. Beyond the established adverse effect profile of protracted corticosteroid use, this analysis substantiates the request for prudent and restricted use of corticosteroids in glioblastoma.
Introduction
Dexamethasone (DEX) is a potent synthetic corticosteroid and is considered the ‘gold standard’ for managing cerebral oedema (Raslan and Bhardwaj, 2007). The exact mechanism underlying the anti-oedema effects of DEX is not known, although it is widely believed that DEX suppresses inflammation and decreases vasogenic oedema through partial restoration of blood–brain barrier integrity (Rovit and Hagan, 1968; Eisenberg et al., 1970; Kotsarini et al., 2010). It is also not clear whether DEX influences the effectiveness of standard DNA-damaging therapy for glioblastoma such as radiotherapy or alkylating agents. There are conflicting studies in vitro (Grasso et al., 1977; Weller et al., 1997) and in vivo (Wang et al., 2004) that show antagonism, no interaction, or even synergy with chemotherapy (reviewed in Piette et al., 2006). Despite uncertainty regarding biological efficacy in glioblastoma, DEX is effective in controlling many radiotherapy- and chemotherapy-induced side effects (i.e. nausea and vomiting) and is therefore frequently continued throughout the duration of radiotherapy. More recently, it has been observed that vascular endothelial growth factor (VEGF) antagonists such as the antibody, bevacizumab, or the tyrosine kinase inhibitor, cediranib, have profound anti-oedema effects, commonly obviating the need for steroid co-medication if administered to patients.
In the present study, we confirmed the use of corticosteroids early in the course of disease, during radiotherapy without or with chemotherapy, as an independent predictor of poor outcome in three independent patient cohorts. We also used disease-relevant genetically engineered mouse models (GEMMs) of murine glioblastoma (Hambardzumyan et al., 2009) to show that DEX pretreatment significantly decreases survival in irradiated glioma-bearing mice. Finally, we report that replacing DEX with short-term VEGF antagonism may be the preferred alternative over corticosteroids in the initial management of glioblastoma.
Materials and methods
Retrospective clinical analyses
Three independent patient cohorts were analysed. Details are provided in the Supplementary material.
Generation of RCAS/Tva system-based PDGFB-driven gliomas
Six to eight-week-old Ntv-a/ink4a-arf−/−, Gli-luc;Ntv-a;Ink4a-Arf−/− and N-tva/Ef-Luc mice were used to generate gliomas via introduction of RCAS-PDGFB-HA and RACS-shp53 (Uhrbom et al., 2004; Hambardzumyan et al., 2009). Details are provided in the Supplementary material.
MRI scans
T2-weighted and T1 contrast-enhanced MRI scans of tumour-bearing mice were performed as described (Koutcher et al., 2002).
Tissue processing, immunohistochemistry and immunofluorescence
Tissue processing, immunohistochemistry and immunofluorescence assays were performed according to previously published protocols (Becher et al., 2008; Hambardzumyan et al., 2008) and antibody information is provided in the Supplementary material.
Bioluminescence imaging
Ef-Luc mice were used for bioluminescence imaging (BLI) studies according to published protocols (Uhrbom et al., 2004). Additional methodological details are provided in the Supplementary material.
Results
Retrospective clinical analyses of glioblastoma patients
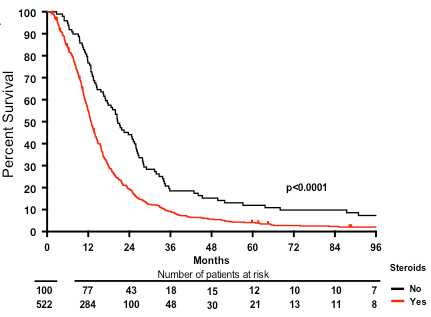
To investigate an association of corticosteroid use with the efficacy of therapy and outcome, we first performed a retrospective clinical analysis of 622 patients treated at Memorial Sloan Kettering Cancer Center (MSKCC). Expectedly (Stupp et al., 2005), patients receiving temozolomide (TMZ) had a significantly longer median survival (15.9 versus 12.8 months; P = 0.0023). Patients not on DEX at the start of radiotherapy had a median survival of 20.6 months whereas patients on DEX had a survival time of 12.9 months (P < 0.0001, Fig. 1A). There were no significant differences in age, gender, duration of symptoms, or TMZ use between patients that did or did not receive steroids at the start of therapy. Steroid use was significantly more common in patients with lower Karnofsky Performance Score, altered mental status, altered neurological function, less extensive surgery, and lower radiation dosing. Accordingly, steroid use was significantly more common in the higher recursive partitioning analysis groups (Supplementary Table 1A). Yet, multivariate COX regression analysis revealed that overall survival was independently associated with recursive partitioning analysis class, TMZ use and steroid use at the start of radiotherapy (Supplementary Table 1B).
Figure 1.

Corticosteroid use at the start of radiotherapy without or with TMZ is an independent marker of poor prognosis in human glioblastoma patients from three independent cohorts. Overall survival of (A) MSKCC; (B) EORTC 26981/22981 NCIC CE.3; and (C) GGN patient cohorts. RT = radiotherapy. O = total observed events; n = total number of patients.
Second, we also explored the association of baseline steroid use with outcome in 573 patients from the pivotal EORTC NCIC trial that established temozolomide and radiotherapy treatment followed by temozolomide (TMZ/RT→TMZ) as the new standard of care (Gorlia et al., 2008; Stupp et al., 2014). The use of steroids was correlated with the extent of resection at initial surgery (P < 0.0001, Fisher test); 91% of patients who underwent biopsy, 74% of patients with partial resection, but only 59% of patients with complete resection had steroids at baseline (Supplementary Table 2A). The median dose administered was 12 mg for biopsied patients and 6 mg for patients with partial or complete resection (P > 0.0001, Kruskal-Wallis test). Patients with baseline steroids had a lower median progression-free survival [5.3 versus 6.4, P < 0.0001, hazard ratio (HR) = 1.39]. After adjustment for the extent of surgery (partial or complete resection versus biopsy), age (continuous), WHO performance status (>0 versus 0), this effect remained significant (P = 0.03) stratified by treatment (Supplementary Table 3A). This effect was borderline, non-significant in the radiotherapy arm (P = 0.06, HR = 1.33) whereas it was not significant in the TMZ/RT→TMZ arm (P = 0.23, HR = 1.17). The prognostic value of baseline steroid dose (split by tercile) was significantly overall stratified by treatment (P = 0.03, HR = 1.14) but not in multivariate analysis (P = 0.45). Steroid dose was significant in the radiotherapy arm (P = 0.004, HR = 1.28) but was borderline, non-significant in multivariate analysis (P = 0.08) and was not significant in the TMZ/RT→TMZ arm (P = 0.8, HR = 1.02; P = 0.88 in multivariate analysis). Altogether, patients with baseline steroids had a lower median overall survival (12 versus 17 months, P < 0.0001, HR = 1.56) (Fig. 1B). This effect remained significant after adjustment for age, extent of surgery, and WHO performance status (P = 0.003) and was significant in the radiotherapy arm (P = 0.004, HR = 1.52) but not in the TMZ/RT→TMZ arm (P = 0.2, HR = 1.2). The prognostic value of steroid dose at baseline, split by tercile, was significantly when stratified by treatment (P = 0.002, HR = 1.2) but not significant in multivariate analysis (P = 0.23). Steroid dose was significant in the radiotherapy arm (P = 0.002, HR = 1.28) and in multivariate analysis (P = 0.048). It was not significant in the TMZ/RT→TMZ arm (P = 0.22, HR = 1.11, also not significant in the multivariate model, P = 0.73). Thus, steroids at baseline was a prognostic factor for both progression-free survival and overall survival in the EORTC NCIC trial and higher doses of steroids were a negative prognostic factor in patients treated with radiotherapy alone more than in patients treated with TMZ/RT→TMZ.
Third, an association between steroid administration at the start of radiotherapy and outcome was examined in a cohort of 832 glioblastoma patients enrolled in the German Glioma Network (GGN) (Supplementary Table 2B). Progression-free survival and overall survival were inferior in steroid-exposed patients in all patients pooled (Figs 1C and 2A, and Supplementary Table 3B) as well as in patients treated with radiotherapy plus chemotherapy, although not patients treated initially with radiotherapy alone (Fig. 2B–E and Supplementary Table 3B). The association between steroids and inferior outcome was prominent in patients who had received a gross total resection, notably in those treated with radiotherapy plus chemotherapy (Fig. 3 and Supplementary Table 3B). Multivariate analysis confirmed that steroid administration was an independent negative prognostic factor when adjusting for extent of resection, initial treatment, age and Karnofsky Performance Score (Supplementary Table 3B).
Figure 2.

Association between steroid administration and outcome in the GGN cohort. Progression-free survival by steroid use in all patients (A), progression-free survival (B) and overall survival (C) for patients with radiotherapy only as initial treatment. Progression-free survival (D) and overall survival (E) for patients with radiotherapy (RT)/CT as initial treatment. O = total observed events; n = total number of patients.
Figure 3.

Association between steroid administration and outcome in the GGN cohort by extent of resection. Progression-free survival (A) and overall survival (B) by steroid use in German Glioma Network (GGN) patients with gross total resection. progression-free survival (C) and overall survival (D) by steroid use in GGN patients without gross total resection (GTR). O = total observed events; n = total number of patients.
Dexamethasone, but not anti-VEGFA antibodies, compromise radiotherapy efficacy in murine glioma models in vivo
To support potential adverse effects of DEX on survival, we used a murine PDGFB-driven glioblastoma model, based on the RCAS/Tva system, a somatic cell specific gene transfer (Hambardzumyan et al., 2009). DEX alone had no impact on survival (P = 0.32, Fig. 4A) whereas pretreatment with single doses of DEX for three consecutive days profoundly decreased the survival advantage afforded by a single 10 Gy dose of irradiation (P = 0.03, Fig. 4B). This effect was even more pronounced when a fractionated irradiation schedule was used (P = 0.002, Fig. 4C).
Figure 4.

DEX but not anti-VEGFA antibody treatment decreases survival after radiotherapy. Mice with PDGF-driven gliomas were randomized into either DEX or untreated cohorts, and on the third day were further randomized into either (A) non-irradiated (No IR), (B) single dose, or (C) fractionated radiotherapy groups. DEX alone had no impact on survival, but decreased the efficacy of both single dose and fractionated radiotherapy. The cartoon at the bottom of each survival plot depicts the treatment schedule. (D) Kaplan-Meier survival curves for mice randomized into vehicle, B20-4.1.1 treatment, radiotherapy alone, or radiotherapy + B20.4.1.1 groups. Schematic illustrations of treatment paradigms for A–C are below and for D on the right together with experimental groups. P-values were calculated using a Log-rank (Mantel-Cox) test, *P < 0.05, ***P < 0.01, ****P < 0.0001.
Because of the deleterious effects of DEX on the survival benefit afforded by radiotherapy, we next focused on identifying an alternative to DEX to decrease oedema. Inhibition of VEGF signalling, either with neutralizing antibodies or VEGFR-targeted kinase inhibitors, leads to decreased oedema, which can result in improved survival of brain tumour-bearing mice despite persistent tumour growth (Gerstner et al., 2009; Kamoun et al., 2009). To assess whether the anti-VEGF antibody B20-4.1.1, a murine surrogate for bevacizumab, also interferes with the response to radiotherapy, we allocated tumour-bearing mice into different treatment groups based on gender and age. Compared to vehicle treatment, both B20-4.1.1 and radiotherapy independently prolonged survival (Fig. 4D). However, in contrast to DEX, there was no significant difference between B20-4.1.1 + radiotherapy-treated animals compared to radiotherapy alone, demonstrating that B20-4.1.1 treatment does not interfere with radiotherapy efficacy (P = 0.86, Fig. 4D).
Dexamethasone-responsive gene signature in gliomas
To broadly assess the genes and pathways that are responsive to DEX and identify pathways associated with treatment resistance, we used microarray analyses from untreated and DEX-treated mouse glioma samples. The data were first analysed by principle component analyses (PCA) (Fig. 5A). We identified 19 genes that were significantly altered (all downregulated) in response to DEX treatment, seven of which were subsequently validated by quantitative real-time polymerase chain reaction (Fig. 5B and C). These genes were significant components of the cell cycle and mitotic machinery (Supplementary Table 4).
Figure 5.

DEX-treated gliomas have downregulation of cell cycle genes, which correlates with poor prognosis in the TCGA dataset. (A) 3D-PCA plot showing control- (black) and DEX- (red) treated samples (n = 4 per each group). The axes of the plot denote the variation accounted for by each component. (B) Heat map and supervised clustering of arrays based on the most significant 25 probes, representing 19 genes. All of the genes are strongly downregulated in DEX-treated samples. (C) Validation of seven DEX downregulated genes by quantitative reverse transcription polymerase chain reaction (qRT-PCR). All samples are normalized to TBP expression levels. ****P < 0.0001 for all comparisons, determined using one-way ANOVA. (D) Representative distribution of TCGA-samples by abnormal spindle-like, microcephaly-associated (ASPM) gene expression, based on z-scores normalized to diploid samples. Patients with elevated expression levels >2 SD above the mean were considered minimally DEX-responsive. This process was repeated for the most significantly DEX-regulated genes. (E) Distribution of patients based on classifications as described above, showing the number of patients with high expression of 0–15 DEX-regulated genes. The graph (right) does not include the 423 (87%) patients who had no genes expressed above our threshold. This more clearly shows the distribution of minimally DEX-responsive patients. (F) Minimally DEX-responsive patients have significantly better overall survival rates (18.3 versus 13.7 months). P-values were calculated using a Log-rank (Mantel-Cox) test, **P < 0.01, *P < 0.05. OS = overall survival.
We generated a DEX-responsive signature based on the arrays described above and queried The Cancer Genome Atlas (TCGA) patient survival and expression data. Because DEX is given to essentially all patients prior to surgery, we predicted that they would have expression signatures reflecting DEX treatment. We further identified a subset of patients with a comparatively high expression of genes downregulated by DEX as having tumours that are minimally responsive to DEX treatment (Fig. 5D). High expression was defined as > 2 standard deviations (SD) above the mean, which classified 13% of patients as minimally responsive (‘Untreated-Like’; Fig. 5E). These patients had the highest expression levels of one or more of the DEX downregulated genes and had a significantly longer median survival (P = 0.008, Fig. 5F). Our TCGA analysis was suggestive of an improved survival for patients whose tumours reflected our untreated mouse samples compared to DEX-treated samples.
Dexamethasone treatment effects on glioma growth in vivo and in vitro
One mechanistic explanation of drugs compromising radiotherapy efficacy is based on the phenomenon that, in general, cells are more radiosensitive when they are in G2/M and more radio-resistant when they are in G1. Therefore if a drug induces a decrease in proliferation, it can potentially reduce the radio-sensitivity of a population via redistribution of the cell cycle, leading to accumulation in G1 and decreased fractions in G2/M (Powell and Abraham, 1994). We have previously documented that DEX induces the cell cycle inhibitor p21 (Glaser et al., 2001), and overexpression of p21 is associated with radioresistance in human gliomas (Kokunai and Tamaki, 1999). Accordingly, we next examined DEX effects on proliferation of tumour cells in vivo. We first set out to assess the effect of DEX on proliferation of mouse gliomas using a non-invasive bioluminescence (BLI) reporter. We have previously described a Ntv-a;Ef-Luc transgenic mouse that expresses firefly luciferase driven by the E2f1 promoter (Ef-Luc) (Uhrbom et al., 2004). The E2F1 transcription factor is normally activated in cell cycle progression during the G1/S transition and is highly active in gliomas (Parr et al., 1997). The dose of DEX used in the clinic for patients with high grade gliomas is variable, ranging from 0.5 to 16 mg daily (discussed in detail in Kostaras et al., 2014). Considering the toxicity associated with higher doses of DEX and the wide range of doses used in human patients, we used 10 mg/kg (Kamoun et al., 2009). First, we confirmed that 10 mg/kg DEX was not toxic in non-tumour-bearing mice when given at that dose for 7 days and followed for 45 days (data not shown). Next, tumour-bearing mice were imaged by BLI and then randomized into either vehicle or DEX-treated cohorts (daily DEX intraperitoneally at 10 mg/kg) and followed for 48 h (Fig. 6A). Vehicle-treated mice showed an increase in BLI signal (P = 0.35, Fig. 6B) while there was a decrease in BLI with DEX treatment (P = 0.0006, Fig. 6B). In our model, glioma cells express high levels of Olig2, which can be used to distinguish the bulk tumour from stromal cells (Helmy et al., 2012). To specifically address the anti-proliferative effect of DEX on tumour cells, we utilized immunofluorescence double staining for Olig2 and PCNA (Fig. 6C) and showed a specific decrease in tumour cell proliferation in DEX-treated mice (P < 0.0001, Fig. 6D), which was also confirmed with Ki67 staining, a second marker of proliferation (Supplementary Fig. 1A). Comparing areas of necrosis and levels of the pro-apoptotic cleaved caspase 3 revealed no significant differences between groups, confirming that the decrease in proliferation was due to a cytostatic effect and not due to overall cell loss (Supplementary Fig. 1B).
Figure 6.

DEX suppresses proliferation of glioma cells in PDGF-driven murine gliomas. Representative bioluminescence images (BLI) of individual Ntv-a/Ef-Luc mice with PDGF-driven gliomas treated with either vehicle or with DEX at 10 mg/kg (B). DEX-treated mice received three total doses: at Day 0 (immediately after BLI), Day 1, and Day 2 (1 h pre-BLI). The bar graph shows that DEX treatment significantly decreased BLI output whereas vehicle treatment did not. Each group had n = 3 mice, values were normalized to day 0. (C) Representative tumour sections and (D) quantification of glioma proliferation using Olig2 expression as a glioma cell marker and PCNA as a proliferative marker. Data in upper panel are presented as the % of PCNA positive cells in total (based on nuclei count by DAPI per field) and the lower panel is the % of PCNA/Olig2 double positive cells in total PCNA positive cells per field. P-values were calculated by paired ANOVA analyses within each group, **P < 0.01, ***P < 0.001 for B by unpaired Student’s t-test, ****P < 0001 for D. Scale bars = 50 μm in C.
To further characterize the effects of DEX on proliferation in vitro, we generated mouse primary glioma cell cultures. When different doses of DEX were tested in brain tumour patients, the average concentration of DEX in tumour tissue was ∼225 ng/g (∼225.27 ng/ml) (Nestler et al., 2002). We chose three concentrations of DEX (39.5 ng/ml = 0.1 μM, 395 ng/ml = 1 μM and 3950 ng/ml = 10 μM). In contrast to the results of tumours in vivo, none of the murine cell lines tested showed a significant decrease in growth or viability (Supplementary Fig. 2A). Next, we compared the cell cycle profile of primary cultures treated with or without DEX (Supplementary Fig. 2B). There was a marginal increase in G1 and a marginal decrease in G2, which may explain why we did not observe a difference in growth or viability. Taken together, these assays suggest that the in vivo effects of DEX are not recapitulated in vitro, highlighting the importance of using an in vivo system with an intact tumour microenvironment. These data imply that the in vivo effect of DEX on tumour cell proliferation is probably indirect.
Effects of dexamethasone and anti-VEGFA antibody on oedema and in vivo glioma growth
It has been well-documented that inhibition of VEGF signalling, either with neutralizing antibodies or VEGFR-targeted kinase inhibitors decreases oedema, resulting in improved survival of brain tumour-bearing mice despite persistent tumour growth (Gerstner et al., 2009; Kamoun et al., 2009). Therefore, we examined the tumour size at onset of neurological signs, such as seizure, lethargy, or weight loss, of vehicle-, DEX-, and B20-4.1.1-treated animals, reasoning that effective oedema treatment would permit larger tumours at the time of onset of signs. Based on MRI-determined tumour volume, asymptomatic glioma-bearing mice were randomized into vehicle, DEX or B20-4.1.1 treatment groups. Animals were treated with their respective agent and subsequently reimaged at time of sign development. Although DEX treatment alone offered no survival advantage, DEX-treated animals had significantly larger tumours at the time of sign onset (Fig. 7B). Despite controlling oedema and permitting mice to remain with no signs with larger tumours, there was no survival advantage to DEX treatment alone (Fig. 7A). Interestingly, in B20-4.1.1-treated animals we saw a significant survival advantage as well as significantly larger tumours at the end point of survival than vehicle-treated mice (Fig. 7C and D). This was also true when quantified by T1-contrast enhancement (Supplementary Fig. 3B).
Figure 7.

Effects of anti-VEGF versus DEX treatment on blood vessel density, leakiness, tumour size and survival. (A and C) Kaplan-Meier survival curves for (A) DEX treatment versus vehicle (n = 7 and 7 for vehicle and DEX, respectively) and (C) B20-4.1.1 versus vehicle (n = 14 and 15, respectively). Groups were matched based on initial asymptomatic T2-MRI tumour volume. (B and D) Representative images and quantification of T2-weighted MRI scans at time of symptom development for (B) vehicle- versus DEX treated mice (n = 6 and 6, respectively) and (D) vehicle- and B20-4.1.1-treated mice (n = 4 and 5, respectively). (E) Representative images and quantification (F) of CD31 staining of a tumour region after the administration of vehicle or DEX. (G) Representative images and quantification (H) of CD31 staining of a tumour region after the administration of vehicle or B20-4.1.1 (I) Illustration and representative images of a Hoechst dye leakage assay and functional vessel labelling with circulating FITC-conjugated lectin in vehicle- and a B20-4.1.1-treated tumours. (F) Corresponding quantification of Hoechst-positive area in B20-4.1.1-treated tumours compared to vehicle-treated tumours. P-values were calculated using an unpaired Student’s t-test, *P < 0.05 **P < 0.01, ****P < 0.0001: Scale bars = 50 μm for A and C and 100 μm for E. OS = overall survival.
The effects of dexamethasone and anti-VEGFA antibody on tumour vasculature and associated myeloid cell infiltration
As both DEX and B20-4.1.1 were effective in controlling oedema, we next investigated their effects on tumour vasculature by immunohistochemistry. Sections of mouse gliomas treated with vehicles and either B20-4.1.1 or DEX were stained with anti-CD31 antibody, which specifically labels endothelial cells of glioblastoma and normal blood vessels outside of the tumour area. In DEX-treated tumours we saw no changes either in total vessel area or average vessel size compared to vehicle (Fig. 7E and F). B20-4.1.1 tumours had decreases in total vessel area (P < 0.0001) and average vessel size (P = 0.0076), while no differences were observed in non-tumour cortical areas (Fig. 7G and H, and Supplementary Fig. 3A). Decrease in total vessel area and average vessel size induced by B20-4.1.1 was transient as vessels returned to their initial morphology when B20-4.1.1 was stopped (data not shown). We further investigated the effect of B20-4.1.1 on tumour vasculature using Hoechst dye leakage assays and functional vessel labelling with circulating FITC-conjugated lectin in vehicle- and B20-4.1.1-treated tumours (Fig. 7I and J). B20-4.1.1 decreased leaky areas compared to vehicle (Fig. 7J) (P = 0.002).
DEX was suggested to be immunosuppressant, as are radiotherapy and TMZ (Ellsworth and Grossman, 2015), and a recent correlative study suggested that DEX treatment-induced immune suppression could interfere with clinical efficacy of standard therapy in recurrent glioblastoma (Wong et al., 2015). Microglia/macrophages are the major immune infiltrates in both murine and human glioblastoma, accounting for up to 20–30% of the total tumour mass (Feng et al., 2015; Hambardzumyan et al., 2015).
To investigate potential immunosuppressive characteristics of DEX we looked at the numbers of infiltrated tumour-associated microglia/macrophages in response to either DEX or VEGFA antibody treatment using the myeloid lineage marker Iba1. No difference was observed between DEX-treated and vehicle-treated tumours (Supplementary Fig. 3C), whereas we observed a significant increase in Iba1 positive microglia/macrophage infiltration in B20-4.1.1 treated mice when compared to vehicle (Supplementary Fig. 3C).
Anti-VEGFA antibody B20-4.1.1 decreases glioma proliferation and induces tumour cell death
In our mouse model, DEX treatment alone offers no survival advantage whereas B20-4.1.1 alone consistently prolonged survival (Figs 4A, D and 7A). To examine potential effects on proliferation, we treated tumour-bearing mice with vehicle or B20-4.1.1 for 2 weeks (5 mg/kg B20-4.1.1 twice a week) and compared the proliferation of tumour cells by double staining for Olig2 and PCNA. We observed a significant decrease in proliferation of tumour cells in B20-4.1.1-treated samples (P < 0.0001, Fig. 8A and B). Similarly, we confirmed this reduction in both Ki67 staining and in pH3 staining (data not shown).
Figure 8.

B20-4.1.1 decreases proliferation and increases cell death in murine PDGF-driven gliomas. (A) Representative tumour sections and (B) quantification bar graphs of glioma proliferation using Olig2 expression as a glioma cell marker and PCNA as a proliferative marker. There was a significant reduction in the proliferation of glioma cells after B20-4.1.1 treatment. (C) Representative images of Olig2 staining in tumour sections showing decreased staining in B20-4.1.1-treated mice compared to vehicle-treated mice. (D) Representative images of TUNEL staining in vehicle- and B20-4.1.1-treated tumours and (E) corresponding quantification of Olig2-positive area (F) and TUNEL-positive cells in response to B20-4.1.1 treatment. P-values were calculated by an unpaired Student’s t-test, *P < 0.05, ***P < 0.001, ***P < 0.0001. Scale bars = 50 μm for A, C and D.
Next, we examined whether B20-4.1.1 promotes cell death in gliomas. B20-4.1.1 treated tumours had a decrease in the Olig2-positive tumour area (P = 0.02, Fig. 8C and E) and an increase in apoptotic cells, as evidence by increased terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL)-positivity (P = 0.02, Fig. 8D and F). To address whether it is only tumour cells or other non-neoplastic cells in the tumour microenvironment that are affected by B20-4.1.1 treatment, we performed immunofluorescence double staining for TUNEL and the tumour-specific marker Olig2, the endothelial cell-specific marker CD31, the microglia/macrophage-specific marker Iba1, or the reactive astrocyte-specific marker GFAP. In vehicle-treated tumours, 1.01% of TUNEL-positive cells were also Olig2-positive, which was significantly increased to 3.55% in B20-4.1.1-treated mice (P = 0.03) (Supplementary Fig. 4A and B). We did not observe significant changes in the number of TUNEL-positive cells from the tumour microenvironment (Supplementary Fig. 4A and B, and data not shown)
Discussion
The administration of steroids to control neurological morbidity associated with brain tumours has been established as a standard of care decades ago. Except for primary CNS lymphoma where steroids exert direct cytotoxic effects, amelioration of brain tumour-association oedema has been proposed to underlie these symptomatic effects of steroids. However, these undisputed beneficial effects of steroids have to be weighed against a plethora of acute and long-term side effects.
In the current study, we identified the use of corticosteroids early in the disease course, during radiotherapy without or with alkylating chemotherapy, as an independent predictor of poor outcome in three independent patient cohorts. Although higher steroid doses are commonly given to patients with larger tumours and more prominent neurological deficits, we tried to control for such unfavourable confounders as feasible. Yet, we cannot exclude that detrimental effects of steroids other than direct interference with radiotherapy contributed to the overall inferior outcome of patients exposed to steroids. Thus, steroids can contribute to morbidity and mortality through their direct toxicity, including steroid myopathy, impaired immune function, adrenal insufficiency, and bowel perforation. Our findings with three large independent datasets were further supported with a recent correlative retrospective analysis of 73 patients with glioblastoma showing that DEX use during radiotherapy with concurrent TMZ correlated with reduced overall survival and progression-free survival (Shields et al., 2015).
In support of a direct detrimental effect interfering with the activity of radiotherapy, we report that pretreatment with DEX decreased the survival benefit afforded by radiotherapy in murine gliomas. In these tumours, DEX decreased proliferation and the expression of many cell cycle-related genes. Expression of these genes inversely correlated with survival in the TCGA glioblastoma patient dataset. Of note, these genes are primarily known or predicted to be involved in proliferation, either via cell mitotic assembly, cycle checkpoints, DNA damage response and ATM signalling. Radiotherapy sensitivity varies with the position of the cells in the cell cycle, and in general, tumours with high cell turnover rates are the most radiosensitive (Kempf et al., 2013). Specifically, the p21 protein is induced by DEX in glioma cells, slows cell cycle progression and may confer cytoprotection (Naumann et al., 1998; Glaser et al., 2001; Ueda et al., 2004). Further studies are warranted to more precisely establish interactions between DEX and these cell types and how these might influence the disease course.
In pursuit of alternatives to DEX, we demonstrated that short-term treatment with a low dose of VEGF antibody results in decrease in the total vessel area and the average vessel size, decreased leakage, and control of oedema signs, permitting mice to be with no signs from intracranial tumors with larger tumor volumes. Moreover, VEGF antibody treatment did not interfere with the efficacy of radiotherapy. Our data support the use of anti-VEGF agents as an alternative oedema management strategy for glioblastoma patients. Of note, although the VEGF antibody bevacizumab did not prolong survival in newly diagnosed glioblastoma patients when added to standard of care, it nevertheless strongly prolonged progression-free survival which has been attributed in part to a confounding effect on imaging (Verhoeff et al., 2009). Yet, these data altogether provide ample evidence that VEGF antagonism does not compromise the efficacy of radiotherapy in human patients in vivo. Moreover, a retrospective analysis of the AVAglio data demonstrated that patients with IDH1 wild-type proneural glioblastoma might even derive a survival benefit from bevacizumab in that setting (Sandmann et al., 2015).
In conclusion, given that controlled clinical trials to address the steroid question in glioblastoma are unlikely to be ever performed, we believe that our retrospective clinical data and corresponding data from animal models provides the strongest evidence so far against the traditional, often uncritical use of steroids in brain tumour patients.
Supplementary Material
Acknowledgements
We would like to thank Paul Gambon and Dr Xi Feng for technical assistance. We are grateful to Dr Judy Drazba from the Imaging Core at the Lerner Research Institute for technical advices and to Dr Grahame Kidd for confocal imaging. We are also grateful to the staff at the small-animal imaging core at CWRU. We are grateful to Dr Jaekeun Park at Small Animal Imaging Core at Emory University School of Medicine for assistance with MRI scans. We thank Genentech for B20-4.1.1 supply and Dave Schumick BS CMI for graphical abstract. We acknowledge the support of all staff involved in EORTC 26981-22981 NCIC CE.3 and at the clinical sites of the German Glioma Network.
Glossary
Abbreviations
- BLI =
bioluminescence imaging
- DEX =
dexamethasone
- TMZ =
temozolomide
- TMZ/RT→TMZ =
temozolomide and radiotherapy treatment followed by temozolomide
- TUNEL =
terminal deoxynucleotidyl transferase dUTP nick end labelling
- VEGF =
vascular endothelial growth factor
Funding
NIH grants (E.C.H.) R01 CA100688, U01 CA160882 (D.H.), U54 CA163167, U54 CA143798, U01 CA141502-01; (T.S.J.) K08 CA163765, (K.L.P.) MSTP GM07739, F31 NS076028. M.C. was supported by a MSKCC Brain Tumor Center Medical Student Summer Fellowship.
Supplementary material
Supplementary material is available at Brain online.
References
- Becher OJ, Hambardzumyan D, Fomchenko EI, Momota H, Mainwaring L, Bleau AM, et al. Gli activity correlates with tumor grade in platelet-derived growth factor-induced gliomas. Cancer Res 2008; 68: 2241–9. [DOI] [PubMed] [Google Scholar]
- Eisenberg HM, Barlow CF, Lorenzo AV. Effect of dexamethasone on altered brain vascular permeability. Arch Neurol 1970; 23: 18–22. [DOI] [PubMed] [Google Scholar]
- Ellsworth S, Grossman SA. Comment on ‘Dexamethasone exerts profound immunologic interference on treatment efficacy for recurrent glioblastoma'. Br J Cancer 2015; 113: 1632–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng X, Szulzewsky F, Yerevanian A, Chen Z, Heinzmann D, Rasmussen RD, et al. Loss of CX3CR1 increases accumulation of inflammatory monocytes and promotes gliomagenesis. Oncotarget 2015; 6: 15077–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstner ER, Duda DG, di Tomaso E, Ryg PA, Loeffler JS, Sorensen AG, et al. VEGF inhibitors in the treatment of cerebral edema in patients with brain cancer. Nat Rev Clin Oncol 2009; 6: 229–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser T, Wagenknecht B, Weller M. Identification of p21 as a target of cycloheximide-mediated facilitation of CD95-mediated apoptosis in human malignant glioma cells. Oncogene 2001; 20: 4757–67. [DOI] [PubMed] [Google Scholar]
- Gorlia T, van den Bent MJ, Hegi ME, Mirimanoff RO, Weller M, Cairncross JG, et al. Nomograms for predicting survival of patients with newly diagnosed glioblastoma: prognostic factor analysis of EORTC and NCIC trial 26981-22981/CE.3. Lancet Oncol 2008; 9: 29–38. [DOI] [PubMed] [Google Scholar]
- Grasso RJ, Johnson CE, Boler RK, Moore NA. Combined growth-inhibitory responses and ultrastructural alterations produced by 1,3-bis(2-chloroethyl)-1-nitrosourea and dexamethasone in rat glioma cell cultures. Cancer Res 1977; 37: 585–94. [PubMed] [Google Scholar]
- Hambardzumyan D, Amankulor NM, Helmy KY, Becher OJ, Holland EC. Modeling adult gliomas using RCAS/t-va technology. Transl Oncol 2009; 2: 89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambardzumyan D, Becher OJ, Rosenblum MK, Pandolfi PP, Manova-Todorova K, Holland EC. PI3K pathway regulates survival of cancer stem cells residing in the perivascular niche following radiation in medulloblastoma in vivo. Genes Dev 2008; 22: 436–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambardzumyan D, Gutmann DH, Kettenmann H. The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci 2015; 19: 20–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmy K, Halliday J, Fomchenko E, Setty M, Pitter K, Hafemeister C, et al. Identification of global alteration of translational regulation in glioma in vivo. PloS One 2012; 7: e46965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamoun WS, Ley CD, Farrar CT, Duyverman AM, Lahdenranta J, Lacorre DA, et al. Edema control by cediranib, a vascular endothelial growth factor receptor-targeted kinase inhibitor, prolongs survival despite persistent brain tumor growth in mice. J Clin Oncol 2009; 27: 2542–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempf H, Hatzikirou H, Bleicher M, Meyer-Hermann M. In silico analysis of cell cycle synchronisation effects in radiotherapy of tumour spheroids. PLoS Comp Bio 2013; 9: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokunai T, Tamaki N. Relationship between expression of p21WAF1/CIP1 and radioresistance in human gliomas. Jpn J Cancer Res 1999; 90: 638–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostaras X, Cusano F, Kline GA, Roa W, Easaw J. Use of dexamethasone in patients with high-grade glioma: a clinical practice guideline. Curr Oncol 2014; 21: e493–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotsarini C, Griffiths PD, Wilkinson ID, Hoggard N. A systematic review of the literature on the effects of dexamethasone on the brain from in vivo human-based studies: implications for physiological brain imaging of patients with intracranial tumors. Neurosurgery 2010; 67: 1799–815; discussion 815. [DOI] [PubMed] [Google Scholar]
- Koutcher JA, Hu X, Xu S, Gade TP, Leeds N, Zhou XJ, et al. MRI of mouse models for gliomas shows similarities to humans and can be used to identify mice for preclinical trials. Neoplasia 2002; 4: 480–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naumann U, Durka S, Weller M. Dexamethasone-mediated protection from drug cytotoxicity: association with p21WAF1/CIP1 protein accumulation? Oncogene 1998; 17: 1567–75. [DOI] [PubMed] [Google Scholar]
- Nestler U, Winking M, Boker DK. The tissue level of dexamethasone in human brain tumors is about 1000 times lower than the cytotoxic concentration in cell culture. Neurol Res 2002; 24: 479–82. [DOI] [PubMed] [Google Scholar]
- Parr MJ, Manome Y, Tanaka T, Wen P, Kufe DW, Kaelin WG, Jr., et al. Tumor-selective transgene expression in vivo mediated by an E2F-responsive adenoviral vector. Nat Med 1997; 3: 1145–9. [DOI] [PubMed] [Google Scholar]
- Piette C, Munaut C, Foidart JM, Deprez M. Treating gliomas with glucocorticoids: from bedside to bench. Acta Neuropathol 2006; 112: 651–64. [DOI] [PubMed] [Google Scholar]
- Powell SN, Abraham HE. The Biology of radioresistance: similarities, differences and interactions with drug resiastance In: Clynes M, editor. Multiple drug resistance in cancer: cellular, molecular and clinial approaches. 1994. p. 325–47. [DOI] [PubMed] [Google Scholar]
- Raslan A, Bhardwaj A. Medical management of cerebral edema. Neurosurg Focus 2007; 22: E12. [DOI] [PubMed] [Google Scholar]
- Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J 2008; 22: 659–61. [DOI] [PubMed] [Google Scholar]
- Rovit RL, Hagan R. Steroids and cerebral edema: the effects of glucocorticoids on abnormal capillary permeability following cerebral injury in cats. J Neuropathol Exp Neurol 1968; 27: 277–99. [PubMed] [Google Scholar]
- Sandmann T, Bourgon R, Garcia J, Li C, Cloughesy T, Chinot OL, et al. Patients with proneural glioblastoma may derive overall survival benefit from the addition of bevacizumab to first-line radiotherapy and temozolomide: retrospective analysis of the AVAglio trial. J Clin Oncol 2015; 33: 2735–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields LB, Shelton BJ, Shearer AJ, Chen L, Sun DA, Parsons S, et al. Dexamethasone administration during definitive radiation and temozolomide renders a poor prognosis in a retrospective analysis of newly diagnosed glioblastoma patients. Radiat Oncol 2015; 10: 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stupp R, Hegi ME, Gorlia T, Erridge SC, Perry J, Hong YK, et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071-22072 study): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol 2014; 15: 1100–8. [DOI] [PubMed] [Google Scholar]
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005; 352: 987–96. [DOI] [PubMed] [Google Scholar]
- Ueda S, Mineta T, Nakahara Y, Okamoto H, Shiraishi T, Tabuchi K. Induction of the DNA repair gene O6-methylguanine-DNA methyltransferase by dexamethasone in glioblastomas. J Neurosurg 2004; 101: 659–63. [DOI] [PubMed] [Google Scholar]
- Uhrbom L, Nerio E, Holland EC. Dissecting tumor maintenance requirements using bioluminescence imaging of cell proliferation in a mouse glioma model. Nat Med 2004; 10: 1257–60. [DOI] [PubMed] [Google Scholar]
- Verhoeff JJ, van Tellingen O, Claes A, Stalpers LJ, van Linde ME, Richel DJ, et al. Concerns about anti-angiogenic treatment in patients with glioblastoma multiforme. BMC Cancer 2009; 9: 444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Li M, Rinehart JJ, Zhang R. Pretreatment with dexamethasone increases antitumor activity of carboplatin and gemcitabine in mice bearing human cancer xenografts: in vivo activity, pharmacokinetics, and clinical implications for cancer chemotherapy. Clin Cancer Res 2004; 10: 1633–44. [DOI] [PubMed] [Google Scholar]
- Weller M, Schmidt C, Roth W, Dichgans J. Chemotherapy of human malignant glioma: prevention of efficacy by dexamethasone? Neurology 1997; 48: 1704–9. [DOI] [PubMed] [Google Scholar]
- Wong ET, Lok E, Gautam S, Swanson KD. Dexamethasone exerts profound immunologic interference on treatment efficacy for recurrent glioblastoma. Br J Cancer 2015; 113: 1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}