

Abstract
Almost 20 incurable neurodegenerative disorders are caused by trinucleotide repeat (TNR) expansion beyond a certain threshold, with disease time of onset and severity positively correlating with repeat length. Typically, long TNRs display a bias toward further expansion and repeats continue to expand not only during germline transmissions from parents to offspring, but also remain highly unstable in somatic tissues of patients. Hence, understanding TNR instability mechanisms sheds light on underlying disease pathology. Recently, we showed that activated ATR is the major signal for convergent-transcription-induced cell death at CAG repeats and is regulated by the mismatch repair (MMR) pathway. Additionally, components of other DNA repair pathways such as transcription-coupled nucleotide excision repair (TC-NER) and R-loop resolution by RNaseH reduce cell death. Because activated ATR signals the Fanconi anemia (FA) pathway of interstrand crosslink DNA repair, we asked whether the FA pathway also modulates convergent-transcription-induced cell death at expanded CAG repeats. We show here that siRNA knockdown of FA components—FANCI, FANCJ, FANCM, FANCA, and FANCD2—decreases cell death, suggesting that FA proteins, like MMR proteins, are activators of cell death during convergent transcription.
Keywords: Fanconi anemia, convergent transcription, trinucleotide repeat instability, neurodegenerative diseases, cell death
Introduction
Trinucleotide repeats (TNRs) are hypermutable, microsatellite sequences, capable of gaining or losing repeat units at a high frequency [1]. TNRs are distributed throughout genes, in exons, introns, and 5′ and 3′-UTRs [2]. Normally, variations in TNR tract length—repeat instability—acts to fine tune gene expression with attendant evolutionary benefits [3, 4]. However, their continued expansion beyond a certain threshold results in neurodegenerative disorders [5]. Repeat instability occurs in both the germline and somatic tissues of affected individuals, where their continued instability in different tissues exacerbates disease symptoms [6-10]. Disease-associated TNRs tend to form intramolecular secondary structures during DNA metabolic processes, and these structures then become the substrates for toxic DNA damage responses. Therefore, understanding the underlying molecular mechanisms that drive instability at expanded repeats will shed light on disease pathogenesis.
In the past, studies in model organisms have suggested that a range of DNA metabolic processes such as replication, transcription, DNA repair, genome wide demethylation, and rereplication modulate TNR instability [7, 11-17]. Additionally, we showed that both transcription and convergent-transcription across long CAG repeats induce repeat instability in human cells, where DNA repair pathways played a vital modulatory role [12, 18]. More importantly, convergent transcription across expanded repeats was also found to cause a synergistic increase in cell death, which we have termed DNA toxicity to emphasize the role of the repeat DNA sequences in cell death. Convergent transcription also induces DNA toxicity across other expanded TNRs, including GAA, CGG, and CCTG, signifying the commonality of repeat-dependent, convergent-transcription-induced cell death [19]. Because convergent transcription is common in the human genome, including many TNR disease genes [20-24], DNA toxicity may contribute to neuronal cell death in neurodegenerative patients.
Previously, we showed that during convergent transcription at long CAG repeats, ATR (ataxia-telangiectasia mutated [ATM] and Rad3 related) DNA damage response is activated and is the key mediator of cell death [18]. Additionally, we found that TC-NER and R-loop resolution enzymes lower DNA toxicity of convergent transcription, whereas MMR components increase cell death by activating ATR DNA damage response during convergent transcription [25-28]. Because activated ATR is an important trigger for the Fanconi anemia (FA) pathway to repair interstrand DNA crosslinks [29], we asked whether FA components could also modulate convergent-transcription-induced cell death at CAG repeats.
FA pathway comprises 19 distinct functional complementation groups that collaborate to repair interstrand DNA crosslinks [30]. Mutations in FA genes result in a chromosomal instability disorder, which in patients is characterized by developmental deformities, bone marrow failure, and cancer predisposition [31]. In response to crosslinking agents such as diepoxybutane (DEB), mitomycin C (MMC), and cisplatin, the FA core complex (FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, FANCL, FANCM and FANCT) forms at the damage site [30]. This complex enhances ATR activation by localizing ATRIP to the damage site [32]. Activated ATR phosphorylates FANCI protein, which is the major switch for the FA pathway [33]. Phosphorylated FANCI enables monoubiquitination of FANCD2, which in turn maintains FANCI monoubiquitination [34]. Together, the FANCI-D2 complex enables incision and recruitment of downstream recombinational and nucleolytic proteins (FANCD1, FANCJ, FANCN, FANCO, FANCP, FANCQ, FANCR and FANCS), along with a translesion synthesis enzyme to complete the repair [35-37].
In this study, we show that knockdown of FA components—FANCI, FANCJ, FANCM, FANCA, and FANCD2—during convergent transcription across CAG repeats suppresses cell death. This novel result suggests that the FA pathway normally is involved in enhancing convergent-transcription-induced cell death at CAG repeat tracts.
Materials and Methods
Cell Lines and Culture
DIT7 cells used in this study were derived from HT1080 via the intermediate RS11 cell line, as described previously [18]. Briefly, DIT7 cells carry a single integrated copy of an HPRT minigene that carries a CAG95 tract in its intron and is flanked by the promoters pTRE-CMVmini and pNERB-X1, which drive sense and antisense transcription, respectively. The doxycycline-inducible pTRE-CMVmini promoter contains a downstream binding site for the rtTA protein, which is a fusion construct of reverse tetracycline repressor and the HSV VP16 transcription activation domain. The pNERB-X1 promoter is inducible by RSL1 (Rheoswitch ligand 1, NEB) (Figure 1). DIT7-R103 containing 15 repeat units was derived from DIT7 by spontaneous contraction of its 95 repeat units. The cells were grown at 37°C with 5% CO2 in DMEM/F-12 medium (Gibco) supplemented with 10% fetal bovine serum (Hyclone, Thermo Scientific) and 1% MEM nonessential amino acids (Gibco). 0.25% trypsin-EDTA (Gibco) was used for passaging.
Figure 1. Schematics of the HPRT minigenes in the DIT7 and DIT7-R103 cell lines.
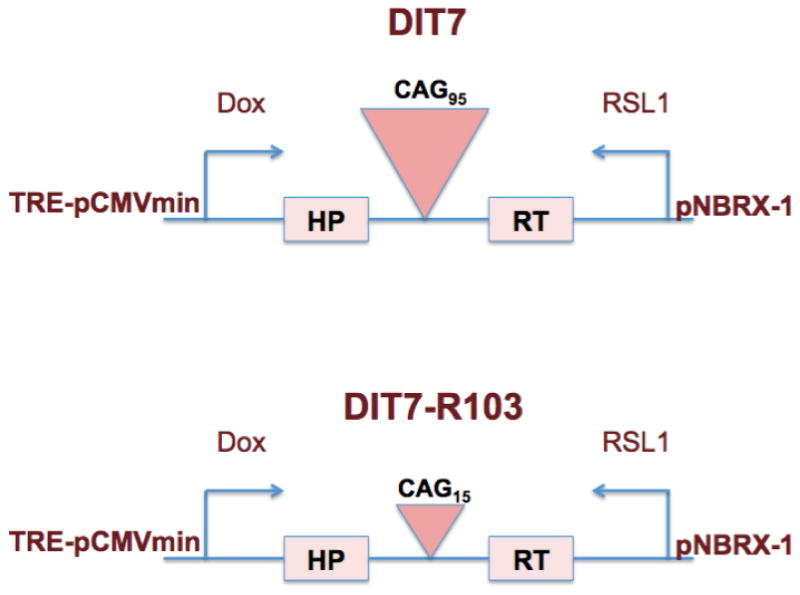
The HPRT minigene in DIT7 cells and DIT7-R103 cells carry 95 and 15 units of CAG repeat tract respectively within the 2.1 kb long intron. The promoters pTRE-CMVmini in the sense direction and pNEBR-X1 in the antisense direction are regulated by doxycycline and RSL1, respectively.
Experimental Outline and siRNA treatments
To test the effects of convergent transcription on CAG repeats, 100,000 DIT7 or DIT7-R103 cells were plated on day -1 in a 6-well plate. At day 0, (24 hours after plating) the inducers doxycycline and RSL1 were added to the medium at a concentration of 2 μg/mL and 500 nM, respectively. Doxycycline was added daily at a concentration of 1 μg/mL because the half-life of doxycycline is 24 hours. No additional RSL1 was added after day 1. On day 5, viable and dead cells were counted.
For siRNA treatments, on day -3, 100,000 DIT7 or DIT7-R103 cells were plated. Then on day -2, siRNAs at a final concentration of 200 nM were transfected using Oligofectamine (Invitrogen), per the manufacturer's protocol. For single gene knockdowns, a target-specific siRNA and a control vimentin siRNA (which has no effect on cells) were added to a final concentration of 100 nM each. For double knockdowns, 100 nM of each target-specific siRNA was added to a final concentration of 100 nM. In all cases, the final siRNAs concentrations were 200 nM. The siRNA sequences (Dharmacon Thermo Scientific) used in this study are listed in Table 1. A second round of siRNA treatment was administered after 48 hours (Day 0) and inducers—doxycycline and RSL1—were added at 2 μg/mL and 500 nM, respectively. From day 1 through day 4, additional doxycycline was added at a concentration of 1 μg/mL. Knockdown efficiencies were evaluated by isolating RNA on day 1 and measuring percentage knockdown by real time RT-PCR, as described previously [38]. At least 70% knockdown of gene expression was achieved by targeted siRNA treatment.
Table 1. Sequences of siRNAs used for gene knockdown.
| Gene | siRNA sequence |
|---|---|
| Vimentin | GAAUGGUACAAAUCCAAGU |
| FANCI | CTGGCTAATCACCAAGCTTAA |
| FANCJ | GUACAGUACCCCACCUUAU |
| FANCA | GGAAGATATCCTGGCTGGCACTCTT |
| FANCM | AAGCTCATAAAGCTCTCGGAA |
| FANCD2 | AATAGACGACAACTTATCCATCACC |
Dead Cell Measurements
Dead cell percentages were calculated by dividing nonadherent or floating cells, by the total number of cells (the sum of adherent and nonadherent cells). Adherent cells are defined as viable cells, and nonadherent cells are the dead cells [18]. Previously, we confirmed the viability of adherent cells by using propidium iodide (a dye retained in dead cells only), which was incorporated in <4% of adherent cells, indicating the >96% of the adherent cells were viable [18]. The very small percentage of dead cells present in the adherent population was ignored in the final calculations. Both floating and adherent cells were counted on day 5.
Statistics
Student's t test was used to calculate statistical significance of means and standard deviations.
Results and Discussion
In this study we used two human cell lines—one carrying an HPRT minigene with 95 CAG units in its intron (DIT7), and the other, with 15 CAG units (DIT7-R103)—to test the effects of convergent transcription on CAG repeat tracts. The modified HPRT minegene has a doxycycline-inducible cytomegalovirus (CMV) promoter pTRE-CMVmini that controls sense transcription from 5′ side and a RSL1-inducible pNEBRX1 promoter controls antisense transcription from 3′ side (Figure 1). Transcription induction at both the sense and antisense promoters causes a synergistic increase in cell death at diverse TNRs, triggered by an activated ATR DNA damage response [18, 19, 39]. DNA repair pathways modulate this DNA toxicity effect, with an additional role of mismatch repair (MMR) pathway in regulating ATR activation during convergent transcription [13, 28].
Typically, ATR activation entails the presence of RPA coated ssDNA or unligated nicks, along with activating factors such as ATRIP, the 9-1-1 complex, and TopBP1. Once activated, ATR phosphorylates downstream proteins—Chk1, Nbs1, SMC1, and p53, thus enabling repair, cell cycle arrest, and apoptosis [40-43]. In addition, activated ATR is also the major switch activating FANCI by phosphorylation in instances of interstrand crosslink DNA damage [33]. Phosphorylated FANCI triggers FANCD2 monoubiquitination and together, as the FANCI-D2 complex, initiate the Fanconi anemia repair pathway.
Because activated ATR could potentially activate the FA cascade at CAG repeats, we sought to establish the role of FA pathway during convergent-transcription-induced cell death at CAG repeats, using siRNA-mediated knockdowns of key FA components. Initially, we tested the effects of knockdowns of FANCI and FANCD2, the two components of the FANCI-D2 complex, which instigates downstream repair, following assembly of the FA core complex. We found that FANCI knockdown and FANCD2 knockdown each significantly reduced the level of cell death induced by convergent transcription in DIT7 cells and in DIT7-R103 cells (Figure 2 and 3; Table 2). These results indicate that the FANCI-D2 complex normally acts to induce cell death during convergent transcription across CAG repeats. It seems likely that convergent transcription activates ATR kinase, which then phosphorylate FANCI, activating the FA pathway; however, this hypothesis must be tested in future studies.
Figure 2. The effect of knockdown of FA pathway genes in DIT7 cells on cell death induced by convergent transcription.
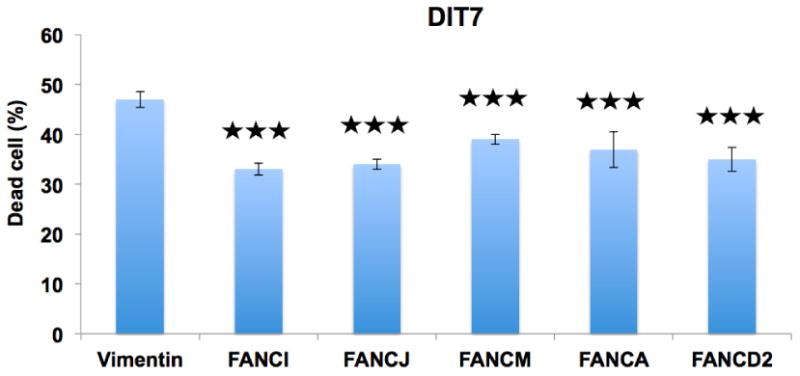
The graph shows the percentage of dead DIT7 cells (95 CAG repeat units) generated by convergent transcription following siRNA-mediated knockdowns. The frequencies of dead cells in each case were: vimentin (47%), FANCI (33%), FANCD2 (35%), FANCA (37%), FANCM (39%), and FANCJ (34%). The siRNAs sequences are shown in Table 1. Data are the average of 2 independent siRNA knockdown experiments each with 3 replicates. Error bars represent standard deviations. P values are indicated: ★★★P<0.001.
Figure 3. The effect of knockdown of FA pathway genes in DIT7-R103 cells on cell death induced by convergent transcription.
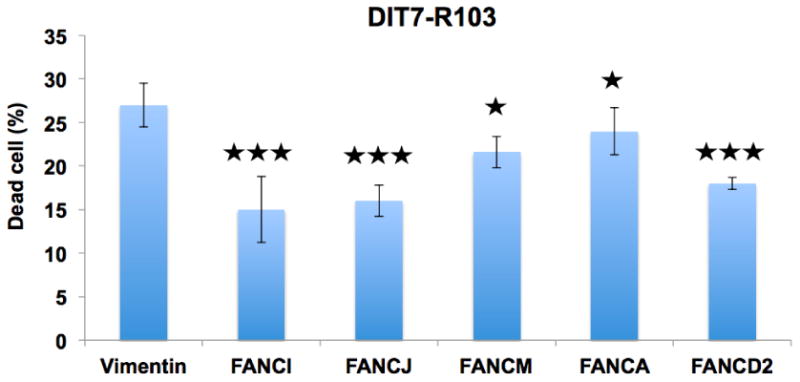
The graph shows the percentage of dead DIT7-R103 cells (15 CAG repeat units) generated by convergent transcription following siRNA-mediated knockdowns. The frequencies of dead cells in each case were: vimentin (27%), FANCI (15%), FANCD2 (18%), FANCA (24%), FANCM (22%), and FANCJ (16%). The siRNAs sequences are shown in Table 1. Data are the average of 2 independent siRNA knockdown experiments each with 3 replicates. Error bars represent standard deviations. P values are indicated: ★P<0.05, ★★★P<0.001.
Table 2. Decrease in percentage of dead cells after siRNA treatment.
| siRNA treatment | Decrease in cell death %a | |
|---|---|---|
| DIT7 | DIT7-R103 | |
| Vimentin | 0 | 0 |
| FANCI | 30 | 44 |
| FANCJ | 28 | 42 |
| FANCM | 16 | 20 |
| FANCA | 21 | 12 |
| FANCD2 | 25 | 37 |
The percentage change in cell death after specific siRNA treatment was calculated as {[(% dead cells after vimentin siRNA) – (% dead cells after specific siRNA)] / (% dead cells after vimentin siRNA )} (100%)
Next, we tested the knockdown of two critical FA core components—FANCA and FANCM—to determine whether the FA core complex could also modulate convergent-transcription-induced cell death. It is known that FANCA of the FA core complex enhances binding and localization of ATRIP, which together with RPA allows activation of ATR-TOPBP1 [32]. Thus, if FANCA were to potentiate the ATR pathway, then its knockdown should result in a reduction in cell death from convergent transcription. Similarly, FANCM has a direct role in accumulating RPA and TOPBP1 following DNA damage and thus has a role in ATR activation [44, 45]. Moreover, FANCM, in complex with FAAP24 can signal ATR activation independent of the FA core complex [45, 46]. In this study, we found that knockdown both FANCA and FANCM in DIT7 and DIT7-R103 cells reduced convergent-transcription-induced cell death (Figure 2 and 3; table 2). These results suggest that the FA core complex normally acts to induce cell death during convergent transcription across CAG repeats.
Finally, we tested knockdown of one of the downstream FA proteins involved in interstrand cross link repair, FANCJ, which is a helicase involved in opening the helix so that other proteins can repair the damage [47]. FANCJ knockdown in both DIT7 and DIT7-R103 cells causes a reduction in cell death from convergent transcription induced cell death (Figure 2 and Figure 3; Table 2), suggesting that the helicase activity of FANCJ normally acts to increase DNA toxicity at CAG repeats during convergent transcription. Whether other proteins in the FA pathway are also involved in enhancing cell death at CAG repeats remains to be tested.
Conclusion
In this study, we have shown that the FA pathway proteins, including the FANCI-D2 complex (FANCI and FANCD2), the FA core complex (FANCA and FANCM), and the downstream repair protein (FANCJ), contribute to convergent-transcription-induced cell death at CAG repeats. This novel result suggests that the FA DNA repair pathway is involved in processing aberrant CAG repeat structures that arise during convergent transcription. Mechanistically, we speculate that MMR-mediated ATR DNA damage response triggers the FA pathway via FANCI, but ATR itself may also influence the FA core proteins—FANCA and FANCM. Once the FA pathway is activated via the FANCI-D2 complex, the downstream proteins such FANCJ may aid in processing the CAG repeats and enhance convergent transcription induced cell death.
References
- 1.Richard GF, Kerrest A, Dujon B. Comparative genomics and molecular dynamics of DNA repeats in eukaryotes. Microbiol Mol Biol Rev. 2008;72(4):686–727. doi: 10.1128/MMBR.00011-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kozlowski P, de Mezer M, Krzyzosiak WJ. Trinucleotide repeats in human genome and exome. Nucleic Acids Res. 2010;38(12):4027–39. doi: 10.1093/nar/gkq127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chatterjee N, S B, Wilson J. Microsatellite repeats: canaries in the coal mine in Stress- induced mutagenesis. 2013 [Google Scholar]
- 4.Vinces MD, et al. Unstable tandem repeats in promoters confer transcriptional evolvability. Science. 2009;324(5931):1213–6. doi: 10.1126/science.1170097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Annu Rev Neurosci. 2007;30:575–621. doi: 10.1146/annurev.neuro.29.051605.113042. [DOI] [PubMed] [Google Scholar]
- 6.La Spada AR, Taylor JP. Repeat expansion disease: progress and puzzles in disease pathogenesis. Nat Rev Genet. 2010;11(4):247–58. doi: 10.1038/nrg2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McMurray CT. Mechanisms of trinucleotide repeat instability during human development. Nat Rev Genet. 2010;11(11):786–99. doi: 10.1038/nrg2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dion V. Tissue specificity in DNA repair: lessons from trinucleotide repeat instability. Trends Genet. 2014;30(6):220–9. doi: 10.1016/j.tig.2014.04.005. [DOI] [PubMed] [Google Scholar]
- 9.Pearson CE, Nichol Edamura K, Cleary JD. Repeat instability: mechanisms of dynamic mutations. Nat Rev Genet. 2005;6(10):729–42. doi: 10.1038/nrg1689. [DOI] [PubMed] [Google Scholar]
- 10.Mirkin SM. Expandable DNA repeats and human disease. Nature. 2007;447(7147):932–40. doi: 10.1038/nature05977. [DOI] [PubMed] [Google Scholar]
- 11.Liu G, et al. Replication-dependent instability at (CTG) × (CAG) repeat hairpins in human cells. Nat Chem Biol. 2010;6(9):652–9. doi: 10.1038/nchembio.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin Y, Dion V, Wilson JH. Transcription promotes contraction of CAG repeat tracts in human cells. Nat Struct Mol Biol. 2006;13(2):179–80. doi: 10.1038/nsmb1042. [DOI] [PubMed] [Google Scholar]
- 13.Lahue RS, Slater DL. DNA repair and trinucleotide repeat instability. Front Biosci. 2003;8:s653–65. doi: 10.2741/1107. [DOI] [PubMed] [Google Scholar]
- 14.Liu Y, Wilson SH. DNA base excision repair: a mechanism of trinucleotide repeat expansion. Trends Biochem Sci. 2012;37(4):162–72. doi: 10.1016/j.tibs.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Concannon C, Lahue RS. Nucleotide excision repair and the 26S proteasome function together to promote trinucleotide repeat expansions. DNA Repair (Amst) 2014;13:42–9. doi: 10.1016/j.dnarep.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 16.Chatterjee N, et al. Environmental stress induces trinucleotide repeat mutagenesis in human cells. Proc Natl Acad Sci U S A. 2015;112(12):3764–9. doi: 10.1073/pnas.1421917112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dion V, et al. Genome-wide demethylation promotes triplet repeat instability independently of homologous recombination. DNA Repair (Amst) 2008;7(2):313–20. doi: 10.1016/j.dnarep.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin Y, et al. Convergent transcription through a long CAG tract destabilizes repeats and induces apoptosis. Mol Cell Biol. 2010;30(18):4435–51. doi: 10.1128/MCB.00332-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin WY, Lin Y, Wilson JH. Convergent transcription through microsatellite repeat tracts induces cell death. Mol Biol Rep. 2014;41(9):5627–34. doi: 10.1007/s11033-014-3432-y. [DOI] [PubMed] [Google Scholar]
- 20.Wilburn B, et al. An antisense CAG repeat transcript at JPH3 locus mediates expanded polyglutamine protein toxicity in Huntington's disease-like 2 mice. Neuron. 2011;70(3):427–40. doi: 10.1016/j.neuron.2011.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moseley ML, et al. Bidirectional expression of CUG and CAG expansion transcripts and intranuclear polyglutamine inclusions in spinocerebellar ataxia type 8. Nat Genet. 2006;38(7):758–69. doi: 10.1038/ng1827. [DOI] [PubMed] [Google Scholar]
- 22.Ladd PD, et al. An antisense transcript spanning the CGG repeat region of FMR1 is upregulated in premutation carriers but silenced in full mutation individuals. Hum Mol Genet. 2007;16(24):3174–87. doi: 10.1093/hmg/ddm293. [DOI] [PubMed] [Google Scholar]
- 23.He Y, et al. The antisense transcriptomes of human cells. Science. 2008;322(5909):1855–7. doi: 10.1126/science.1163853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chung DW, et al. A natural antisense transcript at the Huntington's disease repeat locus regulates HTT expression. Hum Mol Genet. 2011;20(17):3467–77. doi: 10.1093/hmg/ddr263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin Y, Wilson JH. Transcription-induced CAG repeat contraction in human cells is mediated in part by transcription-coupled nucleotide excision repair. Mol Cell Biol. 2007;27(17):6209–17. doi: 10.1128/MCB.00739-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin Y, et al. R loops stimulate genetic instability of CTG.CAG repeats. Proc Natl Acad Sci U S A. 2010;107(2):692–7. doi: 10.1073/pnas.0909740107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin Y, Wilson JH. Nucleotide excision repair, mismatch repair, and R-loops modulate convergent transcription-induced cell death and repeat instability. PLoS One. 2012;7(10):e46807. doi: 10.1371/journal.pone.0046807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chatterjee N, L Y, Wilson J. Mismatch repair enhances convergent transcriptioon indiced cell death by activating ATR. DNA Repair (Amst) 2016 doi: 10.1016/j.dnarep.2016.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shigechi T, et al. ATR-ATRIP kinase complex triggers activation of the Fanconi anemia DNA repair pathway. Cancer Res. 2012;72(5):1149–56. doi: 10.1158/0008-5472.CAN-11-2904. [DOI] [PubMed] [Google Scholar]
- 30.Michl J, Zimmer J, Tarsounas M. Interplay between Fanconi anemia and homologous recombination pathways in genome integrity. EMBO J. 2016 doi: 10.15252/embj.201693860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim H, D'Andrea AD. Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev. 2012;26(13):1393–408. doi: 10.1101/gad.195248.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tomida J, et al. A novel interplay between the Fanconi anemia core complex and ATR-ATRIP kinase during DNA cross-link repair. Nucleic Acids Res. 2013;41(14):6930–41. doi: 10.1093/nar/gkt467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang W. A major switch for the Fanconi anemia DNA damage-response pathway. Nat Struct Mol Biol. 2008;15(11):1128–30. doi: 10.1038/nsmb1108-1128. [DOI] [PubMed] [Google Scholar]
- 34.Kennedy RD, D'Andrea AD. The Fanconi Anemia/BRCA pathway: new faces in the crowd. Genes Dev. 2005;19(24):2925–40. doi: 10.1101/gad.1370505. [DOI] [PubMed] [Google Scholar]
- 35.Boisvert RA, Howlett NG. The Fanconi anemia ID2 complex: dueling saxes at the crossroads. Cell Cycle. 2014;13(19):2999–3015. doi: 10.4161/15384101.2014.956475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang J, Walter JC. Mechanism and regulation of incisions during DNA interstrand cross-link repair. DNA Repair (Amst) 2014;19:135–42. doi: 10.1016/j.dnarep.2014.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haynes B, et al. Crosstalk between translesion synthesis, Fanconi anemia network, and homologous recombination repair pathways in interstrand DNA crosslink repair and development of chemoresistance. Mutat Res Rev Mutat Res. 2015;763:258–66. doi: 10.1016/j.mrrev.2014.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin Y, Wilson JH. Diverse effects of individual mismatch repair components on transcription-induced CAG repeat instability in human cells. DNA Repair (Amst) 2009;8(8):878–85. doi: 10.1016/j.dnarep.2009.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin Y, Wilson JH. Transcription-induced DNA toxicity at trinucleotide repeats: double bubble is trouble. Cell Cycle. 2011;10(4):611–8. doi: 10.4161/cc.10.4.14729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fanning E, Klimovich V, Nager AR. A dynamic model for replication protein A (RPA) function in DNA processing pathways. Nucleic Acids Res. 2006;34(15):4126–37. doi: 10.1093/nar/gkl550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cortez D, et al. ATR and ATRIP: partners in checkpoint signaling. Science. 2001;294(5547):1713–6. doi: 10.1126/science.1065521. [DOI] [PubMed] [Google Scholar]
- 42.Yang XH, Zou L. Recruitment of ATR-ATRIP, Rad17, and 9-1-1 complexes to DNA damage. Methods Enzymol. 2006;409:118–31. doi: 10.1016/S0076-6879(05)09007-5. [DOI] [PubMed] [Google Scholar]
- 43.Kumagai A, et al. TopBP1 activates the ATR-ATRIP complex. Cell. 2006;124(5):943–55. doi: 10.1016/j.cell.2005.12.041. [DOI] [PubMed] [Google Scholar]
- 44.Huang M, et al. The FANCM/FAAP24 complex is required for the DNA interstrand crosslink-induced checkpoint response. Mol Cell. 2010;39(2):259–68. doi: 10.1016/j.molcel.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schwab RA, Blackford AN, Niedzwiedz W. ATR activation and replication fork restart are defective in FANCM-deficient cells. EMBO J. 2010;29(4):806–18. doi: 10.1038/emboj.2009.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Collis SJ, et al. FANCM and FAAP24 function in ATR-mediated checkpoint signaling independently of the Fanconi anemia core complex. Mol Cell. 2008;32(3):313–24. doi: 10.1016/j.molcel.2008.10.014. [DOI] [PubMed] [Google Scholar]
- 47.Wu Y, Shin-ya K, Brosh RM., Jr FANCJ helicase defective in Fanconia anemia and breast cancer unwinds G-quadruplex DNA to defend genomic stability. Mol Cell Biol. 2008;28(12):4116–28. doi: 10.1128/MCB.02210-07. [DOI] [PMC free article] [PubMed] [Google Scholar]