

Abstract
Insulin biosynthesis is an essential β-cell function and inappropriate insulin secretion and biosynthesis contribute to the pathogenesis of diabetes mellitus type 2. Previous studies showed that the dual leucine zipper kinase (DLK) induces β-cell apoptosis. Since β-cell dysfunction precedes β-cell loss, in the present study the effect of DLK on insulin gene transcription was investigated in the HIT-T15 β-cell line. Downregulation of endogenous DLK increased whereas overexpression of DLK decreased human insulin gene transcription. 5′- and 3′-deletion human insulin promoter analyses resulted in the identification of a DLK responsive element that mapped to the DNA binding-site for the β-cell specific transcription factor MafA. Overexpression of DLK wild-type but not its kinase-dead mutant inhibited MafA transcriptional activity conferred by its transactivation domain. Furthermore, in the non-β-cell line JEG DLK inhibited MafA overexpression-induced human insulin promoter activity. Overexpression of MafA and DLK or its kinase-dead mutant into JEG cells revealed that DLK but not its mutant reduced MafA protein content. Inhibition of the down-stream DLK kinase c-Jun N-terminal kinase (JNK) by SP600125 attenuated DLK-induced MafA loss. Furthermore, mutation of the serine 65 to alanine, shown to confer MafA protein stability, increased MafA-dependent insulin gene transcription and prevented DLK-induced MafA loss in JEG cells. These data suggest that DLK by activating JNK triggers the phosphorylation and degradation of MafA thereby attenuating insulin gene transcription. Given the importance of MafA for β-cell function, the inhibition of DLK might preserve β-cell function and ultimately retard the development of diabetes mellitus type 2.
Keywords: Diabetes mellitus, Insulin gene transcription, MafA, DLK
1. Introduction
The decline in β-cell function and mass leads to the progression from a prediabetic state characterized by insulin resistance with impaired glucose tolerance to clinically overt diabetes mellitus type 2 with elevated fasting glucose levels [1–4]. Previous studies demonstrated that overexpression of the dual leucine zipper kinase (DLK) resulted in apoptosis in neuronal cells and in the β-cell line HIT-T15 [5,6]. In addition, downregulation of cellular DLK reduced cyclosporine A-induced β-cell apoptosis [5]. DLK is expressed in many cells including HIT-T 15 β-cells, murine primary pancreatic islets of Langerhans, adipocytes and keratinocytes [7–9]. In addition, DLK is present in the neuronal system where it has been shown to regulate neuronal migration, axon growth, axon degeneration and neuronal apoptosis [10] (and references therein). Mice lacking DLK die perinatal [11,12], underlining the importance of this kinase for survival.
DLK belongs to the class of the mixed-lineage kinases within the mitogen-activated kinase family. This family is characterized by a catalytic domain that resembles in its primary structure both serine/threonine and tyrosine kinases. Functionally, these kinases are serine/threonine kinases [13–15]. Acting as a mitogen-activated 3 kinase DLK via activation of the dual-specificity kinases MKK4/7 and MKK3/6 stimulates the enzymatic activity of c-Jun N-terminal kinase (JNK) and p38, respectively [13,14,16].
In previous studies it was demonstrated that overexpression of DLK interferes with CREB-dependent gene transcription at different levels [7,17] in a β-cell line. The transcription factor CREB seems to play a pivotal role for the survival and the function of β-cells. In mice, overexpressing a dominant-negative CREB mutant specifically in β-cells, the mice became diabetic due to β-cell apoptosis [18]. Cytokine induced β-cell apoptosis was prevented by overexpression of CREB while overexpression of dominant-negative CREB mutants in human islets lead to apoptosis [19,20]. In β-cells CREB-dependent gene transcription was enhanced in response to elevated glucose levels leading to membrane depolarization and calcium influx through the voltage gated L-type calcium channels and exendin-4, stimulating the G-protein coupled GLP-1 receptor and enhancing the intracellular concentration of cAMP, thereby mimicking feeding stimuli [21]. Furthermore, CREB binds to and stimulates insulin gene transcription in HIT cells and in isolated primary murine islets after membrane depolarization and an increase in intracellular cAMP [22,23]. Therefore, in the present study the effect of DLK on human insulin gene transcription was investigated. The present study provides evidence, that DLK by activation of its downstream kinase JNK triggers the phosphorylation and degradation of the β-cell specific transcription factor MafA thereby inhibiting human insulin gene transcription.
2. Material and methods
2.1. Plasmids
The plasmids containing the 5′- and 3′-deleted fragments of the human insulin gene promoter (−258/+113 hInsLuc; −222/+113 hInsLuc; −193/+113 hInsLuc; −140/+113 hInsLuc; −93/+113 hInsLuc; −56/+113 hInsLuc; +18/−336 hInsLuc; −57/−336 hInsLuc; −94/−336 hInsLuc; −141/−336 hInsLuc; −223/−336 hInsLuc; −259/−336 hInsLuc), the CRE2 (CRE2mut) and C1 (C1mut) mutated human insulin gene promoters, the human insulin gene promoter itself (−336hInsLuc) and the 4xhInsC1 have been described before [24]. The plasmid G5E1Bluc has been described [7], and the expression vectors for GAL4 MafA fusion proteins have been described previously [25]. The expression vectors for DLK and its kinase dead mutant DLK K185A are described in Holzman et al. [15], those for MafA and its DNA binding deficient mutant used for reporter gene assays are described in Harmon et al. [26], and those for MafA and the MafA S65A mutants used for immunoblot analysis are described in Guo et al. [25]. The expression vectors for MafA wild-type, MafA S65A and MafA 4A, used in reporter gene assays are described by Rocques et al. [27]. The expression vectors expressing MafA siRNA (MafA RNAi 64–82, MafA RNAi 82–102 and MafA RNAi 1054–1074) are described in Zhao et al. [28].
2.2. Cell culture and transient transfection assays
HIT-T15 cells [29] were grown in RPMI 1640 medium supplemented with 10% FCS, 5% horse serum, penicillin (100 U/ml) and streptomycin (100 μg/ml). JEG-3 choriocarcinoma cells [30] and HEK293 cells were grown in DMEM supplemented with 10% FCS, penicillin (100 U/ml) and streptomycin (100 μg/ml). For the reporter gene assays HIT and JEG cells were transiently transfected with 2 μg/6 cm-dish of the luciferase reporter gene and cotransfected with 2 μg/dish of the expression plasmids by metafectene according to the manufacturer’s protocol (Biontex, Munich, Germany). When indicated 50 pmol of small interfering RNA against DLK (5′-GACUCAGACUGUGACAGCACUGAAU-3′) or unspecific RNAi with medium GC content (Invitrogen, Karlsruhe, Germany) was cotransfected by oligofectamine (Invitrogen, Karlsruhe, Germany) according to the manufacturer’s protocol. Cotransfection of 1 μg of the plasmid CMV-GFPtpz served to control for transfection efficiency. Cells were harvested 48 h after transfection. Luciferase assays and GFP measurement were performed as described [24]. To check the efficiency of down regulation of MafA by the MafA siRNA, expression vectors for MafA and the MafA siRNAs (1 μg/well, each) were transiently cotransfected into HEK293 cells by metafectene. For the measurement of insulin secretion HIT-T15 cells were grown on 6-well-plates and transiently transfected with 2.1 μg/well of the expression plasmids by K2 according to the manufacturer’s protocol (Biontex, Munich, Germany).
2.3. Immunoblot analysis
JEG-3 cells were transiently transfected with 2.5 μg of the expression vectors for MafA wild-type or its mutant S65A [25] or with 2.5 μg of pBluescript vector (Stratagene, La Jolla, CA, USA) as indicated. The expression vectors for DLK wild-type or DLK K185A mutant [15] were cotransfected (2 μg per well in a 6-well dish) by metafectene. Forty-eight hours after transfection, cells were harvested into 50 μl of lysis buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 10% glycerol, 1% Triton X-100, 1% Nonidet P-40 and protease inhibitors). The cell suspension was passed five times through a 26 gauge needle (or frozen and thawed four times with vortexing in between) and centrifuged at 4 °C for 10 min at 21,130 g. Loading buffer (0.5 M Tris, 20% SDS, 1% bromophenol blue, glycerol, β-mercaptoethanol) was added to the supernatant which was then boiled for 5 min at 95 °C, subjected to SDS-PAGE (10% gel) and transferred to a nitrocellulose membrane. The membrane was incubated in 5% fat-free dried milk in TBST (25 mM Tris–HCl, pH 7.4; 137 mM NaCl; 5 mM KCl; 0.7 mM CaCl2; 0.1 mM MgCl2; 0.1% Tween 20) for 1 h at room temperature and then overnight at 4 °C with fresh TBST supplemented with an antibody against MafA (1:2,000; Bethyl Laboratories, Montgomery, TX, USA), DLK (1:3,000) [15] and GAPDH (1:10,000; Santa Cruz, Heidelberg, Germany). Before and after the incubation with the secondary antibody the membrane was washed three times for 10 min with TBST. The antibody–antigen complex was detected by chemiluminescence reagents (GE Healthcare, Little Chalfont, Bucks, UK) in a ChemiGenius2 Bio imaging system (Syngene, UK) The optical density of the respective bands was evaluated by Quantity one software (Bio-Rad, Munich, Germany).
2.4. Insulin secretion
Transiently transfected HIT-T15 cells were incubated for 48 h, washed twice with modified Krebs buffer (129 mM NaCl 129; 5 mM NaHCO3; 4.7 mM KCl 4.7; 1.2 mM KH2PO4; 1.2 mM MgSO4; 10 mM HEPES, pH 7.4; 2 mM CaCl2; 0.2% bovine serum albumin) and incubated in 1 ml of the same buffer supplemented with 5 mM glucose for 2 h. The supernatant was harvested, centrifuged twice and secreted insulin was measured by the Ultrasensitive Mouse Insulin ELISA according to the manufacturer’s protocol (Mercodia, Uppsala, Sweden) using a Tecan Safire2 photometer. For the determination of the protein content the cells were harvested into 50 μl of lysis buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 10% glycerol, 1% Triton X-100, 1% Nonidet P-40 and protease inhibitors). The cell suspension was passed five times through a 26 gauge needle (or frozen and thawed four times with vortexing in between) and centrifuged at 4 °C for 10 min at 21,130 g. Protein concentration was measured by Bradford assay.
2.5. Statistical analysis
All results are expressed as means ± SEM. Statistical significance was calculated with ANOVA, followed by Student’s t test. A value of p < 0.05 was considered significant.
3. Results
3.1. Effect of DLK on human insulin gene transcription in a β-cell line
In a first approach to investigate the effect of DLK on human insulin gene transcription, a luciferase reporter gene containing the human insulin promoter from −336 to +113 bp [24] was transiently transfected into the insulin producing pancreatic β-cell line HIT. Fig. 1A shows that small interfering RNA mediated reduction of DLK stimulated human insulin gene transcription 2.5 fold. This increase in human insulin gene transcription is similar to that reached by treatment with KCl or glucose leading to membrane depolarization with calcium influx and the adenylate cyclase activator forskolin in HIT cells and in primary mature islets [22,23]. In contrast, overexpression of DLK wild-type reduced human insulin gene promoter transcriptional activity by 50%; overexpression of a kinase-dead DLK mutant in which the lysine 185 is exchanged for alanine, thus preventing ATP binding, had no effect on insulin gene transcription [15] (Fig. 1B). In addition, overexpression of DLK wild-type decreased insulin secretion from HIT cells by approx. 54% (Fig. 1C). These data suggest that DLK can exert an inhibitory effect on insulin gene transcription and insulin secretion under basal conditions in the β-cell. DLK’s inhibitory effect depends on its enzymatic activity.
Fig. 1.
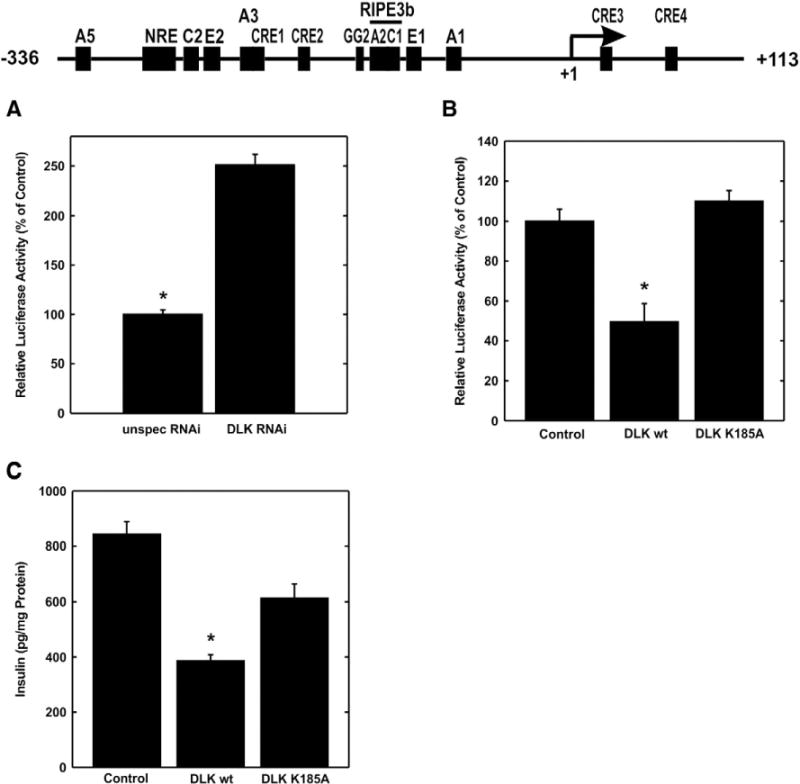
Effect of DLK human insulin gene transcription. A, the plasmid −336 hInsLuc (2 μg) and an RNAi against DLK or an unspecific RNAi was transiently transfected into HIT cells as indicated. Cells were harvested after 48 h and luciferase activity was measured. Luciferase activity is expressed relative to the mean value measured in cells transfected with unspecific RNAi in each experiment. Values are means ± SEM of three independent experiments each done in duplicate. *p < 0.05 vs. unspecific RNAi. B, the plasmids −336 hInsLuc (2 μg) was transiently transfected in HIT cells together with expression plasmids (2 μg, each) for DLK wild-type (DLK wt) or its kinase-dead mutant (DLK K185A) or pBluescript (Control) as indicated. Luciferase activity is expressed relative to the mean value measured in the control in each experiment. Values are means ± SEM of three independent experiments each done in duplicate. *p < 0.05 vs. control. C, expression vectors for DLK wild-type (DLK wt) or its kinase-dead mutant (DLK K185A) or pBluescript (Control) (2.1 μg) were transiently transfected into cells. Insulin was determined in the supernatant of the cells and corrected to the protein content of the cells. Values are means ± SEM of three independent experiments each done in triplicate. *p < 0.001 vs. control and vs. DLK K185A.
The human insulin gene promoter contains several enhancer-like sequences among them the CREB binding CRE and the C1 element bound by MafA [23,24,31–34]. To identify the region within the human insulin gene promoter which confers responsiveness to DLK, 5′- and 3′-deletion promoter analysis was conducted. As shown in Fig. 2A, overexpression of DLK but not its kinase-dead mutant decreased insulin gene transcription by 40%. This inhibitory effect was still present in a promoter construct containing −193 bp, but attenuated upon further 5′ deletion (Fig. 2A). The 3′-deleted insulin promoter fragments were fused to the minimal thymidine kinase promoter (−81 to +52 bp) of the herpes simplex virus (Fig. 2B). Truncation up to −94 bp did not interfere with the inhibitory action of DLK, whereas the −141 bp mutant was no longer decreased by DLK in a mutation sensitive manner (Fig. 2B). Taken together, 5′- and 3′ promoter analyses suggest that the C1 element confers DLK-responsiveness to the human insulin gene promoter. CREB binding to the CRE2 confers the stimulation by glucose, by KCl-induced membrane depolarization and by cAMP to the human insulin gene promoter [22,23]; and DLK inhibits stimulated CREB transcriptional activity [7]. To determine whether CRE2 or C1 confers the inhibitory effect of DLK within the promoter, luciferase reporter genes under control of the human insulin gene promoter containing either a mutated CRE2 or a mutated C1 element within the promoter were transiently transfected. Treatment of the cells with KCl leading to membrane depolarization and the adenylate cyclase activator forskolin enhanced the transcriptional activity of the wild-type and the C1 mutated but not that of the CRE2 mutated human insulin gene promoter (Supplementary Fig. 1). Overexpression of DLK reduced basal and stimulated human insulin gene transcription but did not interfere with the transcriptional activities conferred by the CRE2 or the C1 mutated human insulin gene promoters (Supplementary Fig. 1). Of note, mutation of C1 decreased basal human insulin gene by approx. 40% (Supplementary Fig. 1). These data suggest that C1 mediates DLK responsiveness to the human insulin gene promoter under basal conditions, whereas CRE2 confers DLK responsiveness under stimulated conditions.
Fig. 2.
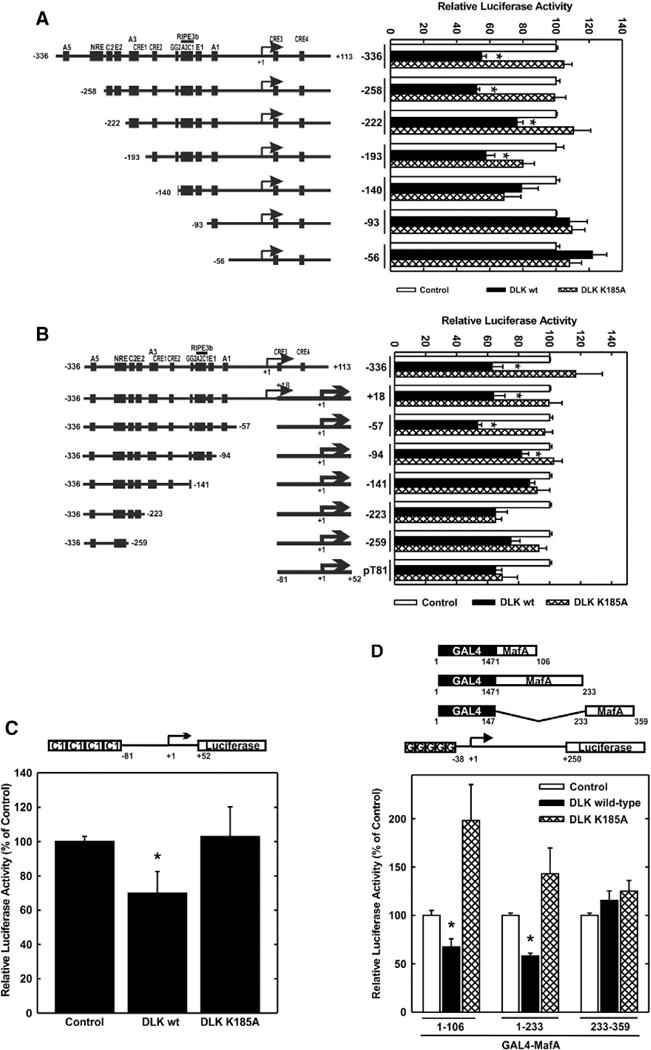
Identification of a DLK responsive element within the human insulin gene promoter. A, 5′-deletion analysis. The plasmids (2 μg) depicted on the left were transiently transfected with Bluescript (2 μg) (Control), the expression vector for DLK wild-type (2 μg) (DLK wt) or its kinase-dead mutant (2 μg) (DLK K185A) as indicated. Luciferase activity is expressed relative to the mean value measured in the control in each experiment. Values are means ± SEM of three independent experiments each done in duplicate. *p < 0.05 vs. control. B, 3′- deletion analysis. The plasmids (2 μg) depicted on the left were transiently transfected with Bluescript (2 μg) (Control), the expression vector for DLK wild-type (2 μg) (DLK wt) or its kinase-dead mutant (2 μg) (DLK K185A) as indicated. Luciferase activity is expressed relative to the mean value measured in the control in each experiment. Values are means ± SEM of three independent experiments each done in duplicate. *p < 0.05 vs. control. C, effect of DLK on C1-dependent transcriptional activity. The plasmid 4xhInsC1 (2 μg) was transiently transfected with Bluescript (Control) (2 μg), the expression plasmid for DLK wild-type (DLK wt) (2 μg) or its kinase-dead mutant (DLK K185A) (2 μg). Luciferase activity is expressed relative to the mean value measured in the control in each experiment. Values are means ± SEM of three independent experiments each done in duplicate. *p < 0.05 vs. control. D, effect of DLK on MafA transcriptional activity. The plasmid G5E1Bluc (2 μg) was transiently cotransfected with expression vectors for the GAL4 DNA binding domain fused to MafA containing the amino acids from 1 to 106, 1 to 233, or 233 to 359, respectively, as depicted in the upper panel. Plasmids for Bluescript (Control) (2 μg), the expression plasmid for DLK wild-type (DLK wt) (2 μg) or its kinase-dead mutant (DLK K185A) (2 μg) were transiently cotransfected. Luciferase activity is expressed relative to the mean value measured in the control in each experiment. Values are means ± SEM of three independent experiments each done in duplicate. *p < 0.05 vs. control.
To investigate whether the transcriptional activity of the isolated C1 element is decreased by DLK, a luciferase reporter gene under control of four copies of the C1 element fused to the minimal thymidine kinase promoter was transiently transfected into HIT cells [24]. Overexpression of DLK reduced C1-dependent transcriptional activity by 30% whereas the kinase-dead DLK mutant exerted no inhibitory effect (Fig. 2C). The transcription factor binding to the C1 element within the insulin promoter is MafA [31–34]. MafA was shown to stimulate insulin gene transcription in response to glucose and to play an overall essential role in the regulation of insulin gene transcription [32,35].
To investigate whether MafA transcriptional activity is inhibited by DLK, the GAL4 system was employed. A luciferase reporter gene under control of five copies of the binding-site for the yeast transcription factor GAL4 fused to the minimal adenoviral E1B promoter was transiently cotransfected with expression vectors for the DNA binding domain of GAL4 and MafA N- and C-terminal deleted fragments [28]. As shown in Fig. 2D, overexpression of DLK inhibited the transcriptional activity of the transactivation domain spanning constructs of MafA, encompassing the amino acids 1 to 106 and 1 to 223, respectively. Overexpression of the kinase-dead DLK mutant did not reduce the transcriptional activity conferred by the MafA transactivation domain but rather stimulated it (Fig. 2D). This suggests that the kinase-dead DLK mutant acts as a dominant-negative DLK mutant, interacting via its leucine zipper with endogenous DLK thereby attenuating the enzymatic activity of endogenous DLK. Taken together, these data show that DLK inhibits human insulin gene transcription and MafA transcriptional activity.
3.2. Dependence of DLK inhibitory action on MafA
To investigate whether the inhibitory effect of DLK on human insulin gene transcription is mediated by MafA, the luciferase reporter gene under control of the human insulin gene promoter was transiently transfected into the human choriocarcinoma JEG cell line together with expression vectors for DLK, its kinase-dead mutant, MafA or a MafA mutant carrying a dysfunctional mutation within the DNA binding domain (MafA R265A) [28]. Overexpression of MafA robustly increased human insulin gene transcription in comparison to overexpression of the inactive MafA R265A mutant, consistent with previous data [24]. Additional overexpression of DLK decreased MafA-dependent transcriptional activity of the human insulin gene promoter by 57% (Fig. 3A). In addition, down regulation of MafA protein content in HIT cells by overexpression of MafA RNAi containing plasmids decreased human insulin gene transcription and prevented the inhibitory effect of DLK (Fig. 3B).
Fig. 3.
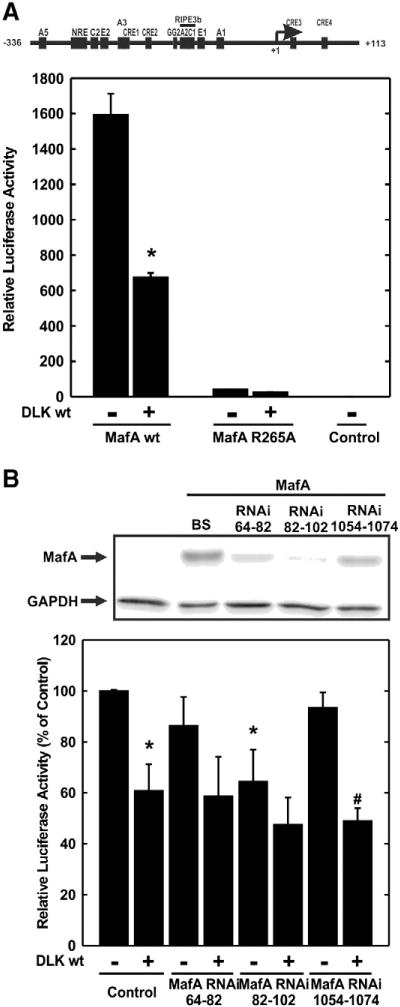
Effect of DLK on MafA dependent human insulin gene transcription. A, the plasmid −336hInsLuc (2 μg) was transiently cotransfected with pBluescript (Control) (2 μg), an expression vector for MafA wild-type (MafA wt) (2 μg) or its DNA binding deficient mutant (MafA R265A) into JEG cells. When indicated the expression plasmids for DLK wild-type (DLK wt) (2 μg) was cotransfected. Luciferase activity is expressed to the mean value in the respective controls (no MafA, no DLK) in each experiment. Values are means ± SEM of three independent experiments each done in duplicate. *p < 0.05 vs. control. B, upper panel, expression vectors for MafA and MafA RNAi 64–82, MafA 82–102 and MafA 1054–1074 (1 μg, each) were transiently cotransfected into HEK293 cells. After 48 h cells were harvested and immunoblot analysis was performed. The arrow labeled “MafA” points to the MafA representing band, the arrow labeled “GAPDH” points to the GAPDH representing band. Lower panel, the plasmid −336hInsLuc (1 μg) was transiently cotransfected with pBluescript (Control) (1 μg), expression vectors for MafA RNAi 64–82, MafA 82–102 and MafA 1054–1074 (1 μg, each) and for DLK wild-type (DLK wt) (1 μg) as indicated into HIT cells. Luciferase activity is expressed to the mean value in the control (no MafA RNAi, no DLK) in each experiment. Values are means ± SEM of three independent experiments each done in duplicate. *p < 0.05 vs. control, #p < 0.05 vs. MafA RNAi 1054–1074.
3.3. Effect of DLK on MafA protein content
MafA-dependent transcriptional activity is regulated at the transcriptional and posttranscriptional level with a diverse set of posttranslational modifications influencing MafA protein level (phosphorylation, sumoylation and ubiquitination with proteasomal degradation [24, 36–48] (and references therein)). To investigate whether DLK reduces MafA protein content, expression vectors for MafA, DLK and its mutant were transiently transfected into JEG cells. Overexpression of DLK reduced overexpressed MafA protein by 40%. Since the kinase-dead DLK mutant exerted no such effect, DLK-induced MafA protein reduction depends on its enzymatic activity (Fig. 4). Additional treatment of the cells with SP600125, an inhibitor of JNK [39], a downstream kinase of DLK [16], prevented DLK-induced reduction of MafA protein, but had itself no effect on MafA protein level (Fig. 4). These data indicate that DLK acts through JNK to decrease MafA protein content. Since MafA expression here was directed by the cytomegalovirus promoter, DLK might act via a posttranslational mechanism to reduce MafA protein content.
Fig. 4.
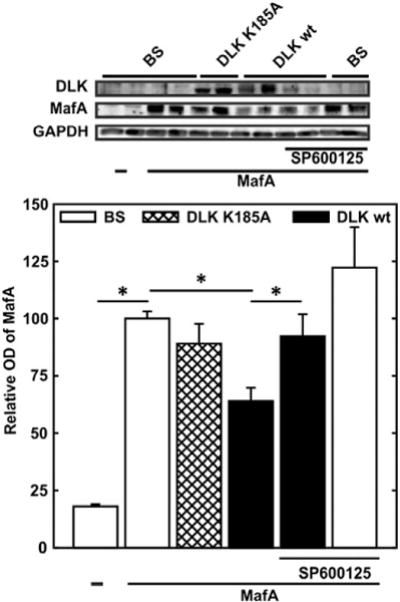
Effect of DLK on MafA protein content. Upper panel – typical immunoblot, lower panel – quantitative evaluation. JEG cells were transiently cotransfected with an expression vector for MafA (2.5 μg) together with expression vectors for DLK wild-type (DLK wt) (2.5 μg) or its kinase-dead mutant (DLK K185A) (2.5 μg) as indicated. In addition, when indicated, cells were treated with the JNK inhibitor SP600125 (25 μM) 16 h before harvest. Cell extracts were subjected to immunoblot analysis and the optical density of the respective bands was evaluated. Optical density (OD) of MafA is expressed relative to the mean value of the optical density of overexpressed MafA in the absence of DLK wt or DLK K185A in each experiment. Values are means ± SEM of three independent experiments each done in duplicate. *p < 0.05 vs. control.
Phosphorylation of serine 65 within the transactivation domain of MafA has been shown to regulate MafA stability and transcriptional activity [25,27,40,41]. In a first approach to investigate whether the serine 65 is a target of DLK action, the luciferase reporter gene under control of the human insulin gene promoter was transiently transfected into JEG cells together with expression vectors for wild-type or a MafA Ser65Ala mutant and DLK wild-type or the kinase dead mutant. Insulin gene transcription was enhanced approx. 3 fold when the MafA Ser65Ala mutant was recruited to the promoter (Fig. 5A). Neither the overexpression of DLK wild-type nor its kinase-dead mutant reduced the transcriptional activity of MafA Ser65Ala-dependent insulin gene transcription (Fig. 5A), suggesting that the phosphorylation of this amino acid residue by JNK triggers the DLK-induced inhibition of MafA-dependent insulin gene transcription. Indeed, overexpression of DLK did not reduce the protein content of overexpressed MafA Ser65Ala (Fig. 5B).
Fig. 5.
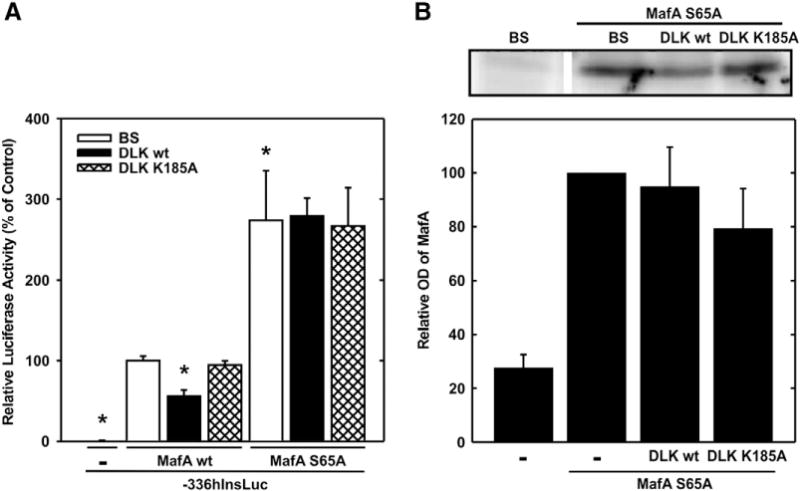
Effect of DLK on MafA S65A mutant. A, the plasmid −336hInsLuc (1.6 μg) was transiently transfected into JEG cells together with expression vectors for MafA wild-type (MafA wt), MafA S65A (MafA S65A) and DLK wild-type (DLK wt) or its kinase-dead mutant (DLK K185A) (1.6 μg, each) as indicated. Luciferase activity is expressed relative to the mean value measured in the presence of hInsLuc and MafA wild-type without DLK or its mutant in each experiment. Values are means ± SEM of three independent experiments each done in duplicate. *p < 0.05 vs. control. B, upper panel – typical immunoblot, lower panel – quantitative evaluation. JEG cells were transiently cotransfected with an expression vector for MafA S65A (1 μg) together with pBluescript (1 μg) or expression vectors for DLK wild-type (DLK wt) or the kinase-dead mutant (DLK K185A) (1 μg, each). Cell extracts were subjected to immunoblot analysis. The optical density is expressed relative to the value measured of the MafA S65A representing band in the absence of DLK or its kinase dead mutant. Values are means ± SEM of four independent experiments.
4. Discussion
The preservation of the integrity of the β-cells seems to be a key to prevent the development of diabetes mellitus type 2 under conditions of peripheral insulin resistance [1,2,4]. Appropriate insulin secretion and insulin biosynthesis in response to metabolic demands are the main features of the β-cells [1,42]. Hence, signals attenuating insulin secretion or insulin biosynthesis promote the pathogenesis of diabetes mellitus type 2. The present study shows that DLK inhibits the transcriptional activity of the human insulin gene promoter; overexpression of DLK but not its kinase-dead mutant inhibited insulin gene transcription whereas the downregulation of endogenous DLK in HIT cells stimulated the transcriptional activity of the human insulin gene promoter. In addition, overexpression of DLK reduced insulin secretion from HIT cells. Taken together with our previous studies demonstrating that reduction of cellular DLK attenuates cyclosporin A-induced β-cell apoptosis and overexpression of DLK results in β-cell apoptosis [5], these data suggest that activation of DLK activity contributes to the decline in β-cell function and mass and ultimately to diabetes mellitus.
DLK acts as a MAP3K and stimulates the dual specificity kinase MKK4/7 and MKK3/6 which in turn stimulate their downstream MAPKs JNK and p38 [13,16,43]. Within the cell under basal conditions monomeric, catalytically inactive DLK is associated with the scaffold proteins JIP1/IB1 (JNK interacting protein 1/islet-brain-1) and POSH (plenty of SH3) [44–47]. Phosphorylation of JIP1 on tyrosine residues by the Src family kinases strengthens the interaction between DLK and the scaffold protein [48] whereas the phosphorylation of JIP1 on Thr-103 by JNK promotes the dissociation of DLK from JIP1 [45,47]. DLK then homodimerizes via its leucine zipper and presumably by autophosphorylation becomes catalytically active [47,49,50]. Hence, overexpression of DLK is sufficient to stimulate the catalytic activity of DLK. In addition, phosphorylation of DLK itself by JNK prevents the ubiquitination and proteasomal degradation of DLK, resulting in a feed-forward loop [51,52]. These studies imply that signals activating the downstream kinase JNK promote the activation of DLK thereby perpetuating JNK and DLK activation. Oxidative stress and proinflammatory cytokines, considered as prediabetic signals, have been shown to activate the JNK pathway in β-cells [53] (and references therein) and inhibition of JNK restored oxidative stress-mediated suppression of insulin expression [54]. Furthermore, a missense mutation within the IB1 encoding gene MAPKIP1 is associated with diabetes mellitus type 2 [55]. These studies support the notion that activation of the JNK pathway contributes to the development of diabetes mellitus at least in part by interfering with β-cell function.
The present study shows that DLK inhibits human insulin gene transcription, and the DLK responsive element was mapped to the DNA binding-site of MafA. Furthermore, DLK reduced MafA transcriptional activity and MafA-dependent insulin gene transcription and diminished MafA protein content. Inhibition of JNK or mutation of MafA Ser 65 to Ala prevented DLK-caused reduction of MafA protein content. These data suggest that DLK through activation of JNK decreases MafA protein levels thereby inhibiting insulin gene transcription. Given that Ser 65 of MafA corresponds to Ser 70 in MafB, DLK might inhibit MafB-dependent gene transcription and subsequently MafA and MafB-dependent gene transcription as well [25].
MafA transcriptional activity is regulated at various levels like transcription, translation and posttranslational modifications [24,36–38] (and references therein). Phosphorylation of Ser 65 within the transactivation domain of MafA (and of Ser 70 in MafB) seems to play a crucial role for the stability and the transactivation and transforming potential of these transcription factors [25,27,40,41]. Phosphorylated Ser 65 provides the recognition site for glycogen synthase kinase-3 beta (GSK-3β), which in turn phosphorylates sequentially Ser 61, Thr 57, Thr 53 and Ser 49. Thus phosphorylated MafA undergoes either proteasomal degradation [27,40] or recruits the transcriptional coactivator P/CAF [27]. Although it has been shown that ERK2 phosphorylates MafA Ser 65 in vitro the kinase phosphorylating Ser 65 in vivo remains unknown [25,27,40,41]. The present study suggests that JNK activated by DLK is the kinase phosphorylating MafA on Ser 65, since inhibition of JNK prevented DLK-induced loss of MafA protein. MafA seems to be constitutively phosphorylated by GSK-3β in diverse β-cell and non-β-cell lines, as judged by its mobility in immunoblot analysis [25,27,40,56] (this study). In the present study recruitment of the Ser 65 Ala MafA mutant to the human insulin gene promoter enhanced its transcriptional activity approx. 3 fold in comparison to wild-type MafA. Recruitment of a MafA mutant, in which the GSK-3β phosphorylation sites were changed to alanine, to the insulin gene promoter induced a 4 fold increase in transcriptional activity and prevented the inhibitory action of DLK on insulin gene transcription (Supplementary Fig. 2). In addition, the protein content of overexpressed MafA Ser 65 Ala was not reduced by DLK as demonstrated by immunoblot analysis. These data support the notion that the phosphorylation of MafA on Ser 65 through DLK-induced JNK activation, presumably as priming event for the consecutive phosphorylation by GSK-3β, triggers the degradation of MafA thereby terminating MafA-dependent gene transcription. Given that MafA controls the transcription of many genes important to maintain β-cell function [36,57,58] and that MafA deficient mice develop age-dependent glucose intolerance and diabetes [59], the impairment of MafA function and loss of MafA caused by DLK might promote the pathogenesis of diabetes mellitus type 2. Indeed, immunohistological analysis of pancreatic slides revealed that in humans suffering from diabetes mellitus type 2 the content of MafA in the nuclei of β-cells was severely diminished when compared to non-diabetic individuals [60]. In addition, loss of MafA and/or MafB was shown to be an early indicator of β-cell inactivity and β-cell dysfunction in diabetic mice and in type 2 diabetic human islets [56].
Our previous study showed that DLK induces β-cell apoptosis [5] presumably by inhibiting CREB-dependent gene transcription [7,17] and the present study demonstrates that DLK diminished insulin gene transcription presumably by decreasing MafA protein content. Thus, activation of DLK leads to a reduction in β-cell mass and function. It is unknown which signals stimulate DLK enzymatic activity in β-cells. Considering that prediabetic signals like oxidative stress and proinflammatory cytokines stimulate JNK activity in β-cells [5] (and references therein) and JNK promotes the activation of DLK starting the perpetuation of JNK and DLK activation [45–47,51,52], the inhibition of DLK would interrupt this vicious cycle thereby protecting β-cell function and mass from the deleterious effects of the prediabetic signals and preventing the development of diabetes mellitus. Hence, the inhibition of DLK in β-cells provides a valuable drug target for the therapy of diabetes mellitus.
5. Conclusions
In the present study the effect of DLK on human insulin gene transcription as an important β-cell function was investigated. This study shows that (i) DLK inhibits insulin gene transcription through activation of its downstream kinase JNK followed by a reduction of MafA protein content. (ii) The Ser 65 within MafA is a target of DLK–JNK action. Considering that activation either of DLK or of JNK results into a forward-loop thereby perpetuating the β-cell function impairing signals, the inhibition of DLK might preserve MafA and presumably MafB protein content and thereby β-cell function under prediabetic conditions.
Acknowledgments
The present study was supported by a grant from the Deutsche Forschungsgemeinschaft (OE 181/3-1) to EO and by a fellowship for MJS from the University Medical Center Göttingen. In addition, this work was supported by NIH ROI DK90570 to RS.
Abbreviations
- CRE
cAMP responsive element
- CREB
CRE binding protein
- DLK
dual leucine zipper kinase
- JIP/IB-1
JNK interacting protein/islet-brain-1
- JNK
c-Jun N-terminal kinase
- MAPK
mitogen-activated protein kinase
Footnotes
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.cellsig.2014.04.006.
Contributors
MJS and CD performed the main part of the experiments, MJS helped writing the manuscript, SS performed the immunoblots with the overexpressed MafA mutant and insulin secretion assay, DK performed the immunoblots with overexpressed MafA mutant and helped writing the manuscript, RB performed the DLK-RNAi experiments, RS contributed the expression vectors for MafA wild type, MafA S65A, MafA265A, MafA RNAi and for the GAL4-MafA fusion proteins and commented on the manuscript, CP contributed the expression vectors for quail MafA wild-type, MafA S65A and MafA 4A (depicted in Fig. 5A and in Supplementary Fig. 2) and commented on the manuscript, and EO conceived the strategy and prepared the manuscript.
Disclosure
The authors state no actual or potential conflict of interest including any financial, personal or other relationships with other people or organizations. All authors have approved the final manuscript.
References
- 1.Kahn SE, Zraika S, Utzschneider KM, Hull RL. Diabetologia. 2009;52(6):1003–1012. doi: 10.1007/s00125-009-1321-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lazar MA. Science. 2005;307(5708):373–375. doi: 10.1126/science.1104342. [DOI] [PubMed] [Google Scholar]
- 3.Rhodes CJ. Science. 2005;307(5708):380–384. doi: 10.1126/science.1104345. [DOI] [PubMed] [Google Scholar]
- 4.Stumvoll M, Goldstein BJ, van Haeften TW. The Lancet. 2005;365(9467):1333–1346. doi: 10.1016/S0140-6736(05)61032-X. [DOI] [PubMed] [Google Scholar]
- 5.Plaumann S, Blume R, Borchers S, Steinfelder HJ, Knepel W, Oetjen E. Mol Pharmacol. 2008;73(3):652–659. doi: 10.1124/mol.107.040782. [DOI] [PubMed] [Google Scholar]
- 6.Xu Z, Maroney AC, Dobrzanski P, Kukekov NV, Greene LA. Mol Cell Biol. 2001;21(14):4713–4724. doi: 10.1128/MCB.21.14.4713-4724.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oetjen E, Lechleiter A, Blume R, Nihalani D, Holzman L, Knepel W. Diabetologia. 2006;49(2):332–342. doi: 10.1007/s00125-005-0087-1. [DOI] [PubMed] [Google Scholar]
- 8.Couture JP, Blouin R. Biochem J. 2011;438(1):93–101. doi: 10.1042/BJ20101840. [DOI] [PubMed] [Google Scholar]
- 9.Germain L, Fradette J, Robitaille H, Guignard R, Grondin R, Nadeau A, Blouin R. J Invest Dermatol. 2000;115(5):860–867. doi: 10.1046/j.1523-1747.2000.00024.x. [DOI] [PubMed] [Google Scholar]
- 10.Tedeschi A, Bradke F. EMBO Rep. 2013;14(7):605–614. doi: 10.1038/embor.2013.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hirai S-i, Feng Cui D, Miyata T, Ogawa M, Kiyonari H, Suda Y, Aizawa S, Banba Y, Ohno S. J Neurosci. 2006;26(46):11992–12002. doi: 10.1523/JNEUROSCI.2272-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sengupta Ghosh A, Wang B, Pozniak CD, Chen M, Watts RJ, Lewcock JW. J Cell Biol. 2011;194(5):751–764. doi: 10.1083/jcb.201103153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gallo KA, Johnson GL. Nat Rev Mol Cell Biol. 2002;3(9):663–672. doi: 10.1038/nrm906. [DOI] [PubMed] [Google Scholar]
- 14.Cuevas BD, Abell AN, Johnson GL. Oncogene. 2007;26(22):3159–3171. doi: 10.1038/sj.onc.1210409. [DOI] [PubMed] [Google Scholar]
- 15.Holzman LB, Merritt SE, Fan G. J Biol Chem. 1994;269(49):30808–30817. [PubMed] [Google Scholar]
- 16.Fan G, Merritt SE, Kortenjann M, Shaw PE, Holzman LB. J Biol Chem. 1996;271(40):24788–24793. doi: 10.1074/jbc.271.40.24788. [DOI] [PubMed] [Google Scholar]
- 17.Phu DT, Wallbach M, Depatie C, Fu A, Screaton RA, Oetjen E. Cell Signal. 2011;23(2):344–353. doi: 10.1016/j.cellsig.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 18.Jhala US, Canettieri G, Screaton RA, Kulkarni RN, Krajewski S, Reed J, Walker J, Lin X, White M, Montminy M. Genes Dev. 2003;17(13):1575–1580. doi: 10.1101/gad.1097103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jambal P, Masterson S, Nesterova A, Bouchard R, Bergman B, Hutton JC, Boxer LM, Reusch JEB, Pugazhenthi S. J Biol Chem. 2003;278(25):23055–23065. doi: 10.1074/jbc.M212450200. [DOI] [PubMed] [Google Scholar]
- 20.Sarkar SA, Gunter J, Bouchard R, Reusch JEB, Wiseman A, Gill RG, Hutton JC, Pugazhenthi S. Diabetologia. 2007;50(8):1649–1659. doi: 10.1007/s00125-007-0707-z. [DOI] [PubMed] [Google Scholar]
- 21.Jansson D, Ng AC-H, Fu A, Depatie C, Al Azzabi M, Screaton RA. Proc Natl Acad Sci. 2008;105(29):10161–10166. doi: 10.1073/pnas.0800796105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oetjen E, Baun D, Beimesche S, Krause D, Cierny I, Blume R, Dickel C, Wehner S, Knepel W. Mol Pharmacol. 2003;63(6):1289–1295. doi: 10.1124/mol.63.6.1289. [DOI] [PubMed] [Google Scholar]
- 23.Oetjen E, Grapentin D, Blume R, Seeger M, Krause D, Eggers A, Knepel W. Naunyn Schmiedebergs Arch Pharmacol. 2003;367(3):227–236. doi: 10.1007/s00210-003-0694-7. [DOI] [PubMed] [Google Scholar]
- 24.Oetjen E, Blume R, Cierny I, Schlag C, Kutschenko A, Krätzner R, Stein R, Knepel W. Diabetologia. 2007;50(8):1678–1687. doi: 10.1007/s00125-007-0712-2. [DOI] [PubMed] [Google Scholar]
- 25.Guo S, Burnette R, Zhao L, Vanderford NL, Poitout V, Hagman DK, Henderson E, Özcan S, Wadzinski BE, Stein R. J Biol Chem. 2009;284(2):759–765. doi: 10.1074/jbc.M806314200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harmon JS, Stein R, Robertson RP. J Biol Chem. 2005;280(12):11107–11113. doi: 10.1074/jbc.M410345200. [DOI] [PubMed] [Google Scholar]
- 27.Rocques N, Abou Zeid N, Sii-Felice K, Lecoin L, Felder-Schmittbuhl M-P, Eychène A, Pouponnot C. Mol Cell. 2007;28(4):584–597. doi: 10.1016/j.molcel.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 28.Zhao L, Guo M, Matsuoka T-a, Hagman DK, Parazzoli SD, Poitout V, Stein R. J Biol Chem. 2005;280(12):11887–11894. doi: 10.1074/jbc.M409475200. [DOI] [PubMed] [Google Scholar]
- 29.Santerre RF, Cook RA, Crisel RM, Sharp JD, Schmidt RJ, Williams DC, Wilson CP. Proc Natl Acad Sci U S A. 1981;78(7):4339–4343. doi: 10.1073/pnas.78.7.4339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kohler PO, Bridson WE. J Clin Endocrinol Metab. 1971;32(5):683–687. doi: 10.1210/jcem-32-5-683. [DOI] [PubMed] [Google Scholar]
- 31.Harrington RH, Sharma A. J Biol Chem. 2001;276(1):104–113. doi: 10.1074/jbc.M008415200. [DOI] [PubMed] [Google Scholar]
- 32.Kataoka K, Han SI, Shioda S, Hirai M, Nishizawa M, Handa H. J Biol Chem. 2002;277(51):49903–49910. doi: 10.1074/jbc.M206796200. [DOI] [PubMed] [Google Scholar]
- 33.Matsuoka TA, Artner I, Henderson E, Means A, Sander M, Stein R. Proc Natl Acad Sci U S A. 2004;101(9):2930–2933. doi: 10.1073/pnas.0306233101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Olbrot M, Rud J, Moss LG, Sharma A. Proc Natl Acad Sci U S A. 2002;99(10):6737–6742. doi: 10.1073/pnas.102168499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Artner I, Hang Y, Guo M, Gu G, Stein R. J Endocrinol. 2008;198(2):271–279. doi: 10.1677/JOE-08-0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aramata S, Han S-i, Kataoka K. Endocr J. 2007;54(5):659–666. doi: 10.1507/endocrj.kr-101. [DOI] [PubMed] [Google Scholar]
- 37.Vanderford NL. Islets. 2011;3(1):35–37. doi: 10.4161/isl.3.1.14032. [DOI] [PubMed] [Google Scholar]
- 38.Eychene A, Rocques N, Pouponnot C. Nat Rev Cancer. 2008;8(9):683–693. doi: 10.1038/nrc2460. [DOI] [PubMed] [Google Scholar]
- 39.Bennett BL, Sasaki DT, Murray BW, O’Leary EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, Bhagwat SS, Manning AM, Anderson DW. Proc Natl Acad Sci. 2001;98(24):13681–13686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Han S-i, Aramata S, Yasuda K, Kataoka K. Mol Cell Biol. 2007;27(19):6593–6605. doi: 10.1128/MCB.01573-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Benkhelifa S, Provot S, Nabais E, Eychène A, Calothy G, Felder-Schmittbuhl MP. Mol Cell Biol. 2001;21(14):4441–4452. doi: 10.1128/MCB.21.14.4441-4452.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ashcroft Frances M, Rorsman P. Cell. 2012;148(6):1160–1171. doi: 10.1016/j.cell.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Merritt SE, Mata M, Nihalani D, Zhu C, Hu X, Holzman LB. J Biol Chem. 1999;274(15):10195–10202. doi: 10.1074/jbc.274.15.10195. [DOI] [PubMed] [Google Scholar]
- 44.Xu Z, Kukekov NV, Greene LA. EMBO J. 2003;22(2):252–261. doi: 10.1093/emboj/cdg021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mooney LM, Whitmarsh AJ. J Biol Chem. 2004;279(12):11843–11852. doi: 10.1074/jbc.M311841200. [DOI] [PubMed] [Google Scholar]
- 46.Nihalani D, Meyer D, Pajni S, Holzman LB. EMBO J. 2001;20(13):3447–3458. doi: 10.1093/emboj/20.13.3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nihalani D, Wong HN, Holzman LB. J Biol Chem. 2003;278(31):28694–28702. doi: 10.1074/jbc.M304212200. [DOI] [PubMed] [Google Scholar]
- 48.Nihalani D, Wong H, Verma R, Holzman LB. Mol Cell Biol. 2007;27(7):2431–2441. doi: 10.1128/MCB.01479-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Leung IWL, Lassam N. J Biol Chem. 2001;276(3):1961–1967. doi: 10.1074/jbc.M004092200. [DOI] [PubMed] [Google Scholar]
- 50.Nihalani D, Merritt S, Holzman LB. J Biol Chem. 2000;275(10):7273–7279. doi: 10.1074/jbc.275.10.7273. [DOI] [PubMed] [Google Scholar]
- 51.Huntwork-Rodriguez S, Wang B, Watkins T, Ghosh AS, Pozniak CD, Bustos D, Newton K, Kirkpatrick DS, Lewcock JW. J Cell Biol. 2013;202(5):747–763. doi: 10.1083/jcb.201303066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu Z, Kukekov NV, Greene LA. Mol Cell Biol. 2005;25(22):9949–9959. doi: 10.1128/MCB.25.22.9949-9959.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kaneto H, Matsuoka TA, Nakatani Y, Kawamori D, Matsuhisa M, Yamasaki Y. Curr Diabetes Rev. 2005;1(1):65–72. doi: 10.2174/1573399052952613. [DOI] [PubMed] [Google Scholar]
- 54.Kaneto H, Xu G, Fujii N, Kim S, Bonner-Weir S, Weir GC. J Biol Chem. 2002;277(33):30010–30018. doi: 10.1074/jbc.M202066200. [DOI] [PubMed] [Google Scholar]
- 55.Waeber G, Delplanque J, Bonny C, Mooser V, Steinmann M, Widmann C, Maillard A, Miklossy J, Dina C, Hani E H, Waeber G, Delplanque J, Vionnet N, Nicod P, Boutin P, Froguel P. Nat Genet. 2000;24(3):291–295. doi: 10.1038/73523. [DOI] [PubMed] [Google Scholar]
- 56.Guo S, Dai C, Guo M, Taylor B, Harmon JS, Sander M, Robertson RP, Powers AC, Stein R. J Clin Invest. 2013;123(8):3305–3316. doi: 10.1172/JCI65390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Matsuoka T, Kaneto H, Stein R, Miyatsuka T, Kawamori D, Henderson E, Kojima I, Matsuhisa M, Hori M, Yamasaki Y. Mol Endocrinol. 2007;21(11):2764–2774. doi: 10.1210/me.2007-0028. [DOI] [PubMed] [Google Scholar]
- 58.Wang H, Brun T, Kataoka K, Sharma AJ, Wollheim CB. Diabetologia. 2007;50(2):348–358. doi: 10.1007/s00125-006-0490-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang C, Moriguchi T, Kajihara M, Esaki R, Harada A, Shimohata H, Oishi H, Hamada M, Morito N, Hasegawa K, Kudo T, Engel JD, Yamamoto M, Takahashi S. Mol Cell Biol. 2005;25(12):4969–4976. doi: 10.1128/MCB.25.12.4969-4976.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Butler AE, Robertson RP, Hernandez R, Matveyenko AV, Gurlo T, Butler PC. Diabetologia. 2012;55(11):2985–2988. doi: 10.1007/s00125-012-2666-2. [DOI] [PMC free article] [PubMed] [Google Scholar]