

Abstract
The peroxisome proliferator-activated receptor gamma (PPARγ) regulates osteoblast and osteoclast differentiation, and is the molecular target of thiazolidinediones (TZDs), insulin sensitizers that enhance glucose utilization and adipocyte differentiation. However, clinical use of TZDs has been limited by side effects including a higher risk of fractures and bone loss. Here we demonstrate that the same post-translational modifications at S112 and S273, which influence PPARγ pro-adipocytic and insulin sensitizing activities, also determine PPARγ osteoblastic (pS112) and osteoclastic (pS273) activities. Treatment of either hyperglycemic or normoglycemic animals with SR10171, an inverse agonist that blocks pS273 but not pS112, increased trabecular and cortical bone while normalizing metabolic parameters. Additionally, SR10171 treatment modulated osteocyte, osteoblast, and osteoclast activities, and decreased marrow adiposity. These data demonstrate that regulation of bone mass and energy metabolism shares similar mechanisms suggesting that one pharmacologic agent could be developed to treat both diabetes and metabolic bone disease.
Keywords: Bone, PPARγ, Insulin sensitizers, Osteoblast, Osteoclast, Osteocyte, Adipocyte, Post-translational modifications
Graphical abstract
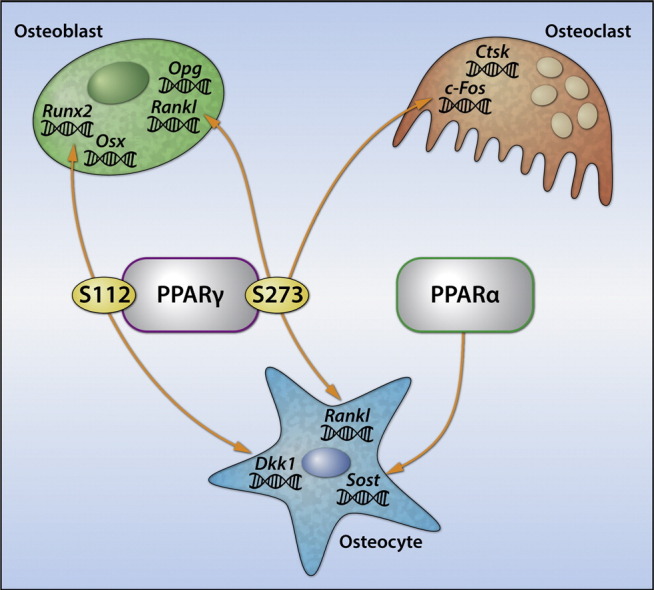
Highlights
-
•
PPARγ S273 regulates osteoclast differentiation and insulin sensitivity
-
•
PPARγ S112 regulates osteoblast and adipocyte differentiation
-
•
PPARγ and PPARα regulate osteocyte activities of bone formation and turnover
-
•
SR10171, a PPARγ inverse agonist and PPARα weak agonist, is anabolic for bone
Diabetes is a condition with compromised energy balance and is associated with bone fractures. Some treatment options for diabetes sensitize the patient to insulin via targeting the transcription factor PPARγ. PPARγ is also key regulator of bone formation and bone resorption. Anti-diabetic drugs TZDs target PPARγ protein and this leads to bone loss and increase in fractures in postmenopausal women. Bone mass and energy metabolism share similar regulating pathways, and here we demonstrate a new class of insulin sensitizers that is a selective modulator of PPARγ activity; resulting in a pharmacologic agent that can be beneficial for both diabetes and metabolic bone disease.
1. Introduction
Maintenance of bone mass and quality across a lifetime is a direct function of bone remodeling. Three types of cells are essential for this process and their role is coordinated in a highly sequenced manner. Osteoclasts remove old bone, osteoblasts form new bone, and osteocytes orchestrate this process through production of cytokines including receptor activator of nuclear factor kappa-B ligand (RANKL), sclerostin and dickkopf-related protein 1 (DKK1), which balance bone resorption and formation directly at the remodeling site. Animal studies have shed light on a close relationship between bone and energy metabolism, demonstrating that bone remodeling is closely linked to the osteoblastic response to insulin. Insulin induces osteoblastogenesis and RANKL production leading to high bone mass that is associated with increased bone turnover (Fulzele et al., 2010, Ferron et al., 2010). In contrast, in models of insulin resistance, induced by either high-fat diet feeding or genetic manipulation, bone turnover is decreased (Wei et al., 2014, Lecka-Czernik et al., 2015).
Numerous epidemiologic studies have shown that skeletal fragility in individuals with Type 2 diabetes (T2D) is increased at least 2-fold despite normal or high bone mineral density (BMD) and greater body mass index (BMI), the factors which are considered protective for most fractures in non-diabetic adults. The lack of the association between higher BMD and lower incidence of fractures indicates that in T2D bone biomechanical properties are compromised. Indeed, diabetic bone, particularly in the appendicular skeleton, has a number of structural characteristics which predispose to fractures, including greater cortical porosity, smaller cortical area, decreased bone material strength, and high bone marrow adiposity (Lecka-Czernik and Rosen, 2015).
Histomorphometric studies in humans indicate that bone turnover in older T2D patients is compromised, which may result in higher BMD but decreased bone quality (Krakauer et al., 1995). Furthermore, patients with T2D have decreased circulating levels of bone turnover markers, higher levels of sclerostin, a negative regulator of bone formation, and lower numbers of circulating osteoprogenitors. Moreover, highly reactive glucose metabolites (AGEs) are increased in T2D. These may impact bone ‘quality’ by slowing bone turnover and increasing bone stiffness and fragility through aberrant cross-links between collagen fibers (Lecka-Czernik and Rosen, 2015).
Skeletal homeostasis is linked to insulin sensitivity through the nuclear receptor PPARγ. PPARγ regulates lineage commitment of mesenchymal cells toward osteoblasts and adipocytes, and recruitment of osteoclasts from a pool of hematopoietic cells. Activation of PPARγ with the full agonist rosiglitazone suppresses osteoblastogenesis and induces adipocyte differentiation. This results in decreased bone formation and accumulation of adipocytes in the marrow cavity (Lazarenko et al., 2007). PPARγ activation also supports osteoclast differentiation through direct and indirect mechanisms. In monocytes, rosiglitazone stimulates osteoclast differentiation through a PPARγ coactivator 1-beta (PGC-1β)-dependent mechanism, while in mesenchymal cells it increases support for osteoclastogenesis by stimulating RANKL production (Wei et al., 2010, Lazarenko et al., 2007).
Thiazolidinediones (TZDs), full agonists of PPARγ, have consistently afforded robust efficacy for treatment of T2D. However, safety concerns including bone loss and increased fracture rates, particularly in postmenopausal women (Kahn et al., 2008), have restricted their use in T2D. Previously we demonstrated that the insulin sensitization provided by full and partial PPARγ agonists correlates with the ability of these drugs to block phosphorylation of PPARγ at serine 273 (pS273) (Choi et al., 2010), and that PPARγ anti-osteoblastic and insulin-sensitizing activity can be separated using selective modulators which block pS273 but have no activity toward phosphorylated serine 112 (pS112) (Kolli et al., 2014). Recently, we reported that the PPARγ inverse agonist SR2595 promoted osteoblastogenesis when tested in cultured human mesenchymal stem cells (MSCs) (Marciano et al., 2015). Synthetic optimization led to the identification of 2-phenoxy propanoic acids that exhibited superior pharmacokinetic properties over compounds in the biphenyl-2-carboxylic acid series (e.g., SR1664 and SR2595).
Here, we demonstrate that optimized 2-phenoxy propanoic acid modulator SR10171, with inverse agonist activity (repression of target gene expression) for PPARγ and modest agonist activity (activation of target gene expression) for PPARα, increases bone mass by increasing bone formation and bone turnover; two processes affected in diabetic bone. We show that SR10171 targets osteoblast, osteocytes, and osteoclasts through mechanisms overlapping those regulating whole body energy metabolism. We demonstrate that the same posttranscriptional modifications (PTMs) which determine adipocytic (pS112) and insulin sensitizing (pS112) activities also determine PPARγ osteoblastic and osteoclastic activities, respectively. The elucidation of such overlap is a prerequisite for development of efficient anti-diabetic therapy that will simultaneously treat diabetic bone disease.
2. Material and methods
2.1. Animal experiments
C57BL/6 mice, 12 week old males, were purchased from the Jackson Laboratories (Bar Harbor, ME). The animal treatment and care protocols conformed to the NIH Guidelines and were performed under the University of Toledo Health Science Campus Institutional Animal Care and Utilization Committee protocol.
2.1.1. Lean mice experiment
Animals were fed for 8 weeks either non-supplemented regular diet (RD) providing 12 kcal% from fat (2016 Teklad Global, Harlan Laboratories, Indianapolis, IN)., or RD supplemented with either rosiglitazone or SR10171 at the dose of 15 mg/kg/d of each drug (n = 5 per group). This dose of rosiglitazone and duration of treatment was previously established as decreasing bone mass by 20%. The matching dose of SR10171 was calculated based on the following reasoning. SR10171 binding affinity to PPARγ is 10-fold lower and S273 phosphorylation blocking activity is 50% lower than rosiglitazone (Table 1). This together with 2.5-fold higher than rosiglitazone retention levels in plasma of animals treated for 4 days (Table 2) resulted in the decision to treat animals with the same dose of each drug. Food intake was monitored during the entire experiment to calculate an ingested dose of SR10171 (12.7 mg/kg/d) and rosiglitazone (11.7 mg/kg/d).
Table 1.
Characteristics of SR10171 and rosiglitazone.
| Compound | IC50 (nM)a PPARγ | EC50 (nM) PPARγ GAL-4 | EC50 (nM) PPARγ PPRE | % inhibition S273pPPARγ (20 μM) | IC50 (μM)a PPARα | EC50 (μM) PPARα GAL-4 |
|---|---|---|---|---|---|---|
| Rosiglitazone | 18 | 7.4 (100%) | 10 | 100 | No binding | No activity |
| SR10171 | 220 | 7260 (4%) | Inverse agonist | 54 | 1.1 | 1.5 |
LanthaScreen; IC50 — half maximal inhibitory concentration; EC50 — half maximal effective concentration.
Table 2.
Plasma levels of SR10171 and rosiglitazone at the end of experiments.
| Treatment | Plasma levels |
|
|---|---|---|
| SR10171 (μM) | Rosi (μM) | |
| C57BL6 on RD 4 days treatment: SR10171 – 5 mg/kg/d; Rosi – 5 mg/kg/d |
4.01 | 1.67 |
| DIO model on HFD 4 wks treatment: SR10171 – 22.1 mg/kg/d; Rosi – 24.2 mg/kg/d |
14.25 | 0.10 |
| C57BL/6 on RD 8 wks treatment: SR10171 – 12.7 mg/kg/d; Rosi – 11.7 mg/kg/d |
15.97 | 6.28 |
2.1.2. DIO mice experiment
Animals were fed ad libitum for 11 weeks high fat diet (HFD) providing 45 kcal% from fat (D12451; Research Diets, New Brunswick, NJ) to develop diet induced obesity (DIO). DIO mice were randomized according to body weight and were separated into three groups (n = 7 per group) and fed for 4 weeks as follows: HFD (control), HFD supplemented with rosiglitazone at the dose 25 mg/kg/d, and HFD supplemented with SR10171 at the dose 25 mg/kg/d. The dose of each drug for this experiment was decided based on our experience that a dose of rosiglitazone between 20 and 25 mg/kg/day administered for 4 weeks to DIO animals results in significant loss of trabecular bone in both, axial and appendicular skeleton (Kolli et al., 2014). Ingested doses of SR10171 (22.1 mg/kg/d) and rosiglitazone (24.2 mg/kg/d) were calculated at the end of treatment based on food intake monitored throughout duration of entire experiment. In parallel, a group of sex- and age-matched control (n = 4) was fed RD. Testing of metabolic parameters is described in the Supplemental data.
2.2. Bone analysis using microCT and histomorphometry
Details of methods to measure bone and marrow fat using mCT and static and dynamic histomorphometry are described in the Supplemental data.
2.3. Cell culture experiments
MLO-A5 cells represent an osteocytic cell line (gift from Dr. Bonewald, University of Missouri, Kansas City, MO), AD2 and U33 cells represent spontaneously immortalized marrow pre-adipocytes and pre-osteoblasts, respectively, whereas U33/γ2 cells represent U33 cells stably transfected with PPARγ2 expression construct, and were previously described (Lecka-Czernik et al., 1999). RAW264.7 cells represented a murine monocyte/macrophage cell line. Cell cultures, differentiation assays, gene silencing using lentiviral infection with shRNA specific for PPARγ or PPARα, and gene expression analysis using real time PCR are described in detail in the Supplemental data.
2.4. Analysis of S112 and S273 phosphorylation
AD2 cells representing marrow preadipocytes were pre-treated with vehicle, 1 μM rosiglitazone, or 5 μM SR10171 for 1 h followed by treatment with 50 ng/ml TNF-α for 1 h followed by Western blot analysis for pS273 and pS112 of PPARγ (Choi et al., 2010). Details are in the Supplemental data.
2.5. Extraction of osteoblast- and osteocyte-enriched fractions
Cell fractions enriched in either osteoblasts or osteocytes were isolated by sequential collagenase digestion of fresh femora bone, according to a previously described protocol (Kramer et al., 2010), and immediately lysed for analysis of gene expression.
2.6. Analysis of SR10171 and SR9525 pharmacology
Methods for the LanthaScreen TR-FRET Competitive Binding Assay, hydrogen/deuterium exchange mass spectrometry (HDX-MS), NR box peptide interaction assay, and in silico docking are described in detail in the Supplemental data.
2.7. Statistical analysis
Data are presented as the means ± SD and were analyzed by One Way Anova using statistical software package of SigmaPlot (version 13.0). P value less than 0.05 was considered statistically significant.
3. Results
3.1. pS112 and pS273 determine PPARγ skeletal activities
PPARγ pro-adipogenic and insulin sensitizing activities in cells of mesenchymal lineage correlate respectively with the phosphorylation status of serines S112 and S273 (Hinds et al., 2011, Choi et al., 2011). To test whether these PTMs correlate with PPARγ osteoblastic and osteoclastic activities we transfected cells of either osteoblastic lineage (U33 cells) or osteoclastic lineage (RAW264.7) with mutated PPARγ expression constructs in which phosphorylation at either S112 (S112A mutant) or S273 (S273A mutant) was blocked. As shown in Fig. 1a, in U33 cells S112A but not S273A mutation decreased extracellular matrix mineralization and expression of runt-related transcription factor 2 (Runx2) and osterix (Osx), and increased expression of Dkk1, an inhibitor of Wnt signaling pathway. Thus, induction of adipocytic differentiation with PPARγ dephosphorylated at S112 simultaneously suppresses osteoblastic phenotype, perhaps as a result of reciprocal relationship between PPARγ and RUNX2 activities (Ge et al., 2016). In contrast, blocking phosphorylation at S273 increased PPARγ pro-osteoclastic activity in RAW264.7 cells (Fig. 1b) including increased number of TRAP + multinucleated osteoclasts and expression of c-Fos and catepsin K (Ctsk) markers of maturating osteoclasts (Fig. 1b). In addition, blocking phosphorylation at S273 increases support for osteoclastogenesis by increasing Rankl and decreasing osteoprotegerin (Opg) expression in mesenchymal U33 cells (Fig. 1a). Thus, PPARγ anti-osteoblastic action requires blocking the same PTM as is required for adipogenesis, whereas pro-osteoclastic action requires blocking the PTM as is required for insulin-sensitizing activity.
Fig. 1.

Effect of pS112 and pS273 of PPARγ on adipocyte, osteoblast and osteoclast differentiation.
(a) The effect of S112A and S273A mutations on osteoblast differentiation. U-33 cells were transfected with either WT or mutated PPARγ expression constructs and grown in pro-osteoblastic conditions followed by measurement of extracellular calcium or RNA isolation. (b) Effect of PPARγ S112A and S273A mutations on osteoclast differentiation. RAW264.7 was transfected with either WT or mutated PPARγ expression constructs followed by culture in medium supplemented with 50 ng/ml M-CSF and 50 ng/ml RANKL for 4 days, then either stained for TRAP expression (left panel) or RNA was isolated for analysis of gene expression (right panel). (c) Chemical structure of SR10171 ((S)-2-(3-((5-((1-(4-(tert-butyl)phenyl)ethyl)carbamoyl)-2,3-dimethyl-1H-indol-1-yl)methyl)phenoxy)-2-methylpropanoic acid) and rosiglitazone ((RS)-5-[4-(2-[methyl(pyridin-2-yl)amino]ethoxy)benzyl]thiazolidine-2,4-dione). (d) The effect of SR10171 and rosiglitazone on blocking of S273 phosphorylation in AD2 cells. (e) The effect of ligands on blocking of S112 phosphorylation in AD2 cells. Protein levels quantification was performed using Image J software and displayed as graphs below Western blot renderings. (f)–(j) cell treatment consisted of 10 μM SR10171 (71), or 1 μM rosiglitazone (R), or DMSO as vehicle control (V). (f) Left and middle panels — the effect on adipocyte-specific gene marker expression in marrow MSCs treated for 6 days, as above. Right panel — the effect on transcriptional activity of PPARγ measured in U33/γ2 cells transfected with PPRE promoter construct and treated for 24 h. (g) Osteoblast differentiation as measured in CFU-OB assay. (h) Expression of osteoblast-specific gene markers in marrow MSCs. (i) The effect on osteoclast differentiation. Non-adherant bone marrow cells were cultured as described in experimental procedures. TRAP + multinucleated cells (> 4 nuclei) were enumerated and average calculated from triplicates. (j) Expression of osteoclasts gene markers in cultures described in (i). Data represent the mean ± SD (n = 3–6 experiments). *P < 0.05; **P < 0.01; ***P < 0.001 One Way Anova.
We then examined whether these activities can be manipulated pharmacologically. SR10171 compound (Fig. 1c) is an optimized analog of SR1664 (Choi et al., 2011) which demonstrates high-affinity binding to PPARγ and unlike SR1664, exhibits partial inverse agonism of PPRE-mediated transcriptional activity (Table 1) and demonstrates modest PPARα agonist activity (EC50 = 1.5 μM). The structure activity relationship (SAR) of SR10171 is consistent with other examples from this chemical series (Asteian et al., 2015, Marciano et al., 2015). Importantly, SR10171 effectively blocks phosphorylation of S273 (pS273), which has been shown to correlate with PPARγ insulin sensitizing activity; however, in contrast to full agonist rosiglitazone, SR10171 inverse agonism actively suppresses PPARγ basal transcriptional activity (Table 1).
As shown in Fig. 1d and e, both SR10171 and rosiglitazone block phosphorylation of S273, yet only rosiglitazone alters phosphorylation of S112. Next, we investigated whether SR10171 changes marrow MSC allocation toward osteoblast and adipocyte lineage. Consistent with inverse agonist activity, SR10171 decreased basal expression of fatty acid binding protein 4 (Fabp4) and adiponectin (Adipoq) in marrow MSC culture, while the full agonist rosiglitazone increased their expression substantially (Fig. 1f). This correlated with the expected changes in transcriptional activity of PPARγ, as measured in a reporter gene assay (Fig. 1f). A combination of a colony forming units of osteoblasts (CFU-OB) assay and expression levels of osteoblast-specific gene markers in primary bone marrow cultures demonstrated that SR10171 treatment did not alter MSC commitment toward the osteoblast lineage, while rosiglitazone attenuated CFU-OB formation and expression of osteoblast-specific gene markers (Fig. 1g and h).
Results from an assay for osteoclast differentiation from a pool of non-adherant hematopoietic stem cells (HSCs) confirmed that both SR10171 and rosiglitazone induce pro-osteoclastic activity of PPARγ where treatment with either compound increased the number of multi-nucleated TRAP-positive osteoclast-like cells ex vivo and increased expression of c-Fos and Ctsk (Fig. 1i and j). These observations were consistent with PTM status and suggest that the mechanism by which PPARγ increases osteoclast differentiation from a pool of HSCs is different than PPRE-mediated transcriptional activity regulating adipocyte/osteoblast differentiation from MSCs.
3.2. Pharmacological repression of PPARγ is anabolic for bone
Given that the full agonists rosiglitazone and pioglitazone decrease bone mass and increase marrow adiposity, and inverse agonists (Marciano et al., 2015) suppress adipogenesis and promote osteogenesis in vitro, we sought to determine if the above findings can be recapitulated in vivo following chronic administration in mice. SR10171 was chosen for testing in long-term bone studies due to its excellent pharmacokinetic properties enabling formulation in feed to facilitate chronic daily administration of drug coupled with its unique pharmacological profile compared to SR2595. While both SR2595 and SR10171 (Fig. S1a) are inverse agonists that suppress PPARγ transcriptional activity (Fig. S1b), they display subtle differences in their binding poses (Fig. S1c), distinct conformational dynamics as determined by HDX (Fig. S1d), and differential affinity for peptides containing NR box binding motifs derived from transcriptional coregulators (Fig. S1e). These distinct molecular interactions result in different effects on RUNX2 activity (Fig. S1f) and expression of markers of Wnt and BMP signaling pathways activity (Fig. S1g).
To test the longitudinal effects of SR10171 administration on the skeleton of lean, normoglycemic C57BL/6 mice were treated with compound at a dose of 15 mg/kg/d for 8 weeks. Rosiglitazone treated animals (15 mg/kg/d) served as a control of PPARγ-mediated metabolic and skeletal effects. The dose of each drug used in these studies was based on compilation of SR10171 and rosiglitazone pharmacokinetics, binding affinities, and activities toward blocking of PPARγ pS273. After 8 weeks of treatment, we observed a robust positive effect of SR10171 on bone. Trabecular bone mass in proximal tibia was increased by 26% as compared to non-treated controls mainly due to increased number of trabeculae (Fig. 2a). As expected, rosiglitazone decreased trabecular bone mass by decreasing trabeculae thickness. Bone formation was markedly increased in the proximal tibia of mice receiving SR10171. Histomorphometric measurements confirmed that an increase in bone mass with SR10171 was due to an increase in mineral apposition rate (MAR) and bone formation rate (BFR), as calculated from the distance between two calcein-labeled osteoid layers (Fig. 2b), and from an increased number of osteoblasts (Fig. 2c). As expected, rosiglitazone attenuated MAR and BFR and decreased the number of osteoblasts. Interestingly, both compounds increased the number of osteoclasts (Fig. 2c). The calculated ratio of osteoblasts to osteoclasts was 2.5 for control, 1.4 for rosiglitazone and 2.8 for SR10171 indicating higher bone formation for SR10171 and higher resorption for rosiglitazone, as compared to control (Fig. 2c). SR10171, consistent with its inverse agonist activity, decreased the number of marrow adipocytes by 50%, whereas full agonist rosiglitazone increased adipocyte number by 5-fold (Fig. 2c). Combined, these results indicate that the increased trabecular bone mass in lean animals receiving SR10171 resulted from high bone formation associated with increased bone turnover, whereas bone loss in animals receiving rosiglitazone resulted from suppressed bone formation and increased bone resorption.
Fig. 2.

Effect of SR10171 and rosiglitazone on trabecular and cortical bone of lean and DIO C57BL/6 mice.
(a)–(c) Analysis of trabecular bone in proximal tibia of lean mice. (a) mCT renderings (upper panels) and measurements of trabecular parameters. Bar — 0.1 mm. (b) Dynamic histomorphometry of bone double-labeled with calcein. Microphotographs of entire proximal tibia labeled with calcein (upper panels) (magnification 5 ×; bar — 0.2 mm) and bone surface labeled with calcein (lower left panels) (magnification 40 ×; bar — 0.02 mm). Quantification of mineral apposition rate (MAR) and bone formation rate (BFR) (lower right panels). (c) Number of osteoblasts per bone perimeter (N.Ob/B.Pm), osteoclasts per bone perimeter (N.Oc/B.Pm), osteoblasts to osteoclasts ratio (N.Ob/N.Oc), and number of adipocytes (N.Ad) per region of interest (ROI). (d) Glucose tolerance in DIO mice at the end of experiment. (e)–(f) Analysis of trabecular bone in tibia of DIO mice. (e) mCT measurements of trabecular parameters. (f) MAR and BFR quantified from bone double-labeled with calcein. (g) mCT renderings of fat stained with osmium tetroxide (left panels, fat appears as yellow, bar — 1.0 mm) and its volumetric quantification (right panel). (h) Cortical bone of lean animals. (i) Cortical bone of DIO animals. Representative microphotographs of calcein-labeled endosteal and periosteal bone surface (upper panels) (magnification 5 ×; bar — 0.2 mm), histomorphometry of labeled periosteal bone perimeter (MS/BS) and osteocyte lacunae density (lower panels). TV — tissue volume; BV — bone volume; BV/TV — bone mass; Tb.N — trabecular number; Tb.Th — trabecular thickness. Data represent the mean ± SD (n = 3–7 per group). *P < 0.05; **P < 0.01; ***P < 0.001 One Way Anova.
Next we tested the efficacy of SR10171 in mice with diet-induced obesity (DIO) and impaired glucose tolerance. Mice were administered either high fat chow, or the same chow supplemented with either SR10171 or rosiglitazone at the dose of 25 mg/kg/d for 4 weeks. The dose was increased as the duration of treatment was decreased; however, similar effects on metabolic parameters as described below were observed with dose as low as 5 mg/kg given once-a-day orally (data not shown). SR10171 normalized glucose tolerance in DIO animals to the same degree as rosiglitazone (Fig. 2d). SR10171 increased both bone tissue volume and trabecular bone mass, as well as both MAR and BFR (Fig. 2e and f). For marrow adiposity, the effect of SR10171 resulted in almost 5-fold decrease in fat volume, while fat volume in the tibia of mice treated with rosiglitazone increased almost 100-fold, as compared to no drug control animals (Fig. 2g). Not surprisingly, rosiglitazone caused significant bone loss across all bone sites and was associated with high volumes of marrow adipose tissue, during regular and high fat diets, whereas SR10171 had a positive effect on the appendicular skeleton but was neutral for the axial skeleton and showed virtually no marrow adiposity at either site (Fig. S2a, Table S1).
Positive effects on cortical bone mass were also observed following SR10171 treatment. Calcein labeling of the periosteal surface of the midshaft tibia in both lean and DIO animals was increased, as compared to control mice (Fig. 2h and i). Accordingly, in both models the labeled surface of periosteum was larger in bone of animals that received SR10171, and the labeling was more intense. In lean SR10171 treated animals in which periosteal labeling was only 1.5 times larger than in the control (Fig. 2h), the cortical area was not affected (Fig. S2b). However, in DIO mice the periosteal labeling was 9 times larger in SR10171 treated than in control, which had almost completely attenuated periosteal activity (Fig. 2i). This increase in activity correlated with increased cortical area (Fig. S2b). SR10171 also increased osteocyte lacunae density in cortical bone of both models providing further evidence of increased bone turnover induced by this compound (Fig. 2h and i). Analysis of the organization of collagen fibers by second-harmonic generation confocal microscopy showed a highly organized fiber structure in bone adjacent to mineralization zone of the periosteum (Fig. S3) indicating that a newly formed bone is of normal structure. The beneficial effect of SR10171 on bone was also reflected in changes in cortical tissue mineral density (TMD, mg HA/cm3) (control: 954.6 ± 17.6; rosiglitazone: 954.5 ± 17.9; SR10171: 978.6 ± 10.4, P < 0.05 vs. control and rosiglitazone).
3.3. Treatment of DIO mice with SR10171 normalizes energy metabolism
Even though SR10171 and rosiglitazone exhibited the same actions on glucose tolerance (Fig. 2d), these compounds had contrasting effects on body weight, fat content, and energy metabolism. SR10171 decreased body weight in DIO mice maintained on HFD, as compared to controls or rosiglitazone-treated group on the same diet, and had no impact on body weight in lean animals (Fig. 3a and b). Interestingly, the decrease in body weight in DIO mice was observed as early as one week after initiation of SR10171 treatment, and was stabilized to the level of control animals two weeks post dosing. Food intake was not affected indicating that both administered drugs did not cause taste aversion (Fig. S4a). The observed decrease in the body mass in SR10171 treated DIO mice was accompanied by a decrease in fat content (Fig. S4b), and whole body composition measured as lean/fat ratio was similar to mice fed regular chow (Fig. S4b). Finally, SR10171 treatment resulted in a decrease in the mass of both epididymal white adipose tissue (eWAT) and interscapular brown adipose tissue (iBAT) in obese mice (Fig. S4c).
Fig. 3.

Effect of SR10171 and rosiglitazone on fat and metabolic parameters of DIO and lean C57BL/6 mice.
(a) Body weight change in animals fed HFD and RD for 8 weeks before initiation of feeding medicated diet for the next 4 weeks. Arrow indicates start of administration of medicated diet. Asterisks indicate significant differences between control RD and HFD + 71. (b) Body weight of lean animals at the end of an 8 week feeding medicated diet. (c) Calorimetric measurements of respiratory parameters in DIO mice at the end of experiment. Linear plots show measurements made every 20 min within 24 h period, whereas bar plots show average of measurements collected during either 12 h light or 12 h dark period. VO2 — oxygen consumption (ml/h/kg); VCO2 — carbon dioxide production (ml/h/kg); RER — respiratory exchange ratio. (d) Gene expression analysis of adipocytic markers in eWAT of DIO mice. (e) Calorimetric measurements of respiratory parameters in lean mice at the end of experiment. (f) Gene expression analysis of adipocytic markers in eWAT of lean mice. Data represent the mean ± SD (n = 4–5 per group). *P < 0.05; **P < 0.01; ***P < 0.001 One Way Anova.
Changes in body composition in SR10171 treated DIO mice corresponded to increased energy expenditure. Oxygen consumption, carbon dioxide release, calculated respiratory exchange ratio (RER), heat production and locomotion were significantly higher in DIO mice receiving SR10171, as compared to control and rosiglitazone-treated group (Fig. 3c, Tables S2 and S3). Treatment with SR10171 increased the expression of adiponectin and fatty acid transporter FABP4 in eWAT of DIO mice to that observed in age-matched lean controls (Fig. 3d), and normalized the expression of regulators of futile metabolism in adipocytes (forkhead box protein C2 (FoxC2) and bone morphogenic protein 4 (Bmp4)), and beige fat markers (T-box transcription factor 1 (Tbx1) and homeobox protein C9 (HoxC9)). That was accompanied with a modest increase in uncoupling protein 1 (Ucp1) expression (Fig. 3d).
Consistent with no effect on body weight in lean animals (Fig. 3b), SR10171 had no impact on body composition (not shown) and had no effect on eWAT weight; however, a modest decrease in the weight of iBAT was observed (Fig. S4d). In lean animals, indirect respiratory calorimetry showed no effect of SR10171 on any measured parameter, while rosiglitazone increased respiration and heat production indicating active modulation of metabolism even in non-pathologic conditions (Fig. 3e, Tables S2 and S3). Consequently, in lean non-diabetic mice, SR10171 afforded only a modest decrease in expression of Fabp4 and Ucp1 whereas rosiglitazone increased the expression of Adipoq, Fabp4, Bmp4, HoxC9, and Ucp1 (Fig. 3f). Thus, the normalizing effect of SR10171 under conditions of impaired energy metabolism (obesity) and the neutral effect in conditions of efficient energy metabolism (lean) suggest that SR10171 modulates metabolism in an “on-demand” manner to which increased insulin sensitivity may contribute.
3.4. PPARγ and PPARα regulate osteocyte activities
Since SR10171 increased bone formation and osteoblast number in animals (Fig. 2) but was neutral for osteoblast differentiation in cultured marrow MSCs (Fig. 1g and h), we analyzed the phenotype of osteoblasts and osteocytes freshly-isolated from femora of treated animals. Isolated cell populations reflect the status of osteoblasts and osteocytes at the time of animal sacrifice as it is analyzed immediately after liberation from either endosteal bone surface (fraction 3, OB) or cortical mineralized matrix (fraction 6, OT). The enrichment of each fraction was assessed by comparing levels of expression of genes known to be more abundantly expressed in either osteoblasts (Osx and Ibsp) or osteocytes (Dmp1, Sost, Rankl, Dkk1, E11, and FGF23). As shown in Fig. S5, the expression level of cell type specific markers was 3–100 folds different between OB and OT fractions, thus providing reassurance of high enrichment. In the osteoblast fraction isolated from SR10171-treated lean animals, the expression of Osx and matrix extracellular phosphoglycoprotein regulating phosphate metabolism (Mepe), was increased indicating that osteoblasts were highly activated (Fig. 4a).
Fig. 4.

PPARγ and PPARα activities regulate osteoblast and osteocyte functions.
(a) Relative mRNA expression of gene markers in isolated osteoblasts from femora endosteal surface. (b) The effect of SR10171 and rosiglitazone on transcriptional activity of TCF/LIF/β-catenin complex measured in U33/γ2 cells transfected with Top-Flash promoter construct and treated for 72 h with tested ligands. (c) Relative mRNA expression of gene markers in isolated osteocytes from femora cortical bone. Expression of Dmp1, E11, and Fgf23, served as a purity control of osteocyte isolates and confirmed equivalent enrichment in all samples analyzed. (d) Relative mRNA expression of Sost, Dkk1, and Rankl in primary osteocytes treated with either 10 μM SR10171, or 1 μM rosiglitazone, or 50 μM WY14643 in ex vivo conditions for 3 days. (e) Protein levels after shRNA knocked-down (KD) of either PPARα or PPARγ. KD was introduced in MLO-A5 cells using lentiviral shRNA constructs. Protein levels of either PPARγ or PPARα were assessed by Western blots, quantitated and normalized to the levels of HSP90, a housekeeping protein. (f) The effect of PPARγ (γKD) or PPARα (αKD) silencing on Sost and Dkk1 gene expression. Data represent the mean ± SD (n = 3 per group, repeated twice). *P < 0.05; **P < 0.01; ***P < 0.001 One Way Anova.
Previously it has been shown that networks of signaling pathways regulate the activity of osteoblasts. The Wnts and BMPs are the predominant ligands for local autocrine/paracrine effects in bone, including control of Osx and Mepe expression (Cho et al., 2012, Tan et al., 2014). Indeed, osteoblasts from SR10171-treated animals showed increased expression of Wnt10b ligand and decreased expression of Axin2, a member of β-catenin degradation complex, but no change in expression of BMP ligands (Fig. 4a). In contrast, osteoblasts from rosiglitazone-treated animals have an opposite pattern of these gene expression consistent with suppressive effect on Wnt and BMP signaling activities and osteoblastogenesis (Shockley et al., 2009). Results from a luciferase reporter gene assay using Top-Flash construct confirmed that SR10171 activated β-catenin transcriptional activity, whereas rosiglitazone decreased it (Fig. 4b). Paradoxically, SR10171 decreased Opg expression which is positively regulated by the canonical Wnt pathway and contributes to the anti-osteoclastic effect of this network (Fig. 4a). Regardless, as shown in Fig. 1a, Opg expression is directly dependent on pS273, the same PPARγ PTM which regulates osteoclastogenesis and RANKL expression and insulin sensitivity. Recently it was demonstrated that pioglitazone (a TZD) had a negative effect on Opg expression (Avtanski et al., 2016) supporting the conclusion that PPARγ is dominant over the Wnt pathway with respect to regulation of this network. Although both compounds have pro-osteoclastic effect and both decreased Opg expression, it is interesting that only rosiglitazone decreased expression of Wnt16 (Fig. 4a). Wnt16 was recently identified as a negative regulator of resorption in cortical bone (Moverare-Skrtic et al., 2014) and a positive regulator of periosteal bone formation (Wergedal et al., 2015). Thus, SR10171 and rosiglitazone exhibit differential regulation of Wnt16 expression which in turn may contribute to their different effects on cortical bone (Fig. 2h and i; Fig. S2b).
Osteoblasts activity is regulated by the secretory action of osteocytes, including sclerostin, encoded by the Sost gene, and DKK1 proteins, which antagonize Wnt pathway activity by binding to LRP5/6 receptors (Baron and Kneissel, 2013). Consistent with higher osteoblasts function with increased bone formation rates, Sost and Dkk1 expression were decreased in osteocytes of animals receiving SR10171 (Fig. 4c). In contrast, Sost expression was unaffected and Dkk1 was increased in osteocytes derived from animals receiving rosiglitazone. Osteocytes also regulate bone resorption by producing RANKL (Xiong et al., 2011). Both compounds increased expression of Rankl in osteocytes (Fig. 4c). This together with decreased Opg expression in endosteal osteoblasts correlated with a greater number of osteoclasts in vivo in bone of treated animals (Fig. 2c). Thus, in contrast to the opposing effects of SR10171 and rosiglitazone on osteoblast and osteocyte function that support bone formation both compounds positively regulate osteoclast differentiation via two mechanisms, one consisting of enhanced hematopoietic osteoclastogenic differentiation and the other modifying mesenchymal cell support for osteoclast differentiation.
To address the question whether Sost, Dkk1, and Rankl transcriptions are directly regulated by PPARs in osteocytes, the expression level of each gene was measured in primary osteocytes treated ex vivo with SR10171, rosiglitazone, and WY14643 which is a PPARα full agonist. As expected, SR10171 decreased the expression of Sost and Dkk1, whereas rosiglitazone not only increased expression of Dkk1, but also Sost, which was not observed in vivo (Fig. 4d). In contrast to the effect on Rankl expression in vivo, when applied directly on primary osteocytes SR10171 did not increase its expression, while rosiglitazone had a sustained positive effect (Fig. 4d). Treatment with WY14643 decreased expression of Sost, but did not have an effect on expression of Dkk1 and Rankl (Fig. 4d). To analyze which actions of SR10171 in osteocytes are mediated through PPARγ or PPARα, we knocked-down (KD) the expression of each receptor separately in MLO-A5 cells using lentiviral delivery of PPAR-specific shRNA. Decrease in PPARα protein levels by 65% (Fig. 4e) resulted in a 5-fold increase in basal Sost expression (Fig. 4f), while a 50% decrease in PPARγ protein levels had no effect suggesting that Sost is under negative control of PPARα. In contrast, PPARγ KD, but not PPARα, abrogated Dkk1 expression (Fig. 4f) suggesting that Dkk1 is under positive control of PPARγ. In conclusion, SR10171 regulates osteocyte activities via combined effects on PPARγ and PPARα.
4. Discussion
Currently there are no drugs that prevent insulin resistance resulting from diet-induced obesity while simultaneously being anabolic for bone despite the need for such agents to combat the ongoing epidemic of T2D and its deleterious effect on the skeleton. Approved agents that are used to help control obesity and insulin sensitization are either neutral on bone or more troublesome and have negative effects on the skeleton. Drugs that enhance sympathetic outflow and thereby reduce obesity by increasing energy expenditure cause bone loss by uncoupling formation from resorption (Motyl et al., 2015). The TZDs which are robust insulin sensitizers, cause bone loss in mice and result in increased risk for fractures in humans (Kahn et al., 2008). SGLT2 inhibitors effectively control plasma glucose levels, but negatively affect bone by altering calcium and phosphate homeostasis (Taylor et al., 2015). Underlying the negative side effects of these therapies is the increased bone fragility typical in T2D patients that is associated with decreased bone mechanical properties caused by impaired bone turnover (Lecka-Czernik and Rosen, 2015). Drugs which restore coupled bone remodeling and that simultaneously restore insulin sensitivity would be ideal.
Here we present compelling evidence that polypharmacological modulation of both PPARγ (repression) and PPARα (mild activation) with SR10171, a prototype chemical probe which fulfills the “one drug, multiple targets” concept, is anabolic for bone independent of its insulin sensitizing activity as supported by the following observations. First, we demonstrated that the same PTMs that determine adipocytic and insulin sensitizing properties, also define PPARγ osteoblastic and osteoclastic activities. We showed that a reduction in pS273 correlated with pro-osteoclastic and a reduction in pS112 correlated with anti-osteoblastic activity. Importantly, we demonstrated that the same PTM at S273 regulates both osteoclast differentiation from a pool of hematopoietic cells, and mesenchymal cell support by regulating RANKL and OPG expression. Since SR10171 reduced pS273, it simultaneously enhanced insulin sensitivity while increasing osteoclastogenesis. In contrast to rosiglitazone, SR10171 did not reduce pS112, blocked adipogenesis and increased β-catenin and RUNX2 transcriptional activity when acting directly on cells of the mesenchymal lineage. Therefore, SR10171 can be categorized as an agonist for insulin sensitizing and bone resorbing activities, and an inverse agonist for adipocytic and bone forming activities. The phenotype of mice treated with SR10171 was one of insulin sensitization and increased bone turnover. Although bone resorption was increased with SR10171, there was no break on osteoblastogenesis, hence coupling was restored within the bone remodeling unit as formation was maintained in spite of higher turnover.
Second, we demonstrated that two members of the PPAR subfamily regulate osteocytic activity that in turn supports bone formation and bone turnover. SR10171 was shown to target PPARγ and/or PPARα in osteocytes, which results in decreased expression of two inhibitors of Wnt signaling, DKK1 and sclerostin, while increasing RANKL. This would result in increased bone turnover with the balance shifted toward bone formation. Indeed, high expression of Osterix and Mepe in endosteal osteoblasts, and the lack of an SR10171 effect on these transcript levels in vitro, suggest an overall increase in osteocyte activity. Considering that the expression of all three factors, sclerostin, DKK1, and RANKL in osteocytes, the most abundant cells in bone, highly exceeds their expression in osteoblasts (Fig. S4), suggests that osteocytes are the principal cell target for the anabolic effect of SR10171 on bone.
A potential mechanism underlying the skeletal effects of SR10171 could be modulation of the Wnt/β-catenin signaling pathway. Wnt signaling is crucial to all bone cell types and constitutes a complex communication network between different components and modes of signaling (e.g. canonical vs. non-canonical) which operate in the context of specific cell types (osteoblast, osteocyte and osteoclast) and bone marrow milieu (Baron and Kneissel, 2013). It has previously been established that rosiglitazone inhibits Wnt canonical pro-osteoblastic and anti-osteoclastic activities in bone, including degradation of β-catenin and suppression of multiple members of this signaling pathway (Shockley et al., 2009, Rahman et al., 2012). Here we showed that SR10171 uncouples Wnt pro-osteoblastic (β-catenin activity; Osx and Mepe expression) from anti-osteoclastic activities (Opg expression) and correlated these activities to the distinctive effects on pS112 and pS273 of PPARγ.
Third, we observed a positive effect of SR10171 on cortical bone mass, an important skeletal compartment as it represents 80% of skeletal mass and is affected by high cortical porosity in T2D (Patsch et al., 2013), and most fractures in T2D patients occur in the cortical compartment. Increased cortical thickness in DIO mice treated with SR10171 was associated with extensive activation of the periosteal surface, which was relatively inactive in DIO and lean animals. As such, the magnitude of periosteal activation induced by SR10171 (9-fold in DIO vs. 1.5-fold in lean) may explain the positive effect on cortical thickness in the context of obesity. Although it is unclear at this point whether these effects result from direct activation of PPARs in periosteal osteoblasts, or were mediated through signaling derived from osteocytes, this observation has clinical relevance since the periosteum actively facilitates fracture healing. Since T2D patients have impaired fracture healing in general, understanding periosteal function in response to treatment with PPAR modulators is essential. Similarly, the role of PPARα in the maintenance of bone mass is not well understood. It has been shown that PPARα deficiency increased the size of the bone marrow cavity, but did not have an effect on cortical thickness (Wu et al., 2000). Interestingly, two reports demonstrated that PPARα activation in rodents with fibrates prevented bone loss induced by either estrogen deficiency or TZD treatment, and induced periosteal activity (Still et al., 2008, Syversen et al., 2009). Here, we clearly demonstrate that PPARα is a negative regulator of sclerostin expression. Recently, anti-sclerostin antibodies have been described as potential anti-osteoporotic therapies, thus targeting of PPARα directly in osteocytes may offer alternative pharmacological possibilities to regulate sclerostin levels.
Interestingly, SR10171 had an anabolic effect on the appendicular but not the axial skeleton, whereas rosiglitazone had a negative effect in both skeletal sites. There are several possible explanations for this divergence. Firstly, osteocytes were shown to be a major anabolic target of SR10171. These cells are more abundant in cortical bone of appendicular skeleton as compared to cortical bone in axial skeleton (Qing et al., 2012), and they are present at higher density in cortical versus trabecular bone (Lai et al., 2015). Thus, appendicular skeleton, which mostly consists of compact bone, may provide for a dominant target of SR10171 action. Secondly, major differences were observed in marrow adipose volume between the vertebrae and the long bones when comparing rosiglitazone and SR10171 treated mice (Table S1). Increased marrow adipose volume has been associated with bone loss in several model systems including rosiglitazone treated mice. With the exception of rosiglitazone treated mice, it is difficult to detect adipocytes in the vertebrae at the age of the mice used here. As such, the adipogenic cell population in the vertebra may not constitute a sufficient target pool of cells for SR10171 inverse agonist activity to show an effect. Therefore suppression of the adipocytic activity in the vertebra may not be able to increase the pool of potential osteoblasts recruited for bone formation. Thirdly, discordance between axial and appendicular bone mass is common with DXA measurements in humans. Moreover, the skeletal response to osteoporosis treatment varies by site, suggesting that determinants of axial bone mass may differ from those of the appendicular skeleton.
In conclusion, SR10171 represents a polypharmacological PPAR modulator that is insulin sensitizing and normalizes impaired adipocyte metabolic function. Unlike the TZDs, SR10171 does not affect metabolism during normal adipose function. Most striking, SR10171 has a positive effect on bone formation and bone turnover, processes that are attenuated in diabetes, and that are associated with decreased bone quality and increased fractures. Determining that SR10171 directly regulates osteocyte function opens an approach for the development of anti-osteoporotic therapies targeting both PPARγ (repressive) and PPARα (activation) in osteocytes, while at the same time improving insulin sensitivity. Finally, PTMs of PPARγ may provide a unifying explanation for the paradox of low bone formation, high bone resorption and greater fracture risk in T2D.
Author contributions
B.L-C and P.R.G. conceived and designed experiments, L.A.S., P.J.C., Z.C.R., F.N.T., C.A.C., D.P.M., A.A., J.Z., J.B.B., and T.M.K performed experiments and generated key reagents/compounds. All authors analyzed and discussed data. B.L-C and P.R.G. wrote the manuscript. D.P.M., P.J.C., and C.J.R. contributed to the writing.
Conflict of interest
None of the authors have conflict of interest.
Acknowledgments
We thank M. Cameron for providing support for the pharmacokinetic studies and D. Kuruvilla for performing reporter gene assays on SR10171 and rosiglitazone. The work was supported to B.L-C. by a research grant from the American Diabetes Association, Award 7-13-BS-089, and to P.R.G. by Awards from the National Institutes of Health DK080261 (PI:Spiegelman), the Abrams Charitable Trust (D.P.M.), and the Klorfine Family Fellowship (C.A.C.), and to P.R.G. and B.L-C by grant from the National Institutes of Health DK105825 (PI:Griffin; PI for Consortium: Lecka-Czernik).
Footnotes
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.ebiom.2016.06.040.
Contributor Information
P.R. Griffin, Email: pgriffin@scripps.edu.
B. Lecka-Czernik, Email: beata.leckaczernik@utoledo.edu.
Appendix A. Supplementary data
Supplementary material.
References
- Asteian A., Blayo A.L., He Y., Koenig M., Shin Y., Kuruvilla D.S., Corzo C.A., Cameron M.D., Lin L., Ruiz C., Khan S., Kumar N., Busby S., Marciano D.P., Garcia-Ordonez R.D., Griffin P.R., Kamenecka T.M. Design, synthesis, and biological evaluation of indole biphenylcarboxylic acids as PPARgamma antagonists. ACS Med. Chem. Lett. 2015;6:998–1003. doi: 10.1021/acsmedchemlett.5b00218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avtanski D., Hirth Y., Babushkin N., Sy V., Sharma D., Poretsky L., Seto-Young D. In vitro effects of pioglitazone on the expression of components of Wnt signaling pathway and markers of bone mineralization. Horm. Metab. Res. 2016;48(7):468–475. doi: 10.1055/s-0042-101027. [DOI] [PubMed] [Google Scholar]
- Baron R., Kneissel M. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat. Med. 2013;19:179–192. doi: 10.1038/nm.3074. [DOI] [PubMed] [Google Scholar]
- Cho Y.D., Kim W.J., Yoon W.J., Woo K.M., Baek J.H., Lee G., Kim G.S., Ryoo H.M. Wnt3a stimulates Mepe, matrix extracellular phosphoglycoprotein, expression directly by the activation of the canonical Wnt signaling pathway and indirectly through the stimulation of autocrine Bmp-2 expression. J. Cell. Physiol. 2012;227:2287–2296. doi: 10.1002/jcp.24038. [DOI] [PubMed] [Google Scholar]
- Choi J.H., Banks A.S., Estall J.L., Kajimura S., Bostrom P., Laznik D., Ruas J.L., Chalmers M.J., Kamenecka T.M., Bluher M., Griffin P.R., Spiegelman B.M. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by Cdk5. Nature. 2010;466:451–456. doi: 10.1038/nature09291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J.H., Banks A.S., Kamenecka T.M., Busby S.A., Chalmers M.J., Kumar N., Kuruvilla D.S., Shin Y., He Y., Bruning J.B., Marciano D.P., Cameron M.D., Laznik D., Jurczak M.J., Schurer S.C., Vidovic D., Shulman G.I., Spiegelman B.M., Griffin P.R. Antidiabetic actions of a non-agonist PPARgamma ligand blocking Cdk5-mediated phosphorylation. Nature. 2011;477:477–481. doi: 10.1038/nature10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferron M., Wei J., Yoshizawa T., Del Fattore A., Depinho R.A., Teti A., Ducy P., Karsenty G. Insulin signaling in osteoblasts integrates bone remodeling and energy metabolism. Cell. 2010;142:296–308. doi: 10.1016/j.cell.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulzele K., Riddle R.C., Digirolamo D.J., Cao X., Wan C., Chen D., Faugere M.C., Aja S., Hussain M.A., Bruning J.C., Clemens T.L. Insulin receptor signaling in osteoblasts regulates postnatal bone acquisition and body composition. Cell. 2010;142:309–319. doi: 10.1016/j.cell.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge C., Cawthorn W.P., Li Y., Zhao G., Macdougald O.A., Franceschi R.T. Reciprocal control of osteogenic and adipogenic differentiation by ERK/MAP kinase phosphorylation of Runx2 and PPARgamma transcription factors. J. Cell. Physiol. 2016;231:587–596. doi: 10.1002/jcp.25102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinds T.D., Jr., Stechschulte L.A., Cash H.A., Whisler D., Banerjee A., Yong W., Khuder S.S., Kaw M.K., Shou W., Najjar S.M., Sanchez E.R. Protein phosphatase 5 mediates lipid metabolism through reciprocal control of glucocorticoid and PPAR{gamma} receptors. J. Biol. Chem. 2011;286:42911–42922. doi: 10.1074/jbc.M111.311662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn S.E., Zinman B., Lachin J.M., Haffner S.M., Herman W.H., Holman R.R., Kravitz B.G., Yu D., Heise M.A., Aftring R.P., Viberti G. Rosiglitazone-associated fractures in type 2 diabetes: an analysis from A Diabetes Outcome Progression Trial (ADOPT) Diabetes Care. 2008;31:845–851. doi: 10.2337/dc07-2270. [DOI] [PubMed] [Google Scholar]
- Kolli V., Stechschulte L.A., Dowling A.R., Rahman S., Czernik P.J., Lecka-Czernik B. Partial agonist, telmisartan, maintains PPARgamma serine 112 phosphorylation, and does not affect osteoblast differentiation and bone mass. PLoS One. 2014;9:e96323. doi: 10.1371/journal.pone.0096323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krakauer J.C., Mckenna M.J., Buderer N.F., Rao D.S., Whitehouse F.W., Parfitt A.M. Bone loss and bone turnover in diabetes. Diabetes. 1995;44:775–782. doi: 10.2337/diab.44.7.775. [DOI] [PubMed] [Google Scholar]
- Kramer I., Halleux C., Keller H., Pegurri M., Gooi J.H., Weber P.B., Feng J.Q., Bonewald L.F., Kneissel M. Osteocyte Wnt/beta-catenin signaling is required for normal bone homeostasis. Mol. Cell. Biol. 2010;30:3071–3085. doi: 10.1128/MCB.01428-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai X., Price C., Modla S., Thompson W.R., Caplan J., Kirn-Safran C.B., Wang L. The dependences of osteocyte network on bone compartment, age, and disease. Bone Res. 2015;3 doi: 10.1038/boneres.2015.9. (pii:15009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarenko O.P., Rzonca S.O., Hogue W.R., Swain F.L., Suva L.J., Lecka-Czernik B. Rosiglitazone induces decreases in bone mass and strength that are reminiscent of aged bone. Endocrinology. 2007;148:2669–2680. doi: 10.1210/en.2006-1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecka-Czernik B., Rosen C.J. Energy excess, glucose utilization, and skeletal remodeling: new insights. J. Bone Miner. Res. 2015;30:1356–1361. doi: 10.1002/jbmr.2574. [DOI] [PubMed] [Google Scholar]
- Lecka-Czernik B., Gubrij I., Moerman E.A., Kajkenova O., Lipschitz D.A., Manolagas S.C., Jilka R.L. Inhibition of Osf2/Cbfa1 expression and terminal osteoblast differentiation by PPAR-gamma 2. J. Cell. Biochem. 1999;74:357–371. [PubMed] [Google Scholar]
- Lecka-Czernik B., Stechschulte L.A., Czernik P.J., Dowling A.R. High bone mass in adult mice with diet-induced obesity results from a combination of initial increase in bone mass followed by attenuation in bone formation; implications for high bone mass and decreased bone quality in obesity. Mol. Cell. Endocrinol. 2015;410:35–41. doi: 10.1016/j.mce.2015.01.001. [DOI] [PubMed] [Google Scholar]
- Marciano D.P., Kuruvilla D.S., Boregowda S.V., Asteian A., Hughes T.S., Garcia-Ordonez R., Corzo C.A., Khan T.M., Novick S.J., Park H., Kojetin D.J., Phinney D.G., Bruning J.B., Kamenecka T.M., Griffin P.R. Pharmacological repression of PPARgamma promotes osteogenesis. Nat. Commun. 2015;6:7443. doi: 10.1038/ncomms8443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motyl K.J., Demambro V.E., Barlow D., Olshan D., Nagano K., Baron R., Rosen C.J., Houseknecht K.L. Propranolol attenuates risperidone-induced trabecular bone loss in female mice. Endocrinology. 2015;156:2374–2383. doi: 10.1210/en.2015-1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moverare-Skrtic S., Henning P., Liu X., Nagano K., Saito H., Borjesson A.E., Sjogren K., Windahl S.H., Farman H., Kindlund B., Engdahl C., Koskela A., Zhang F.P., Eriksson E.E., Zaman F., Hammarstedt A., Isaksson H., Bally M., Kassem A., Lindholm C., Sandberg O., Aspenberg P., Savendahl L., Feng J.Q., Tuckermann J., Tuukkanen J., Poutanen M., Baron R., Lerner U.H., Gori F., Ohlsson C. Osteoblast-derived WNT16 represses osteoclastogenesis and prevents cortical bone fragility fractures. Nat. Med. 2014;20:1279–1288. doi: 10.1038/nm.3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patsch J.M., Burghardt A.J., Yap S.P., Baum T., Schwartz A.V., Joseph G.B., Link T.M. Increased cortical porosity in type 2 diabetic postmenopausal women with fragility fractures. J. Bone Miner. Res. 2013;28:313–324. doi: 10.1002/jbmr.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qing H., Ardeshirpour L., Pajevic P.D., Dusevich V., Jahn K., Kato S., Wysolmerski J., Bonewald L.F. Demonstration of osteocytic perilacunar/canalicular remodeling in mice during lactation. J. Bone Miner. Res. 2012;27:1018–1029. doi: 10.1002/jbmr.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman S., Czernik P.J., Lu Y., Lecka-Czernik B. Beta-catenin directly sequesters adipocytic and insulin sensitizing activities but not osteoblastic activity of PPARgamma2 in marrow mesenchymal stem cells. PLoS One. 2012;7:e51746. doi: 10.1371/journal.pone.0051746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shockley K.R., Lazarenko O.P., Czernik P.J., Rosen C.J., Churchill G.A., Lecka-Czernik B. PPARγ2 nuclear receptor controls multiple regulatory pathways of osteoblast differentiation from marrow mesenchymal stem cells. J. Cell. Biochem. 2009;106:232–246. doi: 10.1002/jcb.21994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Still K., Grabowski P., Mackie I., Perry M., Bishop N. The peroxisome proliferator activator receptor alpha/delta agonists linoleic acid and bezafibrate upregulate osteoblast differentiation and induce periosteal bone formation in vivo. Calcif. Tissue Int. 2008;83:285–292. doi: 10.1007/s00223-008-9175-9. [DOI] [PubMed] [Google Scholar]
- Syversen U., Stunes A.K., Gustafsson B.I., Obrant K.J., Nordsletten L., Berge R., Thommesen L., Reseland J.E. Different skeletal effects of the peroxisome proliferator activated receptor (PPAR)alpha agonist fenofibrate and the PPARgamma agonist pioglitazone. BMC Endocr. Disord. 2009;9:10. doi: 10.1186/1472-6823-9-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan S.H., Senarath-Yapa K., Chung M.T., Longaker M.T., Wu J.Y., Nusse R. Wnts produced by Osterix-expressing osteolineage cells regulate their proliferation and differentiation. Proc. Natl. Acad. Sci. U. S. A. 2014;111:E5262–E5271. doi: 10.1073/pnas.1420463111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor S.I., Blau J.E., Rother K.I. Possible adverse effects of SGLT2 inhibitors on bone. Lancet Diabetes Endocrinol. 2015;3:8–10. doi: 10.1016/S2213-8587(14)70227-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W., Wang X., Yang M., Smith L.C., Dechow P.C., Wan Y. PGC1beta mediates PPARgamma activation of osteoclastogenesis and rosiglitazone-induced bone loss. Cell Metab. 2010;11:503–516. doi: 10.1016/j.cmet.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J., Ferron M., Clarke C.J., Hannun Y.A., Jiang H., Blaner W.S., Karsenty G. Bone-specific insulin resistance disrupts whole-body glucose homeostasis via decreased osteocalcin activation. J. Clin. Invest. 2014;124:1–13. doi: 10.1172/JCI72323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wergedal J.E., Kesavan C., Brommage R., Das S., Mohan S. Role of WNT16 in the regulation of periosteal bone formation in female mice. Endocrinology. 2015;156:1023–1032. doi: 10.1210/en.2014-1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X., Peters J.M., Gonzalez F.J., Prasad H.S., Rohrer M.D., Gimble J.M. Frequency of stromal lineage colony forming units in bone marrow of peroxisome proliferator-activated receptor-alpha-null mice. Bone. 2000;26:21–26. doi: 10.1016/s8756-3282(99)00238-0. [DOI] [PubMed] [Google Scholar]
- Xiong J., Onal M., Jilka R.L., Weinstein R.S., Manolagas S.C., O'brien C.A. Matrix-embedded cells control osteoclast formation. Nat. Med. 2011;17:1235–1241. doi: 10.1038/nm.2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material.