

Establishment of single-molecule pull-down in plants allows rapid, sensitive, and quantitative interrogation of protein interactions and reveals tetrameric HD-ZIPIII:ZPR complexes.
Abstract
Deciphering complex biological processes markedly benefits from approaches that directly assess the underlying biomolecular interactions. Most commonly used approaches to monitor protein-protein interactions typically provide nonquantitative readouts that lack statistical power and do not yield information on the heterogeneity or stoichiometry of protein complexes. Single-molecule pull-down (SiMPull) uses single-molecule fluorescence detection to mitigate these disadvantages and can quantitatively interrogate interactions between proteins and other compounds, such as nucleic acids, small molecule ligands, and lipids. Here, we establish SiMPull in plants using the HOMEODOMAIN LEUCINE ZIPPER III (HD-ZIPIII) and LITTLE ZIPPER (ZPR) interaction as proof-of-principle. Colocalization analysis of fluorophore-tagged HD-ZIPIII and ZPR proteins provides strong statistical evidence of complex formation. In addition, we use SiMPull to directly quantify YFP and mCherry maturation probabilities, showing these differ substantially from values obtained in mammalian systems. Leveraging these probabilities, in conjunction with fluorophore photobleaching assays on over 2000 individual complexes, we determined HD-ZIPIII:ZPR stoichiometry. Intriguingly, these complexes appear as heterotetramers, comprising two HD-ZIPIII and two ZPR molecules, rather than heterodimers as described in the current model. This surprising result raises new questions about the regulation of these key developmental factors and is illustrative of the unique contribution SiMPull is poised to make to in planta protein interaction studies.
INTRODUCTION
The remarkable complexity of biological processes is nicely captured in the network-level information provided by the -omic revolution. Unraveling this complexity will rely on direct, mechanistic insights into the underlying biomolecular interactions. For example, the interaction partners of a given protein can provide important information about its function and position within regulatory networks (Gingras et al., 2005, 2007); as such, assaying protein-protein interaction is integral to understanding any given biological process. A wide variety of techniques are available to assess interactions between proteins, including coimmunoprecipitation, bimolecular fluorescence complementation (BiFC), Forster resonance energy transfer (FRET), surface plasmon resonance, and yeast two-hybrid (Y2H) assays. The specifics of these techniques have been reviewed elsewhere (Hecker et al., 2015; Nguyen et al., 2015; Xing et al., 2016), but it is important to note that each technique, and its associated detection method, has specific advantages and disadvantages. For instance, methods such as BiFC, FRET, and Y2H mostly sample direct pairwise protein interactions, whereas the principal advantage of coimmunoprecipitation is an ability to capture a more complete picture of in vivo-assembled complexes, maximizing the likelihood of capturing biologically relevant interactions (Berggård et al., 2007; Gingras et al., 2007). However, this technique is also known to capture nonspecific proteins, and as coimmunoprecipitation is a protracted, multistep process, it is biased against the detection of weak or transient interactions (Berggård et al., 2007). In addition, traditional methods for detecting coimmunoprecipitation, such as immunoblotting or mass spectrometry, suffer from poor sensitivity and provide an averaged representation of protein complexes that can obscure the true composition of individual physiological complexes (reviewed in Aggarwal and Ha, 2014). Moreover, most of these methods, as commonly used, provide a nonquantitative readout, and none yield information on the stoichiometry of detected protein complexes.
Single-molecule pull-down, or SiMPull, uses single-molecule total internal reflection fluorescence (TIRF) microscopy to mitigate the disadvantages of coimmunoprecipitation, while preserving and augmenting most of its advantages (Jain et al., 2011). In brief, antibodies tethered to the surface of a passivated microscope slide are used to capture a protein of interest, along with its physiological interaction partners, from applied cell extracts. After washing away unbound components, immunoprecipitated protein complexes are probed with multicolor, single-molecule TIRF microscopy. Captured proteins may be visualized with genetically encoded fluorescent tags or via immunofluorescence labeling (Figure 1; Jain et al., 2011). SiMPull is minimally invasive, sensitive (∼100-fold more sensitive than detection by immunoblotting), and rapid, allowing direct visualization of immunoprecipitated proteins at single-complex resolution in under thirty minutes (Jain et al., 2011, 2014). The simplicity of this protocol also preserves weak or transient interactions that are sensitive to lengthy, invasive procedures. Once immobilized, colocalization analysis can be used to quantitatively assess whether coimmunoprecipitated proteins are indeed part of the same complex, and stoichiometries of these complexes can be directly determined from the stepwise pattern of fluorophore photobleaching (Jain et al., 2011, 2014; Yeom et al., 2011; Lee et al., 2013; Giri et al., 2015). As SiMPull is compatible with FRET (Jain et al., 2011; Krishnan et al., 2013; Hwang et al., 2014a), dynamic interactions between immunoprecipitated proteins and their binding partners can easily be tested. Finally, SiMPull is not limited solely to protein-protein interactions but provides single-molecule resolution data on interactions between proteins and other compounds, such as nucleic acids, small molecule ligands, and lipids (Jain et al., 2011, 2014; Krishnan et al., 2013; Rodgers et al., 2015; Arauz et al., 2016).
Figure 1.

Workflow of SiMPull.
Biotinylated antibodies against, e.g., GFP, RFP, or HA, are tethered to slides using NeutrAvidin (Jain et al., 2012). Plant lysate containing fluorophore-tagged prospective partners is serially diluted, applied to antibody-coated slides, and imaged in appropriate fluorescence channels (e.g., YFP and mCherry) via TIRF microscopy. IP spots: Proteins immunoprecipitated by antibodies against their tagged fluorophore imaged in the matching fluorescence channel (e.g., protein-YFP on α-GFP slide imaged in the YFP channel). Co-IP spots: Proteins immunoprecipitated via their fluorophore-tagged interacting partner (e.g., protein-mCherry on α-GFP slides imaged in the mCherry channel). Background: Signal on control antibody slides (e.g., protein-YFP on α-HA slides imaged in the YFP channel). Serial dilutions are screened using TIRF imaging to identify the optimal lysate dilution, which meets the two indicated criteria. Coimmunoprecipitation is apparent as a significant enrichment of co-IP spots above background. Overlaid TIRF images of YFP (green) and mCherry (magenta) channels show colocalization of protein-YFP and protein-mCherry (white spots in Overlay panels). Bars = 2 µm.
Single-molecule techniques are transforming the study of mammalian biological processes (reviewed in Joo et al., 2008; Liu et al., 2015), and this prompted us to develop the technique of SiMPull in plants. Using the HOMEODOMAIN LEUCINE ZIPPER III (HD-ZIPIII) and LITTLE ZIPPER (ZPR) interaction as proof-of-principle (Wenkel et al., 2007; Kim et al., 2008), we find that relative protein levels and degree of coexpression are important parameters in SiMPull and establish generalizable strategies through which to realize these in transient systems. Under these conditions, quantitative statistical evidence of complex formation was obtained by colocalization analysis of fluorophore-tagged HD-ZIPIII and ZPR proteins. Moreover, by directly quantifying YFP and mCherry maturation probabilities and performing fluorophore photobleaching on over 2000 individual HD-ZIPIII:ZPR complexes, we find that these have a heterotetrameric stoichiometry rather than being heterodimers as previously envisioned (Wenkel et al., 2007; Kim et al., 2008). Importantly, this empirical result could not have been predicted from secondary structure analyses and is demonstrative of the substantial impact SiMPull is poised to make to plant protein interaction studies.
RESULTS
Relative Protein Level Is a Parameter in Single-Complex Detection by SiMPull
To establish SiMPull for the detection of protein-protein interactions in plants, we turned to the Nicotiana benthamiana transient transformation system, which allows critical parameters to be rapidly determined and optimized. As a proof-of-principle, we focused on the interaction between HD-ZIPIII and ZPR proteins (Wenkel et al., 2007; Kim et al., 2008). HD-ZIPIII transcription factors regulate such fundamental processes as vascular development, adaxial-abaxial patterning, and meristem maintenance (McConnell et al., 2001; Prigge et al., 2005; Carlsbecker et al., 2010). Perhaps reflective of this, their activity is subject to multiple levels of regulation, including posttranscriptionally via miR166-mediated mRNA turnover and posttranslationally via protein-protein interactions (Juarez et al., 2004; Mallory et al., 2004; Chandler et al., 2007; Wenkel et al., 2007; Kim et al., 2008). Important to this study, HD-ZIPIII activity in Arabidopsis thaliana is modulated by interaction with ZPR proteins. These small proteins consist primarily of a single leucine zipper domain similar to the one found in HD-ZIPIII proteins. As HD-ZIPIII proteins must homodimerize in order to bind DNA (Sessa et al., 1998), it is thought that dimerization occurs via their leucine zipper and that ZPR interaction abrogates HD-ZIPIII activity through the formation of nonfunctional heterodimers (Wenkel et al., 2007; Kim et al., 2008). Interestingly, ZPR expression itself is directly induced at the transcriptional level by HD-ZIPIII proteins, thus creating a negative feedback loop that intricately links expression of these proteins and fine-tunes HD-ZIPIII activity critical to normal development (Wenkel et al., 2007).
Using Agrobacterium tumefaciens infiltration, we coexpressed fluorescent protein fusions of the HD-ZIPIII family member PHABULOSA (PHB) and ZPR3, both of which retain functionality upon reporter fusion (Wenkel et al., 2007; Kim et al., 2008; Dello Ioio et al., 2012; Müller et al., 2016). Imaging of N. benthamiana leaves expressing PHB-YFP and ZPR3-mCherry from the 35S promoter showed mCherry fluorescence is noticeably more intense than YFP fluorescence 24 h postinfiltration (hpi; Figure 2A), which may suggest ZPR3-mCherry accumulates to higher levels than PHB-YFP. This was true for leaves infiltrated with a single Agrobacterium strain carrying both binary vectors (Figure 2A, upper panel) or leaves infiltrated with an equal mixture of two Agrobacterium strains each carrying a single binary vector (Figure 2A, lower panel). As expected, the PHB-YFP transcription factor accumulates predominantly in the nuclei of epidermal cells, whereas the ZPR3-mCherry signal is also detected in the cytoplasm (Figure 2A). Leaves infiltrated with a single Agrobacterium strain carrying both binary vectors showed a higher frequency of coexpression, with over 93% of epidermal cells showing both PHB-YFP and ZPR3-mCherry signal (Figure 2A, upper panel), compared with 85% in doubly infiltrated leaves (Figure 2A, lower panel). These results, which are in line with recently published observations (Hecker et al., 2015), indicate coexpression efficiency improves when binary vectors are contained within the same Agrobacterium strain. As such, this approach was used for the development of SiMPull in plants.
Figure 2.

Relative Protein Level Is a Key Parameter in Single Complex Detection by SiMPull.
(A) Fluorescence images of leaves infiltrated with pro35S:PHB-YFP and pro35S:ZPR3-mCherry show more intense mCherry than YFP fluorescence when infiltrated with a single Agrobacterium strain carrying both binary vectors (upper row) or two Agrobacterium strains carrying each vector separately (lower row). Normal exposure: YFP imaged at the same exposure settings as mCherry. Overexposure of the YFP channel reveals most cells accumulate both PHB-YFP and ZPR3-mCherry (gray hatched bars), though some cells accumulate either PHB-YFP (green) or ZPR3-mCherry (magenta). Arrows, examples of bifluorescent cells; arrowheads, examples of unifluorescent cells. Coexpression frequency improves when both vectors are contained within a single Agrobacterium strain (upper row).
(B) Representative TIRF images of PHB-YFP and ZPR3-mCherry SiMPull show that when PHB-YFP IP and co-IP spots are at single-spot resolution, ZPR3-mCherry IP spots are not.
(C) Histogram shows PHB-YFP IP and co-IP spots are enriched over background. ZPR3-mCherry IP spots are saturated (sat) and not quantifiable, while the ZPR3-mCherry co-IP spot number is indistinguishable from its background. Green, PHB-YFP; magenta, ZPR3-mCherry. Spot densities (mean ± se) are calculated from at least three independent biological replicates.
Bars = 150 µm in (A) and 5 µm in (B).
For SiMPull, lysates containing fluorescently tagged proteins are serially diluted and applied to antibody-coated slides that either specifically immunoprecipitate the tagged protein of interest (α-GFP and α-RFP; Figure 1) or serve as a negative control (α-HA; Figure 1). Serial dilutions are used to determine optimal lysate dilution (Supplemental Figure 1; Jain et al., 2012). Here, it is important to note that relative protein concentrations within a given lysate are unaffected during this step. The function of the optimal lysate dilution is to ensure that fluorophores are quantifiable, for which it must meet two criteria. First, proteins in the lysate must be concentrated enough to yield a high signal-to-noise ratio after immunoprecipitation. Ideally, the number of immunoprecipitated fluorescent spots (IP spots; e.g., PHB-YFP on a α-GFP slide) should be 8- to 15-fold higher than signal from negative control slides (background; e.g., PHB-YFP on a α-HA slide). Second, proteins in the lysate must be dilute enough that IP spot density is quantifiable at single-spot resolution. This corresponds to 600 to 800 spots per 5000 μm2 imaging area. At the optimal dilution, coimmunoprecipitation of a protein is then visualized as fluorescent spots on slides coated with antibodies against its prospective partner (co-IP spots; e.g., ZPR3-mCherry on a α-GFP slide). Coimmunoprecipitation is considered successful when the number of such co-IP spots is significantly higher than the background signal from negative control slides. Finally, colocalization analysis of IP and co-IP spots provides direct, statistical evidence that coimmunoprecipitated proteins are indeed found in the same complex (Figure 1; Jain et al., 2011, 2012).
Following these principles, lysate from leaves infiltrated with Agrobacterium carrying both binary vectors was optimally diluted for IP of PHB-YFP on α-GFP slides (Figures 2B and 2C). However, at this dilution, the density of ZPR3-mCherry IP spots on α-RFP slides was too high for single-spot resolution and, as such, not quantifiable (Figures 2B and 2C). This finding indicates ZPR3-mCherry protein accumulates to much higher levels than PHB-YFP in coinfiltrated leaves. Higher ZPR3-mCherry protein levels also resulted in substantially higher background on α-HA control slides; indeed, this signal was indistinguishable from PHB-YFP IP signal (Figures 2B and 2C). This confounds detection of potential co-IP of ZPR3-mCherry with PHB-YFP, as the number of ZPR3-mCherry co-IP spots can never exceed that of background. Thus, while these data demonstrate SiMPull can rapidly (see Methods) and specifically immunoprecipitate proteins from leaf extracts, assaying potential coimmunoprecipitation will require both proteins to be at concentrations yielding single-spot resolution with minimal background.
Modulating Infiltration Densities Impacts Both Protein Accumulation and Degree of Coexpression
In an attempt to balance relative protein levels, we next coinfiltrated leaves with pro35S:ZPR3-mCherry and pro35S:PHB-YFP Agrobacterium strains at optical densities of 0.3 and 1, respectively. Imaging of infiltrated leaves showed YFP and mCherry fluorescence are both evident 24 hpi, with the latter possessing a somewhat patchy distribution (Figure 3A). At lysate dilutions optimal for SiMPull of PHB-YFP, ZPR3-mCherry background on α-HA slides was reduced to well below the number of PHB-YFP IP spots (Figures 3B and 3C), indicating that lowering Agrobacterium density leads to reductions in ZPR3-mCherry levels in the lysate. The fact that ZPR3-mCherry background falls below the number of PHB-YFP IP spots also permits analysis of potential coimmunoprecipitation of ZPR3 with PHB. However, ZPR3-mCherry co-IP spot density on α-GFP slides was only marginally higher than background (Figures 3B and 3C), suggesting little to no coimmunoprecipitation with PHB-YFP. One potential explanation for this follows from the observation that far fewer cells accumulate both PHB-YFP and ZPR3-mCherry when the pro35S:ZPR3-mCherry Agrobacterium strain is infiltrated at a lower optical density (compare Figures 3A and 2A). This indicates that while reducing Agrobacterium density does adjust protein levels in the lysate, it does so at the expense of the number of cells accumulating both proteins and, thus, the efficacy of SiMPull.
Figure 3.

Modulating Agrobacterium Infiltration Densities Impacts Both Accumulation Levels and Degree of Coexpression.
(A) Fluorescence images of leaves infiltrated with pro35S:PHB-YFP and pro35S:ZPR3-mCherry Agrobacterium strains at optical densities of 1 and 0.3, respectively, show relative PHB-YFP fluorescence is increased, but the frequency of cells accumulating both PHB-YFP and ZPR3-mCherry (gray hatched bar) is decreased, with more cells expressing exclusively PHB-YFP (green) or ZPR3-mCherry (magenta). Arrows, examples of bifluorescent cells; arrowheads, examples of unifluorescent cells.
(B) Representative TIRF images of PHB-YFP and ZPR3-mCherry SiMPull show that when PHB-YFP IP and co-IP spots are at single-spot resolution, ZPR3-mCherry IP spots are not.
(C) Histogram shows PHB-YFP IP spots are enriched over background, while the number of ZPR3-mCherry IP spots remains saturated (sat). Co-IP spot numbers for both ZPR3-mCherry and PHB-YFP are only marginally higher than background, suggestive of minimal coimmunoprecipitation. Green, PHB-YFP; magenta, ZPR3-mCherry. Spot densities (mean ± se) are calculated from at least three independent biological replicates.
Bars = 150 µm in (A) and 5 µm in (B).
Transient Protein Accumulation Correlates with Protein Size
Despite ZPR3-mCherry accumulating in fewer cells than PHB-YFP (Figure 3A), overall ZPR3-mCherry protein levels in the lysate were higher than those of PHB-YFP (Figures 3B and 3C), indicating that cotransfected cells accumulate far more ZPR3-mCherry than PHB-YFP. Uncovering the basis of this disparity might provide a means through which to equalize protein production while maintaining robust coexpression. Differential protein accumulation may result from inherent properties of PHB and ZPR3, but in transient systems, where protein levels are not at steady state, may also reflect properties that are more general, such as protein size, subcellular localization, or potentially the attached fluorophore.
To assess these possibilities, a set of proteins ranging in length from 67 to 920 amino acids was tagged with YFP or mCherry and placed under control of the single 35S promoter. This set contains proteins with predominantly nuclear localization (ZPR3, PHB, ASYMMETRIC LEAVES2 [AS2; Iwakawa et al., 2002], and AUXIN RESPONSE FACTOR3 [ARF3; Chitwood et al., 2009], as well as those predicted to act outside the nucleus (ACTIN2 [ACT2; Kim et al., 2005] and DYNAMIN-RELATED PROTEIN 2B [DRP2B; Fujimoto et al., 2010]. ZPR3, ARF3, ACT2, and DRP2B were fused to mCherry, while PHB and AS2 were fused to YFP. Leaves infiltrated with these constructs were imaged every 2 h, starting at 18 hpi, using the time of first appearance of fluorescence as a proxy for protein accumulation. The time at which fluorescence first appeared strongly correlated with protein size (R2 = 0.86), whereas the identity of the fluorophore seemed to have little to no effect (Figure 4A). Subcellular localization did have a small but noticeable effect, as the correlation coefficient for first fluorescence detection and protein size was even higher for nuclear-localized proteins (R2 = 0.99 versus R2 = 0.86). This likely reflects the fact that without nuclear concentration, more protein is required to elevate signal above the threshold of detection. Thus, in transient systems, size is a key determinant of protein accumulation, with smaller proteins accumulating faster than larger proteins (Figure 4A). This finding may be explained by the fixed kinetics of transcription and translation, which predicts that the time required to transcribe and subsequently translate a given transcript scales with length (Singh and Padgett, 2009; Shah et al., 2013).
Figure 4.

Transient Protein Accumulation Correlates with Protein Size.
(A) Fluorescence imaging of leaves infiltrated with Agrobacterium strains carrying pro35S:ZPR3-mCherry, pro35S:AS2-YFP, pro35S:ACT2-mCherry, pro35S:ARF3-mCherry, pro35S:PHB-YFP, or pro35S:DRP2B-mCherry reveals a positive, linear correlation between protein size and the time of first detectable fluorescence (solid regression line; R2 = 0.86). This correlation is even stronger when only nuclear-localized proteins are considered (dotted regression line; R2 = 0.99). The time to first detectable fluorescence for 2xpro35S:PHB-YFP (red dot) is substantially shorter. Note: This data point was omitted from regression analyses. Protein size including fluorophore is indicated. Values (mean ± se) are calculated from 10 to 20 leaves and at least three independent infiltrations.
(B) Immunoblot analysis of leaves coinfiltrated with 2xpro35S:PHB-YFP and pro35S:AS2-YFP Agrobacterium strains shows PHB-YFP is first detectable 22 hpi, and its levels increase subtly at 24 and 26 hpi. By contrast, AS2-YFP is weakly detectable 24 hpi and levels increase substantially by 26 hpi, consistent with a higher rate of accumulation. Both proteins migrate at the expected size. Incubation with α-tubulin confirms even loading of samples.
As ZPR3-mCherry is much smaller than PHB-YFP (305 versus 1089 amino acids), increasing the rate of PHB-YFP accumulation to match that of ZPR3-mCherry may align protein production within a given cell. This could be accomplished, in part, by elevating the amount of PHB-YFP transcripts available for translation via use of a stronger promoter. To this end, we cloned PHB-YFP behind the double 35S promoter (2xpro35S), which produces ∼10-fold more transcripts than the single 35S promoter (Kay et al., 1987). Indeed, leaves infiltrated with Agrobacterium carrying a 2xpro35S:PHB-YFP binary vector showed visible fluorescence by 20 hpi, much earlier than infiltrations performed with pro35S:PHB-YFP (25 hpi; Figure 4A). Thus, promoter strength may be a means by which to compensate for the effect protein size has on accumulation.
To test this possibility, we assayed relative abundance of a large and a small protein at various time points postinfiltration using immunoblotting. N. benthamiana leaves were infiltrated with 2xpro35S:PHB-YFP and pro35S:AS2-YFP binary vectors, and lysates collected at 2-h intervals from 18 to 26 hpi. PHB-YFP protein was first detectable via α-GFP immunoblot at 22 hpi, and its levels rose subtly over the rest of the time course (Figure 4B). AS2-YFP protein, on the other hand, was not detected until 24 hpi (Figure 4B), consistent with a weaker single 35S promoter driving AS2-YFP transcription (Kay et al., 1987). However, its levels then rose dramatically, surpassing those of PHB-YFP by 26 hpi (Figure 4B). This reinforces the notion that the transient accumulation rate of a given protein scales with its size and also explains the dramatic mismatch in protein levels seen in pro35S:PHB-YFP and pro35S:ZPR3-mCherry coinfiltrations (Figure 2). Importantly, these findings show that cellular levels of large and small proteins can be adjusted by tuning transcription through the use of strong and weak promoters, respectively, and collecting tissue at an appropriate time point.
Detection of PHB and ZPR3 Coimmunoprecipitation via SiMPull
The above experiments suggest a strategy that balances PHB-YFP and ZPR3-mCherry levels while maintaining their robust coexpression. Specifically, leaves were infiltrated with a single Agrobacterium strain carrying binary vectors for both 2xpro35S:PHB-YFP and pro35S:ZPR3-mCherry and tissue was collected at 25 hpi, when protein levels are predicted to be near-equivalent (Figure 4B). Images of leaf epidermal cells reveal 98% of nuclei expressed both PHB-YFP and ZPR3-mCherry proteins (Figure 5A). In addition, both proteins accumulated to similar levels, as SiMPull showed comparable numbers of PHB-YFP and ZPR3-mCherry IP spots on α-GFP and α-RFP slides, respectively (Figure 5B). At this optimal lysate dilution, we obtained single PHB:ZPR3 complexes, as the number of ZPR3-mCherry co-IP spots was significantly enriched over background (Figures 5C and 5D). SiMPull on lysates containing equalized levels of noninteracting proteins, AS2-YFP and ZPR3-mCherry (Figure 5B), did not produce this enrichment (Figures 5C and 5D), demonstrating specific coimmunoprecipitation of ZPR3-mCherry with PHB-YFP. To confirm that the captured PHB:ZPR3 complexes represent a true physiological state, and are not assembled during lysate preparation, we also performed SiMPull on in vitro-mixed lysates prepared from separate infiltrations with either pro35S:PHB-YFP or pro35S:ZPR3-mCherry. Lysates were first optimally diluted and calibrated to yield roughly equivalent numbers of IP spots on α-GFP or α-RFP slides (Figure 5E) and then mixed at ratios of 1:1, 1:2, and 1:3. SiMPull using these in vitro-mixed lysates did not lead to ZPR3-mCherry coimmunoprecipitation on α-GFP slides (Figure 5F), demonstrating postlysis association is not responsible for PHB:ZPR3 complex formation. Together, these data show that SiMPull can be used for sensitive and specific detection of coimmunoprecipitation in plants and identifies parameters that are critical for experimental success.
Figure 5.

Detection of PHB-YFP and ZPR3-mCherry Coimmunoprecipitation via SiMPull.
(A) Fluorescence images of leaves infiltrated with a single Agrobacterium strain carrying 2xpro35S:PHB-YFP and pro35S:ZPR3-mCherry show roughly equivalent levels and near-perfect colocalization (gray hatched bar) of PHB-YFP and ZPR3-mCherry fluorescence 25 hpi. Arrowhead indicates unifluorescent cell.
(B) Representative TIRF images and IP spot counts from SiMPull using lysates coexpressing PHB-YFP and ZPR3-mCherry (top) or AS2-YFP and ZPR3-mCherry (bottom) show proteins are at single-spot resolution and accumulate to near-equivalent levels.
(C) and (D) SiMPull using PHB-YFP + ZPR3-mCherry lysate shows PHB-YFP IP and ZPR3-mCherry co-IP spot numbers are comparably enriched over background, whereas in SiMPull using AS2-YFP + ZPR3-mCherry negative control lysate, ZPR3-mCherry co-IP spots are indistinguishable from background.
(E) SiMPull of PHB-YFP (top) and ZPR3-mCherry (bottom) on lysates from separate Agrobacterium infiltrations with pro35S:PHB-YFP and pro35S:ZPR3-mCherry, respectively, identifies dilutions that yield equivalent IP spot numbers.
(F) SiMPull using these calibrated PHB-YFP and ZPR3-mCherry lysates mixed in vitro at different ratios demonstrates PHB-YFP does not coimmunoprecipitate ZPR3-mCherry under these conditions. Green, PHB-YFP or AS2-YFP; magenta, ZPR3-mCherry. Spot densities (means ± se) are calculated from at least three independent biological replicates. Student’s t test: ***P < 0.001.
Bars = 150 µm in (A) and 5 µm in (B) and (C).
Colocalization and Stoichiometric Analysis of PHB:ZPR3 Complex Formation
Traditional methods for detecting coimmunoprecipitation typically provide qualitative or semiquantitative readouts and cannot differentiate between specific protein-protein interactions and nonspecific contamination (reviewed in Aggarwal and Ha, 2014). SiMPull, on the other hand, directly scores immobilized fluorescent protein and can quantitatively assess the extent to which coimmunoprecipitated proteins physically colocalize (Jain et al., 2011; Giri et al., 2015). This provides direct, statistical evidence that coimmunoprecipitated proteins are found in the same complex. With this in mind, we assayed the colocalization frequency of coimmunoprecipitated PHB-YFP and ZPR3-mCherry by overlaying images of PHB-YFP IP spots and ZPR3-mCherry co-IP spots obtained from an α-GFP slide. Overlaid images showed 22% of ZPR3-mCherry co-IP spots colocalized with PHB-YFP spots (Figures 6A, left, and 6B). However, as the colocalization criterion was set to two pixels, which corresponds to a diffraction limited spot size of ∼300 nm for the TIRF microscope, single PHB-YFP and ZPR3-mCherry spots in close proximity could, at some particular frequency, be miscalled as colocalized. To evaluate this, we overlaid images taken from two random, nonoverlapping slide regions (Figure 6A, middle) and found that PHB-YFP and ZPR3-mCherry spots falsely colocalize with a frequency of ∼9% (Figure 6B). Similarly, image overlays from SiMPull using AS2-YFP and ZPR3-mCherry lysates showed a false colocalization frequency of ∼9% (Figures 6A, right, and 6B). Importantly, both controls are highly significantly different from the 22% PHB-YFP and ZPR3-mCherry colocalization frequency (P < 10−24; Figure 6B), supporting the conclusion that PHB and ZPR3 are specifically immunoprecipitated as a single complex by SiMPull.
Figure 6.

PHB and ZPR3 Form Tetrameric Complexes.
(A) Overlaid TIRF images of YFP (green) and mCherry (magenta) channels taken from the same region of the slide (left column) show pronounced colocalization of PHB-YFP and ZPR3-mCherry (white spots in Overlay panels), compared with overlaid TIRF images obtained from random, nonoverlapping regions of the slide (middle column) or the same region of an AS2-YFP + ZPR3-mCherry SiMPull slide (right column). Bars = 5 µm.
(B) The frequency of PHB-YFP and ZPR3-mCherry colocalization is significantly higher (P < 10−24) for overlaid TIRF images taken from the same region of a PHB-YFP + ZPR3-mCherry SiMPull slide compared with controls. Colocalization frequencies (means ± se) are calculated from at least 30 images, from three independent biological replicates. Student’s t test: ***P < 0.001.
(C) Photobleaching analysis of 2006 single PHB-YFP:ZPR3-mCherry complexes reveals photobleaching step distributions occur at frequencies expected for a tetrameric complex comprised of two YFP and two mCherry fluorophores (black dots). Values (means ± se) are calculated from six independent biological replicates.
The colocalization frequency of PHB-YFP and ZPR3-mCherry (∼22%) is lower than values commonly reported for interacting proteins in animal systems (Jain et al., 2011, 2014; Giri et al., 2015). Colocalization frequency is known to be strongly influenced by fluorophore maturation probabilities; for instance, as ∼80% of YFP and 40% of mCherry molecules are fluorescently active in mammalian systems, measurements of colocalization never reach 100% (reviewed in Aggarwal and Ha, 2014; Ulbrich and Isacoff, 2007, 2008; Jain et al., 2014; Rodgers et al., 2015). Our findings thus hint at differences in fluorophore maturation probabilities between animal and plant systems, prompting us to experimentally determine these properties for YFP and mCherry in infiltrated N. benthamiana leaves. Fluorophore maturation probabilities can be quantitatively determined by performing SiMPull of tandem dimers (td) of a given fluorophore and subsequently analyzing their photobleaching step distributions (Jain et al., 2014). As photobleaching of a fluorophore is marked by a single, abrupt decrease in fluorescence intensity, dimers possessing two active fluorophores appear as two-step photobleaching events (Jain et al., 2011; Supplemental Figures 2A and 2B; see Methods). The maturation probability of the fluorophore is then obtained by fitting the observed frequency of these two-step photobleaching events to expectations generated by a binomial distribution (see Methods). Following this strategy, we performed SiMPull on lysates from leaves infiltrated with 2xpro35S:tdYFP or 2xpro35S:tdmCherry Agrobacterium strains on α-GFP and α-RFP slides, respectively. Twenty-five percent of YFP and 38% of mCherry IP spots showed two-step photobleaching (Supplemental Figures 2A and 2B), corresponding to maturation probabilities of 0.4 and 0.55, respectively (Supplemental Figure 2C). Thus, YFP and mCherry maturation probabilities differ substantially between plants and animals, which neatly explains why colocalization of PHB-YFP and ZPR3-mCherry in N. benthamiana is approximately half that seen for interacting proteins in animal systems (Jain et al., 2011, 2014; Giri et al., 2015).
One of the most powerful features of SiMPull is its ability to directly determine the stoichiometry of a given complex using photobleaching analysis of fluorophore-tagged members, provided the maturation probabilities of those fluorophores have been experimentally determined (Jain et al., 2011). ZPR proteins are proposed to abrogate HD-ZIPIII DNA binding by forming nonfunctional HD-ZIPIII:ZPR heterodimers at the expense of HD-ZIPIII homodimers (Wenkel et al., 2007; Kim et al., 2008). However, the actual stoichiometry of HD-ZIPIII:ZPR complexes remains unknown. To resolve this, we performed single-molecule photobleaching analysis on over 2000 single PHB-YFP:ZPR3-mCherry complexes (Supplemental Figure 3B). Intriguingly, YFP-mCherry photobleaching step distributions of 1-1, 1-2, 2-1, and 2-2 were observed (e.g., a fluorescent spot showing two photobleaching steps for YFP and one step for mCherry would be called as 2-1). This indicates that higher-order complexes between PHB and ZPR3 are possible (Figure 6C). However, it is important to note that the observed step distributions cannot be used as direct proxies for PHB-YFP:ZPR3-mCherry complex stoichiometry. As fluorophores do not mature with 100% efficiency (Supplemental Figure 2), observed step distributions of 1-1, 1-2, and 2-1 will be observed even for complexes consisting solely of YFP:mCherry tetramers. We therefore characterized the relationship between observed photobleaching step distributions and true PHB-YFP:ZPR3-mCherry complex stoichiometry. To do this, we used a binomial distribution (see Methods) to generate expected frequencies of photobleaching steps for a range of hypothetical YFP:mCherry complexes, using experimentally determined fluorophore maturation probabilities of 0.4 and 0.55, respectively (Supplemental Figure 2). Expected frequencies for hypothetical YFP:mCherry complexes with 1:1, 1:2, 2:1, or 2:2 stoichiometries were then overlaid on observed frequencies to determine which, if any, best fit our observations. Complexes with 1:1, 1:2, and 2:1 stoichiometries all fail to predict at least one category of observed step distributions and show poor overall agreement (Supplemental Figure 3A). Expected frequencies for YFP:mCherry complexes with 2:2 stoichiometry, on the other hand, are in excellent agreement with observed step distributions (Supplemental Figure 3A; Figure 6C), consistent with a model in which two molecules of PHB-YFP interact with two molecules of ZPR3-mCherry. Further supporting this, the vast majority of randomly colocalized spots from nonoverlapping imaging areas (Figure 6B, middle) displayed a 1-1 photobleaching step distribution (∼85%), demonstrating higher-order step distributions rarely occur by chance (Supplemental Figure 3B). These data provide strong evidence that, under these experimental conditions, HD-ZIPIII and ZPR proteins form heterotetrameric, rather than heterodimeric, complexes. Importantly, this novel stoichiometric information could not have been easily obtained with traditional methods used to detect protein-protein interactions.
DISCUSSION
Here, we establish SiMPull to enable detection of single-protein complexes in plants and identify relative protein levels and the degree of coexpression as important parameters. The abundance of a protein depends on its rate of accumulation, which we demonstrate scales with size in transient systems. This effect can be compensated for by modulating transcriptional output and, as such, promoter strength is an important consideration, particularly for interaction partners that are severely mismatched in size. Importantly, these insights are also applicable to other systems commonly used to study protein-protein interactions. Applying the above criteria, and in keeping with the known relationship between these developmental regulators (Wenkel et al., 2007; Kim et al., 2008), PHB and ZPR3 protein levels were balanced, allowing single PHB:ZPR3 complexes to be successfully detected via SiMPull, with subsequent colocalization analysis providing direct, statistical evidence of complex formation.
Our study thus positions SiMPull as a powerful, quantitative alternative to current methods of examining directed protein-protein interactions in plants, including one of the most popular methods of choice: BiFC. This approach, as it is commonly used, suffers from a variety of disadvantages, including nonquantitative readouts, lack of statistical power, false positives due to fluorophore “self-assembly,” and lack of stoichiometric information (Horstman et al., 2014; Xing et al., 2016). SiMPull resolves these issues, though it does not yield information on subcellular localization of interactions nor permit the de novo discovery of interactors. SiMPull, as performed here in this proof-of-principle analysis, may also be subject to the potential caveats of transient expression systems. However, it is important to note that SiMPull is ∼100-fold more sensitive than detection of protein-protein interactions via traditional immunoblots (Jain et al., 2014). Proteins at concentrations as low as 100 to 250 pM are detectable (Jain et al., 2011), and SiMPull has been used successfully in animal systems to probe interactions between protein partners at endogenous levels in their native context (Jain et al., 2011, 2014; Lee et al., 2013). Small amounts of tissue expressing PHB and ZPR3 were likewise used in these experiments (equivalent to ∼0.5 mm2 per assay; see Methods for details), and tissue amounts can easily be scaled up to further facilitate capture of lower abundance proteins. Through selection of the optimal lysate dilution, SiMPull adjusts for protein abundance as well as for variation in antibody affinities (Supplemental Figure 1). Given these advantages, SiMPull may prove to be a valuable tool for plant researchers conducting interaction studies with endogenous proteins as well as those limited by antibodies with low affinities. In addition, unlike most assays, SiMPull is not limited solely to protein-protein interactions but can also quantitatively interrogate interactions between proteins and compounds, such as nucleic acids, small molecule ligands, and lipids (Jain et al., 2011, 2014; Krishnan et al., 2013; Hwang et al., 2014a, 2014b; Rodgers et al., 2015; Arauz et al., 2016).
One of the most potent features of SiMPull is an ability to directly determine stoichiometries of immunoprecipitated complexes. Interestingly, our stoichiometric analysis supports a model in which PHB and ZPR3 form tetramers, consisting of two molecules of PHB and two molecules of ZPR3, rather than heterodimers as described in the current model (Wenkel et al., 2007; Kim et al., 2008). This finding implies that interaction with ZPR proteins does not necessarily disrupt HD-ZIPIII dimerization. This surprising result challenges prior assumptions regarding HD-ZIPIII:ZPR interactions and raises new questions about the regulation of these key developmental factors. For instance, how might ZPR proteins prevent HD-ZIPIII DNA binding, if not through disrupting their dimerization? In light of our observations that these proteins can form higher-order complexes, could this be accomplished via recruitment of additional interaction partners? Perhaps the repression of ARFs via the Aux/IAA-mediated recruitment of TOPLESS (Szemenyei et al., 2008) provides a complementary paradigm for the regulation of HD-ZIPIII activity by ZPR proteins? Moreover, does ZPR interaction always lead to a complete loss of DNA binding activity, or could higher-order complex formation also permit ZPR proteins to modulate, rather than abolish, HD-ZIPIII function? These potentially interesting avenues of research, suggested by measurements of complex stoichiometry, nicely illustrate the value of SiMPull to studies of protein-protein interactions. In sum, with its successful establishment in plants, SiMPull is now poised to have the same broad transformative effects on plant protein interaction studies as it did for animal systems.
METHODS
Plant Materials and Growth Conditions
Nicotiana benthamiana plants were grown at 22°C under long-day conditions on soil.
Molecular Biology
cDNAs corresponding to ACT2, ARF3, DRP2B, and ZPR3 were amplified using cDNA template synthesized from 10 d-old Arabidopsis thaliana seedling RNA (SuperScript III First-Strand Synthesis; Life Technologies) and cloned into pCR8/GW/TOPO in preparation for Gateway recombination (Life Technologies). pro35S:ACT2-mCherry, pro35S:ARF3-mCherry, pro35S:DRP2B-mCherry, and pro35S:ZPR3-mCherry binary vectors were generated via LR recombination into destination vector pFGC5941-mCherry, a modified version of pFGC5941 (Kerschen et al., 2004) in which the chsA intron was replaced with a Gateway attL1-attL2 cassette upstream of mCherry. An AS2-YFP entry clone, created via overlapping PCR and BP recombination into pDONR207 (Life Technologies), was already available (Lodha et al., 2013), and identical methods were used to create a PHB-YFP entry clone. pro35S:AS2-YFP and pro35S:PHB-YFP binary vectors were generated via LR recombination of AS2-YFP and PHB-YFP entry clones into destination vector pH2GW7 (Laboratory of Plant Systems Biology, Ghent, Belgium). The 2xpro35S:PHB-YFP binary vector was created via LR recombination of the PHB-YFP entry clone into destination vector 502Ω (Nakagawa et al., 2007). Tandem fluorophore cDNAs in a pCR8/GW/TOPO backbone were assembled using Gibson Assembly (NEB) and recombined into destination vector 502Ω to yield 2xpro35S:tdYFP and 2xpro35S:tdmCherry binary vectors. Cloning primers are listed in Supplemental Table 1.
Transient Transfection and Imaging of N. benthamiana Leaves
Leaves of N. benthamiana were transiently transfected with Agrobacterium tumefaciens via syringe-mediated infiltration (Sparkes et al., 2006). In brief, overnight cultures of Agrobacterium were centrifuged, resuspended in 2 to 5 mL of room temperature infiltration medium (10 mM MgCl2, 10 mM MES-KOH, pH 5.6, 150 mM acetosyringone [Sigma-Aldrich], and 1% DMSO), and diluted to a working optical density of 1, unless otherwise indicated. Diluted Agrobacterium cultures were incubated for 2 h at room temperature and then infiltrated into third and fourth leaves of 3- to 4-week-old N. benthamiana plants as described previously (Sparkes et al., 2006). Abaxial surfaces of infiltrated leaves were imaged using a Nikon SMZ1500 stereomicroscope with P-FLA2 epifluorescence attachment and NIS Elements software (Nikon). All images were taken at an exposure of 600 ms using a 1.4× digital gain. Unless otherwise indicated, all imaging was done 24 h postinfiltration.
Immunoblot Analysis
Approximately 1- to 2-cm2 pieces of infiltrated leaf tissue were flash frozen and then ground in 100 μL freshly prepared lysis buffer (25 mM Tris HCl, pH 8, 150 mM NaCl, 1% SDS, 1× cOmplete ULTRA protease inhibitor [Roche], and 1× PhosSTOP [Roche]). Lysates were cleared via 14,000g centrifugation at 4°C, mixed with 100 μL 2× Laemmli sample buffer, and boiled for 1 min. Proteins were resolved via SDS-PAGE, blotted to Hybond ECL membrane (GE Healthcare), and blocked in 20% SlimFast. Antibodies were used at the following dilutions: anti-GFP (Rockland Immunochemicals), 1:500; tubulin alpha chain antibodies (Agrisera), 1:2500; anti-rabbit horseradish peroxidase-conjugated secondary antibodies (Jackson Immunoresearch), 1:5000. Detection of secondary antibodies was performed with SuperSignal West Pico Chemiluminescent Substrate (ThermoFisher Scientific).
Single-Molecule TIRF Imaging and Spot Counting
Single-molecule images were acquired using a prism-type TIRF microscope equipped with an electron-multiplying CCD camera (EM-CCD). Quartz slides and glass cover slips were passivated with 5000 MW methoxy polyethylene glycol (mPEG) doped with 2 to 5% 5000 MW biotinylated PEG (Laysan Bio; Jain et al., 2012). Each passivated slide and cover slip was assembled into six flow chambers using double-sided tape (Scotch 3M), allowing multiple pull-down experiments to be performed simultaneously. Biotinylated antibodies against GFP (Rockland Immunochemicals), RFP (Abcam), or HA (Abcam), all at a dilution of 1:300, were immobilized in flow chambers using NeutrAvidin (Thermo Fisher Scientific). Cell lysates used in SiMPull were prepared using ∼1- to 2-cm2 pieces of infiltrated leaf tissue that were flash frozen, ground in 200 μL freshly prepared lysis buffer (40 mM Tris HCl, pH 8.0, 150 mM NaCl, 0.3% [v/v] IGEPAL CA-630 [Sigma-Aldrich], and 1× cOmplete ULTRA protease inhibitor [Roche]), and rotated at 4°C for 10 min. Lysates were cleared via 14,000g centrifugation at 4°C in two consecutive steps for 12 min each. Lysates were serially diluted using T50-BSA (20 mM Tris HCl, pH 8.0, 50 mM NaCl, and 0.1 mg mL−1 BSA) to obtain optimal single molecule density on the surface (600 to 800 molecules in a 5000 μm2 imaging area; Supplemental Figure 1). Fifty microliters of each dilution was incubated for 10 min on antibody-tethered slides and washed out with an excess of T50-BSA before imaging; this corresponds to ∼0.5 mm2 of tissue per SiMPull assay, assuming an optimal dilution of 1:50. YFP and mCherry proteins were excited with lasers of wavelength 488 and 568 nm (Coherent Sapphire), respectively; a YFP excitation wavelength of 488 nm was chosen to prevent mCherry cross-excitation. Band-pass filters of 535/30 nm for YFP (Chroma Technology) and 607/36 nm for mCherry (Semrock) were used to image emission signal. Sixteen frames (100 ms each) were recorded for each imaging area of ∼5000 μm2, and isolated single-molecule peaks were identified by fitting a Gaussian profile to the average intensity from the first 10 frames, followed by background subtracting using IDL scripts. Mean spot count per image for YFP or mCherry was calculated by averaging 20 or more imaging areas using MATLAB scripts. All pull-down experiments were performed at room temperature (22 to 25°C).
Single-Molecule Colocalization Analysis
Colocalization data were acquired by taking two separate movies of the same region of a slide using YFP and mCherry excitation. Colocalization criterion was set to a distance of two pixels or less between the centers of the YFP and mCherry spots, corresponding to a diffraction limited region of ∼300 nm for this TIRF setup. The number of coaligned molecules, divided by the total number of mCherry molecules, was presented as the colocalization percentage. Images of YFP and mCherry taken from two random, nonoverlapping slide regions were used to determine the frequency with which false colocalization occurs due to random spatial overlap of single molecules. Data were collected from a minimum of 30 individual slide areas, from three independent biological replicates, and statistical significance was calculated with a Student’s t test.
Photobleaching Step-Counting Analysis
Stoichiometric analysis was performed by counting the number of photobleaching steps for each YFP or mCherry spot. A single photobleaching step corresponds to an abrupt decrease in fluorescence intensity (see Supplemental Figures 2A and 2B for representative traces). Longer images of 600 frames (100 ms each) were acquired to ensure bleaching of all fluorescent molecules. Bleaching steps were counted manually and each molecule was classified as having either one or two bleaching steps, or discarded if no clear bleaching steps could be identified. Intensity histograms of the first five frames of one- and two-step photobleaching molecules corroborated the step-counting analysis. Images of PHB-YFP:ZPR3-mCherry complexes were acquired by sequentially photobleaching mCherry and YFP fluorophores (in that order) and counting all possible xYFP-xmCherry photobleaching step distributions (e.g., 1-1, 1-2, 2-1, and 2-2; see Supplemental Figure 3C for representative traces). All spots with no fluorescent signal from either or both fluorophores were rejected, i.e., 0-0, 1-0, 2-0, 0-1, and 0-2 photobleaching step possibilities were not counted.
Binomial Distribution for Determining Maturation Probabilities and Stoichiometry
SiMPull was performed with a 1:4000 dilution of lysate from leaves infiltrated with 2xpro35S:tdYFP or 2xpro35S:tdmCherry Agrobacterium strains and the frequency of two-step photobleaching events was measured for tandem YFP (25%) and tandem mCherry (38%) IP spots. In general, for an oligomeric protein with N copies, the probability P(x) of x photobleaching steps follows a binomial distribution below, where m is the maturation probability:
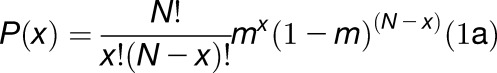 |
where x = 0, 1, 2 for a dimeric protein. Since P(0) could not be obtained experimentally, as it corresponds to the probability of zero fluorescence, the binomial probability distribution was renormalized, resulting in maturation probabilities of 0.40 and 0.55 for YFP and mCherry, respectively. YFP and mCherry maturation probabilities were then used to obtain the stoichiometry of the PHB:ZPR3 complex using binomial distribution 1a, where 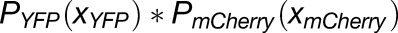 describes the probability of observing various PHB-YFP:ZPR3-mCherry photobleaching step distributions, xYFP-xmCherry. As above, binomial probability distributions were renormalized since spots lacking either or both fluorophores were not counted during the photobleaching analysis.
describes the probability of observing various PHB-YFP:ZPR3-mCherry photobleaching step distributions, xYFP-xmCherry. As above, binomial probability distributions were renormalized since spots lacking either or both fluorophores were not counted during the photobleaching analysis.
Accession Numbers
Sequence data from this article can be found in the Arabidopsis Genome Initiative database under the following accession numbers: ACT2, AT3G18780; ARF3, AT2G33860; AS2, At1g65620; DRP2B, AT1G59610; PHB, AT2G34710; ZPR3, AT3G52770.
Supplemental Data
Supplemental Figure 1. Determining the optimal lysate dilution.
Supplemental Figure 2. Resolving YFP and mCherry fluorophore maturation probabilities using photobleaching analysis and a binomial probability model.
Supplemental Figure 3. Binomial distributions support the formation of tetrameric PHB:ZPR3 complexes.
Supplemental Table 1. Primer sequences.
Supplementary Material
Acknowledgments
We thank Ankur Jain, Benjamin Leslie, Christopher Grefen, and members of the Ha and Timmermans labs for helpful ideas and comments that have strengthened the manuscript. We also thank the laboratory of David Jackson for the modified pFGC5941-mCherry construct. A.Y.H. thanks Michael Regulski for SlimFast diet tips. Work on leaf polarity in the Timmermans lab is supported by grants from the National Science Foundation (IBN-0615752 and IOS-1355018) and an Alexander von Humboldt Professorship. T.H. is funded by the Howard Hughes Medical Institute.
AUTHOR CONTRIBUTIONS
A.Y.H., V.A., T.H., and M.C.P.T. designed the project and experiments. A.Y.H. and V.A. performed the experiments. A.Y.H. and M.C.P.T. wrote the manuscript.
Glossary
- BiFC
bimolecular fluorescence complementation
- Y2H
yeast two-hybrid
- FRET
Forster resonance energy transfer
- TIRF
total internal reflection fluorescence
- hpi
hours postinfiltration
Footnotes
Articles can be viewed without a subscription.
References
- Aggarwal V., Ha T. (2014). Single-molecule pull-down (SiMPull) for new-age biochemistry: methodology and biochemical applications of single-molecule pull-down (SiMPull) for probing biomolecular interactions in crude cell extracts. BioEssays 36: 1109–1119. [DOI] [PubMed] [Google Scholar]
- Arauz E., Aggarwal V., Jain A., Ha T., Chen J. (2016). Single-molecule analysis of lipid-protein interactions in crude cell lysates. Anal. Chem. 88: 4269–4276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berggård T., Linse S., James P. (2007). Methods for the detection and analysis of protein-protein interactions. Proteomics 7: 2833–2842. [DOI] [PubMed] [Google Scholar]
- Carlsbecker A., et al. (2010). Cell signalling by microRNA165/6 directs gene dose-dependent root cell fate. Nature 465: 316–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler J.W., Cole M., Flier A., Grewe B., Werr W. (2007). The AP2 transcription factors DORNROSCHEN and DORNROSCHEN-LIKE redundantly control Arabidopsis embryo patterning via interaction with PHAVOLUTA. Development 134: 1653–1662. [DOI] [PubMed] [Google Scholar]
- Chitwood D.H., Nogueira F.T., Howell M.D., Montgomery T.A., Carrington J.C., Timmermans M.C. (2009). Pattern formation via small RNA mobility. Genes Dev. 23: 549–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dello Ioio R., Galinha C., Fletcher A.G., Grigg S.P., Molnar A., Willemsen V., Scheres B., Sabatini S., Baulcombe D., Maini P.K., Tsiantis M. (2012). A PHABULOSA/cytokinin feedback loop controls root growth in Arabidopsis. Curr. Biol. 22: 1699–1704. [DOI] [PubMed] [Google Scholar]
- Fujimoto M., Arimura S., Ueda T., Takanashi H., Hayashi Y., Nakano A., Tsutsumi N. (2010). Arabidopsis dynamin-related proteins DRP2B and DRP1A participate together in clathrin-coated vesicle formation during endocytosis. Proc. Natl. Acad. Sci. USA 107: 6094–6099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras A.C., Gstaiger M., Raught B., Aebersold R. (2007). Analysis of protein complexes using mass spectrometry. Nat. Rev. Mol. Cell Biol. 8: 645–654. [DOI] [PubMed] [Google Scholar]
- Gingras A.C., Aebersold R., Raught B. (2005). Advances in protein complex analysis using mass spectrometry. J. Physiol. 563: 11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giri S., Aggarwal V., Pontis J., Shen Z., Chakraborty A., Khan A., Mizzen C., Prasanth K.V., Ait-Si-Ali S., Ha T., Prasanth S.G. (2015). The preRC protein ORCA organizes heterochromatin by assembling histone H3 lysine 9 methyltransferases on chromatin. eLife 4: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecker A., Wallmeroth N., Peter S., Blatt M.R., Harter K., Grefen C. (2015). Binary 2in1 vectors improve in planta (co)localization and dynamic protein interaction studies. Plant Physiol. 168: 776–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horstman A., Tonaco I.A., Boutilier K., Immink R.G. (2014). A cautionary note on the use of split-YFP/BiFC in plant protein-protein interaction studies. Int. J. Mol. Sci. 15: 9628–9643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang H., Opresko P., Myong S. (2014a). Single-molecule real-time detection of telomerase extension activity. Sci. Rep. 4: 6391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang H., Kreig A., Calvert J., Lormand J., Kwon Y., Daley J.M., Sung P., Opresko P.L., Myong S. (2014b). Telomeric overhang length determines structural dynamics and accessibility to telomerase and ALT-associated proteins. Structure 22: 842–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwakawa H., Ueno Y., Semiarti E., Onouchi H., Kojima S., Tsukaya H., Hasebe M., Soma T., Ikezaki M., Machida C., Machida Y. (2002). The ASYMMETRIC LEAVES2 gene of Arabidopsis thaliana, required for formation of a symmetric flat leaf lamina, encodes a member of a novel family of proteins characterized by cysteine repeats and a leucine zipper. Plant Cell Physiol. 43: 467–478. [DOI] [PubMed] [Google Scholar]
- Jain A., Arauz E., Aggarwal V., Ikon N., Chen J., Ha T. (2014). Stoichiometry and assembly of mTOR complexes revealed by single-molecule pulldown. Proc. Natl. Acad. Sci. USA 111: 17833–17838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain A., Liu R., Ramani B., Arauz E., Ishitsuka Y., Ragunathan K., Park J., Chen J., Xiang Y.K., Ha T. (2011). Probing cellular protein complexes using single-molecule pull-down. Nature 473: 484–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain A., Liu R., Xiang Y.K., Ha T. (2012). Single-molecule pull-down for studying protein interactions. Nat. Protoc. 7: 445–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo C., Balci H., Ishitsuka Y., Buranachai C., Ha T. (2008). Advances in single-molecule fluorescence methods for molecular biology. Annu. Rev. Biochem. 77: 51–76. [DOI] [PubMed] [Google Scholar]
- Juarez M.T., Kui J.S., Thomas J., Heller B.A., Timmermans M.C.P. (2004). MicroRNA-mediated repression of rolled leaf1 specifies maize leaf polarity. Nature 428: 84–88. [DOI] [PubMed] [Google Scholar]
- Kay R., Chan A., Daly M., McPherson J. (1987). Duplication of CaMV 35S promoter sequences creates a strong enhancer for plant genes. Science 236: 1299–1302. [DOI] [PubMed] [Google Scholar]
- Kerschen A., Napoli C.A., Jorgensen R.A., Müller A.E. (2004). Effectiveness of RNA interference in transgenic plants. FEBS Lett. 566: 223–228. [DOI] [PubMed] [Google Scholar]
- Kim H., Park M., Kim S.J., Hwang I. (2005). Actin filaments play a critical role in vacuolar trafficking at the Golgi complex in plant cells. Plant Cell 17: 888–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y.S., Kim S.G., Lee M., Lee I., Park H.Y., Seo P.J., Jung J.H., Kwon E.J., Suh S.W., Paek K.H., Park C.M. (2008). HD-ZIP III activity is modulated by competitive inhibitors via a feedback loop in Arabidopsis shoot apical meristem development. Plant Cell 20: 920–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan R., Blanco M.R., Kahlscheuer M.L., Abelson J., Guthrie C., Walter N.G. (2013). Biased Brownian ratcheting leads to pre-mRNA remodeling and capture prior to first-step splicing. Nat. Struct. Mol. Biol. 20: 1450–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H.W., et al. (2013). Real-time single-molecule co-immunoprecipitation analyses reveal cancer-specific Ras signalling dynamics. Nat. Commun. 4: 1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z., Lavis L.D., Betzig E. (2015). Imaging live-cell dynamics and structure at the single-molecule level. Mol. Cell 58: 644–659. [DOI] [PubMed] [Google Scholar]
- Lodha M., Marco C.F., Timmermans M.C. (2013). The ASYMMETRIC LEAVES complex maintains repression of KNOX homeobox genes via direct recruitment of Polycomb-repressive complex2. Genes Dev. 27: 596–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallory A.C., Reinhart B.J., Jones-Rhoades M.W., Tang G., Zamore P.D., Barton M.K., Bartel D.P. (2004). MicroRNA control of PHABULOSA in leaf development: importance of pairing to the microRNA 5′ region. EMBO J. 23: 3356–3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell J.R., Emery J., Eshed Y., Bao N., Bowman J., Barton M.K. (2001). Role of PHABULOSA and PHAVOLUTA in determining radial patterning in shoots. Nature 411: 709–713. [DOI] [PubMed] [Google Scholar]
- Müller C.J., Valdés A.E., Wang G., Ramachandran P., Beste L., Uddenberg D., Carlsbecker A. (2016). PHABULOSA mediates an auxin signaling loop to regulate vascular patterning in Arabidopsis. Plant Physiol. 170: 956–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T., Kurose T., Hino T., Tanaka K., Kawamukai M., Niwa Y., Toyooka K., Matsuoka K., Jinbo T., Kimura T. (2007). Development of series of gateway binary vectors, pGWBs, for realizing efficient construction of fusion genes for plant transformation. J. Biosci. Bioeng. 104: 34–41. [DOI] [PubMed] [Google Scholar]
- Nguyen H.H., Park J., Kang S., Kim M. (2015). Surface plasmon resonance: a versatile technique for biosensor applications. Sensors (Basel) 15: 10481–10510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prigge M.J., Otsuga D., Alonso J.M., Ecker J.R., Drews G.N., Clark S.E. (2005). Class III homeodomain-leucine zipper gene family members have overlapping, antagonistic, and distinct roles in Arabidopsis development. Plant Cell 17: 61–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers M.L., Paulson J., Hoskins A.A. (2015). Rapid isolation and single-molecule analysis of ribonucleoproteins from cell lysate by SNAP-SiMPull. RNA 21: 1031–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sessa G., Steindler C., Morelli G., Ruberti I. (1998). The Arabidopsis Athb-8, -9 and -14 genes are members of a small gene family coding for highly related HD-ZIP proteins. Plant Mol. Biol. 38: 609–622. [DOI] [PubMed] [Google Scholar]
- Shah P., Ding Y., Niemczyk M., Kudla G., Plotkin J.B. (2013). Rate-limiting steps in yeast protein translation. Cell 153: 1589–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J., Padgett R.A. (2009). Rates of in situ transcription and splicing in large human genes. Nat. Struct. Mol. Biol. 16: 1128–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparkes I.A., Runions J., Kearns A., Hawes C. (2006). Rapid, transient expression of fluorescent fusion proteins in tobacco plants and generation of stably transformed plants. Nat. Protoc. 1: 2019–2025. [DOI] [PubMed] [Google Scholar]
- Szemenyei H., Hannon M., Long J.A. (2008). TOPLESS mediates auxin-dependent transcriptional repression during Arabidopsis embryogenesis. Science 319: 1384–1386. [DOI] [PubMed] [Google Scholar]
- Ulbrich M.H., Isacoff E.Y. (2007). Subunit counting in membrane-bound proteins. Nat. Methods 4: 319–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulbrich M.H., Isacoff E.Y. (2008). Rules of engagement for NMDA receptor subunits. Proc. Natl. Acad. Sci. USA 105: 14163–14168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenkel S., Emery J., Hou B.H., Evans M.M., Barton M.K. (2007). A feedback regulatory module formed by LITTLE ZIPPER and HD-ZIPIII genes. Plant Cell 19: 3379–3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing S., Wallmeroth N., Berendzen K.W., Grefen C. (2016). Techniques for the analysis of Protein-protein interactions in vivo. Plant Physiol. 171: 727–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeom K.H., Heo I., Lee J., Hohng S., Kim V.N., Joo C. (2011). Single-molecule approach to immunoprecipitated protein complexes: insights into miRNA uridylation. EMBO Rep. 12: 690–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.