

ABSTRACT
The potential of a tumor cell to metastasize profoundly depends on its microenvironment, or “niche” interactions with local components. Tumor-associated-macrophages (TAMs) are the most abundant subpopulation of tumor stroma and represent a key component of tumor microenvironment. The dynamic interaction of cancer cells with neighboring TAMs actively drive cancer progression and metastatic transformation through intercellular signaling networks that need better elucidation. Thus, current study was planned for discerning paracrine communication networks operational between TAMs, and breast cancer cells with special reference to cancer cell invasion and dissemination to distant sites. Here, we report role of MIP-1β in enhancing invasive potential of metastatic breast cancer MDA-MB-231 and MDA-MB-468 cells. In addition, the poorly metastatic MCF-7 cells were also rendered invasive by MIP-1β. The MIP-1β-driven cancer cell invasion was dependent on upregulated expression levels of MYO3A gene, which encodes an unconventional myosin super-family protein harboring a kinase domain. Ex ovo study employing Chick-embryo-model and in vivo Syngenic 4T1/BALB/c mice-model further corroborated aforementioned in vitro findings, thereby substantiating their physiological relevance. Concordantly, human breast cancer specimen exhibited significant association between mRNA expression levels of MIP-1β and MYO3A. Both, MIP-1β and MYO3A exhibited positive correlation with MMP9, an established molecular determinant of cancer cell invasion. Higher expression of these genes correlated with poor survival of breast cancer patients. Collectively, these results point toward so far undisclosed MIP-1β/MYO3A axis being operational during metastasis, wherein macrophage-derived MIP-1β potentiated cancer cell invasion and metastasis via up regulation of MYO3A gene within cancer cells. Our study exposes opportunities for devising potential anti-metastatic strategies for efficient clinical management of breast cancer.
KEYWORDS: Invadopodia, invasion, migration, MIP-1β, MYO3A, MMP-9
Abbreviations
- CAM
Chick chorioallantoic membrane
- ECM
Extracellular matrix
- H&E
Hematoxylin and Eosin
- IHC
Immunohistochemistry
- RIPA
Radio immunoprecipitation Assay
- TAMs
tumor-associated macrophages.
Introduction
Accounting for 90% of breast-cancer-related deaths, the distant metastasis remains a major cause of breast-cancer-related deaths and poses a serious health hazard.1 Owing to poor knowledge regarding biology of metastasis, the clinical intervention becomes extremely challenging making metastatic cancer largely incurable.2 A better understanding of the molecular determinants that regulate onset and progression of metastasis may provide basis for development of targeted therapeutic regimen against metastatic cancer, and result in identification of potential diagnostic and prognostic markers for tracking disease progression.3 So far, the majority of the research toward developing an insight about cancer metastasis has remained focused on malignant epithelial cells. However, the more recent studies demonstrate that potential of a tumor cell to metastasize depends, to a great extent, on its microenvironment or “niche” interactions with local factors/components.4 The complex tumor microenvironment is comprised of not only an expanding population of transformed epithelial cells, but it also includes other non-cancer cells such as smooth muscle cells, fibroblasts and macrophages.5 Collectively, these cells are termed as stromal cells and they play a crucial role during cancer progression.6 The dynamic interactions between tumor cells and the surrounding stromal cells actively promote cancer progression as well as response to therapy by contributing toward tumor-neovasculogenesis, metastatic transformation and drug resistance by driving reparative mechanisms.7,8
Tumor-associated macrophages (TAMs) represent the most abundant subpopulation of these non-cancer stromal cells.5,9 Although, cytotoxicity of macrophages during the early immune response contributes to tumor killing, in most solid tumors, macrophages inversely affect prognosis by promoting angiogenesis, potentiating tissue remodeling, imparting invasive properties to cancer cells and thereby rendering them metastatic.10 In order to metastasize, cancer cells of epithelial origin need to acquire features which would enable them to (i) degrade the basal membrane and disseminate from primary site, (ii) invade neighboring tissue, intravasate into blood or lymph vessels (iii) extravasate from vessels into distant organs (iv) colonize and thrive in distant tissue to create secondary tumor(s).11 TAMs have been reported to facilitate all the above key steps of cancer metastasis through paracrine signaling networks. At primary tumor site, TAMs promote cancer cell dissemination via upregulation of matrix metalloproteases, resulting in enhanced ECM degradation and cancer cell invasion into neighboring tissue. TAMs facilitate cancer cell intravasation by promoting endothelial cell migration resulting in enhanced angiogenesis. At distant metastatic site, TAMs promote cancer cell extravasation, seeding and persistent growth of tumor cells.12
Although TAMs are important components of tumor stroma and have an established role in promoting metastasis,13 the intercellular paracrine signals that mediate direct crosstalk between TAMs and tumor cells during metastasis need better elucidation. Furthermore, the ensuing molecular events within tumors cells that eventually impart them an ability to invade surrounding tissue and disseminate from primary site during metastasis are poorly understood. In view of this, the current study was planned to elucidate paracrine communication networks operational between TAMs and malignant epithelial cell with special reference to cancer cell invasion and dissemination during metastasis. Here, we report that MIP-1β secreted from macrophages augmented invasiveness and motility of breast cancer cells. Furthermore, we show that MIP-1β-driven cancer cell invasion and metastasis is MYO3A dependent. MIP-1β is a member of chemokine β subgroup of chemokine superfamily with an established role as chemoattractant for macrophages.14 Here, we report a previously undisclosed role for MIP-1β as a mediator of TAMs-assisted metastasis. MYO3A is a myosin family gene that is expressed primarily in retina and cochlea and functionally involved in hearing.15 Our studies reveal a possible new function of MYO3A during cancer metastasis. Collectively, this so-far undisclosed MIP-1β-MYO3A pathway is likely to play a biologically relevant role in cancer metastasis and thus may have possible utility as a diagnostic marker for detecting metastasis at an early stage. It may have potential usage during clinical management of breast cancer as a prognostic marker for tracking progression of breast cancer toward metastasis.
Results
Presence of macrophages correlated with increased invadopodia formation and intensified focal degradation of matrix by invasive breast adenocarcinoma MDA-MB-231 and MDA-MB-468 cells
One of the earliest hallmarks of cancer cell invasion and metastasis is the biogenesis of specialized membrane protrusions called Invadopodia.16 Richly endowed with matrix-degrading activities, these specialized membrane protrusions allow cancer cells to proteolytically degrade extracellular matrix and thus migrate through the three-dimensional interstitial collagen networks.17 Since the focal degradation of extracellular matrix by invadopodia represents the beginning of the process of metastasis, we first set out to study the effect of macrophages on ability of MDA-MB-231 and MDA-MB-468 cancer cells to degrade pericellular matrix through enhanced invadopodia formation. Results revealed that compared to monocultured MDA-MB-231 and MDA-MB-468 cancer cells, the ones that were co-cultured with macrophages exhibited enhanced focal degradation of pericellular matrix (Fig. 1A and B) in a time-dependent manner, detectable dark foci of degradation occurred at as early as 3 h time point, exhibiting an incremental change further upto 6 h and 24 h (Figs. S1 and 3).
Figure 1.

Invasive breast adenocarcinoma MDA-MB-231 and MDA-MB-468 exhibited intensified focal degradation of pericellular matrix, increased invadopodia formation and poorly metastatic breast cancer MCF-7 cells were rendered invasive in presence of THP-1 macrophages. (A and B) Representative images from the in vitro matrix degradation assay. Cells (MDA-MB 231 and MDA-MB-468) were seeded on Alexa Fluor 633 labeled gelatin (Red) in absence or presence of macrophages (housed in 0.4 μm PET transwell hanging cell culture insert) and maintained for 24 h, followed by fixation, staining with Alexa fluor 488 phalloidin (Green) and mounted in aqueous media containing DAPI (Blue). Compared to mono-cultured MDA-MB-231 and MDA-MB-468 cancer cells [C], the ones that were co-cultured with macrophages [C+M] exhibited enhanced focal degradation of pericellular matrix as indicated by dark area of degraded fluorescent matrix underneath that cell. Bars represent mean invadopodia count/cell from 10 fields per experiment ±SE (*p < 0.05). (C) Compared to monocultured MCF-7 cells [C], the co-cultured MCF-7 cells (macrophages housed in 0.4 μm PET transwell hanging cell culture insert) [C+M] exhibited enhanced focal degradation (dark area of degraded fluorescent matrix underneath that cell) of pericellular matrix. Bars represent mean invadopodia count/cell (by dot count software) from 10 fields per experiment ±SE (*p < 0.05). All the experiments were done in triplicates and repeated at least thrice. Abbreviations—C: Respective cancer Cells; C+M: Respective cancer cells co-cultured with macrophages.
Cortactin, an actin nucleation-promoting factor has recently emerged not only as crucial regulator of actin cytoskeleton dynamics, but also a key player in aggressive cancers.18,19 It has a central role in the development and maturation of invadopodia.20 The formation of actin-rich puncta called ‘invadopodia precursors' requires recruitment of cortactin.21,22 Consistent with this, cancer cells that were co cultured with macrophages exhibited enhanced localization of cortactin to peripheral cellular structures (Fig. 2A and B), overlying foci of matrix degradation i.e. dark areas of degraded fluorescent matrix underneath that cell (Figs. S2 and 4) indicating for enhanced formation of functional invadopodia by cancer cells in presence of macrophages.
Figure 2.

Invasive breast adenocarcinoma MDA-MB-231 and MDA-MB-468 and poorly metastatic breast cancer MCF-7 cells exhibited enhanced localization of cortactin to peripheral cellular structures and potentiated invasive activity in presence of THP-1 macrophages. (A, B and C) Macrophages stimulated formation of functional invadopodia in MDA-MB-231, MDA-MB-468 and MCF-7 cells as revealed by enhanced localization of cortactin to peripheral cellular structures and greater number of cortactin positive puncta overlying foci of matrix degradation in cancer cells co-cultured with macrophages [C+M] as compared to cells that were cultured alone [C]. (D, E and F) Representative images from the in vitro cell invasion assay. The matrigel transvasation assay (MDA-MB-231, MDA-MB-468 and MCF-7) revealed a significant increase in the invasive capacity of co-cultured [C+M] (24 h) cancer cells. Bars represent mean invasive cell count ±SE (*p < 0.05.). All the experiments were done in triplicates and repeated at least thrice. Abbreviations—C: Respective cancer Cells; C+M: Respective cancer cells co-cultured with macrophages.
Presence of macrophages correlated with increased cancer cell invasion by breast adenocarcinoma MDA-MB-231 and MDA-MB-468 cells
Having established that key attributes for cancer cell invasion viz. focal degradation of pericellular matrix were intensified in presence of macrophages, we next studied if proximity to macrophages would eventually also culminate into enhanced local invasion by cancer cell. Matrigel invasion assay revealed that co-cultured cancer cells exhibited enhanced invasive potential (Fig. 2D and E). Collectively, these results indicated that cancer cells exhibited intensified focal degradation in response to incubation with macrophages, which eventually culminated into enhanced invasive potential.
Presence of macrophages correlated with acquisition of invasive potential by non-invasive breast adenocarcinoma MCF-7 cells
Having established that macrophages augment matrix protrusive and invasive potential of invasive breast cancer cells, we next decided to evaluate the effect of macrophages on poorly invasive breast cancer MCF-7 cells vis a vis attributes indicative of cancer cell invasion. Results revealed that compared to monocultured MCF-7 cancer cells, the co-cultured cancer cells exhibited enhanced focal degradation of pericellular matrix in a time-dependent manner (Fig. 1C and Fig. S5). Consistent with this, MCF-7 breast cancer cells that were incubated with macrophages exhibited enhanced redistribution of cortactin to peripheral cellular structures overlying foci of matrix degradation (Fig. 2C and Fig. S6), thereby confirming the formation of functionally intact invadopodia. Furthermore, co-cultured MCF-7 cancer cells exhibited enhanced invasion through matrigel (Fig. 2F) after 24 h. These results showed that poorly invasive MCF-7 cells were rendered invasive by macrophages.
Genesis of matrix protrusive activity within cancer cells co-incided with enhanced secretion of MIP-1β, primarily from macrophages
In order to identify the key mediators of macrophage-assisted invadopodia biogenesis and cancer cell invasion, culture supernatant from cancer cell/macrophage co-culture and monocultures of either cell types were profiled for secreted cytokines using antibody-based human cytokine protein membrane array. Results revealed that compared to culture supernatant from monocultures of either cell type, the co-culture supernatant exhibited elevated levels of MCP-1, TIMP-1, MCSF, MIP1-β, I-309, ENA-78, and GCP-2 (Fig. S7A). These marked alterations in secretome were further validated using Quantitative-RTPCR (q-PCR) analysis with an additional aim to decipher the possible source of each of these mediators. Q-RTPCR analysis indicated that possible source of I-309, ENA-78 and GCP-2 could be cancer cells, as mRNA expression levels of these cytokines were primarily upregulated in cancer cells. On the other hand, the macrophages exhibited an upregulated mRNA expression level of MCP-1, TIMP-1, MCSF, and MIP-1β, thereby indicating macrophages as primary source of release of these cytokines (Fig. S7B). Since we aimed at deciphering macrophage-derived mediators of cancer cell invasion and metastasis, we focused on MCP-1, TIMP-1, MCSF, and MIP-1β. Closer inspection of q-RTPCR data revealed that while all the four cytokines showed marked upregulation in MDA-MB-231 cells after 24 h of co-culture, the most prominent upregulation was that in the level of MIP-1β. Interestingly, at 6 h time point while MIP-1β mRNA expression levels were upregulated, the levels of other cytokines remained much lesser. Since considerable matrix protrusive activity was also observed at 6 h time point (Figs. S1, 3 and 5) and with regards to this, only MIP-1β mRNA levels were elevated. Thus, a key role of MIP-1β was hypothesized. A closer look at MCF-7 cell cytokine profiling data strengthened the above possibility. Results revealed that at 6 h time point, when the matrix protrusive activity was minimal, MIP-1β levels were much lower than the levels at 24 h time point where there was exhibition of significant matrix protrusive activity by MCF-7 cells (Fig. S5), which were otherwise non-invasive and had minimal matrix protrusive activity. In light of consistent presence of MIP-1β in particular experimental setting that culminated in significant matrix protrusion by cancer cells, we hypothesized that MIP-1β could be one of the key mediators employed by macrophages for rendering cancer cell invasive.
Blockade of MIP-1β action minimized the pro-metastatic effect of macrophages
In order to ascertain above the role of MIP-1β possibility in metastasis, the effect of MIP-1β blockade was studied in matrix degradation and invasive potential of cancer cells. Blockade of MIP-1β using anti-human MIP-1β goat IgG polyclonal antibody (0.8 ug/uL) impeded the pericellular matrix degradation and compromised invasive potential of MDA-MB-231, MDA-MB-468 and MCF-7 cancer cells even in the presence of macrophages (Fig. 3A and B, Fig. S8). Marked inhibition of matrix degradation and cancer cell invasion in presence of MIP-1β-neutralizing antibody indicated for critical role of MIP-1β in macrophage-assisted cancer cell invasion and metastasis. In agreement with this addition of recombinant MIP-1β (0.2 ug/uL) resulted in enhanced matrix degradation by monocultured cancer cells with a concurrent increase in invasion through matrigel (Fig. 3A and B, Fig. S8). Secreted MIP-1β exerts its effects via its cognate receptor CCR5.23 Additionaly, fewer reports indicate an interaction of MIP-1β with CCR4 as well.24 Therefore, to further confirm the role of MIP-1β as one of the key mediators of macrophage-assisted cancer cell invasion, MIP-1β function was impeded by knocking down cognate receptors within breast cancer cell prior to co-culturing them with THP-1-derived macrophages. The corresponding cancer cells that were transfected with scrambled RNA served as control. As expected, the control cells exhibited enhanced matrix degradation after an incubation of 24 h with THP-1-derived macrophages. However, upon silencing the cognate receptors, the extent of matrix degradation by cancer cells was markedly impeded even in the presence of macrophages. Similarly, CCR5 and CCR4 silencing reversed the macrophage-induced cancer cell invasion (Fig. 4A and B, Fig. S9), thereby confirming the functional role of MIP-1β during macrophage-assisted cancer cell invasion.
Figure 3.

Macrophage-assisted ECM degradation and invasion by cancer cells is mediated by MIP-1β. (A and B) Representative images showing the effect of anti-human MIP-1β blockade by MIP-1β-neutralizing antibody (MIP-1β NA) or addition of MIP-1β-purified cytokine (MIP-1β) with respect to matrix protrusive activity, and invasion in MDA-MB-231 and MCF-7. IgG serve as isotype antibody control for MIP-1β-neutralizing antibody (MIP-1β NA). Cancer cells (MDA-MB-231 and MCF-7) treated with MIP-1β NA showed decreased activity in matrix protrusive activity and diminished invasion compared to cells that were not treated with MIP-1β NA. Cancer cells treated with MIP-1β-purified cytokine (MIP-1β) showed an increase in matrix protrusive activity and invasion compare to cells that did not treated with MIP-1β. Bars represent mean invadopodia count/cell from 10 fields per experiment and mean invasive cell count ±SE (*p < 0.05.). All the experiments were done in triplicates and repeated at least thrice. Abbreviations—C: Respective cancer Cells; C+M: Respective cancer cells co-cultured withMacrophages; C+M(IgG): Respective cancer cells co-cultured with macrophages treated with isotype antibody control IgG; C+M+MIP-1β NA: Respective cancer cells co-cultured with macrophages treated with MIP-1β-neutralizing antibody; MIP-1β: Respective cancer cells treated with MIP-1β-purified cytokine.
Figure 4.
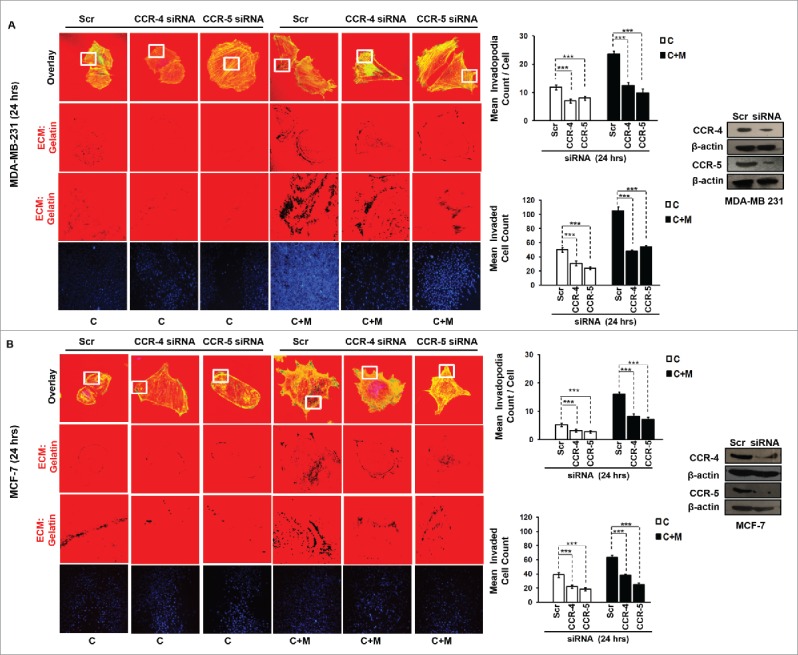
Macrophage-assisted ECM degradation and invasion by cancer cells is mediated by MIP-1β via its receptor. (A and B) Silencing MIP-1β cognate receptors viz. CCR4 and CCR5 abrogated macrophage induced in vitro ECM degradation, and invasion by cancer cell. Cancer cells (MDA-MB-231 and MCF-7) silenced with CCR4 and CCR5 showed decreased ECM degardation and invasive activity compared to cells having scrambled in both the conditions viz. cancer cells co-cultured with macrophages [C+M] and cultured alone [C]. Bars represent mean invadopodia count/cell from 10 fields per experiment and mean invasive cell count ±SE (*p < 0.05.). Silencing was confirmed by protein gel blot. All the experiments were done in triplicates and repeated at least thrice. Abbreviations—C: Respective cancer cells; C+M: Respective cancer cells co-cultured withmacrophages.
Blockade of MIP-1β function minimized ex ovo dissemination of co-cultured cancer cells from chick chorioallantoic membrane to chick brain
In vitro observations were further validated using ex ovo chick chorioallantoic membrane assay for metastasis. Results revealed that compared to monocultured MCF-7 and MDA-MB-231 breast cancer cells, the co-cultured cancer cells exhibited enhanced dissemination from the graft site i.e., chorioallantoic membrane to brain of chick embryo as evidenced by presence of relatively larger sized fluorescent foci in greater numbers in chick brain. Addition of MIP-1β-neutralizing antibody during co-culture and thereafter at graft site resulted in appearance of smaller sized fluorescent foci in fewer numbers with in the brain of chick embryo, indicating impeded dissemination of cancer cells from CAM to brain (Fig. 5A and B). These ex ovo findings substantiated the role of MIP-1β in promoting cancer cell invasion and metastasis, particularly in the capacity of a paracrine signaling mediator originating from proximate macrophages.
Figure 5.

Blockade of MIP-1β function minimized ex ovo dissemination of co-cultured cancer cells from chick chorioallantoic memebrane to chik brain. (A and B) Representatitive results of ex ovo chick chorioallantoic membrane assay for spontaneous metastasis. MIP-1β-neutralizing antibody (MIP-1β NA) mediated in vivo functional blockade of MIP-1β-impeded spontaneous metastasis of co-cultured (with macrophages) breast cancer cells [C + M + MIP-1β NA] compare to cells that were not treated with MIP-1β NA [C + M + IgG and C + M] from chicken chorioallantoic membrane to chicken brain. Bars represent number of fluorescent foci in each group. All the experiments were done in triplicates and repeated at least thrice. Abbreviations—C: Respective cancer cells; C + M: Respective cancer cells co-cultured with macrophages; C + M(IgG): Respective cancer cells co-cultured with macrophages treated with isotype antibody control IgG; C + M + MIP-1β NA: Respective cancer cells co-cultured with macrophages treated with MIP-1β-neutralizing antibody.
MYO3A serves as a key cancer cell intrinsic effector for macrophage-assisted cancer cell invasion via MIP-1β
While TAMs may have a decisive role during metastasis,25 the tumor-intrinsic molecular determinants are still essential for metastasis and thus remain key effector for cancer cell invasion and dissemination. Having established an involvement of MIP-1β in macrophage-assisted cancer cell invasion, we next set out to identify the tumor-intrinsic molecular determinants through which macrophage-derived MIP-1β might potentiate invasive property of cancer cells. Comparision of whole genome microarray analysis (GSE36047) revealed that mRNA expression levels of five genes viz. HGF, SIGLEC8, MYO3A, HLA-DBRS, and PRKY were markedly upregulated in cancer cells following their incubation with macrophages (24 h). The microarray data was further validated through q-RT-PCR analysis. Interestingly, one particular gene viz. MYO3A showed most robust upregulation in cancer cells in response to their co-incubation with macrophages (Fig. 6A–C). Thus, we hypothesized that MYO3A could be a potential key molecular determinant intrinsic to cancer cell for MIP-1β-induced cancer cell invasion and metastasis. In order to address this question, the effect of MIP-1β blockade/addition on the mRNA expression levels of MYO3A and other candidate genes was evaluated. Results revealed that MYO3A exhibited a characteristic MIP-1β-responsive exprerssion pattern. While neutralizing antibody-mediated MIP-1β blockade (0.8 ug/uL) downregulated the macrophage-induced upregulation of all the five traget genes in cancer cells, addition of recombinant MIP-1β (0.2 ug/mL) to cancer cells elicited robust upregulation of only MYO3A gene in cancer cells (Fig. 6D–F) indicating that MYO3A expression levels were regulated by MIP-1β. In agreement with this, functional blockade of MIP-1β through (i) neutralizing antibody treatment (ii) cognate receptor knockdown resulted in downregulation of MYO3A expression levels (Fig. 6G and H). These obsrvations substantiated expression of MYO3A being dependent on MIP-1β and pointed toward potential role of MYO3A as intrinsic determinant through which MIP-1β could enhance an invasiveness of cancer cells. In order to ascertain this, the effect of MYO3A knockdown was evaluated. Results revealed that targeting MYO3A expression levels using MYO3A-directed siRNA impaired the matrix protrusive activity of cancer cells. MYO3A knockdown abrogated pericellular matrix degradation and invasion by cancer cells despite being co-cultured with macrophages (Fig. 7A and B, Fig. S10).To further substantiate the involvement of MYO3A for MIP-1β-induced cancer cell invasion, effect of MYO3A knockdown was evaluated in absence or presence of recombinant MIP-1β. As expected, compared to RNAi control cells (scrambled cells), MYO3A-silenced cells exhibited markedly diminished pericellular matrix degradation and invasion (Fig. 8, Fig. S11).These results confirmed the role of MYO3A as a key molecular determinant for cancer cell invasion partucularly in response to MIP-1β.
Figure 6.

TAMs-assisted cancer cell invasion via MIP-1β is dependent on upregulation of MYO3A gene with in cancer cells. (A, B and C) Quantitative RT-PCR-based validation of selected genes (through c-DNA-based gene-expression analysis) in mono-cultured [C] and co-cultured (with macrophages) cancer cells [C+M]. Bars represent relative fold change in expression levels ±SE (*p < 0.05). (D, E and F) MYO3A gene exhibited a characteristic MIP-1β-responsive mRNA exprerssion profile. MIP-1β-neutralizing antibody (MIP-1β NA) treated cells showed decrease in MYO3A expression compared to cells that were co-cultured with macrophages [C+M] and/or IgG antibody control [C + M + IgG]. MIP-1β-purified cytokine (MIP-1β) enhanced the expression of MYO3A gene in monocultured cancer cells compared to cells that were not treated with MIP-1β-purified cytokine (C). Bars represent Quantitative RT-PCR relative fold change expression ±SE (*p <0.05.). All the experiments were done in triplicates and repeated at least thrice. (G and H) Blockade of MIP-1β with MIP-1β-neutralizing antibody (MIP-1β NA, Fig. 6G) and its cognate receptor CCR4 and CCR5 silencing (CCR4-siRNA and CCR5-siRNA, Fig. 6H)) downregulated expression levels of MYO3A in breast cancer cells (MDA-MB-231, MDA-MB-468 and MCF-7). MIP-1β-purified cytokine (MIP-1β) enhanced the expression of MYO3A gene in monocultured cancer cells compared to cells that were not treated with MIP-1β-purified cytokine (C) Fig. 6G. All the experiments were done in triplicates and repeated at least thrice. Abbreviations—C: Respective cancer cells; C + M: Respective cancer cells co-cultured with macrophages; C+M(IgG): Respective cancer cells co-cultured with macrophages treated with isotype antibody control IgG; C + M + MIP-1β NA: Respective cancer cells co-cultured with macrophages treated with MIP-1β-neutralizing antibody; MIP-1β: Respective cancer cells treated with MIP-1β-purified cytokine.
Figure 7.

MYO3A serves as a key cancer cell intrinsic effector for macrophage-assisted cancer cell invasion via MIP-1β. TAMs-assisted cancer cell invasion via MIP-1β is dependent on upregulation of MYO3A gene within cancer cells. (A and B) Macrophage-induced matrix protrusion and invasion by cancer cells was abrogated upon siRNA-mediated silencing of MYO3A gene compare to scrambled in both C (mon-oculture cancer cells) and C + M (cancer cells co-cultured with macrophages). Bars represent mean invadopodia count/cell from 10 fields per experiment and mean invasive cell count ±SE (*p < 0.05.). All the experiments were done in triplicates and repeated at least thrice. Abbreviations—C: Respective cancer cells; C+M: Respective cancer cells co-cultured withmacrophages.
Figure 8.
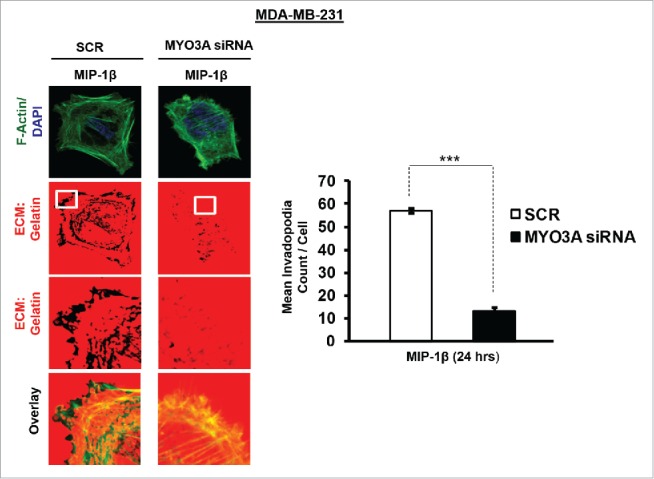
Invasive breast adenocarcinoma MDA-MB-231 silenced with MYO3A exhibited diminished focal degradation of pericellular matrix, diminished invadopodia formation, compared to Scrambled control in presence of MIP-1β-Purified cytokine. Representative images from the in vitro matrix degradation assay. Cells (MDA-MB-231) Scr and silenced with MYO3A siRNA were seeded on Alexa Fluor 633 labeled gelatin (Red) in absence or presence of MIP-1β-purified cytokine (MIP-1β) for 24 h, followed by fixation, staining with Alexa fluor 488 phalloidin (Green) and mounting in aqueous media containing DAPI (Blue). Compared to Scr control MYO3A-directed siRNA, MDA-MB-231 cancer cells did not show any effect of MIP-1β-purified cytokine on focal degradation of pericellular matrix. Bars represent mean invadopodia count/cell from 10 fields per experiment ±SE (*p < 0.05.). All the experiments were done in triplicates and repeated at least thrice. Abbreviations—MIP-1β: Respective cancer cells treated with MIP-1β-purified cytokine.
In vivo blockade of MIP-1β resulted in regressed 4T1 primary tumor volume, downregulated MYO3A and MMP-9 gene expression levels and impeded distant tissue metastasis to lungs and liver
Further confirmation of above findings was done in syngenic 4T1/BALB/c in vivo mouse model of breast cancer. Because the model is syngenic in BALB/c mice, and employs animals that have functionally intact immune system, it allows for studying the role of immune system in tumor progression. The anti-mouse MIP-1β goat IgG polyclonal antibody-treated 4T1 tumors exhibited significantly downregulated expression levels of MYO3A gene (Fig. 9A). Matrix metalloproteinases (MMPs) are regarded as one of the key molecular determinants that actively promote cancer cell invasion by way of carrying ECM degradation.26-29 As an indirect measure of invasive index, the mRNA expression levels of MMP-9 gene were assessed, which were significantly downregulated (Fig. 9B) in response to anti-mouse MIP-1β goat IgG polyclonal antibody. Collectively, these results indicated that in vivo blockade of MIP-1β downregulated the MYO3A expression and prevented 4T1 cells from acquiring an invasive potential. To validate these findings, distant tissue metastasis was evaluated as a direct measure of invasive potential. Metastasis occurs in 4T1/BLAB/c mouse model approximately at day 24th post grafting. Therefore, the remaining five mice/group were continued with the treatment upto day 26th. Tumor volume analysis revealed considerable regression following treatment with MIP-1β-neutralizing antibody (Fig. S12). As expected, the mock control BALB/c mice that were not inoculated with 4T1 cells exhbited uniform tissue architecture, while the tumor vehicle control 4T1/BALB/c mouse that were inoculated with 4T1 cells exhibited increased cellular burden in lungs and perivascular areas of liver (Fig. 9C). As compared to vehicle control or isotype control-antibody treated 4T1/BALB/c mouse model, the MIP-1β-neutralizing anibody-treated 4T1/BALB/c mouse model exhibited diminished cellular burden in lungs and liver at day 26th post grafting. Taken together, our in vivo data corroborated the findings of in vitro studies and substantiated the involvement of MIP-1β during cancer cell invasion and metastasis.
Figure 9.

Diminished expression of MMP-9 and MYO3A upon neutralizing antibody-mediated blockade of MIP-1β function was followed by reduced cellular burden in lungs and diminished presence of metastatic foci in liver of syngenic 4T1/BALB/c mouse model of breast cancer. (A and B) Anti-mouse MIP-1β goat IgG polyclonal antibody-treated 4T1 tumors expressed much lower invasive potential as revealed by significantly downregulated mRNA expression levels of MYO3A. The mRNA expression levels of MMP-9 gene were significantly downregulated. Bars represent Quantitative RT-PCR relative fold change expression ±SE (*p <0.05). (C): On day 26th post grafting, compared to controls (PBS or isotype control antibody (IgG)), the intratumoral administration of MIP-1β-neutralizing antibody (MIP-1β NA) resulted in reduced cellular burden in lungs and perivascular regions of liver from 4T1/BALB/c mouse models. Lung and liver sections obtained from healthy uninoculated mice served as mock control. Bars represent no. of metastatic lesions ±SE (*p < 0.05.). All the experiments were done in triplicates. All the experiments were done in triplicates. Abbreviations—4T1 tumor (PBS): 4T1-induced tumor treated with PBS; 4T1 tumor (IgG): 4T1-induced tumor treated with isotype control antibody; 4T1 Tumor (MIP-1β NA): 4T1-induced tumor treated with MIP-1β-neutralizing antibody.
In vivo knockdown of MYO3A expression in 4T1/BALB/c tumors resulted in diminished metastasis to lungs and liver
To further provide direct evidence that MYO3A is critical for distant cancer metastasis, in vivo knockdown of MYO3A expression by Avalanche in vivo transfection reagent was evaluated in 4T1/BALB/c tumor intratumorally, followed by H&E staining to detecting distant metastasis. MYO3A knockdown was ascertained by immunohistochemistry (IHC) and protein gel blot (Fig. 10A and B). As expected, the mock control i.e. BALB/c mice that were not inoculated with 4T1 cells exhbited uniform tissue architecture, while 4T1/BALB/c mouse exhibited increased cellular burden in lungs and perivascular areas of liver (Fig. 10C); as compared to scrambled 4T1/BALB/c mouse, the MYO3A knockdown 4T1/BALB/c mouse model resulted in diminished cellular burden in lungs and liver at day 26th post grafting. Taken together, our in vivo data corroborated the findings of in vitro studies and substantiated the involvement of MYO3A during cancer cell invasion and metastasis.
Figure 10.

In vivo blockade of MYO3A expression was followed by reduced cellular burden in lungs and diminished presence of metastatic foci in liver of syngenic 4T1/BALB/c mouse model of breast cancer. (A and B) Confirmation of MYO3A silencing by IHC and protein gel blot analysis in mice tumor tissue. (C) On day 26th post grafting, compared to controls (Scr), the intratumoral administration of MYO3A shRNA resulted in regressed tumor volume and reduced cellular burden in lungs and perivascular regions of liver from 4T1/BALB/c mouse models. Lung and liver sections obtained from healthy uninoculated mice served as mock control. Bars represent no. of metastatic lesions ±SE (*p < 0.05.). All the experiments were done in triplicates. Abbreviations—4T1 tumor (SCR): 4T1-induced tumor treated with scrambled shRNA; 4T1 tumor (MYO3A): 4T1-induced tumor treated with MYO3A shRNA.
Human breast cancer specimen exhibited a positive correlation between fold change (w.r.t. matched normal tissue) in expression level of MIP-1β and MYO3A and MMP-9
For clinical validation of our findings, the association between MIP-1β and MYO3A was studied in 50 breast cancer patients. (Their demographic, clinicopathological, hormone receptors and fold expression (ΔΔCt) of genes in the selected 50 patients are summarized in Fig. S13. The mean fold expression of MYO3A and MIP-1β showed 19 and 25 times over expression in cancer specimen as compared to matched normal tissue. Furthermore, frequency distribution showed significant associations between the MIP-1β and MYO3A (p < 0.001). The correlation analysis also revealed a significant and direct correlation between MIP-1β and MYO3A (r = 0.90; p < 0.001) (Fig. 11A). Since our in vitro and in vivo findings pointed toward role of MIP-1β and MYO3A in cancer cell invasion, we decided to extend these observations to human breast cancer specimen. Attempt was made to study the correlation of MIP-1β and MYO3A with markers of cancer cell invasion. MMP-9 mRNA expression levels were assessed as an indirect measure of invasive potential and attempt was made to study its association with MIP-1β and MYO3A mRNA expression level. MMP-9 mRNA expression level exhibited 22-fold overexpression in breast cancer specimen as compared to matched normal tissue. Frequency distribution showed significant associations of MMP-9 mRNA expression levels with MIP-1β as well as MYO3A (MIP-1β vs. MMP-9, p = 0.0111; MYO3A vs. MMP-9, p < 0.001) (Fig. S14A). The correlation analysis also revealed a significant and direct association of MMP-9 with these key variables (MIP-1β and MMP-9, r = 0.78, p < 0.001; MYO3A and MMP-9, r = 0.69, p <0.001) (Fig. 11B), thereby substantiating potential involvement in cancer cell invasion.
Figure 11.

Association between fold change (tumor tissue vs. matched normal) in mRNA expression level of MIP-1β, MYO3A, and MMP-9 in human breast cancer specimen. (A and B) Distribution of breast cancer patients (n = `50) with respect to fold change in expression of selected markers and correlation between fold expressions of marker genes of breast cancer patients (n = 50). (C) Association of MYO3A and MIP-1β expression with overall survival. RT-PCR analysis was done in triplicates and repeated at least thrice.
Overall survival
Cancer cell invasion and subsequent distant metastases is the major cause of 90% of breast-cancer-related deaths. Owing to strong correlation of metastasis with diminished survival of breast cancer patients and significant association of MYO3A and MIP-1β with cancer cell invasion emerging in our study, we studied association of these two variables with overall survival. Survival analysis is summarized graphically in Fig. 11C and Fig. S14B. During the 4-y follow-up period, 9 (18.0%) patients died due to disease and 41 (82.0%) were alive (alive = 28, lost to follow-up = 13). The Logrank test showed significant association of MYO3A expression with overall survival. The patient with high expression (ΔΔCt >15) had lower survival as compared to patient with low expression (ΔΔCt ≤ 15) (Logrank test: χ2 = 4.52, p = 0.034; Hazard ratio: ratio = 0.22, 95% CI=0.06 to 0.89). MIP-1β mRNA expression levels did not (p > 0.05) correlate well with survivals, though patients with high expression exhibited poor survival.
Discussion
Macrophages are the most abundant subpopulations of immune cells infiltrating into tumor microenvironment. They exhibit tremendous degree of plasticity. Taking cue from the tissue microenvironment, they undergo phenotype switching to acquire functionally distinct phenotype viz. classically activated M1 polarized or alternatively activated M2 polarized phenotype.30,31 M1 macrophages possess strong antigen-presenting property and affect microbicidal and tumoricidal feedback by (a) generating proinflammatory mediators such as nitric oxide, ROS and (b) inducing Th1 immunity by cytokine production such as IL-12 and IL- 23. M2 macrophages have poor antigen-presenting efficiency. They effectively abolish T cell activation and exhibit less or no cytotoxicity for tumor cells because of their limited capacity to produce NO and proinflammatory cytokines. Rather they exhibit pro-tumor functions such as promoting tumor neo-angiogenesis, metastasis and tissue remodeling.8 Although both the subtypes of macrophages (M1/M2 phenotype) have been observed in tumor microenvironment but the majority of TAMs exhibit M2 specific attributes as directed by tumor microenvironmental cues.32,33 Cancer cells stimulate macrophages to acquire predominantly M2 polarized phenotype (Fig. S16) and derive vital growth support from these pro-tumoral macrophages for surviving in the hostile tumor microenvironment.5,33 While role of M2-polarized macrophages/TAMs in cancer cell invasion and metastasis is established,34 the substratal intercellular communication networks, particularly those accountable for initiation and onset of metastasis, need better characterization. Here, we demonstrate that TAMs-derived MIP-1β not only markedly enhanced the invasive potential of metastatic breast cancer MDA-MB-231 and MDA-MB-468 cells, but the poorly metastatic breast cancer cells MCF-7 were also rendered invasive by MIP-1β. Furthermore, through the microarray and real-time PCR-based mRNA expression analysis, we demonstrated that that MIP-1β-driven cancer cell invasion and metastasis is dependent on MYO3A gene (Fig. 12).
Figure 12.

Schematic representation of the study. Macrophages-derived cytokine MIP-1β enhances the expression of MYO3A in cancer cells that markedly increase ECM degradation and invasive potential of breast cancer cells. These cascade of events culminate into distant metastasis (Fig. 12).
In view of an emerging crucial role of invadopodia in cancer cell invasion and onset of metastasis,35 we set out to determine if TAMs may promote invadopodia formation by cancer cells. Using ECM degradation assay in conjunction with cortactin localization assay, we successfully demonstrate enhanced formation of functionally intact invadopodia by metastatic breast cancer MDA-MB-231 and MDA-MB-468 cells. A follow-up on invasive potential was carried out using matrigel transmigration assay and results validated the findings of ECM degradation assay. Interestingly, while metastatic breast cancer cells MDA-MB-231 and MDA-MB-468 exhibited enhanced invasive potential, the non-metastatic breast cancer MCF-7 cells were also rendered invasive by the presence of macrophages, thereby implicating macrophages in earlier stages of metastatic transformation.
Having established that proximity to macrophages not only increases invasive potential of breast cancer cells, but also imparts an invasive phenotype to otherwise poorly metastatic breast cancer cells, we next set out to decipher the intercellular mediators of TAMs-assisted cancer cell invasion and dissemination. Collective analysis of the cytokine profile of respective culture supernatants in conjunction with mRNA expression levels of selected cytokines in monocultured vs. co-cultured cancer cells and macrophages pointed toward MIP-1β as one of the key mediators of macrophage-assisted cancer cell invasion. Subsequent neutralizing antibody and cognate receptor knockdown studies strengthened our conclusion. Results of ex ovo CAM assay for metastasis further validated the role of MIP-1β in TAMs-assisted metastasis. The physiological relevance of these observations was substantiated by in vivo studies conducted on 4T1/BALB/c mice model of metastasis, wherein functional blockade of MIP-1β by injecting MIP-1β-neutralizing antibody intratumorally not only resulted in tumor regression, but also resulted in diminished intra-tumoral mRNA expression levels of MMP-9, a vital molecular tool for cancer cell invasion indicating that MIP-1β blockade could possibly minimize the in vivo invasive and metastatic potential of cancer cells. Blockade of MIP-1β function resulted in reduced cellular burden within lungs and perivascular regions in liver of BALB/c female mice, thereby substantiating the role of MIP-1β in cancer cell invasion and metastasis. Interestingly, in the human breast cancer specimen, the fold change (tumor vs. matched normal) in mRNA expression level of MIP-1β exhibited a significant and direct correlation with one of the key invasive attribute viz. MMP-9, thereby confirming the clinical relevance of our findings. Although MIP-1β mRNA expression levels did not correlate well with survivals, patients with high expression exhibited poor survival. This could be attributed to the lower sample size. While MIP-1β is known to preferentially recruit T lymphocytes, dendritic cells and NK cells and in its capacity to act as a chemoattractant for cells of immune system, it could possibly elicit an antitumor response,36 its role in cancer as a tumor-promoting agent is not documented so far. In addition, there have been no studies so far implicating MIP-1β in cancer cell invasion, particularly at such an early step as invadopodia formation. Our study for the first time highlights role of MIP-1β as one of the earlier stages of cancer cell invasion and metastasis, particularly as an effector employed by TAMs. This was at variance with studies conducted by Luo et al.,36 wherein they emphasize antitumor property of MIP-1β. One possible explanation for this could be the fact that while their study relies on ectopic expression of MIP-1β with in tumor cells itself, our study is focused on extracellular MIP-1β primarily secreted by macrophages and thus acting via an altogether different mechanism involving cognate receptors.
With an aim to identify molecular determinants of invasion that are intrinsic to cancer cells, we used c-DNA microarray analysis to search for genes whose expression would correlate with cancer cell invasion, particularly in response to signals emanating from macrophages. The analysis identified MYO3A as a possible candidate gene involved in macrophage-assisted cancer cell invasion. Detailed investigations revealed that mRNA expression levels of MYO3A gene were characteristically responsive to MIP-1β function. Similarly, in 4T1/BALB/c mouse model of breast cancer, the neutralizing antibody-mediated functional blockade of MIP-1β resulted in diminished mRNA expression levels of MYO3A in 4T1/BALB/c tumor specimen, thereby corroborating the findings of in vitro studies. Furthermore, quantitative RT-PCR analysis of human breast cancer specimen and matched normal tissue also revealed a significant and direct association between MIP-1β and MYO3A. The characteristic MIP-1β responsive nature of MYO3A gene indicated that it could possibly be involved in macrophage-assisted invadopodia formation and cancer cell invasion. The protein product of MYO3A gene is an unconventional myosin harboring a kinase domain N-terminal to the conserved motor domains. Expression of this gene is highly restricted, with the strongest expression in retina and cochlea.15 While protein encoded by MYO3A plays an important role in hearing in humans, so far there are no reports linking it to breast cancer metastasis. On the contrary, the other members of myosin family are known to play pivotal role during cytoskeletol reorganization, which inturn is an important prerequisite for sucessful formation of functionaly intact invadopodia and commencement of mataststic transformation. For instance, MYO10, an unconventional myosin with MyTH4-FERM domains, is best known for its marked localization to the tips of filopodia and its ability to induce filopodia formation.37 Our in vitro studies for the first time implicate MYO3A in cancer cell invasion and metastasis. In agreement with this, the fold change (tumor vs. matched normal) in mRNA expression level within human breast cancer specimen revealed a significant and direct association of MYO3A with MMP-9, one of the key molecular determinants for cancer cell invasion. Cancer cell invasion and distant metastasis is the leading cause for breast-cancer-related death. In accordance, higher MYO3A mRNA expression levels correlated with diminished survival among patients recruited in our study, thereby substantiating the clinical relevance of our findings and prognostic significance of this gene. What could be the possible mechanism by way of which MYO3A may promote cancer cell invasion? Owing to intrinsic ser/thr kinase activity, it could possibly be involved in phosphorylation of key structural protein instrumental in invadopodia biogenesis and ensuing cancer cell invasion. Considering that expression of MYO3A is highly restricted and its expression in response to stimuli that signals cell invasion, it appears plausible that MYO3A could be a molecular signature of cancer cell invasion and its expression could be a key event marking earlier stages of metastasis. An in-depth investigation in this direction to develop a clearer understanding may have significant implication, particularly during clinical management of breast cancer patients since it may lead to identification of MYO3A as a possible prognostic marker for tracking progression of breast cancer toward metastasis. A better understanding in this context will not only unravel the molecular intricacies underlying TAMs-assisted cancer cell invasion and metastasis, but may possibly also lead to identification of MIP-1β/MYO3A axis as a potential target for devising anti-metastasis strategy.
Material and methods
Antibodies and reagents
Recombinant human MIP-1β (271-BME/CF) and anti-human MIP-1β goat IgG polyclonal antibody (AB-271-NA) and anti-mouse MIP-1β goat IgG polyclonal antibody (AB-451-NA) were from R&D systems (USA). Anti-human CCR4(sc-7936), anti-human CCR-5 (sc-13950), anti-human CD206 (551135), anti-human CD68(sc-20060), anti-human CD86 (MHCD8601) anti-human β-actin (sc-8432), anti-human MYO3A (sc-163105), IgG (sc-2025), antibody and CCR-4 (sc-39886), CCR-5 (sc-35062), directed siRNA and MYO3A-directed shRNA (sc-149758-SH) were from Santacruz (USA). Human cytokine antibody array (V) (AAH-CYT-5) was from Ray Biotech (USA). DAPI (P-36931), Alexa fluor 633 (A20170), Alexa fluor 488 Phalloidin (A12379), Q-tracker 525 cell labeling kit (Q25041MP), lipofectamine (11668-019) and MYO3A (AM16708) directed siRNA were from Invitrogen (USA). Alexa Fluor 555 conjugate of anti-Cortactin (P80/85) clone 4F11 (16–229) was from Millipore (USA). Phorbol 12-myristate 13-acetate (PMA) (524400-5MG) was from Calbiochem (USA). Matrigel™ Basement Membrane Matrix (356234) was from BD Biosciences (Europe). Agilent's Quick-Amp labeling Kit (p/n5190-0442), Qiagen's RNeasy minikit (Cat#74104), Agilent's In situ Hybridization kit (5188–5242), Avalanche – in vivo Transfection Reagent (EZT-VIVO-1).
Cell culture, in vitro differentiation
Human leukemia monocyte THP-1 cells, human mammary cancer-derived cells (MCF-7 cells, MDA-MB-231 and MDA-MB-468 cells) were procured from ATCC. Cells were maintained in RPMI 1640 or DMEM, respectively, using standard mammalian cell culture methods. THP-1 cells were differentiated to macrophages using 30 ηM Phorbol 12-myristate 13-acetate (PMA) according to Daigneault et al.38 Differentiation was ascertained by evaluating the expression/appearance of macrophage specific markers viz. CD16 and myeloid cell leukemia sequence-1(Mcl-1) using RT-PCR (Fig. S15 A and B) and CD68 by flow cytometry (Fig. S15C).
Flow cytometry analysis of M2 phenotype-specific marker was also assessed using anti-Human CD206 FITC conjugated antibody (Fig. S16).
Animals
All procedures with mice were done under an Institutional Animal Care and Use Committee-approved protocol (#IAEC/2013/44). Female BALB/c mice of 8-weeks age were procured from institutional laboratory animal facility. Mice were housed in polypropylene cages in a group of 5/cage. They were maintained on pellet diet, water ad libitum, and regular alternate cycles of 12 h light and darkness. Prior to tumor initiation, animals were acclimatized for 7 d.
ECM degradation assay
2 × 104 cancer cells (MCF-7, MDA-MB-231 and MDA-MB-468) were seeded on sterile 10-mm round glass coverslips coated with Alexa fluor 633 conjugated gelatin according to Artym et al.39 Simultaneously, the 0.4 μm PET hanging transwell cell culture insert housing 2 × 104 THP-1-derived macrophages were introduced into the wells seeded with cancer cells. These co-cultures were incubated for 3 h, 6 h and 24 h under standard cell culture environment. Thereafter, inserts were removed, cancer cells growing on coverslips were fixed using paraformaldehyde/5% sucrose (pH7.2) for 20 min at 37°C, permeabilized with triton-x100 for 5 min and probed for F-actin using Alexa flour 488 conjugated Phalloidin for 30 min at room temperature in dark. Thereafter, cancer cells were mounted in Prolong Gold Antifade Mountant with DAPI (Invitrogen, US) and visualized in LSM510 META confocal laser scanning microscope (Carl Zeiss) with 63X, 1.4 oil-emersion objectives. The extent of ECM degradation was quantified by counting dark areas of degraded fluorescent matrix underneath that cell in three replica sets.
Cortactin localization/expression
2 × 104 cancer cells and 2 ×104 THP-1-derived macrophages were co-cultured for varying time periods (3 h, 6 h and 24 h) as described above. After designated duration, cancer cells were fixed using paraformaldehyde/5% sucrose (pH7.2) for 20 min at 37°C, permeabilized with triton-x 100 for 5 min and probed for cortactin using Alexa fluor 555 conjugated anti-cortactin antibody. F-actin was probed using Alexa flour 488 conjugated Phalloidin for 30 min at room temperature in dark. Thereafter, cancer cells were mounted in Prolong Gold Antifade Mountant with DAPI (Invitrogen, US) and visualized in LSM510 META confocal laser scanning microscope (Carl Zeiss) with 63X, 1.4 oil-emersion objectives.
Cell invasion assay
For assessing invasive potential, the 2 × 104 cancer cells were seeded onto matrigel-coated 8 μm standing PCF transwell cell culture inserts. These inserts were introduced into standard 12-well cell culture plates. Thereafter, 2 × 104 THP-1-derived macrophages housed in 0.4 μm PET hanging cell culture insert were introduced into 8 μm PCF standing cell culture insert harboring cancer cells. The co-cultures were incubated for 3 h, 6 h, and 24 h under standard cell culture environment (5% CO2, 37°C). After 3 h, 6 h and 24 h, the 8 μm PCF inserts harboring cancer cells were collected, the non-invaded cells at the top side were wiped off using sterile cotton swabs. Invaded cells adhered to the underside of matrigel-coated membrane were fixed and mounted in DAPI mounting media (nuclear stain) for qualitative visualization of invasion using Leica DCF450C fluorescence microscope. The quantification was carried out in five different fields of three replica sets.
Chemokines/cytokines profiling of cell culture supernatant
80 different cytokines (Fig. S17) were detected for their presence in culture supernatant of breast cancer cells co-cultured with and without macrophages using antibody-based human cytokine array (Ray Biotech, USA) according to manufacturer's protocol. The results obtained with antibody-based cytokine array were further validated using quantitative RT-PCR assay.
Protein gel blotting
Total proteins were extracted from cells using radio immunoprecipitation assay (RIPA) buffer containing protease and phosphatase inhibitors (1 mM phenylmethylsulfonyl fluoride, 10 mg/mL aprotinin, and 10 mg/mL leupeptin, 10 µM sodium orthovanadate). Protein content in the supernatants was determined using Lowry's Folin method,40 and separated on sodium dodecyl sulfate-12% polyacrylamide gel electrophoresis. Blots were incubated with corresponding primary and HRP-conjugated secondary antibodies. Peroxidase activity was analyzed with the ECL chemiluminescence substrate system (USA).
Microarray analysis
Total RNA and cRNA purification was done using the Qiagen RNeasy kit. The concentration and purity of the RNA extracted were evaluated using the Nanodrop Spectrophotometer (Thermo Scientific; 1000). The integrity of the extracted RNA was analyzed on the Bioanalyzer (Agilent; 2100). The samples for gene expression were labeled using Agilent Quick-Amp labeling Kit. 500 ng each of control and experimental total RNA were reverse transcribed at 40°C using oligo dT primer tagged to a T7 polymerase promoter and converted to double-stranded cDNA. Synthesized double-stranded cDNA were used as template for cRNA generation. cRNA was generated by in vitro transcription and the dye Cy3 CTP(Agilent) was incorporated during this step. Hybridization was done using the Gene Expression Hybridization kit of (Agilent Technologies, In situ Hybridization kit). Data was normalized and analyzed using GeneSpring GX version 11.5 and Microsoft Excel 2007. Detailed experimental analysis is given in Fig. S18.
Real- time PCR
Total RNA was isolated with TRI reagent (Molecular Research Center), and cDNA was obtained from 2 μg of total RNA using High Capacity cDNA Reverse Transcription Kit. Quantitative PCR was performed on a Light Cycler 480 System (Roche) 96-well plates using SYBR Green qPCR Master Mix in accordance with manufacturer's protocol. Data were analyzed using the Roche Light Cycler 480 software (Version 1.5). Cp and Ct were calculated by the Second Derivate Maximum Method. The amount of the target mRNA was examined and normalized to the β-actin gene mRNA. The relative expression ratio of a target gene was calculated as described by Pfaffi.41 Results represent data from three separate experiments. Forward and reverse primer sequence is given in Fig. S19.
Short interfering RNA (siRNA) and transfections
All siRNA experiments were carried out using prevalidated pool of siRNA using Lipofectamine LTX (2000) reagent according to manufacturer's instruction. Sense and antisense sequences of scrambled and targeted siRNAs is presented in Fig. S20.
Chick chorioallantoic membrane assay
To detect in vivo metastasis, chick chorioallantoic membrane (CAM) assay was carried out according to Wilson et al.42 Fertilized chick eggs were purchased from Chak-Gajariya Poultry farm house, Lucknow, India and transported in an insulated carrier to laboratory. Prior to initiation of the experiment, cancer cells were labeled with Q-tracker 525 nanocrystals. Q-tracker cell labeling technique allows for loading the cells grown in culture with highly fluorescent Q-dot nanocrystals, so that these cells may be traced. Once inside the cells, Q-tracker labels provide intense stable fluorescence that can be traced through several generations, and are not transferred to adjacent cells in a population. Optimal number (2 × 106) of MCF-7/MDA-MB-231 cells that were previously labeled with Q-tracker 525 and then pre-incubated with macrophages in absence or presence of anti-human MIP-1β goat IgG polyclonal antibody 5 ug/mL were grafted onto chorioallantoic membrane through a window cut into the eggshell. Thereafter, the egg windows were sealed. Breast cancer cells that were pre-incubated with macrophages in presence of isotype control antibody (5 ug/mL) and monocultured cancer cells treated in a similar manner served as technical control and mock control, respectively. The eggs harboring Q-dot labeled cancer cells were incubated for 3 d. At day 4th post grafting, MIP-1β-neutralizing antibody (5 ug/mL), isotype control antibody (5 ug/mL) and sterile PBS were re-applied locally at the respective graft sites. Thereafter, at day 8th post grafting, chick embryo was harvested by cutting through the eggshell. Vital organs were dissected out and examined under Leica M205FA stereo zoom fluorescence microscope for presence of fluorescent label.
In vivo tumor experiments
In vivo effect of MIP-1β functional neutralization on breast cancer metastasis
4T1/BALB/c tumors were initiated by injecting 1 × 106 viable 4T1 cells subcutaneously in the pre-shaved skin of the back of the BALB/c mice. After two days, the animals were boosted by injecting another 1 × 106 viable 4T1 cells at the initial injection site. The growth of tumor was monitored throughout the experiment with tumor size being measured daily using Vernier calipers and represented in terms of tumor volume [=4/3π × (Long axis/2) × (Short axis/2)]. After the tumor attained a size of 8–10 mm3, the tumor bearing mice were randomized into three groups based on tumor volume with each group comprising of 10 mice/group. After observing tumor volume for 4 d, mice were treated with anti-mouse MIP-1β goat IgG polyclonal antibody and isotype control antibody at tumor site on day 0, 3 and 6 at the dose of 5 ug/mice. An additional group receiving equal amount of vehicle (PBS) served as control. An additional group of five mice each, wherein 4T1 cells were not grafted, was maintained under similar conditions to serve as mock control. At the end of experiment i.e., at day 9th, animals (5 mice/group) were euthanatized under anesthesia and tumor was excised. The remaining 5 animals/group were continued with the treatment upto day 26th for evaluating lung and liver metastasis. After euthanasia, tumor specimen, lungs and liver were collected. The samples were visualized and photographed using Leica M205FA stereo zoom microscope and processed further for PCR and/or H&E staining.
In vivo effect of MYO3A knockdown on breast cancer metastasis
4T1/BALB/c tumors were initiated by injecting 1 × 106 viable 4T1 cells subcutaneously in the pre-shaved skin of the back of the BALB/c mice. After two days, the animals were boosted by injecting another 1 × 106 viable 4T1 cells at the initial injection site. After the tumor attained a size of 8–10 mm3, the tumor-bearing mice were randomized into two groups (5 mice/group). After observing tumor volume for further 4 d, one group of mice was injected intratumorally with MYO3A-directed shRNA suspended in Avalanche in vivo transfection reagent (three times a week at the dose of 10 ug nucleic acid in 2 uL reagent in 50 uL of 5% glucose solution/mice). The other group receiving equal amount of scrambled shRNA served as control. An additional group of five mice each, wherein 4T1 cells were not grafted, was maintained under similar conditions to serve as mock control. At the end of experiment i.e. at day 26, animals (5 mice/group) were euthanatized under anesthesia and tumor was excised and evaluation of lung and liver metastasis was done by H&E and IHC was done to corroborate MYO3A silencing in tumor tissue.
H&E staining and immunohistochemistry
H&E staining was performed on formalin-fixed, paraffin-embedded tissue sections (8 μm). Tissue architecture and metastatic foci were visualized by examining hematoxylin and eosin stained section of each specimen (200x) using Eclipse BOi-0.90 Dry Bright-Field Microscope [Nikon, Japan]/Leica DCF450C bright field/fluorescence microscope. The immunohistochemistry was carried out to determine MYO3A levels in mice tumor specimen. Briefly, the deparaffinised tissue sections were incubated overnight with primary antibody viz. anti-MYO3A got polyclonal antibody (1:100) after heat-induced epitope retrieval (HIER) with citrate buffer (pH 6.0). Thereafter samples were incubated for 1 hour with HRP conjugated anti-got IgG (1:200). Detection was performed with DAB Peroxidase Substrate Kit (Thermo Fischer Scientific, USA) followed by counterstaining with hematoxylin (Fisher Scientific). Sections were mounted in DPX (Sigma) and visualized at 400X magnification using Eclipse BOi-0.90 Dry Bright-Field Microscope (Nikon, Japan).
Samples from human breast cancer and animal study
Human breast tumor specimens were collected after informed consent from 50 patients who underwent surgery for breast cancer at King George Medical University, Lucknow (India). The ethics committee at King George Medical University, Lucknow (India) approved the study protocol (#ECM IIB/P17), which followed the Declaration of Helsinki. Animal studies were approved by CSIR-CDRI Institutional Animal Ethics Committee (IAEC) vide approval number (#IAEC/2013/15).
Statistical analysis
Data were summarized as Mean ± SD. Groups were compared by independent Student's t test, one-way analysis of variance (ANOVA) followed by Newman–Keuls post hoc test, and Pearson correlation analysis where applicable. Discrete groups were compared by chi-square (χ2) test. Survival analysis was done using Kaplan–Meier method and the difference in survivals was compared by Logrank test. A two-tailed p <0.05 was considered statistically significant.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank the Director CSIR-CDRI for infrastructure/facility support, Division of Sophisticated Analytical Instruments Facility (SAIF) for Confocal Microscopy Analysis and Tissue and cell culture unit for providing cell lines. This manuscript has CDRI Communication Number 9265.
Funding
This work was supported by grants from CSIR India (CSIR-EMPOWER: OLP0006) and CSIR fellowship. The animal models employed in the study were developed through CSIR-Network Project-BSC0103.
References
- 1.Bendre M, Gaddy D, Nicholas RW, Suva LJ. Breast cancer metastasis to bone: it is not all about PTHrP. Clin Orthop Relat Res 2003; (415 Suppl):S39-45; PMID:14600591; http://dx.doi.org/ 10.1097/01.blo.0000093844.72468.f4 [DOI] [PubMed] [Google Scholar]
- 2.Eckert MA, Yang J. Targeting invadopodia to block breast cancer metastasis. Oncotarget 2011; 2(7):562-8; PMID:21725138; http://dx.doi.org/ 10.18632/oncotarget.301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Viadana E, Bross ID, Pickren JW. An autopsy study of the metastatic patterns of human leukemias. Oncology 1978; 35(2):87-96; PMID:274679; http://dx.doi.org/ 10.1159/000225262 [DOI] [PubMed] [Google Scholar]
- 4.Shaffrey ME, Mut M, Asher AL, Burri SH, Chahlavi A, Chang SM, Farace E, Fiveash JB, Lang FF, Lopes MB et al.. Brain metastases. Curr Probl Surg 2004; 41(8):665-741; PMID:15354117; http://dx.doi.org/ 10.1067/j.cpsurg.2004.06.001 [DOI] [PubMed] [Google Scholar]
- 5.Sica A, Schioppa T, Mantovani A, Allavena P. Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: potential targets of anti-cancer therapy. Eur J Cancer 2006; 42(6):717-27; PMID:16520032; http://dx.doi.org/ 10.1016/j.ejca.2006.01.003 [DOI] [PubMed] [Google Scholar]
- 6.Mueller MM, Fusenig NE. Friends or foes - bipolar effects of the tumour stroma in cancer. Nat Rev Cancer 2004; 4(11):839-49; PMID:15516957; http://dx.doi.org/ 10.1038/nrc1477 [DOI] [PubMed] [Google Scholar]
- 7.Wang R, Chadalavada K, Wilshire J, Kowalik U, Hovinga KE, Geber A, Fligelman B, Leversha M, Brennan C, Tabar V. Glioblastoma stem-like cells give rise to tumour endothelium. Nature 2010; 468(7325):829-33; PMID:21102433; http://dx.doi.org/ 10.1038/nature09624 [DOI] [PubMed] [Google Scholar]
- 8.Tripathi C, Tewari BN, Kanchan RK, Baghel KS, Nautiyal N, Shrivastava R, Kaur H, Bhatt ML, Bhadauria S. Macrophages are recruited to hypoxic tumor areas and acquire a pro-angiogenic M2-polarized phenotype via hypoxic cancer cell derived cytokines Oncostatin M and Eotaxin. Oncotarget 2014; 5(14):5350-68; PMID:25051364; http://dx.doi.org/ 10.18632/oncotarget.2110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Welford AF, Biziato D, Coffelt SB, Nucera S, Fisher M, Pucci F, Di Serio C, Naldini L, De Palma M, Tozer GM et al.. TIE2-expressing macrophages limit the therapeutic efficacy of the vascular-disrupting agent combretastatin A4 phosphate in mice. J Clin Invest 2011; 121(5):1969-73; PMID:21490397; http://dx.doi.org/ 10.1172/JCI44562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bingle L, Brown NJ, Lewis CE. The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies. J Pathol 2002; 196(3):254-65; PMID:11857487; http://dx.doi.org/ 10.1002/path.1027 [DOI] [PubMed] [Google Scholar]
- 11.Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer 2002; 2(8):563-72; PMID:12154349; http://dx.doi.org/ 10.1038/nrc865 [DOI] [PubMed] [Google Scholar]
- 12.Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer 2009; 9(4):239-52; PMID:19279573; http://dx.doi.org/ 10.1038/nrc2618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med 2013; 19(11):1423-37; PMID:24202395; http://dx.doi.org/ 10.1038/nm.3394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maurer M, von Stebut E. Macrophage inflammatory protein-1. Int J Biochem Cell Biol 2004; 36(10):1882-6; PMID:15203102; http://dx.doi.org/ 10.1016/j.biocel.2003.10.019 [DOI] [PubMed] [Google Scholar]
- 15.Walsh T, Walsh V, Vreugde S, Hertzano R, Shahin H, Haika S, Lee MK, Kanaan M, King MC, Avraham KB. From flies' eyes to our ears: mutations in a human class III myosin cause progressive nonsyndromic hearing loss DFNB30. Proc Natl Acad Sci USA 2002; 99(11):7518-23; PMID:12032315; http://dx.doi.org/ 10.1073/pnas.102091699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pichot CS, Arvanitis C, Hartig SM, Jensen SA, Bechill J, Marzouk S, Yu J, Frost JA, Corey SJ. Cdc42-interacting protein 4 promotes breast cancer cell invasion and formation of invadopodia through activation of N-WASp. Cancer Res 2010; 70(21):8347-56; PMID:20940394; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-4149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Balassarre M, Pompeo A, Beznoussenko G, Castaldi C, Cortellino S, McNiven MA, Luini A, Buccione R. Dynamin participates in focal extracellular matrix degradation by invasive cells. Mol Biol Cell 2003; 14(3):1074-84; PMID:12631724; http://dx.doi.org/ 10.1091/mbc.E02-05-0308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buday L, Downward J. Roles of cortactin in tumor pathogenesis. Biochim Biophys Acta 2007; 1775(2):263-73; PMID:17292556; http://dx.doi.org/ 10.1016/j.bbcan.2006.12.002 [DOI] [PubMed] [Google Scholar]
- 19.Weaver AM. Cortactin in tumor invasiveness. Cancer Lett 2008; 265(2):157-66; PMID:18406052; http://dx.doi.org/ 10.1016/j.canlet.2008.02.066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.MacGrath SM, Koleske AJ. Cortactin in cell migration and cancer at a glance. J Cell Sci 2012; 125(Pt 7):1621-6; PMID:22566665; http://dx.doi.org/ 10.1242/jcs.093781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Artym VV, Zhang Y, Seillier-Moiseiwitsch F, Yamada KM, Mueller SC. Dynamic interactions of cortactin and membrane type 1 matrix metalloproteinase at invadopodia: defining the stages of invadopodia formation and function. Cancer Res 2006; 66(6):3034-43; PMID:16540652; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-2177 [DOI] [PubMed] [Google Scholar]
- 22.Ayala I, Baldassarre M, Giacchetti G, Caldieri G, Tetè S, Luini A, Buccione R. Multiple regulatory inputs converge on cortactin to control invadopodia biogenesis and extracellular matrix degradation. J Cell Sci 2008; 121(Pt 3):369-78; PMID:18198194; http://dx.doi.org/ 10.1242/jcs.008037 [DOI] [PubMed] [Google Scholar]
- 23.Combadiere C, Ahuja SK, Tiffany HL, Murphy PM. Cloning and functional expression of CC CKR5, a human monocyte CC chemokine receptor selective for MIP-1(α), MIP-1(β), and RANTES. J Leukoc Biol 1996; 60(1):147-52; PMID:8699119 [DOI] [PubMed] [Google Scholar]
- 24.Alexander SPH, Mathie A, Peters JA. Chemokine Br J Pharmacol. 2006; S26-S28; http://dx.doi.org/ 10.1038/sj.bjp.0706562 [DOI] [Google Scholar]
- 25.Lewis C, Murdoch C. Macrophage responses to hypoxia: implications for tumor progression and anti-cancer therapies. Am J Pathol 2005; 167(3):627-35; PMID:16127144; http://dx.doi.org/ 10.1016/S0002-9440(10)62038-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sternlicht MD, Werb Z. How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol 2001; 17:463-516; PMID:11687497; http://dx.doi.org/11990853 10.1146/annurev.cellbio.17.1.463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer 2002; 2(3):161-74; PMID:11990853; http://dx.doi.org/ 10.1038/nrc745 [DOI] [PubMed] [Google Scholar]
- 28.Lynch CC, Matrisian LM. Matrix metalloproteinases in tumor-host cell communication. Differentiation 2002; 70(9-10):561-73; PMID:12492497; http://dx.doi.org/ 10.1046/j.1432-0436.2002.700909.x [DOI] [PubMed] [Google Scholar]
- 29.Fingleton B. Matrix metalloproteinases: roles in cancer and metastasis. Front Bio Sci 2006; 11:479-91; PMID:16146745; http://dx.doi.org/ 10.2741/1811 [DOI] [PubMed] [Google Scholar]
- 30.Gordon GJ, Rockwell GN, Jensen RV, Rheinwald JG, Glickman JN, Aronson JP, Pottorf BJ, Nitz MD, Richards WG, Sugarbaker DJ et al.. Identification of novel candidate oncogenes and tumor suppressors in malignant pleural mesothelioma using large-scale transcriptional profiling. Am J Pathol 2005; 166 (6):1827-40; PMID:15920167; http://dx.doi.org/ 10.1016/S0002-9440(10)62492-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol 2004; 25(12):677-86; PMID:15530839; http://dx.doi.org/ 10.1016/j.it.2004.09.015 [DOI] [PubMed] [Google Scholar]
- 32.Yuan A, Hsiao YJ, Chen HY, Chen HW, Ho CC, Chen YY, Liu YC, Hong TH, Yu SL, Chen JJ et al.. Opposite Effects of M1 and M2 Macrophage Subtypes on Lung Cancer Progression. Sci Rep 2015; 5:14273; PMID:26399191; http://dx.doi.org/ 10.1038/srep14273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Biswas SK, Sica A, Lewis CE. Plasticity of macrophage function during tumor progression: regulation by distinct molecular mechanisms. J Immunol 2008; 180(4):2011-7; PMID:18250403; http://dx.doi.org/ 10.4049/jimmunol.180.4.2011 [DOI] [PubMed] [Google Scholar]
- 34.Shih JY, Yuan A, Chen JJW, Yang PC. Tumor-associated macrophage: its role in cancer invasion and metastasis. J Cancer Mol 2006; 2:101-6 [Google Scholar]
- 35.Paz H, Pathak N, Yang J. Invading one step at a time: the role of invadopodia in tumor metastasis. Oncogene 2014; 33:4193-202; PMID:24077283; http://dx.doi.org/ 10.1038/onc.2013.393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luo X, Yu Y, Liang A, Xie Y, Liu S, Guo J, Wang W, Qi R, An H, Zhang M et al.. Intratumoral expression of MIP-1beta induces antitumor responses in a pre-established tumor model through chemoattracting T cells and NK cells. Cell Mol Immunol 2004; 1(3):199-204; PMID:16219168 [PubMed] [Google Scholar]
- 37.Kerber ML, Cheney RE. Myosin-X: a MyTH-FERM myosin at the tips of filopodia. J Cell Sci 2011; 124(Pt 22):3733-41; PMID:22124140; http://dx.doi.org/ 10.1242/jcs.023549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Daigneault M, Preston JA, Marriott HM, Whyte MK, Dockrell DH. The identification of markers of macrophage differentiation in PMA-stimulated THP-1 cells and monocyte-derived macrophages. PLoS One 2010; 5(1):e8668; PMID:20084270; http://dx.doi.org/ 10.1371/journal.pone.0008668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Artym VV, Yamada KM, Mueller SC. ECM degradation assays for analyzing local cell invasion. Methods Mol Biol 2009; 522:211-9; PMID:19247615; http://dx.doi.org/ 10.1007/978-1-59745-413-1_15 [DOI] [PubMed] [Google Scholar]
- 40.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem 1951; 193(1):265-75; PMID:14907713; http://dx.doi.org/11328886 10.1016/0304-3894(92)87011-4 [DOI] [PubMed] [Google Scholar]
- 41.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 2001; 29(9):e45; PMID:11328886; http://dx.doi.org/ 10.1093/nar/29.9.e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wilson SM, Chambers AF. Experimental metastasis assays in the chick embryo. Curr Protoc Cell Biol 2004; Chapter 19: Unit 19.6; PMID:18228449; http://dx.doi.org/ 10.1002/0471143030.cb1906s21 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.