

ABSTRACT
Toll-like receptor 7 (TLR7) agonists are potent immune stimulants able to overcome cancer-associated immune suppression. Due to dose-limiting systemic toxicities, only the topically applied TLR7 agonist (imiquimod) has been approved for therapy of skin tumors. There is a need for TLR7-activating compounds with equivalent efficacy but less toxicity. SC1, a novel small molecule agonist for TLR7, is a potent type-1 interferon inducer, comparable to the reference TLR7 agonist resiquimod, yet with lower induction of proinflammatory cytokines. In vivo, SC1 activates NK cells in a TLR7-dependent manner. Mice bearing the NK cell-sensitive lymphoma RMA-S are cured by repeated s. c. administrations of SC1 as efficiently as by the administration of resiquimod. No relevant toxicities were observed. Mechanistically, SC1 reverses NK cell anergy and restores NK cell-mediated tumor cell killing in an IFN-α-dependent manner. TLR7 targeting by SC1-based compounds may form an attractive strategy to activate NK cell responses for cancer therapy.
KEYWORDS: Cancer, IFN-α, NK cell, RMA-S, TLR7
Introduction
Toll-like receptors (TLR) are pattern-recognition receptors which recognize conserved molecular structures of viruses, bacteria and fungi.1 Upon binding of their respective ligand, TLR initiate an intracellular signaling cascade, which culminates in the rapid induction of an inflammatory cytokine response and in the activation of cells of the innate and adaptive immune system.
TLR7 is located on the endosomal membrane and senses viral single-stranded RNA.2,3 Synthetic ligands for TLR7 including RNA olignucleotides, guanosine analogs and imidazoquinoline compounds such as imiquimod and resiquimod (R848) have been described and used in a number of applications.4,5 Engagement of these agonists to TLR7 leads to the recruitment of MyD88, interferon regulatory factors and NF-κB and thereafter to rapid induction of a type-1 interferon response together with the secretion of proinflammatory cytokines such as IL-1β, IL-6 and IL-12.6
A hallmark of cancer is tumor-associated immunosuppression, defined by an anti-inflammatory cytokine profile and the accumulation of suppressive cells such as regulatory T cells or myeloid-derived suppressor cells.7 TLR7 agonists have the ability to revert this suppressive milieu and to enable tumor cell killing by the innate and adaptive immune system.8 The potency of TLR7 agonists for cancer immunotherapy is illustrated by imiquimod, the only TLR-agonist approved for clinical use. It induces remissions of basal cell carcinoma, of early stage melanoma or even of breast cancer skin metastases when applied topically.9-12 A major limitation of TLR7 agonists applied systemically is side effects such as fever, fatigue and cardiac toxicity. This has prevented their systemic use at an effective dosage.13 Based on local potency, a compound or a formulation of TLR7 agonist that achieves antitumoral efficacy without limiting systemic cytokine release may form a potent immunotherapeutic approach.
NK cells are innate immune effector cells which exhibit target cell killing in an antigen-unspecific manner and contribute to immunosurveillance and tumor control.14,15 NK cel-mediated lysis of tumor cells is driven by stress-induced surface molecules or the lack of MHC class I expression on cancer cells.16 However, a number of cancer types have included NK cell inhibition as a part of their immunsuppressive network.17,18 A number of strategies aim at harnessing NK cells for cancer therapy, including in vivo activation of NK cells with cytokines such as IL-2, adoptive transfer of NK cells and agonistic antibodies to exploit NK cell antitumoral properties.19-23 However, NK cell-based therapeutic protocols are not yet clinically established, as in vivo activation of NK cells by cytokine administration is limited by systemic cytokine toxicity.24 On the other hand, ex vivo generation, expansion and activation of NK cells need to be established experimentally for optimal in vivo efficacy.25,26 An effective use of NK cells for cancer therapy would need an approach not only mediating enhanced tumor cell killing, but also reversing tumor-associated NK cell anergy.
The small molecule TLR7 agonist SC1 has been hypothesized to activate NK cells and to thereby mediate antitumor efficacy in vivo.27 However, SC1s detailed mode of action has remained unaddressed. We have now performed a detailed analysis of the in vivo mode of action of SC1 with focus on NK cells. We demonstrate that SC1 treatment activates NK cells in a TLR7 and IFN-α dependent manner. SC1 thereby reverses NK cell anergy leading to efficient tumor cell lysis. We provide evidence that SC1 has equal therapeutic efficacy as the prototypical TLR7 agonist resiquimod while exhibiting a more favorable cytokine profile. This lays the basis for further therapeutic development.
Material and methods
Mice and cell lines
C57BL/6 mice were purchased from Janvier (St Berthevin, France). TLR7-deficient mice (C57BL/6 background) were provided by S. Akira (Osaka University, Osaka, Japan) and bred in the animal facility of the Ludwig-Maximilians Universität München. Female β2-microglobulin-deficient mice (B6.129P2-B2mtm1Unc/J) were purchased from Jackson (Bar Harbor, USA). Female IFNAR-deficient mice (B6.129S2-Ifnar1tm1Agt/Mmjax) were a kind gift from T. Brocker (Institute of Immunology, Ludwig-Maximilians Universität München, Munich, Germany) and were later purchased at The Mutant Mouse Resource and Research Center (MMRRC) at Jackson Laboratories (Bar Harbor, USA). Mice were 5 to 12 weeks of age at the onset of experiments. All animal experiments were approved by the local regulatory agency (Regierung von Oberbayern, Munich, Germany). The TAP-deficient T cell lymphoma cell line RMA-S was provided by Dr J. Charo (Berlin, Germany) and was cultured in complete RPMI 1640 (Lonza, Basel, Switzerland) supplemented with 10% FBS, 100 µg/mL streptomycin, 1 IU/mL penicillin and 2 mM L-glutamine (Life Technologies, Darmstadt, Germany). The human TLR7 reporter embryonic kidney cell line HEK-293T containing an NFκB luciferase reporter and transfected with TLR7 was kindly provided by Marc Lamphier (Eisai, Inc., Andover, Massachusetts, USA)(28). SC1 (patent WO 09/118296) was provided by 4SC AG (Martinsried, Germany), R848 was purchased from Enzo Life Sciences (Lörrach, Germany).
Luciferase assay
104 HEK-293T TLR7 reporter cells were stimulated with 10 µM R848 or SC1 for 24 h. The TLR4 ligand LPS (Sigma-Aldrich, Saint Louis, USA) and the TLR9 ligand CpG (ODN 1585, Invivogen, Toulouse, France) served as negative controls. After lysis of cells and addition of substrate, luminescence was measured using a Mithras LB 940 microplate reader (Berthold Technologies, Bad Wildbad, Germany). To analyze TLR specificity of SC-1, 2 × 104 HEKTMhTLR7, HEKTMhTLR8, HEKTMhTLR9 or HEKTMNull cells bearing SEAP as reporter gene (Invivogen, Toulouse, France) were stimulated with SC1 and appropriate controls for 16 h. Expression of target gene was assessed using HEK-BlueTM Detection reagent (Invivogen, Toulouse, France) according to manufacturer's instructions.
In vitro activation assays and cytokine analysis
Murine splenocytes were passed through a 40 µm cell strainer and red blood cell lysis was performed using Bio-Plex Cell Lysis Buffer (Bio-Rad, Hercules, USA). 2 × 105 splenocytes per well were cultured in 96-well plates in RPMI 1640 (Lonza, Basel, Switzerland) supplemented with 10% FCS, 100 µg/mL streptomycin, 1 IU/mL penicillin and 2 mM Lglutamine (Life Technologies). Cells were treated with 1 to 10 µM of SC1, vehicle or 1 ng/mL LPS. Cytokine levels were analyzed 36 h after treatment. IL-6 cytokine ELISA (BD Biosciences, Heidelberg, Germany) of culture supernatant was performed according to the manufacturer's protocol. Premixed 25-Plex Cytokine Array (Merck Millipore, Darmstadt, Germany) was performed according to manufacturer's protocols. IFNγ levels in cytotoxicity assay supernatants were measured by IFNγ ELISA (Biolegend, Fell, Germany) according to the manufacturer's protocol.
In vivo activation assays and flow cytometry
For in vivo activation assays, mice were injected with SC1 (2.6 mg/kg or 10 mg/kg), R848 (2 mg/kg) or vehicle solutions s. c. into the flank. For cytokine ELISA, plasma was collected by centrifugation of whole blood at 400 g for 7 min, 1 to 3 h after treatment. Spleens were harvested 1 to 12 h after treatment and were passed through a 40 µm cell strainer. Erythrocytes were removed and remaining lymphocytes were stained for flow cytometry analysis. Therefore, cells were incubated with fluorochrome-linked antibodies and ZombieAquaTM Fixable Viability Dye (Biolegend) for 30 min at 4°C. After incubation, cells were washed and analyzed on a BD FACSCanto II (BD, Heidelberg, Germany). Where indicated, counting beads (CountBright Absolute Counting Beads, Life Technologies) were added for determination of absolute cell numbers. Fluorochrome-conjugated antibodies against CD3 (clone 145-2C11), CD69 (clone H1.2F3), B220 (clone RA3-6B2), NK1.1 (clone PK136), MHCI (clone KH95), NKG2D (clone CX5), Ly49A (clone YE1/48.10.06), DNAM-1 (clone 10E5), CD96 (clone 3.3) and CD107b (clone M3/84) for flow cytometry were purchased from Biolegend.
Cytotoxicity assays
For in vitro cytotoxicity assays, mice were injected s.c. with 10 mg/kg µg SC1, 2 mg/kg R848 or vehicle solution. 12 h after injection, mice were sacrificed and NK cells were isolated from the spleen by negative selection using magnetic-activated cell sorting (Miltenyi Biotech, Bergisch Gladbach, Germany). NK cells were coincubated with 2 × 104 YAC-1 or B16F10 target cells at different ratios for 4.5 or 6 h. For assessment of direct effects of SC1 on RMA-S cells, RMA-S cells were incubated for 4 h with 4 µg/mL SC1 or vehicle solution prior to coculture with NK cells. Cytotoxicity was assessed by LDH release (CytoTox 96® NonRadioactive Cytotoxicity Assay, Promega, Madison, USA) and calculated using the following formula: %cytotoxicity = [(Compound-treated LDH activity – spontaneous LDH activity)/(Maximum LDH activity – spontaneous LDH activity)] × 100.
For in vivo cytotoxicity assays, mice were injected with 10 mg/kg SC1 or vehicle solution s. c. or in the flank or i. v. Splenocytes from β2-microglobulin-deficient mice were stained with 1 µM of Cell Proliferation Dye eFluor 450 (eBioscience, Frankfurt am Main, Germany). Splenocytes from wild-type mice were stained with 0.1 µM of the same dye. Stained splenocytes were mixed at a 1:1 ratio and 107 splenocytes were injected i. v. into mice 2 h after treatment with SC1. Mice were sacrificed 12 h after i. v. injection and Cell Dye eFluor 450 positive cells in the spleens were analyzed by flow cytometry. Specific lysis was calculated as follows: %specific lysis = 100 – [100 × (eFlour 450high/eFluor 450 low)Treated/(eFluor 450high/eFluor 450low)Control].
Mouse tumor models
Mice were injected with 106 RMA-S lymphoma cells subcutaneously into the right flank. Treatment with 10 mg/kg SC1, 2.6 mg/kg SC1 or 2 mg/kg R848 or vehicle solution was carried out s.c. into the left flank on days 0, 5, 10, 15, 20, 25, 30, 35, 40, 45 and 50 after tumor induction. For i. v. treatment, 10 mg/kg SC1 or vehicle were injected i. v. on days 0, 5, 10, 15 and 20 after tumor induction. Tumor size was measured three times a week and mice were assessed for body condition every week. Criteria for early termination of the experiment were weight loss of >10 %, hunched posture with piloerection or dull fur and severely reduced activity. Mice were sacrificed when tumors had reached a size of 225 mm2 or when tumors were seriously ulcerated. For analysis of tumor-infiltrating lymphocytes, tumors were harvested at a size of 100 mm2, tumor tissue was homogenized with a scalpel and incubated with 1 mg/mL collagenase and DNAse 1 (Sigma-Aldrich) at 37°C for 30 min. Tumors were first passed through a 100 µm cell strainer followed by passing through a 40 µm cell strainer. Tumor tissue-derived lymphocytes were enriched using a gradient of 44% and 67% of EasyColl (d = 1.124, Biochrom Merck Millipore, Darmstadt, Germany) before centrifugation at 800 g for 30 min. Enriched lymphocytes were washed and analyzed by flow cytometry.
NK cell activation assays in tumor bearing mice
For analysis of NK cell activation, RMA-S tumor-bearing mice were injected with 10 mg/kg SC1, 2 mg/kg R848 or vehicle solution on days 15, 20 and 25 after tumor induction. Mice were sacrificed on day 26 and single cell suspensions of spleen and lymph nodes were created by passing organs through a 40 µm cell strainer. Wells of flat-bottom high protein-binding plates (Thermo Scientific) were coated with 0.5 µg of NKp46 (clone 29A1.4, Biolegend, Fell, Germany) mAb or rat IgG2a as isotype control. 2.5 × 105 cells per well were incubated for 5 h in the presence of 100 U recombinant human IL-2 (Peprotec, Hamburg, Germany) and CD107b mAb (clone M3/84, AbD Serotec, Martinsried, Germany) in RPMI medium supplemented with 10% FCS, 1% P/S, 1% L-glutamine, 1% NEAA and 1% sodium pyruvate. CD107b expression was analyzed by flow cytometry.
Statistics
Statistics were calculated with GraphPad Prism software 5.0. Differences between experimental conditions were analyzed using the unpaired, two-tailed Student's t test. p values < 0.05 were considered significant. For in vivo tumor models, differences between groups were calculated using two-way ANOVA with correction for multiple testing.
Results
The novel small molecule SC1 is a TLR7-specific agonist and activates NK cells for tumor cell lysis in vitro
In order to determine the specificity of SC1 for TLR7, we used an HEK-293T cell line stably transfected with TLR7 and a reporter plasmid leading to luciferase activity upon activation of the NFκB promoter region through TLR7. SC1 and the TLR7/8-agonist R848 but not LPS (TLR4 ligand) induced luciferase activity (Fig. 1A). Additionally, SC1 did not induce reporter gene expression in cells bearing human TLR8 or TLR9, indicating specificity for TLR7 (data not shown). SC1 dose dependently increased cytokine secretion from wild-type splenocytes (Fig. 1B). Vice versa, when splenocytes from TLR7-KO mice were incubated with SC1, cytokine secretion, as exemplified by IL-6, was abolished (Fig. 1C). This indicates TLR7-specific activity of SC1 in vitro. To assess the impact of SC1 on in vitro NK cell cytotoxicity, we injected mice with 200 µg SC1 or vehicle solution. 12 h after injection, NK cell cytotoxicity to YAC-1 target cells was measured in vitro. SC1-treatment significantly increased NK cell-mediated target cell lysis at all effector-to-target cell ratios tested, indicating potent effector cell activation (Fig. 1D).
Figure 1.
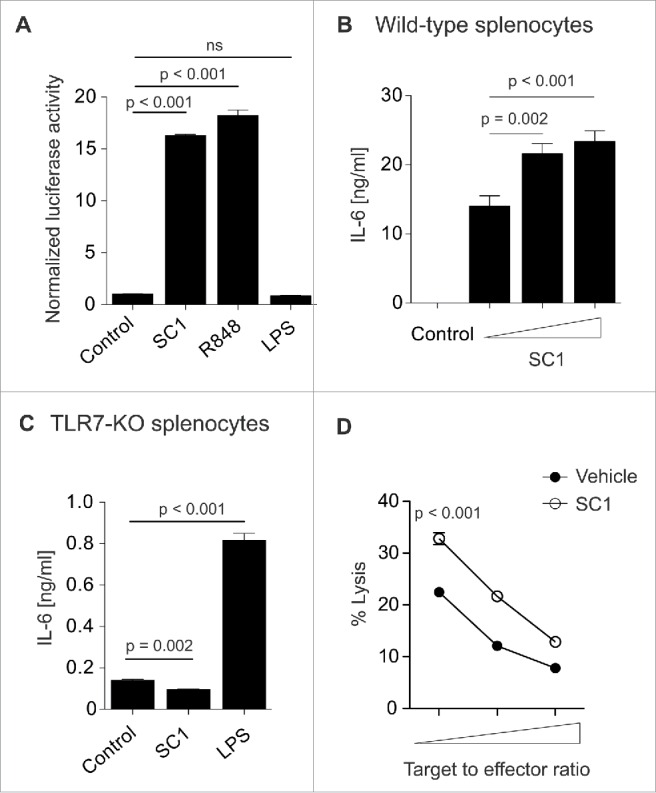
SC1 is a TLR7-specific agonist which enhances NK cell-mediated cytotoxicity in vitro. (A) 104 HEK293T cells, stably transfected with TLR7 and an NFkB reporter plasmid were stimulated over night with 10 µM SC1, R848 or LPS and luciferase activity was measured. (B) Splenocytes from C57Bl/6 mice were stimulated with 1, 5 or 10 µM of SC1 for 36 h. Supernatants were analyzed for IL-6 by ELISA. (C) TLR7-deficient splenocytes were stimulated with 4 µg/mL SC1, 3.1 µg/mL R848 or 1 ng/mL LPS for 36 h. IL-6 in supernatants was detected by ELISA. (D) Mice were treated s. c. with 10 mg/kg SC1 or vehicle solution. 12 h after treatment, NK cells were isolated from spleen and cultured with 2 × 104 YAC-1 target cells at target to effector cell ratios of 1 to 1.25, 1 to 2.5 and 1 to 5. Cytotoxicity was assessed by LDH release assay after 4.5 h of coculture. One representative experiment of three independent experiments is shown. Statistical significance was analyzed by two-tailed unpaired Student's t test. p values < 0.05 were considered significant. Error bars indicate SEM.
SC1 induces type-1 interferon and mediates increased NK cell cytotoxicity in vivo
Systemic treatment with TLR7 agonists results in cytokine induction, leading to dose-limiting systemic adverse effects.13,29 Resiquimod (R848) is a prototypical TLR7 agonist. It has so far failed clinical application in spite of potent antitumoral efficacy in preclinical models.30 We used R848 as a benchmark compound in terms of antitumor efficacy and of systemic cytokine induction. We compared the plasma cytokine profiles 1 to 8 h after s. c. treatment with R848 or SC1. Mice were injected with equimolar amounts of R848 (2 mg/kg) and SC1 (2.6 mg/kg), based on the established therapeutic dosage of R848 in murine tumor therapy.31 Plasma cytokine levels were determined 1, 2, 3, 6 and 12 h after injection. Peak cytokine levels were reached 1 h (IFN-α, IFNγ, IL-1β, IL-6, IL-10, KC, CXCL2, TNF-α), 2 h (G-CSF, GM-CSF, IL-12p40, IL-13, IL-17, CCL3, CCL4, CCL5) or 3 h (IP-10) after injection. Both compounds lead to a rapid systemic cytokine induction, but SC1 consistently induced lower levels of all cytokines as compared to R848 (Fig. 2A). We next injected mice s. c. with 10 mg/kg of SC1, an efficacious dose in antitumoral treatment determined in animal models and compared plasma cytokine levels to injection of a therapeutic dose of R848 (2 mg/kg). Induction of proinflammatory IP-10, TNF- α, CCL4, IL-6 and IL-10 was significantly lower after SC1 treatment compared to R848 treatment, in spite of a 4 times molar excess of SC1 compared to R848. However, IFN-α was the cytokine induced at the highest level by SC1 and did not differ significantly from the levels observed in R848-treated mice (Fig. 2B). To further dissect the functional consequences of the in vivo cytokine induction, we analyzed splenic CD8+ T cells, B and NK cells after SC1 injection. Treatment with SC1 upregulated CD69 on all three cell types (Fig. 2C), indicating early activation of these cells. As a direct consequence of NK cell activation, we could observe a significant increase in NK cell-mediated lysis of β2-microglobulin-deficient splenocytes in SC1-treated mice compared to mice treated with vehicle solution. The observed boost in NK cell cytotoxicity was entirely dependent on the presence of TLR7 in the treated mice, as no selective cytotoxicity was seen in TLR7-KO mice (Fig. 2D). In order to define, if NK cell cytotoxicity upon SC1 treatment was restricted to the rather NK cell-sensitive cell lines YAC-1 and RMA-S, we performed an in vitro cytotoxicity assay on the less immunogenic B16F10 melanoma cell line. We could show that NK cells from mice treated with either SC1 (10 mg/kg) or R848 (2 mg/kg) equally induced specific lysis of B16F10 melanoma cells which was not seen with untreated NK cells (Fig. 2E). Similarly, IFNγ levels measured under the same conditions were equally induced in the SC1 and R848 group (Fig. 2F). These results indicate that NK cell-derived IFNγ induction is comparable in SC1 and R848 treatment, even if systemically IFNγ induction was lower in the SC1 group (Fig. 2A).
Figure 2.

In vivo cytokine profile and immune cell activation after SC1 administration. (A) Mice (n = 4 per group) were injected s.c. with 2 mg/kg R848 or 2.6 mg/kg SC1. Cytokines in blood plasma were measured by ELISA before injection (0), 1, 2, 3 and 8 h after injection. Peak cytokine plasma levels are shown. (B) Mice (n = 3 per group) were injected s.c. with 2 mg/kg R848 or 10 mg/kg SC1. Plasma cytokine levels were analyzed 1, 2 or 3 h after injection by ELISA. Fold induction at the time point of peak cytokine levels is depicted. (C) Mice were injected with 10 mg/kg SC1 s. c. CD69 expression on splenic immune cells was measured by flow cytometry after 6 h and compared to expression in untreated animals. Pooled data from two independent experiments is shown and is representative of a total of five independent experiments. (D) Mice treated s. c. with 10 mg/kg SC1 or vehicle were injected with a 1:1 ratio of wild-type and β2-microglobulin-deficient splenocytes labeled with 0.1 or 1 µM of Cell Dye eFluor450. After 12 h, the ratio of labeled cells in the spleen was determined by flow cytometry. Specific lysis was calculated as follows: specific lysis (%) = 100 − [100 × (β2-microglobulin/wild-type) treated/(β2-microglobulin/wild-type) untreated]. Results of three independent experiments are shown. (E, F) Mice were treated s. c. with 10 mg/kg SC1 or 2 mg/kg R848. 12 h after treatment, NK cells were isolated from spleen and cultured with 2 × 104 B16F10 target cells at target to effector cell ratios of 1 to 1.25, 1 to 5 and 1 to 10. Cytotoxicity was assessed by LDH release assay after 4.5 h of coculture. One representative experiment of two independent experiments is shown (E). IFNγ levels in coculture supernatants were determined by cytokine ELISA (F). Statistical significance was analyzed by two-tailed unpaired Student's t test. p values < 0.05 were considered significant. Error bars indicate SEM.
SC1 cures mice from NK cell-sensitive RMA-S lymphomas in a TLR7- and interferon-α-dependent manner
Efficacy of SC1 in antitumor treatment was assessed in mice bearing s. c. NK cell-sensitive RMA-S lymphomas.32 Mice injected s. c. with 106 RMA-S cells were treated with 10 mg/kg SC1, 2 mg/kg R848 or vehicle solution s. c. every 5th day starting at day 0 (Fig. 3A). SC1 and R848 reduced growth of RMA-S tumors with similar efficacy (Fig. 3B). We could exclude a direct effect of SC1 on RMA-S tumor cells, as pretreatment of tumor cells with SC1 did not alter specific lysis of RMA-S (Fig. S1A). Most mice treated with SC1 or R848 were cured by the treatment (4 out of 6 versus 5 out of 6, respectively). Remarkably, all mice that rejected the tumor upon treatment remained tumor free during at least 150 d, when observation was ended (Fig. 3B). To compare the effect of SC1 in equimolar dosage to R848 (2.6 mg/kg) to the above used four times molar excess (10 mg/kg SC1), we next compared both dosages of SC1. SC1 in the 2.6 mg/kg dosage resulted in a significant reduction of tumor growth, but growth inhibition and cure of mice was less pronounced than in the 10 mg/kg group (Fig. S1B). We thus used the 10 mg/kg dosage for all subsequent tumor experiments. To rule out that the chosen s. c. application route may have deleterious impact on treatment efficacy, we also performed a similar experiment with SC1 (10 mg/kg) using i.v. treatment with the same schedule. SC1 retained similar efficacy against RMA-S lymphoma both in in vivo cytotoxicity assays and in a therapeutic experiment (Fig. S2). We chose to keep the s. c. route for further experiments. Since some TLR7 agonists are known to function through TLR7-independent routes,33 we sought to investigate the role of TLR7 expression on host cells in SC1-based therapy. TLR7-deficient mice bearing RMA-S tumors did not respond to SC1 treatment, supporting the notion that TLR7 is required for SC1-based antitumor efficacy (Fig. 3C). Because type-1 interferon is known to be crucial for the induction of NK cell cytotoxicity upon TLR7 stimulation,5 we hypothesized that IFN-α was the key cytokine for the antitumoral effect of SC1. IFNAR-KO mice bearing RMA-S tumors failed to reject tumors upon SC1 treatment, indicating the dependence on IFN-α effects on host cells (Fig. 3D).
Figure 3.
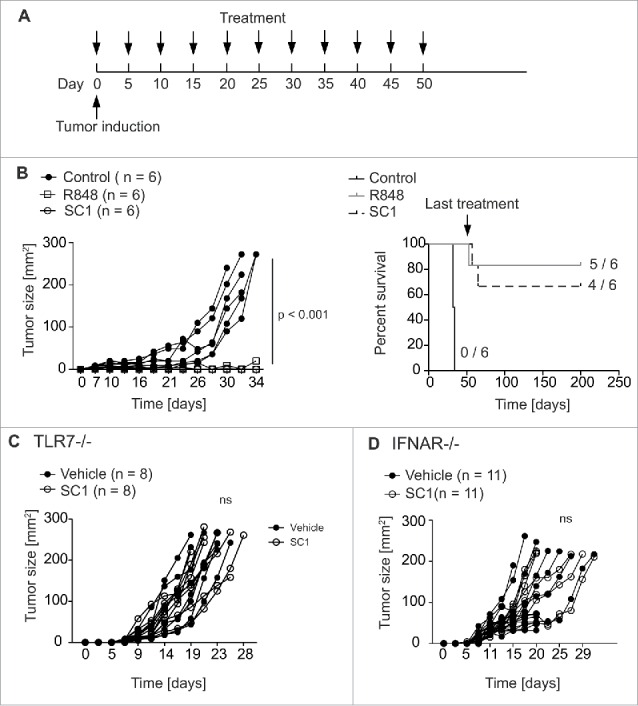
SC1 cures mice bearing RMA-S lymphoma. (A) C57BL/6 mice (n = 6 per group) were injected with 106 RMA-S cells s. c. into the flank. Mice were treated with 2 mg/kg R848, 10 mg/kg SC1 or vehicle s. c. into the contralateral flank (each n = 5) every 5th day beginning on day 0 through day 50. (B) Tumor growth and survival of C57BL/6 wild-type mice bearing s. c. RMA-S tumors. Results are representative of three independent experiments. (C) TLR7-deficient mice (n = 8 per group) were injected with 106 RMA-S cells s. c. in the flank. Contralateral s. c. treatment with 10 mg/kg SC1 or vehicle was administered every 5th day from day 0. Tumor growth over time is shown. (D) IFNAR-deficient mice (n = 11 per group) were injected with 106 RMA-S tumor cells s. c. into the flank. Contralateral s.c. treatment with 10 mg/kg SC1 or vehicle was administered every 5th day from day 0. Tumor growth over time is displayed. C and D show results of two independent experiments. Statistical significance was analyzed by two-way Anova with correction for multiple testing. p values < 0.05 were considered significant.
SC1 induces intratumoral NK cell activation and reverses tumor-induced NK cell anergy
To further explore the mode of action of SC1, we analyzed how NK cells are altered by SC1 to enhance antitumor efficacy. SC1 treatment of RMA-S tumor-bearing mice did not result in an increase of NK cell numbers in tumors or in tumor-draining lymph nodes (TDL) as compared to vehicle solution treated mice (Fig. 4A). In contrast, SC1 treatment significantly induced NK cell activation, as illustrated by enhanced expression of CD69 and NKG2D on NK cells (Fig. 4B,C and Fig. S3). In line with these observations, the NK inhibitory receptors, Ly49A and CD96, were both found to be downregulated on CD69+ activated NK cells after treatment with SC1 (Fig. 4D and E). Similar results were observed for R848 (2 mg/kg) with comparable upregulation of NK cell activation markers in tumors and TDL (Fig. S4A–C), indicating that the observed impact on NK cells is pathway and not compound specific. It has been described that NK cells in a tumor-bearing organism have an exhausted or anergic phenotype, defined by the reduced capacity of intratumoral or TDL NK cells to degranulate upon adequate stimulation, as measured by the surface expression of CD107.17 We investigated whether the ability of NK cells from tumor-bearing mice to degranulate after stimulation with agonistic NKp46 antibodies could be restored by treatment with SC1. As expected, splenic NK cells displayed no anergic phenotype due to the lack of direct tumor cell contact (Fig. 4F). NK cells derived from TDL of RMA-S tumor-bearing mice, however, showed a reduced ability to degranulate upon stimulation, which was restored by treatment with SC1 or R848 (Fig. 4G and Fig. S4D). These results suggest that reversal of anergy by SC1 contributes to the antitumor activity of the compound.
Figure 4.

SC1 activates NK cells and reverts tumor-induced NK cell anergy. (A–E) C57BL/6 mice (n = 7 (vehicle) and n = 8 (SC1) per group) were injected s. c. with 106 RMA-S cells. 10 mg/kg SC1 was injected s. c. in the contralateral flank on days 15, 20 and 25 after tumor induction. Intratumoral and tumor-draining lymph node (TDL) NK cells, defined as MHCI+, Zombie Dye–, CD3–, NK1.1+, were quantified by flow cytometry on day 26. Data is representative of two independent experiments. (F, G) C57BL/6 mice (n = 6 per group) were injected s. c. with 106 RMA-S cells. 10 mg/kg SC1 were injected s. c. in the contralateral flank on days 15, 20 and 25 after tumor induction. NK cells from spleen and TDL were isolated on day 26 and 0.25 × 106 cells were stimulated for 5 h on plates coated with NKp46 antibody. NK cell expression of CD107b was measured by flow cytometry. Results of three independent experiments are shown. Statistical significance was analyzed by two-tailed unpaired Student's t test. p values < 0.05 were considered significant. Error bars indicate SEM.
Discussion
Since the clinical approval of the TLR7 agonist imiquimod for topical treatment of certain cutaneous tumors, a number of clinical trials have investigated TLR7 agonists for local or systemic cancer therapy or as adjuvants in therapeutic vaccination strategies.9 These trials have led to the preliminary conclusion that systemic toxicity limits the use of TLR7 agonists to topical treatment. Other TLR7 agonists such as loxoribin, 852A or TMX-10134-36 have been clinically tested, but showed inadequate efficacy and/or dose-limiting toxicity. It was concluded that clinically feasible systemic TLR7 agonist treatment requires an equally potent immune activation with less systemic cytokine release. IFN-α is the central cytokine responsible for TLR7 agonist antitumor treatment efficacy37 while proinflammatory cytokines, such as IL-1 or IL-6, might predominantly cause their side effects.38 The novel TLR7 agonist SC1 has been designed to more selectively induce IFN-α. Previous work could demonstrate induction of proinflammatory cytokines in human PBMC and the ability to reduce lung metastases in the murine Renca model.27 The mode of action of SC1 and especially the hypothesized more selective IFN-α induction remained unaddressed. Our results corroborate the concept that SC1 activates NFκB signaling and subsequently leads to dose-dependent secretion of IFN-α and of proinflammatory cytokines in immune cells after TLR7 engagement. In vivo, SC1 leads to rapid systemic induction of IFN-α with peak levels 1 h after injection accompanied by early activation of B, T and NK cells as measured by expression of CD69, comparable to the reference compound R848.
We observed similar effects on NK cell activation for i. v. injection compared to s. c. injection in cytotoxicity and in vivo tumor models (Fig. S2), which has already been shown for 852A.39 The s. c. route may be preferable clinically to the i. v. route, in some circumstances, both for safety and applicability purposes, without loss of activity, as suggested for other therapeutic strategies.40 We next compared the plasma cytokine profile induced by SC1 to the cytokine profile of R848. Our results indicate that concentrations of proinflammatory cytokines like IL-1β, IL-6 and TNF-α were significantly lower after treatment with SC1 than after treatment with equimolar amounts of R848. At a dose of SC1 four times higher than the equimolar dose of SC1 to R848, lower levels of IL-6, TNF-α, CCL4 and IP-10 in SC1-treated mice were found. A major obstacle in cancer therapy with systemic administration of TLR7 agonists is the occurrence of dose-limiting systemic adverse effects like fever, fatigue, nausea, dehydration, hepatic and cardiac toxicity—all potentially linked to cytokine release.
The therapeutically critical cytokine after TLR7 engagement is IFN-α.41 Remarkably, SC1 was an equally effective inducer of IFN-α as R848, a finding supported by a comparable therapeutic efficacy in wild-type mice that was absent in IFNAR-KO mice. It has been reported, that IFNγ production by NK cells is critical for efficacy of TLR7/8 agonists.42 In contrast, our in vivo data in IFNAR-deficient mice suggests that systemic type-1 interferon is critical for the antitumoral efficacy of TLR7 agonists. Here, we could, however, demonstrate that NK cell-derived IFNγ levels, but not systemic IFNγ levels, are comparable in R848- and SC1-treated mice, the latter being administered with the four times equimolar doses. A potent systemic IFN-α release may be preferable to induction of other cytokines not only in terms of efficacy, but also of diminished systemic side effects.
The reason of the observed differences in the cytokine profiles induced by SC1 and by R848 are surprising, since both agonists have long been considered to be selective TLR7 agonists in mice.4,27 These differences may be due to discordances in cytokine secretion of TLR7 over TLR8 agonists. Agonists specific for TLR7 preferentially lead to a type-1 interferon response, whereas activation of TLR8 activation culminates in a proinflammatory cytokine response including cytokines like IL-12 and TNF-α.41 SC predominantly activates TLR7 and has no effect on human TLR8 as shown in HEK293 reporter cells,27 whereas R848 is known to bind to both TLR7 and TLR8. The functionality of TLR8 in mice is controversial. Mouse TLR8 is thought to be non-functional due to the lack of a five amino acid motif and missing recognition of ssRNA and other small molecule TLR8 agonists.2,4,43 In contrast, MyD88-dependent cytokine response to TLR8 agonists in murine immune cells has been reported upon addition of polyT ODN to a TLR8 agonist, suggesting at least partial functionality of mouse TLR8.44 In our hands, cytokine induction, NK cell activation as well as antitumoral efficacy of SC1 were completely dependent on TLR7, as they were absent in TLR7-deficient mice. It is thus likely that SC1 does not exert any TLR7-independent effects.
We have shown in previous studies that TLR7 agonist treatment in RMA-S tumor-bearing mice leads to an NK cell-mediated antitumor immune response but the mechanism of increased NK cell killing upon TLR7 activation was not studied.5 NK cells are equipped with a large repertoire of inhibiting and activating receptors. Inhibitory receptors of the Ly49 family bind to the MHC-I complex and inhibit NK cell cytotoxicity upon engagement. This mechanism enables self-tolerance as well as lysis of MHC-I-negative cancer cells.45 Another recently described inhibitory receptor on NK cells is CD96, which binds to CD155, a molecule expressed on dendritic cells and various epithelial cancer cells.46 CD96 seems to be instrumental in cancer cell escape from NK cell killing, as CD96 neutralization mediates cancer regression.46 NKG2D, in contrast, is a NK cell-activating receptor, which recognizes stress-induced ligands on tumor cells and virus-infected cells.47 For efficient target cell killing, a combination of several of these receptors is required.48 Although increased NK cell-mediated tumor cell lysis upon TLR7 agonist treatment has been previously described,27,49 little is known on the effects of TLR7 agonists on NK cell receptor expression and phenotype. In the present study, we could show that SC1 efficiently controls tumor burden of s.c. RMA-S tumors and induces phenotypic changes on NK cells. SC1 also leads to an increased in vitro and in vivo NK cell cytotoxicity toward YAC-1 cells and β2-microglobulin-deficient splenocytes. YAC-1 cell lysis by NK cells is dependent on expression of NKG2D ligands, whereas killing of β2-microglobulin-deficient splenocytes is triggered by lack of MHC-I expression on these cells.50 These results suggest that SC1 leads to a broad activation of NK cells, which is confirmed by our analysis of intratumoral and tumor-draining lymph node NK cells. NK cells did not accumulate in tumor and tumor-draining lymph node of SC1-treated mice, but showed an activated phenotype characterized by the upregulation of activating receptors NKG2D and CD69 and downregulation of inhibitory receptors Ly49A and CD96. Recently, it could be shown that direct contact of NK cells to RMA-S cells induces NK cell anergy.51 In addition, proliferation of NK cells in the presence of tumor cells contributes to NK cell exhaustion.18 We could demonstrate that SC1 treatment reverses NK cell anergy in RMA-S tumor-bearing mice. Treatment with SC1 also upregulated NKG2D expression in NK cells from tumor-draining lymph node, further underlining reversal of NK cell exhaustion.
In summary, we identified SC1 as a small molecule TLR7 agonist with potent immunostimulatory and especially NK cell-activating properties. We describe for the first time a mechanism for TLR7-mediated NK cell activation and the associated phenotypic changes. This may form the basis for the development of TLR7 agonists to harness the therapeutic potential of NK cells in cancer therapy. Because of potent antitumor efficacy and lower inflammatory cytokine induction, SC1 or related compounds may be promising candidates for systemic therapy of NK cell sensitive tumors.
Supplementary Material
Disclosure of potential conflicts of interest
The authors from CIPSM and Division of Clinical Pharmacology have no conflict of interest to declare. The authors SH, SS and RB are employees of 4SC Discovery GmbH and 4SC AG and may have financial interest in the development of novel TLR7/8 agonists for cancer therapies.
Funding
This study was supported by grants from the BMBF-funded leading edge Cluster M4 (to SS, SE and SK), the Wilhelm Sander Stiftung (grant number 2014.018.1 to SE and SK), the international doctoral program “i-Target: Immunotargeting of cancer” funded by the Elite Network of Bavaria (to SK and SE), the Melanoma Research Alliance (grant number N269626 to SE and 409510 to SK), the Graduiertenkolleg 1202 “Oligonucleotides in cell biology and therapy” funded by the Deutsche Forschungsgemeinschaft (to SK, SE and SR), the Marie-Sklodowska-Curie “Training Network for the Immunotherapy of Cancer (IMMUTRAIN)” funded by the H2020 program of the European Union (to SE and SK), the Else Kröner-Fresenius-Stiftung (to SK), the German Cancer Aid (to SK), the CiPSM Women gender funding (Excellence Cluster 114/2, to GMW) and by LMU Munich's Institutional Strategy LMU excellent within the framework of the German Excellence Initiative (to SE and SK).
References
- 1.Uematsu S, Akira S. Toll-like receptors and innate immunity. J Mol Med (Berl) 2006; 84:712-25; PMID:16924467; http://dx.doi.org/ 10.1007/s00109-006-0084-y [DOI] [PubMed] [Google Scholar]
- 2.Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, Lipford G, Wagner H, Bauer S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 2004; 303:1526-9; PMID:14976262; http://dx.doi.org/ 10.1126/science.1093620 [DOI] [PubMed] [Google Scholar]
- 3.Hornung V, Guenthner-Biller M, Bourquin C, Ablasser A, Schlee M, Uematsu S, Noronha A, Manoharan M, Akira S, de Fougerolles A et al.. Sequence-specific potent induction of IFN-alpha by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat Med 2005; 11:263-70; PMID:15723075; http://dx.doi.org/ 10.1038/nm1191 [DOI] [PubMed] [Google Scholar]
- 4.Hemmi H, Kaisho T, Takeuchi O, Sato S, Sanjo H, Hoshino K, Horiuchi T, Tomizawa H, Takeda K, Akira S. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat Immunol 2002; 3:196-200; PMID:11812998; http://dx.doi.org/ 10.1038/ni758 [DOI] [PubMed] [Google Scholar]
- 5.Bourquin C, Schmidt L, Lanz AL, Storch B, Wurzenberger C, Anz D, Sandholzer N, Mocikat R, Berger M, Poeck H et al.. Immunostimulatory RNA oligonucleotides induce an effective antitumoral NK cell response through the TLR7. J Immunol 2009; 183:6078-86; PMID:19890064; http://dx.doi.org/ 10.4049/jimmunol.0901594 [DOI] [PubMed] [Google Scholar]
- 6.Gilliet M, Cao W, Liu YJ. Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nat Rev Immunol 2008; 8:594-606; PMID:18641647; http://dx.doi.org/ 10.1038/nri2358 [DOI] [PubMed] [Google Scholar]
- 7.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science 2011; 331:1565-70; PMID:21436444; http://dx.doi.org/ 10.1126/science.1203486 [DOI] [PubMed] [Google Scholar]
- 8.Kobold S, Wiedemann G, Rothenfusser S, Endres S. Modes of action of TLR7 agonists in cancer therapy. Immunotherapy 2014; 6:1085-95; PMID:25428647; http://dx.doi.org/ 10.2217/imt.14.75 [DOI] [PubMed] [Google Scholar]
- 9.Vacchelli E, Eggermont A, Sautes-Fridman C, Galon J, Zitvogel L, Kroemer G, Galluzzi L. Trial Watch: Toll-like receptor agonists for cancer therapy. Oncoimmunology 2013; 2:e25238; PMID:24083080; http://dx.doi.org/ 10.4161/onci.25238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Adams S, Kozhaya L, Martiniuk F, Meng TC, Chiriboga L, Liebes L, Hochman T, Shuman N, Axelrod D, Speyer J et al.. Topical TLR7 agonist imiquimod can induce immune-mediated rejection of skin metastases in patients with breast cancer. Clin Cancer Res 2012; 18:6748-57; PMID:22767669; http://dx.doi.org/ 10.1158/1078-0432.CCR-12-1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Naylor MF, Crowson N, Kuwahara R, Teague K, Garcia C, Mackinnis C, Haque R, Odom C, Jankey C, Cornelison RL et al.. Treatment of lentigo maligna with topical imiquimod. Br J Dermatol 2003; 149(Suppl 66):66-70; PMID:14616356; http://dx.doi.org/ 10.1046/j.0366-077X.2003.05637.x [DOI] [PubMed] [Google Scholar]
- 12.Schulze HJ, Cribier B, Requena L, Reifenberger J, Ferrandiz C, Garcia Diez A, Tebbs V, McRae S. Imiquimod 5% cream for the treatment of superficial basal cell carcinoma: results from a randomized vehicle-controlled phase III study in Europe. Br J Dermatol 2005; 152:939-47; PMID:15888150; http://dx.doi.org/ 10.1111/j.1365-2133.2005.06486.x [DOI] [PubMed] [Google Scholar]
- 13.Holldack J. Toll-like receptors as therapeutic targets for cancer. Drug Discov Today 2014; 19:379-82; PMID:24012797; http://dx.doi.org/ 10.1016/j.drudis.2013.08.020 [DOI] [PubMed] [Google Scholar]
- 14.Lanier LL. NK cell recognition. Annu Rev Immunol 2005; 23:225-274; PMID:15771571; http://dx.doi.org/ 10.1146/annurev.immunol.23.021704.115526 [DOI] [PubMed] [Google Scholar]
- 15.Moretta L, Pietra G, Montaldo E, Vacca P, Pende D, Falco M, Del Zotto G, Locatelli F, Moretta A, Mingari MC. Human NK cells: from surface receptors to the therapy of leukemias and solid tumors. Front Immunol 2014; 5:87; PMID:24639677; http://dx.doi.org/ 10.3389/fimmu.2014.00087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Long EO, Kim HS, Liu D, Peterson ME, Rajagopalan S. Controlling natural killer cell responses: integration of signals for activation and inhibition. Annu Rev Immunol 2013; 31:227-58; PMID:23516982; http://dx.doi.org/ 10.1146/annurev-immunol-020711-075005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Da Silva IP, Gallois A, Jimenez-Baranda S, Khan S, Anderson AC, Kuchroo VK, Osman I, Bhardwaj N. Reversal of NK-cell exhaustion in advanced melanoma by Tim-3 blockade. Cancer Immunol Res 2014; 2:410-22; PMID:24795354; http://dx.doi.org/ 10.1158/2326-6066.CIR-13-0171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gill S, Vasey AE, De Souza A, Baker J, Smith AT, Kohrt HE, Florek M, Gibbs KD Jr, Tate K, Ritchie DS et al.. Rapid development of exhaustion and down-regulation of eomesodermin limit the antitumor activity of adoptively transferred murine natural killer cells. Blood 2012; 119:5758-68; PMID:22544698; http://dx.doi.org/ 10.1182/blood-2012-03-415364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smyth MJ, Cretney E, Kershaw MH, Hayakawa Y. Cytokines in cancer immunity and immunotherapy. Immunol Rev 2004; 202:275-293; PMID:15546400; http://dx.doi.org/ 10.1111/j.0105-2896.2004.00199.x [DOI] [PubMed] [Google Scholar]
- 20.Krause SW, Gastpar R, Andreesen R, Gross C, Ullrich H, Thonigs G, Pfister K, Multhoff G. Treatment of colon and lung cancer patients with ex vivo heat shock protein 70-peptide-activated, autologous natural killer cells: a clinical phase i trial. Clin Cancer Res 2004; 10:3699-707; PMID:15173076; http://dx.doi.org/ 10.1158/1078-0432.CCR-03-0683 [DOI] [PubMed] [Google Scholar]
- 21.Miller JS, Tessmer-Tuck J, Pierson BA, Weisdorf D, McGlave P, Blazar BR, Katsanis E, Verfaillie C, Lebkowski J, Radford J Jr et al.. Low dose subcutaneous interleukin-2 after autologous transplantation generates sustained in vivo natural killer cell activity. Biol Blood Marrow Transplant 1997; 3:34-44; PMID:9209739 [PubMed] [Google Scholar]
- 22.Rosenberg SA, Mule JJ, Spiess PJ, Reichert CM, Schwarz SL. Regression of established pulmonary metastases and subcutaneous tumor mediated by the systemic administration of high-dose recombinant interleukin 2. J Exp Med 1985; 161:1169-88; PMID:3886826; http://dx.doi.org/ 10.1084/jem.161.5.1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tarek N, Le Luduec JB, Gallagher MM, Zheng J, Venstrom JM, Chamberlain E, Modak S, Heller G, Dupont B, Cheung NK et al.. Unlicensed NK cells target neuroblastoma following anti-GD2 antibody treatment. J Clin Invest 2012; 122:3260-70; PMID:22863621; http://dx.doi.org/ 10.1172/JCI62749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ma C, Armstrong AW. Severe adverse events from the treatment of advanced melanoma: a systematic review of severe side effects associated with ipilimumab, vemurafenib, interferon alfa-2b, dacarbazine and interleukin-2. J Dermatolog Treat 2014; 25:401-8; PMID:23763243; http://dx.doi.org/ 10.3109/09546634.2013.813897 [DOI] [PubMed] [Google Scholar]
- 25.Geller MA, Cooley S, Judson PL, Ghebre R, Carson LF, Argenta PA, Jonson AL, Panoskaltsis-Mortari A, Curtsinger J, McKenna D et al.. A phase II study of allogeneic natural killer cell therapy to treat patients with recurrent ovarian and breast cancer. Cytotherapy 2011; 13:98-107; PMID:20849361; http://dx.doi.org/ 10.3109/14653249.2010.515582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parkhurst MR, Riley JP, Dudley ME, Rosenberg SA. Adoptive transfer of autologous natural killer cells leads to high levels of circulating natural killer cells but does not mediate tumor regression. Clin Cancer Res 2011; 17:6287-97; PMID:21844012; http://dx.doi.org/ 10.1158/1078-0432.CCR-11-1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hamm S, Rath S, Michel S, Baumgartner R. Cancer immunotherapeutic potential of novel small molecule TLR7 and TLR8 agonists. J Immunotoxicol 2009; 6:257-65; PMID:19848448; http://dx.doi.org/ 10.3109/15476910903286733 [DOI] [PubMed] [Google Scholar]
- 28.Lamphier M, Zheng W, Latz E, Spyvee M, Hansen H, Rose J, Genest M, Yang H, Shaffer C, Zhao Y et al.. Novel small molecule inhibitors of TLR7 and TLR9: mechanism of action and efficacy in vivo. Mol Pharmacol 2014; 85:429-40; PMID:24342772; http://dx.doi.org/ 10.1124/mol.113.089821 [DOI] [PubMed] [Google Scholar]
- 29.Dummer R, Hauschild A, Becker JC, Grob JJ, Schadendorf D, Tebbs V, Skalsky J, Kaehler KC, Moosbauer S, Clark R et al.. An exploratory study of systemic administration of the toll-like receptor-7 agonist 852A in patients with refractory metastatic melanoma. Clin Cancer Res 2008; 14:856-64; PMID:18245549; http://dx.doi.org/ 10.1158/1078-0432.CCR-07-1938 [DOI] [PubMed] [Google Scholar]
- 30.Dovedi SJ, Melis MH, Wilkinson RW, Adlard AL, Stratford IJ, Honeychurch J, Illidge TM. Systemic delivery of a TLR7 agonist in combination with radiation primes durable antitumor immune responses in mouse models of lymphoma. Blood 2013; 121:251-9; PMID:23086756; http://dx.doi.org/ 10.1182/blood-2012-05-432393 [DOI] [PubMed] [Google Scholar]
- 31.Ignatz-Hoover JJ, Wang H, Moreton SA, Chakrabarti A, Agarwal MK, Sun K, Gupta K, Wald DN. The role of TLR8 signaling in acute myeloid leukemia differentiation. Leukemia 2015; 29:918-26; PMID:25283842; http://dx.doi.org/ 10.1038/leu.2014.293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karre K, Ljunggren HG, Piontek G, Kiessling R. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature 1986; 319:675-8; PMID:3951539; http://dx.doi.org/ 10.1038/319675a0 [DOI] [PubMed] [Google Scholar]
- 33.Kanneganti TD, Ozoren N, Body-Malapel M, Amer A, Park JH, Franchi L, Whitfield J, Barchet W, Colonna M, Vandenabeele P et al.. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature 2006; 440:233-6; PMID:16407888; http://dx.doi.org/ 10.1038/nature04517 [DOI] [PubMed] [Google Scholar]
- 34.Agarwala SS, Kirkwood JM, Bryant J. Phase 1, randomized, double-blind trial of 7-allyl-8-oxoguanosine (loxoribine) in advanced cancer. Cytokines Cell Mol Ther 2000; 6:171-6; PMID:11565955; http://dx.doi.org/ 10.1080/mccm.6.4.171.176 [DOI] [PubMed] [Google Scholar]
- 35.Falke J, Lammers RJ, Arentsen HC, Ravic M, Pozzi R, Cornel EB, Vergunst H, de Reijke TM, Witjes JA. Results of a phase 1 dose escalation study of intravesical TMX-101 in patients with nonmuscle invasive bladder cancer. J Urol 2013; 189:2077-82; PMID:23206424; http://dx.doi.org/ 10.1016/j.juro.2012.11.150 [DOI] [PubMed] [Google Scholar]
- 36.Weigel BJ, Cooley S, DeFor T, Weisdorf DJ, Panoskaltsis-Mortari A, Chen W, Blazar BR, Miller JS. Prolonged subcutaneous administration of 852A, a novel systemic toll-like receptor 7 agonist, to activate innate immune responses in patients with advanced hematologic malignancies. Am J Hematol 2012; 87:953-6; PMID:22718533; http://dx.doi.org/ 10.1002/ajh.23280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Inglefield JR, Dumitru CD, Alkan SS, Gibson SJ, Lipson KE, Tomai MA, Larson CJ, Vasilakos JP. TLR7 agonist 852A inhibition of tumor cell proliferation is dependent on plasmacytoid dendritic cells and type I IFN. J Interferon Cytokine Res 2008; 28:253-63; PMID:18439103; http://dx.doi.org/ 10.1089/jir.2007.0097 [DOI] [PubMed] [Google Scholar]
- 38.Christian LM, Porter K, Karlsson E, Schultz-Cherry S. Proinflammatory cytokine responses correspond with subjective side effects after influenza virus vaccination. Vaccine 2015; 33(29):3360-6; PMID:26027906; http://dx.doi.org/ 10.1016/j.vaccine.2015.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harrison LI, Astry C, Kumar S, Yunis C. Pharmacokinetics of 852A, an imidazoquinoline Toll-like receptor 7-specific agonist, following intravenous, subcutaneous, and oral administrations in humans. J Clin Pharmacol 2007; 47:962-9; PMID:17660481; http://dx.doi.org/ 10.1177/0091270007303766 [DOI] [PubMed] [Google Scholar]
- 40.Davies A, Merli F, Mihaljevic B, Siritanaratkul N, Solal-Celigny P, Barrett M, Berge C, Bittner B, Boehnke A, McIntyre C et al.. Pharmacokinetics and safety of subcutaneous rituximab in follicular lymphoma (SABRINA): stage 1 analysis of a randomised phase 3 study. Lancet Oncol 2014; 15:343-52; PMID:24521993; http://dx.doi.org/ 10.1016/S1470-2045(14)70005-1 [DOI] [PubMed] [Google Scholar]
- 41.Gorden KB, Gorski KS, Gibson SJ, Kedl RM, Kieper WC, Qiu X, Tomai MA, Alkan SS, Vasilakos JP. Synthetic TLR agonists reveal functional differences between human TLR7 and TLR8. J Immunol 2005; 174:1259-68; PMID:15661881; http://dx.doi.org/ 10.4049/jimmunol.174.3.1259 [DOI] [PubMed] [Google Scholar]
- 42.Hart OM, Athie-Morales V, O'Connor GM, Gardiner CM. TLR7/8-mediated activation of human NK cells results in accessory cell-dependent IFN-gamma production. J Immunol 2005; 175:1636-42; PMID:16034103; http://dx.doi.org/ 10.4049/jimmunol.175.3.1636 [DOI] [PubMed] [Google Scholar]
- 43.Liu J, Xu C, Hsu LC, Luo Y, Xiang R, Chuang TH. A five-amino-acid motif in the undefined region of the TLR8 ectodomain is required for species-specific ligand recognition. Mol Immunol 2010; 47:1083-90; PMID:20004021; http://dx.doi.org/ 10.1016/j.molimm.2009.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gorden KK, Qiu XX, Binsfeld CC, Vasilakos JP, Alkan SS. Cutting edge: activation of murine TLR8 by a combination of imidazoquinoline immune response modifiers and polyT oligodeoxynucleotides. J Immunol 2006; 177:6584-87; PMID:17082568; http://dx.doi.org/ 10.4049/jimmunol.177.10.6584 [DOI] [PubMed] [Google Scholar]
- 45.Belanger S, Tu MM, Rahim MM, Mahmoud AB, Patel R, Tai LH, Troke AD, Wilhelm BT, Landry JR, Zhu Q et al.. Impaired natural killer cell self-education and “missing-self” responses in Ly49-deficient mice. Blood 2012; 120:592-602; PMID:22661698; http://dx.doi.org/ 10.1182/blood-2012-02-408732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chan CJ, Martinet L, Gilfillan S, Souza-Fonseca-Guimaraes F, Chow MT, Town L, Ritchie DS, Colonna M, Andrews DM, Smyth MJ. The receptors CD96 and CD226 oppose each other in the regulation of natural killer cell functions. Nat Immunol 2014; 15:431-8; PMID:24658051; http://dx.doi.org/ 10.1038/ni.2850 [DOI] [PubMed] [Google Scholar]
- 47.Hayakawa Y, Smyth MJ. NKG2D and cytotoxic effector function in tumor immune surveillance. Semin Immunol 2006; 18:176-85; PMID:16675266; http://dx.doi.org/ 10.1016/j.smim.2006.03.005 [DOI] [PubMed] [Google Scholar]
- 48.Bryceson YT, March ME, Ljunggren HG, Long EO. Activation, coactivation, and costimulation of resting human natural killer cells. Immunol Rev 2006; 214:73-91; PMID:17100877; http://dx.doi.org/ 10.1111/j.1600-065X.2006.00457.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bourquin C, Hotz C, Noerenberg D, Voelkl A, Heidegger S, Roetzer LC, Storch B, Sandholzer N, Wurzenberger C, Anz D et al.. Systemic cancer therapy with a small molecule agonist of toll-like receptor 7 can be improved by circumventing TLR tolerance. Cancer Res 2011; 71:5123-33; PMID:21697281; http://dx.doi.org/ 10.1158/0008-5472.CAN-10-3903 [DOI] [PubMed] [Google Scholar]
- 50.Ogawa T, Tsuji-Kawahara S, Yuasa T, Kinoshita S, Chikaishi T, Takamura S, Matsumura H, Seya T, Saga T, Miyazawa M. Natural killer cells recognize friend retrovirus-infected erythroid progenitor cells through NKG2D-RAE-1 interactions In Vivo. J Virol 2011; 85:5423-35; PMID:21411527; http://dx.doi.org/ 10.1128/JVI.02146-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ardolino M, Azimi CS, Iannello A, Trevino TN, Horan L, Zhang L, Deng W, Ring AM, Fischer S, Garcia KC et al.. Cytokine therapy reverses NK cell anergy in MHC-deficient tumors. J Clin Invest 2014; 124:4781-94; PMID:25329698; http://dx.doi.org/ 10.1172/JCI74337 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.