

ABSTRACT
Despite the recent approval of new agents for metastatic melanoma, its treatment remains challenging. Moreover, few available immunotherapies induce a strong cellular immune response, and selection of the correct immunoadjuvant is crucial for overcoming this obstacle. Here, we studied the immunomodulatory properties of arazyme, a bacterial metalloprotease, which was previously shown to control metastasis in a murine melanoma B16F10-Nex2 model. The antitumor activity of arazyme was independent of its proteolytic activity, since heat-inactivated protease showed comparable properties to the active enzyme; however, the effect was dependent on an intact immune system, as antitumor properties were lost in immunodeficient mice. The protective response was IFNγ-dependent, and CD8+ T lymphocytes were the main effector antitumor population, although B and CD4+ T lymphocytes were also induced. Macrophages and dendritic cells were involved in the induction of the antitumor response, as arazyme activation of these cells increased both the expression of surface activation markers and proinflammatory cytokine secretion through TLR4-MyD88-TRIF-dependent, but also MAPK-dependent pathways. Arazyme was also effective in the murine breast adenocarcinoma 4T1 model, reducing primary and metastatic tumor development, and prolonging survival. To our knowledge, this is the first report of a bacterial metalloprotease interaction with TLR4 and subsequent receptor activation that promotes a proinflammatory and tumor protective response. Our results show that arazyme has immunomodulatory properties, and could be a promising novel alternative for metastatic melanoma treatment.
KEYWORDS: 4T1; arazyme, bacterial protease, breast adenocarcinoma, B16F10, immunoadjuvant, melanoma, TLR4
Introduction
In addition to conventional immunostimulation, complementary immunotherapeutic approaches for cancer treatment may also overcome tumor intrinsic mechanisms that prevent a protective immune response, such as induction of regulatory T cells (Tregs), and the suppression of effector immune cell populations and antigen-presenting cells (APCs) in the microenvironment.1,2
Adjuvants may enhance APC function to induce effective immune responses through the activation of innate immunity and consequently the adaptive immune response.3 Toll-like receptors (TLRs) are the best characterized members of the pathogen recognition receptor (PRR) family expressed by APCs, recognizing pathogen-associated molecular patterns (PAMPs) such as lipids, proteins, lipoproteins, and nucleic acids from microbial pathogens, as well those of an endogenous origin. PAMP recognition by TLRs is important for an effective immune response, and several natural and synthetic TLR agonists are being evaluated as adjuvants in preclinical protocols. Of these, Pam3/2Cys, Poly:IC, monophosphoryl lipid A (MPLA), recombinant flagellin, imidazoquinoline analogs, and unmethylated CpG motifs, which interact with TLR2, TLR3, TLR4, TLR5, TLR7/8, and TLR9, respectively,4,5 have shown promising results.6,7
Proteases have numerous biological roles, and are considered virulence factors in several microbial infections. As danger signals of infections and neoplasias, proteases might be recognized by PRRs, and our findings together with those of other groups suggest that proteases could be regarded as a new family of immune response modulators.
Active cysteine proteases from Porphyromonas gingivalis (gingipains) have immunoregulatory properties, acting as virulence factors in periodontitis. Arg- and Lys-gingipains reduced CD14 expression causing macrophage hyporesponsiveness,8 suppressed inflammatory responses by human gingival fibroblasts,9 activated platelets, and cleaved the chemokine RANTES.10 Interestingly, the adhesin, but not the catalytic subunit of gingipains mediated the strong upregulation of proinflammatory cytokines by macrophages.8 The surface-associated subtilisin-like protease (SspA) of Streptococcus suis, an important virulence factor in swine infections, also induced secretion of proinflammatory cytokines by macrophages through the MAPK-signaling pathway after heat-inactivation.9
Exogenous proteases have been evaluated for cancer treatment, and adjuvant treatment with combined exogenous proteases was found to reduce chemotherapy-associated toxicity.11 Fastuosain, a cysteine protease isolated from Bromelia fastuosa, showed a therapeutic effect in a murine metastatic melanoma model, significantly reducing the number of lung nodules after endovenous tumor cell inoculation. This protective response was related to a direct effect on tumor cell migration, an increased number of MHC II+ cells in the lungs, and induction of protease-specific antibodies that recognized cathepsins on the tumor cell surface.12
Arazyme, a 51.5 kDa metalloprotease secreted by Serratia proteamaculans, a symbiotic bacterium from the Nephila clavata spider,13 had a strong antitumor effect in a murine metastatic melanoma B16F10-Nex2 model, mediated by the cleavage of tumor cell surface CD44 and the induction of arazyme-specific antibodies that cross-react with tumor matrix metalloprotease 8 (MMP-8).14
In this study, we show that, in addition to its proteolytic-dependent activity, arazyme has a secondary, non-proteolytic antitumor effect dependent on an intact immune system. The anti-melanoma immune response induced by heat-inactivated arazyme was dependent on IFNγ, and CD8+ T lymphocytes were identified as the main effector cells for tumor rejection. Both macrophages and dendritic cells (DCs) were activated by arazyme, triggering TLR4-MyD88-TRIF- and MAPK-dependent signaling pathways.
Materials and methods
Animals
Inbred male C57BL/6 (WT), BALB/c, TLR4−/−, TLR2−/−, MyD88−/−, MyD88−/−/TRIF−/− double knockout (KO), IFNγ−/−, iNOS−/−, and NSG (NOD SCID gamma null, or NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) mice, 6–8 weeks old, were purchased from the Center for Development of Experimental Models (CEDEME), at UNIFESP. All animal experiments were approved by the Animal Experimentation Ethics Committee at UNIFESP, under protocol number 0288/12.
Cells and reagents
The murine melanoma cell line B16F10-Nex2, syngeneic to C57Bl/6 mice, was established at the Experimental Oncology Unit (UNONEX), EPM-UNIFESP, as described elsewhere.15 Cells were maintained at 37°C and with 5% CO2, in RPMI 1640 medium supplemented with 10 mM HEPES (N-2-hydroxyethylpiperazine-N-2-ethanesulphonic acid), 24 mM sodium bicarbonate, 10% fetal calf serum (FCS, Life Technologies, Carlsbad, CA, USA), and 40 mg/mL gentamicin (Hipolabor Farmaceutica, MG, Brazil). The murine breast adenocarcinoma cell line 4T1 was purchased from Banco de Células do Rio de Janeiro—Universidade Federal do Rio de Janeiro (BCRJ/UFRJ) and maintained as described above.
B3Z cells transduced with retroviral vectors encoding a chimera composed of extracellular portions of CLEC2, DNGR-1, Dectin-1, CLEC12a, or CLEC12b, tagged to hemagglutinin (HA) fused to the CD3ζ chain in tandem with a NFAT-LacZ reporter gene, were a gift from N. Shastri (University of California, Berkeley). Cells were sorted by flow cytometry based on receptor expression.16
Curdlan, CpG, LPS, and murine IFNγ were purchased from Sigma (St Louis, MO, USA). Pepinh-MYD and Pepinh-TRIF inhibitory peptides, and the inhibitors SP600125 (JNK inhibitor), SB203580 (p38 inhibitor), and PD98059 (MAPK—MEK1 and MEK2 inhibitor) were purchased from Invivogen, CA, USA. Peptides PAR1-AP (RLLFT-NH2) and PAR2-AP (LRGILS-NH2) were synthesized as previously described.15
A S. proteamaculans culture medium supernatant, obtained from InsectBiotech, Korea, was purified as previously described.13 Purified arazyme was completely inactivated by incubating at 50°C for 30 min.13
Arazyme treatment in murine models
C57Bl/6, IFNγ−/−, iNOS−/−, or NSG mice were injected in the caudal vein with 5 × 105 B16F10-Nex2 melanoma cells. One day after tumor cell challenge, intraperitoneal injections of active or heat-inactivated arazyme (3 mg/kg) or PBS were administered on alternate days for 2 weeks. Lung metastatic nodules were quantified on the 15th day. For the 4T1 breast cancer model, female BALB/c or NSG mice were inoculated with 1 × 104 tumor cells in mammary fat pads, and mice were treated with heat-inactivated arazyme (1 mg/kg) or PBS on alternate days for 2 weeks. Mammary tumor volumes were measured three times a week using the formula V = 0.52 × D12 × D2, where D1 and D2 are short and long tumor diameters, respectively. Animal survival was also recorded daily, and 4T1 lung metastases were counted 3–4 weeks after tumor challenge.
Quantification of arazyme-specific antibodies
C57Bl/6 mice were treated with heat-inactivated arazyme (3 mg/kg) on alternate days for 3 weeks. On 21st day, mice were sacrificed prior to total blood harvesting. Serum was separated by centrifugation at 500 g for 10 min. Antibodies were detected by ELISA. Briefly, high-binding ELISA plates (Nunc, Thermo Fisher Scientific, NY, USA) were coated with 1 μg of arazyme. After blocking, plates were incubated with serial dilutions of individual sera. Reaction was developed with Horseradish Peroxidase (HRP)-conjugated anti-mouse IgG secondary antibodies (AbCam) and TMB substrate solution (BD Biosciences) and read in a Multiskan ELISA reader at 450 nm, with correction in 570 nm.
Macrophage differentiation from murine bone marrow progenitors
Bone marrow-derived macrophages (BMDMs) were obtained from C57Bl/6 mice. Briefly, all muscles attached to femoral bones were removed, followed by disinfection in 70% alcohol, iodine alcohol, and PBS. The epiphyses were cut and cells were obtained by flushing out the marrow with 10 mL of a solution of PBS/antibiotics (penicillin 100 UmL, streptomycin 100 µg/mL, and gentamicin 40 μg/mL, all from Life Technologies) using a syringe. The progenitor cells were cultivated in RPMI 1640 medium supplemented with 20% FCS, 25% conditioned medium from L929 cells, penicillin 100 U/mL, and streptomycin 100 µg/mL and maintained in 5% CO2 at 37°C for differentiation. The media was changed 4 d later and cells were harvested on the 7th day for experiments.
Dendritic cell differentiation from murine bone marrow progenitors
Bone marrow-derived DCs (BMDCs) were generated as described.17 Briefly, bone marrow cells were obtained as described above, and cultivated in Petri tissue culture dishes (100 mm, Corning, NY), in 10 mL (per each femur) of RPMI 1640 supplemented with 10% of FCS, non-essential amino acids, 50 μM 2-mercaptoethanol (all from Life Technologies), 30 ng/mL murine rGM-CSF, and 10 ng/mL murine rIL-4 (PeproTech, Mexico). Complete medium was added carefully on the 3rd day of culture. After 6–7 d of culture, numerous typical DCs were collected as non-adherent cells. For some assays, an extra purification step was performed using anti-CD11c magnetic beads (Miltenyi Biotec, Bergisch Gladbach, Germany).
Splenic CD11c+ DC isolation
Spleens were digested with liberase (Wünsch units/mL) and DNAse I (0.2 mg/mL) (Roche) for 30 min at 37°C. Spleen cells were enriched for conventional DCs by positive selection with anti-CD11c magnetic MicroBeads (Miltenyi Biotec, Bergisch Gladbach, Germany).
In vitro stimulation of DCs, macrophages, and B3Z cells
For BMDC and BMDM activation assays in vitro, cells were plated at 1–3 × 105 cells/mL in 96-well plates at 37°C in the presence of different stimuli. Cytokine concentration in the supernatant was determined by ELISA or Cytometric Bead Array (CBA).
Transgenic B3Z cells were used to assess the interaction between arazyme and the C-type lectin receptors (CLR). LacZ activity was used as a readout of the aggregation of the extracellular portion of these receptors. Cells were added at 105 cells/well to 96-well UV-irradiated MaxiSorp plates (Thermo Fisher Scientific) and incubated with increasing amounts of active and inactive arazyme, or with anti-HA antibody as control. Ionomycin (1 μg/mL) was used as a positive control. LacZ activity was measured as absorption at 595 nm after lysis in chlorophenolred-β-D-galactopyranoside-containing buffer (Roche Diagnostics, Mannheim, Germany).
Cytokine quantification in serum, lungs, and culture supernatants
Cytokine concentrations were measured in the culture supernatant of activated BMDMs and BMDCs, as well as in serum and lung homogenates, both collected on the 15th day after tumor cell inoculation. Lung homogenates were prepared by tissue digestion with 0.5–1 U/mL collagenase (Sigma, USA) at 37°C for 30 min. IFNγ, IL-12, TNF-α, IL-10, and IL-6 were quantified by ELISA (eBioscience, San Diego, USA).
Measurement of endotoxin activity
The endotoxin activities of arazyme and LPS (Sigma-Aldrich, St. Louis, MO, USA) were determined using the Limulus amebocyte lysate (LAL) assay kit (BioWhittaker, Walkersville, MD, USA), which was performed according to the manufacturer's recommendations. We found that 1 μg/mL of arazyme preparation contained 8.8 endotoxin units (EU)/mL, whereas 2 ng/mL LPS contained 10 EU/mL.
Depletion of CD4+ or CD8+ T cells
C57Bl/6 mice were treated by intraperitoneal injection with Protein A-purified monoclonal antibodies, clones GK1.4 or YTS169, to deplete CD4+ or CD8+ T lymphocytes, respectively. PBS-diluted antibodies (500 μg/dose) were injected 72 h and 24 h before i.v. tumor cell challenge. This resulted in the depletion of 91% of CD4+ and 95% of CD8+ T cells, as observed in FACS analysis of splenocytes (data not shown).
Tumor antigen stimulation of splenocytes
Tumor cell lysates were produced by subjecting viable B16F10-Nex2 cells to 5 freeze/thaw cycles (liquid nitrogen/water bath at 37°C). On the 15th day after tumor cell inoculation, spleens were isolated from arazyme-treated and untreated mice and red blood cells were eliminated by treating with a hemolytic buffer. Splenocytes (2 × 107) were plated in 6-well plates and cultivated for 96 h in the presence or absence of tumor antigens equivalent to 2 × 106 B16F10-Nex2 cells.
Flow cytometry
IFNγ- and IL-10-producing CD4+ and CD8+ T lymphocytes were evaluated by cell surface markers and intracellular cytokine staining (ICCS). Splenocytes were stimulated as described above, and then incubated for 6 h with 10 μg/mL of Brefeldin A (Sigma). Cells were washed twice with PBS and 1% BSA-containing PBS, and incubated for 1 h at 4°C with normal mouse serum (1:30 diluted in PBS with 1% BSA) to block Fc receptors. Cells (106) were then stained with FITC-conjugated anti-CD4+ (clone GK 1.5) or anti-CD8+ (clone 53-6.7) monoclonal antibodies (BD PharMingen, NJ, USA), following washings with PBS and fixation with 2% paraformaldehyde. For ICCS, cells were permeabilized after fixation, with 0.5% saponin in PBS for 10 min at 4°C and stained for 1 h at 4°C with biotinylated anti-IFNγ (clone XMG1.2) or anti-IL-10 (clone SXC-1) monoclonal antibodies (BD PharMingen), diluted in permeabilization buffer. After two washes with permeabilization buffer, cells were incubated with PE-conjugated streptavidin in permeabilization buffer for 1 h at 4°C. Finally, cells were washed and fixed with 500 µL of 2% paraformaldehyde in PBS. All incubations were run protected from light.
BMDM and BMDC phenotypes were evaluated with PE-labeled anti-CD11c (clone N418), PE/Cy7-labeled anti-F4/80 (clone BM8), PerCP/Cy5.5-labeled anti-CD80 (clone 16-0A1), APC-labeled anti-CD86 (clone GL-1), pacific blue-labeled anti I-Ab (clone AF6-120.1), all from BioLegend (San Diego, USA). Data were acquired with a FACSCanto II (BD Biosciences) or FACS LSR II instrument (BD Biosciences). Flow cytometric analyses were performed using FlowJo Software (Tree Star, CA, USA).
Immunohistochemistry (IHC)
Specific monoclonal antibody immunostaining of FOXP3 (diluted 1:100; Santa Cruz, USA) and CD25 (diluted 1:100; Abcam, Cambridge, MA, USA) were assessed in paraffin-embedded tissue sections. After conventional preparation, the slides were incubated with specific primary antibodies or a negative control reagent, and then incubated with the labeled polymer (Dual Link System-HRP; DAKO, Glostrup, Denmark) using two sequential 30-min incubations at room temperature. Staining was completed by performing a 1 min incubation with 3,3-0-diaminobenzidine DAB+ Substrate, chromogen system (DAKO, Glostrup, Denmark). The slides were dehydrated, cleared, counterstained with Harry's hematoxylin, and mounted with cover slips. Immunostaining was then analyzed using a Zeiss light microscope (Germany).
Statistical analysis
Statistical analyses were performed using an unpaired Student´s t test when two groups were compared. Comparisons of three or more groups were performed using the one-way or two-way ANOVA test, followed by Dunnett's or Tukey's multiple comparisons, as described in the Figure legends. In all studies, a p value <0.05 was considered statistically significant.
Results
Anti-metastatic effect of active and heat-inactivated arazyme
Recently, we showed that treatment of melanoma-bearing C57Bl/6 mice with active arazyme, a bacterial metalloprotease secreted by Serratia proteomaculans, induced an expressive reduction in the number of lung metastatic nodules. Arazyme showed a direct cytostatic effect on tumor cells, but also induced arazyme-specific IgGs that cross-reacted to tumor matrix metalloprotease 8 (MMP8). In vitro arazyme-specific antibodies were cytotoxic to tumor cells, an effect increased by complement, and passive transfer to tumor-bearing mice partially inhibited melanoma lung metastasis.14
In order to verify whether the proteolytic activity of the enzyme was responsible for the antitumor effect, arazyme was heat-inactivated at 56°C,13 and used in the same treatment protocol. Melanoma-bearing C57Bl/6 male mice inoculated intraperitoneally with active or inactive arazyme on alternate days for 2 weeks showed a significant reduction in the number of metastatic lung nodules compared to PBS-treated mice (Figs. 1A and B). Active arazyme was slightly more efficient than the heat-inactivated protease, suggesting that the direct effect of arazyme on tumor cells is, at least partially, important for the inhibition of metastasis. However, the metalloprotease can also control melanoma cell growth by other means in vivo, and the possibility of stimulation of an antitumor immune response by the inactivated protease was evaluated.
Figure 1.
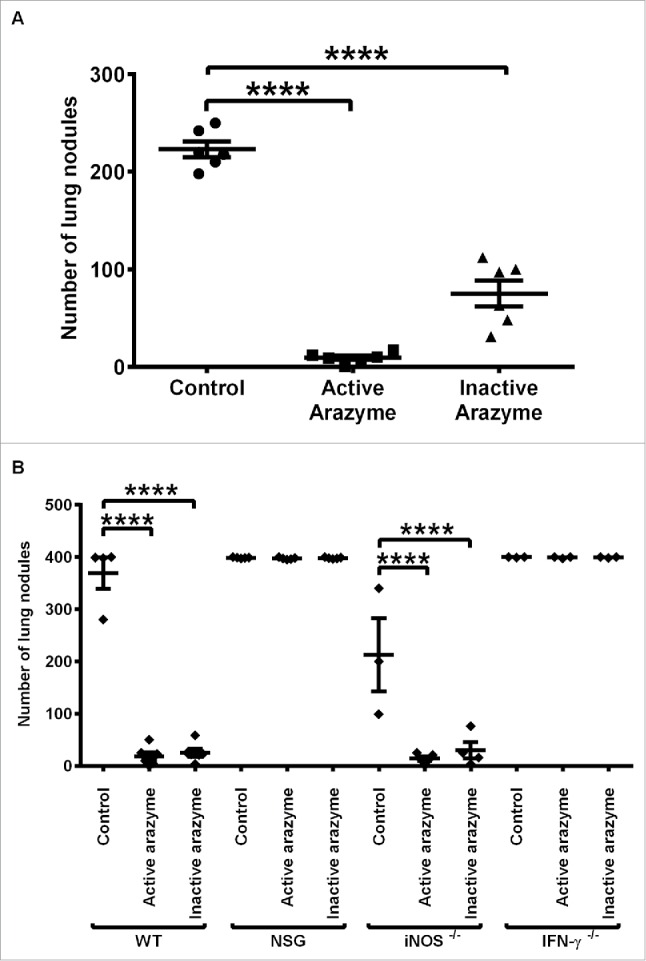
The immune system is involved in the IFNγ dependent antitumor activity of active or heat-inactivated arazyme. (A) C57BL/6 male mice (n = 6 or 7), and (B) C57BL/6 (WT, n = 5), NSG (n = 5), iNOS−/− (n = 5), and IFNγ−/− (n = 3) mice were inoculated intravenously with 5 × 105 B16F10-Nex2 melanoma cells and then treated intraperitoneally with 3 mg/kg of active or heat-inactivated arazyme for 2 weeks on alternate days. The melanotic pulmonary nodules were counted 15 d after tumor cell inoculation. Individual animals are represented, and the average ± SD is shown by central horizontal and vertical lines, respectively. Control, untreated mice. Each experiment was repeated at least twice. ****p < 0.0001, analyzed by one-way ANOVA with Tukey's multiple comparisons.
An intact immune system and IFNγ are necessary for the antitumor effect of active and inactive arazyme
To evaluate the role of the immune system in the inhibition of metastasis by active or inactive arazyme, immunodeficient NSG mice were inoculated with B16F10-Nex2 cells and treated with the protease. NSG mice carry the SCID mutation and a deletion of the IL-2 receptor, and thus lack functional T and B lymphocytes and NK cells, and have deficient cytokine signaling. These mice have been widely used as hosts for both normal and malignant human cells.18,19
Neither active nor inactive arazyme inhibited metastasis in immunodeficient NSG mice, as both the treated and untreated animals had the same number of lung nodules as the C57Bl/6 (WT) control mice (Fig. 1B). This result demonstrates the fundamental importance of an intact immune response for the full protease-mediated antitumor effect.
IFNγ and nitric oxide (NO) play unique roles in the intricate relationship between the immune system and tumor development.20 To verify the importance of these molecules in the protective immune response induced by arazyme treatment, we treated IFNγ and iNOS (inducible NO synthase) genetically deficient (KO) mice with active and inactive arazyme, using the same protocol. In the absence of IFNγ production, arazyme-treated mice could not mount a protective immune response against B16F10-Nex2 melanoma, although this effect was not observed in iNOS KO mice (Fig. 1B).
Active and inactive arazyme treatment induces tumor-specific CD4+ and CD8+ T lymphocytes, and CD8+ T cells are responsible for the inhibition of metastasis
Tumor-specific T lymphocytes from active and inactive arazyme-treated tumor-bearing mice were analyzed. Splenocytes were collected 24 h after the last dose of arazyme, stimulated with melanoma tumor antigens for 96 h ex vivo, and then assessed for phenotype and intracellular cytokines. Treatment with active or inactive arazyme significantly increased the frequency of IFNγ and IL-10-producing CD3+CD4+ or CD3+CD8+ tumor-specific lymphocytes in the spleens of tumor-bearing animals (Fig. 2A). Except for IL-10-producing CD4+ T lymphocytes, all other T lymphocyte types were increased in heat-inactivated arazyme-treated mice compared to those in active arazyme-treated animals.
Figure 2.

CD4+/CD8+-dependent antitumor response and increased production of pro-inflammatory cytokines are induced by active/inactive arazyme treatment. C57Bl/6 mice (n = 6) were challenged endovenously with 5 × 105 B16F10-Nex2 cells and treated intraperitoneally with 3 mg/kg of active/inactive arazyme or PBS (Control) on alternate days for 2 weeks. Spleens were collected 24 h after the last dose, splenocytes were stimulated ex vivo with B16F10-Nex2 tumor lysate, and intracellular cytokine staining (ICCS) was then performed for CD4+ and CD8+ positive T cells. (A) The percentage of splenic IFNγ- or IL10-producing CD4+ or CD8+ T cells was determined by FACS, and is represented by the numbers in the upper right quadrants. Histograms represent six pooled animals. (B) C57BL/6 mice were depleted of CD4+ or CD8+ T lymphocytes by two doses (500 μg each) of specific monoclonal antibodies 72 and 48 h before challenge with tumor cells and treatment with active/inactive arazyme. Animals were sacrificed 24 h after the last dose, and the number of lung nodules is represented individually. (C) IFNγ, (D) TNF-α, and (E) IL-10 were quantified in lung homogenates, (F) IL-12 was quantified in culture supernatant of splenocytes stimulated with tumor lysate, and (G) IFNγ was quantified in 1:2 diluted serum from C57Bl/6 mice challenged and treated as described above. Cytokines were quantified in individual animals, and bars represent the average ± SD of at least three animals. *p < 0.05; ***p < 0.001; and ****p < 0.0001, analyzed by one-way ANOVA with Tukey's multiple comparisons (in B) and one-way ANOVA with Dunnett's multiple comparisons (in C–G).
To investigate the role of CD4+ and CD8+ T lymphocytes in arazyme-induced antitumor activity, we depleted at least 90% (data not shown) of these cells using specific monoclonal antibodies. A reduction in lung nodules was observed in CD4+-depleted, melanoma-bearing untreated mice compared to that in non-depleted untreated controls (Fig. 2B). However, when CD4+-depleted mice were treated with active/inactive arazyme, the number of lung nodules was similar to those in untreated animals. Conversely, CD8+ T lymphocyte depletion completely abrogated the antitumor effect of active or inactive arazyme (Fig. 2B). These results clearly indicate that CD8+ T cells induced by active/inactive arazyme treatment are essential for the observed antitumor effect.
As showed previously for active arazyme,14 treatment of mice with heat-inactivated protease induced high titers of arazyme-specific IgGs (Fig. S1). Heat-inactivation inhibited the protease catalytic site, but maintained the apparent molecular weight, as observed in SDS-PAGE gels, maintaining also the recognition by the rabbit arazyme-specific polyclonal antibody in western blotting assay (data not shown). These results strongly suggest that antibodies produced by heat-inactivated protease were similar to active arazyme-induced antibodies, cross-reacting with tumor MMP-8 and showing cytotoxic activity against tumor cells, and these immunoglobulins may also contribute to the protective effect induced by heat-inactivated arazyme.
Treatment with active and inactive arazyme induces tumor microenvironment and systemic Th1-type cytokine profiles
Systemic and local production of pro-inflammatory cytokines by the immune system are necessary for melanoma growth inhibition, and a protective antitumor response also involves an increased Th1/Th2-type cytokine ratio.21-23
Cytokines present in the lungs of active/inactive arazyme-treated or PBS-treated melanoma-bearing C57Bl/6 mice were measured in pooled lung homogenates (n = 4), prepared 15 d after tumor cell inoculation. IFNγ and TNF-α increased significantly in arazyme-treated groups compared to the untreated group (Figs. 2C–D). Mice treated with inactive arazyme showed a significant reduction in IL-10 (Fig. 2E). Splenocytes were isolated from these mice and stimulated ex vivo with tumor antigens for 96 h. A significant increase in IL-12p70 levels was detected in the splenocyte culture supernatant from active/inactive arazyme-treated mice, compared to that in controls (Fig. 2F). IFNγ was the only cytokine detected in the sera of all mice, and its level in active/inactive arazyme-treated mice measured 15 d after tumor cell inoculation was at least 2-fold higher than in the control group (Fig. 2G). Taken together, these data indicate that treatment with active or inactive arazyme results in increased Th1 cytokine levels both systemically and in the tumor microenvironment.
Arazyme activates BMDCs
The induction of T lymphocytes is dependent on the activation of tumor APCs.24,25 In order to elucidate the mechanisms by which arazyme induces an antimelanoma response, BMDCs were incubated with active and inactive arazyme and cell activation was measured by IL-12p70 production. Active and inactive arazyme induced dose-dependent IL-12p70 production comparable to LPS (Fig. 3A). The optimal dose of active arazyme (0.1 μg/mL) was 10-fold lower than that of inactive arazyme (10 μg/mL). In these conditions, the expression of both CD80 (Fig. 3B) and CD86 (Fig. 3C) increased after incubation with active/inactive arazyme, but only the latter was significant. Similar results were observed after LPS stimulation.
Figure 3.
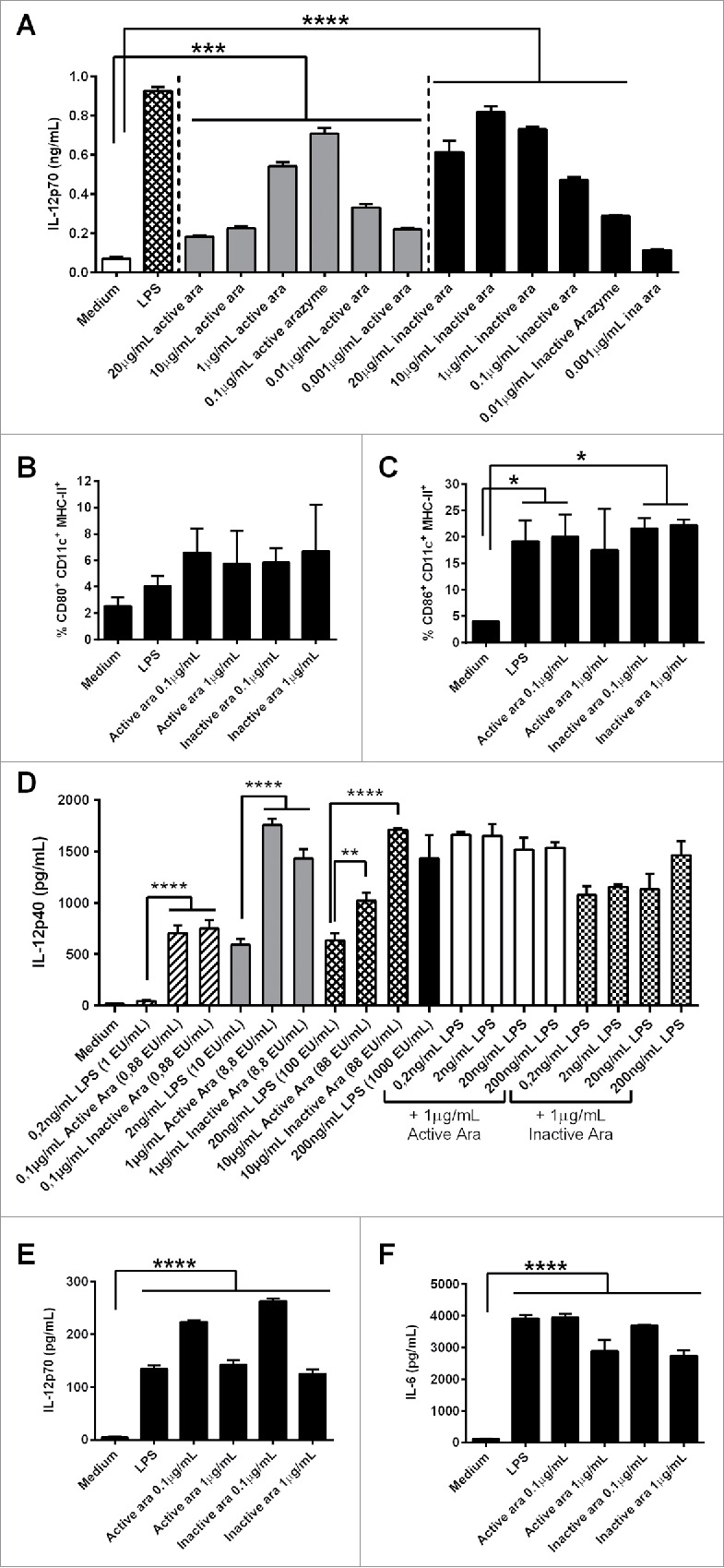
Dendritic cell (DC) activation by active or inactive arazyme. GM-CSF-treated bone marrow-derived DCs (BMDCs) from C57BL/6 mice were incubated in V bottom plates (1 × 105 cells) with lipopolysaccharide (LPS) (200 ng/mL in A, C–F, or indicated concentrations in D) and/or indicated concentrations of active or heat-inactivated arazyme for 24 h. IL-12p70 was quantified in culture supernatants by ELISA (A) and (D) and the expression of CD80 (B) and CD86 (C) was analyzed by FACS in gated CD11c-MHC-II positive cells. The same experiment was performed with BMDCs obtained from BALB/c mice: IL-12p70 (E) and IL-6 (F) were quantified in culture supernatant by ELISA. *p < 0.05; **p < 0.01; ***p < 0.001; and ****p < 0.0001, analyzed by one-way ANOVA with Tukey's multiple comparisons (in A and B) and one-way ANOVA with Dunnett's multiple comparisons (in C–F).
Arazyme is isolated from a gram negative bacteria culture supernatant, and the purification protocol could also isolate some endotoxin contamination. To quantify the endotoxin contamination of arazyme samples, a LAL test was performed, and compared to the LPS used in our assays. When comparable amounts of endotoxin in LPS and arazyme were tested for DCs activation, arazyme always induced a significantly higher effect (Fig. 3D). For example, 1 μg/mL of active or heat-inactivated arazyme, containing 8.8 endotoxin units (EU)/mL, induced a significantly increased IL-12p40 secretion by DCs, as compared to 2 ng/mL of LPS containing 10 EU/mL, Addition of increasing amounts of LPS to 1 μg/mL of active or heat-inactivated arazyme did not induce a synergistic effect (Fig. 3D). Moreover, concentrations of 10 and 20 μg/mL of polymyxin did not inhibited the DC activating effect of 1 and 10 μg/mL of active or inactivated arazyme (containing 8.8 and 88 EU/mL, respectively), but completely abrogated 500 ng/mL of LPS activating effect, containing more than 1000 EU/mL (Fig. S2).
We also demonstrated that BMDC activation by arazyme is not dependent on the genetic background of mice. BMDCs from BALB/c mice were activated by active/inactive arazyme, as demonstrated by the increased concentrations of IL-12p70 (Fig. 3E) and IL-6 (Fig. 3F) and by the increased cell surface expression of CD80 and CD86 (data not shown).
MyD88 and TRIF are essential for APC activation by arazyme
In an attempt to determine the pathway activated by arazyme in APCs, ex vivo assays were performed with GM-CSF-derived BMDCs and with CD11c+-enriched splenocytes obtained from C57Bl/6 (WT) and TRIF and MyD88 double KO mice (MyD88−/−/TRIF−/−). Culture supernatants from MyD88−/−/TRIF−/− and WT BMDCs were tested for cytokine (IL-1α, IL-1β, IL-6, IL-12p70, TNF, and IL-10) secretion and NO production. Cells were incubated for 24 h with active or inactive arazyme, and, as a control, BMDCs were incubated with LPS (with or without IFNγ) or CpG, both activators of the MyD88/TRIF pathway, or with Curdlan (CDL), a dectin-1 and cytosolic NLRP3 inflammasome complex ligand that activates cells in a MyD88/TRIF-independent manner.26 As expected, CDL caused BMDCs from both types of mice to secrete the same levels of cytokines and NO (Figs. 4A and B), and MyD88−/−/TRIF−/− animals showed impaired cytokine secretion and NO production after CpG and LPS activation (Figs. 4A and B). Active and heat-inactivated arazyme stimulated BMDCs from WT mice to produce NO and to secrete all of the evaluated cytokines, while cells from MyD88−/−/TRIF−/− mice were not activated by the metalloprotease, indicating that the MyD88/TRIF pathway is involved in the activation of DCs by arazyme (Figs. 4A and B). Arazyme-induced cytokine and NO production were similar to that promoted by LPS and CpG, which are classical TLR activators. Secretion of the MIP-1α chemokine by BMDCs from both types of mouse after treatment with any of the activation molecules also suggests that arazyme-induced BMDC activation is dependent on the MyD88/TRIF pathway (Fig. S3A). The same experiments were performed with CD11c+-enriched splenocytes from WT and MyD88−/−/TRIF−/− mice, and consistent with the results observed with BMDCs, arazyme induced the secretion of all cytokines, chemokines, and NO in a Myd88/TRIF-dependent pathway (Fig. 4C and Fig. S3B).
Figure 4.

Myd88 and TRIF pathways are involved in the activation of dendritic cells by active and inactive arazyme. GM-CSF-treated bone marrow-derived dendritic cells (BMDCs) from C57BL/6 (white bars) or double MyD88−/−/TRIF−/− knockout (black bars) mice were incubated in V bottom plates (1 × 105 cells/well) for stimulation with indicated concentrations of active or heat-inactivated arazyme, lipopolysaccharide (LPS), Curdlan (CDL), or CPG for 24 h. IL-12p70, IL-1α, TNF-α, IL-1β, IL-6, and IL-10 (A) and NO (B) were quantified in the culture supernatants using the cytometric bead array (CBA) and the Griess method, respectively. (C) CD11c+-enriched splenocytes from either C57BL/6 (white bars) or MyD88−/−/TRIF−/− (black bars) mice were incubated in V bottom plates (1 × 105 cells/well) for stimulation with indicated concentrations of active or heat-inactivated arazyme, LPS, CDL, or CPG for 24 h. IFNγ, IL-1α, IL-6, IL-1β, TNF-α, and IL-10 were quantified in the culture supernatants using CBA. Results represent data from triplicate samples of three (C57BL/6) or two (MyD88−/−/TRIF−/−) individually analyzed mice. Media, unstimulated control.
We next assessed the individual contribution of MyD88 and TRIF molecules in DC activation by arazyme. GM-CSF-derived BMDCs from C57Bl/6, MyD88−/−, and TRIF−/− mice were incubated with different concentrations of active or inactive arazyme and cell activation was measured by NO production. While active and heat-inactivated arazyme induced NO production by BMDCs from WT mice in a dose-dependent manner, BMDCs from MyD88 and TRIF deficient mice were stimulated by neither the protease nor LPS (Fig. 5A).
Figure 5.
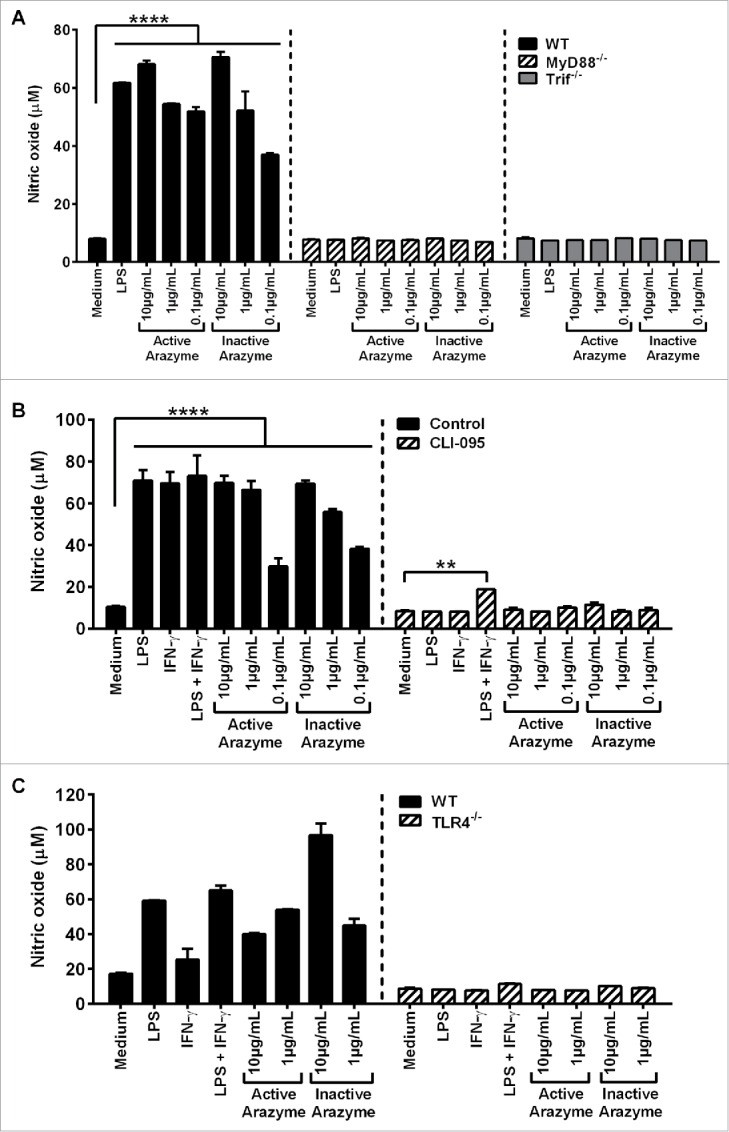
Active and inactive arazyme activates bone marrow-derived dendritic cells (BMDCs) via the TLR-4/Myd88/TRIF/ pathway. (A) GM-CSF-treated BMDCs from C57BL/6, MyD88−/−, or TRIF−/− mice were stimulated for 24 h in V bottom plates (1 × 105 cells/well) with lipopolysaccharide (LPS) (200 ng/mL) or indicated concentrations of active and heat-inactivated arazyme. (B) GM-CSF-derived BMDCs from C57BL/6 mice were incubated as described in (A) and stimulated with LPS (200 ng/mL), IFNγ (100 U/mL), LPS + IFNγ, or indicated concentrations of active and heat-inactivated arazyme, in the presence or absence (Control) of 1 μM CLI-095, a TLR-4 inhibitor. (C) GM-CSF-derived BMDCs from C57BL/6 or TLR-4−/− mice were incubated and stimulated as described in (B). The nitric oxide (NO) released in the supernatant was quantified using the Griess assay in (A), (B), and (C). **p < 0.01 and ****p < 0.0001, analyzed by one-way ANOVA with Dunnett's multiple comparisons.
Finally, the requirement for arazyme in MyD88/TRIF pathway stimulation of APCs was demonstrated by the inhibition of BMDC activation in the presence of inhibitory peptides that block MyD88 or TRIF signaling. Arazyme- and LPS-induced IL-12p70 production were partially and completely blocked by the inhibitory peptides, respectively, and pepinh-MyD and pepinh-TRIF reduced the IL-12p70 level by around 50% and 70% in the culture supernatant of arazyme-activated cells, respectively (Fig. S4). This suggests an important role for MyD88 and TRIF in the stimulation of DCs by arazyme.
TLR4 activates the MyD88/TRIF signaling pathway in APCs
It has been shown that TLR4 interacts with the adapter proteins MyD88 and/or TRIF and regulates the expression of inflammatory mediators.27 To explore possible interactions between arazyme and surface TLR4, BMDCs obtained from C57Bl/6 mice were stimulated with active/inactive arazyme in the presence of CLI-095, a specific inhibitor of TLR4. In the absence of the inhibitor, arazyme activates DCs, inducing NO in a dose-dependent manner, while this effect was not observed in the presence of the TLR4 inhibitor (Fig. 5B). The TLR4-dependent effect of LPS was also abolished, as expected. We also incubated BMDCs from C57Bl/6 or TLR4−/− mice with active or inactive arazyme. Consistent with the results obtained using the TLR4 inhibitor, neither LPS or active or inactive arazyme could induce NO production in DCs from TLR4−/− mice (Fig. 5C). Arazyme activated BMDMs from TLR2−/− mice (data not shown). Together, these results suggest that arazyme activates TLR4 and that signaling through this receptor is dependent on the adapter molecules MyD88 and TRIF.
Interaction of heat-inactivated arazyme with dectin-1 receptor or protease-activated receptors 1 and 2 is not essential for APC activation
Dectin-1 is a CLR which along with dectin-2 and CLEC5a can mediate the innate response to infection.28 To evaluate whether arazyme interactions with CLRs could induce APC activation, we used B3Z cells that stably express the NFAT-reporter gene LacZ ligated in tandem to a chimera composed of different extracellular portions of CLRs and tagged with HA that is fused to the CD3ζ chain. LacZ activity then acts as a readout of extracellular CLR aggregation, and anti-HA acts as a suitable positive control. Curiously, only inactive arazyme showed a dose-dependent interaction with dectin-1, but not with the other CLRs (Fig. S5). However, this interaction alone could not trigger cytokine or NO secretion by APCs, as demonstrated by the complete inhibition of cytokine and NO production by arazyme in MyD88−/−/TRIF−/− mice (Figs. 4 and 5A).
To investigate if protease activated receptor 1 (PAR-1) and protease activated receptor 2 (PAR-2) are involved in arazyme activation of immune cells, we measured the NO production by BMDMs after incubation with 10 µg/mL active arazyme in the presence of reverse peptides of PAR-1 and PAR-2.29 These PAR-1 and PAR-2 antagonists were not able to inhibit NO production by BMDMs stimulated with active arazyme (Fig. S6).
Mitogen-activated protein kinase (MAPK) signaling transduction pathways are required for IL-12p70 production by arazyme-stimulated APCs
The major MAPK pathways (p38, JNK, and ERK) are extremely important for DC maturation and function, as they control the production of IL-12 and other pro-inflammatory cytokines.30,31 To examine the involvement of MAPKs in arazyme-induced IL-12p70 production by BMDCs, cells from C57Bl/6 mice were pre-incubated for 1 h with SP600125, SB203580, or PD98059, which are pharmacological inhibitors of JNK, p38, and MAPK/ERK, respectively. This was followed by incubation for 48 h with active/inactive arazyme, or LPS as a positive control.
IL-12p70 induction by active and inactive arazyme was strongly inhibited by JNK, and MERK inhibitors, and less efficiently by p38 inhibitor (Fig. 6A), while a protein gel blot revealed a significant increase of p-STAT1, p-STAT5, and p-P38 in the lysates of BMDCs incubated with heat-inactivated arazyme or LPS for 2 h (Fig. 6B). These results indicate that MAPK pathways are involved in APC activation by arazyme.
Figure 6.
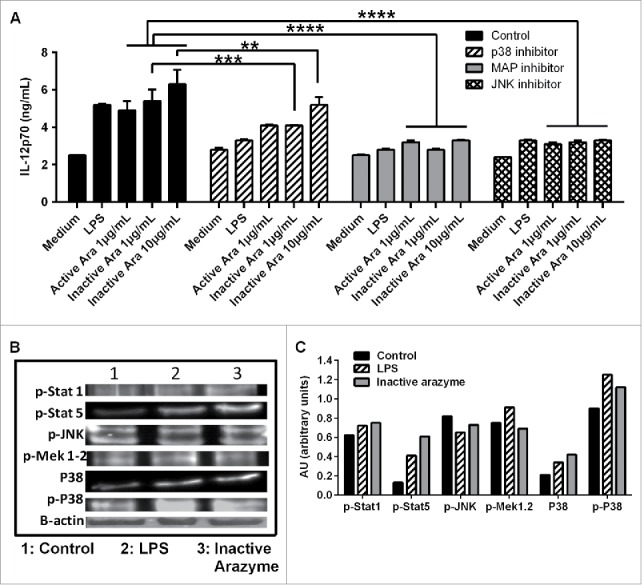
Other signaling pathways are also involved in active or inactive arazyme activation of GM-CSF-derived bone marrow-derived dendritic cells (BMDCs). (A) BMDCs were obtained from C57BL/6 mice, incubated in V bottom plates (1 × 105 cells/well), pretreated for 1 h with SP600125 (JNK inhibitor, 50 µM), SB203580 (P38 inhibitor, 20 µM) or PD98059 (MAPK/ERK inhibitor, 100 µM) and stimulated with lipopolysaccharide (LPS) (200 ng/mL) or with indicated concentrations of active or heat-inactivated arazyme for 48 h. IL-12 was quantified in culture supernatants by ELISA. (B) BMDCs were treated for 2 h with LPS (200 ng/mL) or heat-inactivated arazyme (10 μg/mL) and the levels of p-STAT1, p-STAT5, p-JNK, p-MAPK/ERK, P38, and p-P38 in cell lysates were analyzed by western blotting. (C) Densitometric quantification of bands in (B). Medium, unstimulated cells; Control, stimulated BMDCs in the absence of inhibitors. **p < 0.01 and ****p < 0.0001, analyzed by one-way ANOVA with Dunnett's multiple comparisons.
An intact immune system is required for the antitumor effects of arazyme in breast adenocarcinoma
To investigate whether the antitumor effect of arazyme also occurs in different tumors and genetically distinct mice, we injected BALB/c animals with 4T1 breast adenocarcinoma cells. BMDCs from BALB/c mice are activated in vitro by arazyme (Figs. 3E and F), and treatment with heat-inactivated arazyme (1 mg/kg) delayed the onset and the development of primary tumors in 60% of treated mice (Fig. 7A), as reflected by the tumor weight on day 30 (Fig. 7B). Treatment with inactive arazyme extended the survival of animals compared with PBS-treated controls, completely inhibiting the development of mammary adenocarcinoma in 20% of the mice (Fig. 7C).
Figure 7.

Treatment with inactivated arazyme had protective effects in a 4T1 breast adenocarcinoma preclinical model and requires a competent immune system. (A) Female BALB/c mice (five per group) were inoculated into the mammary fat pad with 104 viable 4T1 murine mammary adenocarcinoma cells. Starting on the 1st day after tumor cell inoculation, heat-inactivated arazyme (1 mg/Kg) or PBS (Control) was inoculated intraperitoneally every other day for 4 weeks. The average ± SD of primary tumor volumes are shown. (B) NSG mice (n = 5) were challenged and treated as described in (A) and primary tumor volumes were measured. (C) Survival plot of BALB/c mice depicted in (A). (D) Primary tumor weights, collected on day 30 from BALB/c mice depicted in (A). (E) Metastatic lung nodules of animals were counted 30 d after tumor cell inoculation, using an inverted microscope. Representative images of each group are shown. (F) Lungs from BALB/c mice were collected after 30 d and tissue sections were stained with hematoxylin/eosin (HE) to show metastasis development, and immunostained with monoclonal anti-CD25 or anti-Foxp3 antibodies. Tissues were visualized microscopically, ×200 magnification. Black bars = 100 μm. T, tumor areas; arrows, immunostained areas. *p < 0.05 and **p < 0.01, analyzed using the unpaired Student's t test (in A, B, D, and E) and the Log-Rank test (in C).
The immune system was also required for the antitumor activity of arazyme in a mammary tumor model. The protective effect of inactive arazyme was completely abolished in immunodeficient NSG mice (Fig. 7D). In addition, arazyme-treated mice had significantly fewer lung nodules compared to PBS-treated controls (Fig. 7E), showing that the antitumor effect of arazyme is effective not only in the primary tumor, but also in metastatic murine breast 4T1 adenocarcinoma.
Finally, while the lungs of untreated mice showed increased numbers of CD25+Foxp3+ Tregs, inactive arazyme-treated mice showed a reduction in the number of tumor-associated Tregs (Fig. 7F), suggesting that the reduction in Tregs in the tumor microenvironment after arazyme treatment facilitates tumor rejection through an antitumor specific immune response.
Discussion
More than a century ago, Willian Coley was the first to observe spontaneous regression of tumors in patients showing post-surgical bacterial infection. Intratumor injection of live bacteria, Streptococcus or Serratia, induced tumor growth control in 10% of tumor-bearing individuals,25,32 and immunotherapy using Mycobacterium bovis vaccine (BCG) is a long established treatment in bladder cancer.33,34 Since the discovery and better characterization of innate immunity receptor families (for example, TLRs), several bacterial components, which are natural ligands to these receptors such as LPS, flagellin, and bacterial DNA, have been studied as immunomodulators and adjuvants in protocols for cancer treatment.35
We previously reported that the active bacterial metalloprotease arazyme, secreted by Serratia proteomaculans, has a direct cytostatic activity on murine B16F10-Nex2 and in human A2058 melanoma cells in vitro by cleavage of the surface adhesion molecule CD44. In vivo inoculation of active arazyme induced the production of arazyme-specific antibodies that cross-react with murine MMP-8. These antibodies were cytotoxic in vitro to B16F10-Nex2 cells, and passive transfer partially inhibited lung metastatic nodule formation.14
In the present study, we show that the anti-melanoma effect of arazyme also involves a strong immune system activation by a mechanism independent of its proteolytic activity.
More recently, it has been observed that bacterial proteolytic enzymes were also found to have immunomodulatory properties,13-15,42 and interestingly the adhesin, but not the catalytic subunits of these enzymes could trigger pro-inflammatory cytokines production, including IL-1β, IL-6, TNF-α, IL-8, and IFNγ.13,14,35,43
Melanoma-bearing mice treated with active or heat-inactivated arazyme both showed a significant reduction in lung metastatic nodules, indicating that the proteolytic activity is not essential for induction of the tumor-protective response in vivo, and the importance of the immune response in this process was demonstrated by its abrogation in immunodeficient mice. Both active and inactive arazyme-induced anti-melanoma response depends on IFNγ. However, the antitumor response was not dependent on NO, as iNOS−/− arazyme-treated mice showed similar tumor growth to WT mice. IFNγ is responsible for the differentiation and function of several types of immune cell, mediates Th1-type induction and activity, and is associated with antitumor protective responses in several tumor types, including melanoma.20,36,37
Tumor-bearing mice treated with active or heat-inactivated arazyme showed a significant increase in IFNγ and TNF-α levels and a decrease in IL-10 levels in the lungs, increased serum IFNγ, and increased secretion of IL-12p70 by splenocytes after tumor cell stimulation in vitro. These results support the hypothesis that arazyme treatment of melanoma-bearing mice induces a local and systemic Th1 cytokine response.
A significant increase in the proportion of tumor-specific T lymphocytes in the spleen was observed in melanoma-bearing mice treated with active/inactive arazyme. Tumor-specific CD4+ and CD8+ T cells generated by arazyme treatment produced increased levels of IFNγ and IL-10. The depletion of CD4+ or CD8+ T cells before melanoma inoculation and arazyme treatment demonstrated that CD8+ T cells are critical effectors of the anti-metastatic effect induced by the protease in vivo. CD8+ and CD4+ T lymphocytes have a major role in specific antitumor responses, wherein CD8+ cytotoxic T lymphocytes (CTL) are considered the most potent antitumor effector cells involved in the recognition of tumor rejection antigens,38 although CD4+ T lymphocytes may also contribute to tumor elimination even in the absence of CD8+ T cells.39
Our results also showed that heat-inactivated arazyme induced high titers of protease-specific IgGs in treated mice. These immunoglobulins can be part of the global antitumor protective immune response induced by the inactive protease, as showed previously for the immunoglobulins induced by active arazyme.14
The priming of CD8+ T cells in draining lymph nodes depends on mature DCs that were properly activated by cytokines and/or TLR ligands.40,41 To evaluate the ability of arazyme to activate APCs, we analyzed the effect of active/inactive arazyme on the activation of BMDCs and macrophages and on CD11c+ cells enriched from splenocytes. These cells produced NO and pro-inflammatory cytokines in a dose-dependent manner after stimulation with arazyme. CD80, CD86, and MIP-1α expression also increased, in both C57Bl/6 and BALB/c mice, indicating that it is not related to the genetic background.
Our results show that arazyme activates the TLR4 receptor. Arazyme could not activate BMDCs from TLR-4 KO mice, or cells from WT mice that were pre-incubated with a TLR-4 inhibitor. Also, arazyme was ineffective when MyD88 or TRIF were absent. This is an important finding, and to our knowledge, the first indication that proteases can serve as a TLR4 agonist. TLR4 is an important PRR that can induce a potent innate immune response and modulate the adaptive immune response. Both MyD88 and TRIF can be recruited after TLR4 activation.42 TRIF plays an important role in allowing TLR4 agonists to have an adjuvant effect on T-cell responses. The defective maturation of DCs and the lack of a T-cell-mediated adjuvant effect after the stimulation of TLR4 in TRIF-deficient mice is mainly due to the loss of IFN-type I production, indicating that this cytokine is central to the adjuvant effects of TLR4.43 Likewise, activation of the TLR4/TRIF and TLR4/MyD88 pathways in APCs induces a strong CD8+ T-cell response,44 and several DAMPs such as high-mobility group box 1 (HMGB-1) and heat shock protein 70 (HSP70) bind to TLR4 inducing a strong antitumor response.45-47
As arazyme is purified from a Gram negative bacteria culture supernatant, a major concern was the possible presence of endotoxins in our preparation that could activate TLR-4, and we determined the endotoxin concentrations in arazyme samples and commercial LPS used in our experiments as a positive control. Activating DCs with similar amounts of endotoxin present in arazyme and LPS showed that arazyme induced a significantly increased IL-12p40 production by these cells, suggesting that the endotoxin present in the protease preparation was not responsible for TLR-4 activation. We also ruled out a possible synergism between arazyme and LPS, as addition of increasing doses of LPS to 1 μg/mL of active or heat-inactivated arazyme did not enhance IL-12p40 production. Moreover, addition of polymyxin B, a cationic cyclic lipopeptide that binds stoichiometrically to the lipid A moiety of LPS and blocks its biological effects, did not interfere with the results of arazyme, while abrogated completely the activation effect of LPS.
Our results also showed that three major MAPKs, JNK, ERK and less efficiently p38, are involved in IL-12p70 secretion by arazyme-activated BMDCs. MAPK pathways are closely involved in TLR signaling and are, therefore, also involved in the activation of immune cells, mediating cytokine production. Other bacterial proteases, such as gingipain and subtilisin-like protease, also activate MAPK pathways.30,48,49
We also evaluated whether alternative receptors could interact with arazyme to activate APCs. PARs are a five-members family of G protein-coupled receptors that harbors a cryptic ligand sequence within their N-terminus, exposed only after proteolytic cleavage by serine proteases, and the role of metalloproteases is unknown. Mature DCs express PARs,50 and PAR2-agonist peptides can activate these cells.49,51,52 Stimulation of BMDCs by active arazyme was not prevented by antagonistic peptides of PAR-1 and PAR-2, suggesting that these receptors are not activated by the protease. We also evaluated the CLRs, another family of pathogen recognizing receptors (PRR) involved in the microbial pattern recognition and responsible for immune cell activation.35 The interaction of arazyme and CLRs was tested, using cells expressing transgenic receptors. Interestingly, only heat-inactivated arazyme interacted with dectin-1. However, DCs from MyD88−/−TRIF−/− mice incubated with Curdlan, a dectin-1 agonist, did not secrete cytokines, suggesting that this interaction is not specific.
Arazyme treatment was also effective in the 4T1 breast adenocarcinoma model, as treatment with heat-inactivated arazyme delayed primary tumor growth, reduced the number of metastatic pulmonary nodules, and extended survival compared to the untreated group. This contrasts with the murine melanoma B16F10-Nex2 model, in which arazyme did not retard primary tumor growth.11 As with the melanoma model, the antitumor effect of arazyme in 4T1 tumors depended on an intact immune system, with the protease showing no protective effect in immunodeficient mice. Mice treated with inactive arazyme had a significantly reduced number of Tregs in the lungs. Our findings are supported by previous reports showing that increased numbers of Tregs are associated with tumor development.53
In conclusion, our findings indicate that arazyme has immunostimulatory effects that are independent of its proteolytic activity. This suggests a number of new directions and applications of arazyme and other exogenous proteases in cancer treatment.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was financed by FAPESP, CAPES and CNPq. EGR, NOCS, LJ, MAJ, ACK, and LRT are recipients of fellowships from CNPq. We thank Dr Caetano Reis e Souza, Immunobiology Laboratory, Cancer Research UK, London Research Institute, London, UK, for kindly supplying the double Myd88/TRIF knockout mice and reagents for cytokine/chemokine quantification in these animals. We also thank Dr Alexandre S. Basso, Department of Microbiology, Immunology and Parasitology, EPM-UNIFESP for reagents and helpful discussions.
References
- 1.Klebanoff CA, Acquavella N, Yu Z, Restifo NP. Therapeutic cancer vaccines: are we there yet? Immunol Rev 2011; 239:27-44; PMID:21198663; http://dx.doi.org/ 10.1111/j.1600-065X.2010.00979.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zigler M, Shir A, Levitzki A. Targeted cancer immunotherapy. Curr Opin Pharmacol 2013; 13:504-10; PMID:23648271; http://dx.doi.org/ 10.1016/j.coph.2013.04.003 [DOI] [PubMed] [Google Scholar]
- 3.Leroux-Roels G. Unmet needs in modern vaccinology: adjuvants to improve the immune response. Vaccine 2010; 28 Suppl 3:C25-36; PMID:20713254; http://dx.doi.org/ 10.1016/j.vaccine.2010.07.021 [DOI] [PubMed] [Google Scholar]
- 4.de Melo FM, Braga CJ, Pereira FV, Maricato JT, Origassa CS, Souza MF, Melo1 Amanda C, Silva P, Tomaz SL, Gimenes KP et al.. Anti-metastatic immunotherapy based on mucosal administration of flagellin and immunomodulatory P10. Immunol Cell Biol 2015; 93:86-98; PMID:25223833; http://dx.doi.org/ 10.1038/icb.2014.74 [DOI] [PubMed] [Google Scholar]
- 5.Casella CR, Mitchell TC. Putting endotoxin to work for us: monophosphoryl lipid A as a safe and effective vaccine adjuvant. Cell Mol Life Sci 2008; 65:3231-40; PMID:18668203; http://dx.doi.org/ 10.1007/s00018-008-8228-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bogunovic D, Manches O, Godefroy E, Yewdall A, Gallois A, Salazar AM, Marie I, Levy DE, Bhardwaj N. TLR4 engagement during TLR3-induced proinflammatory signaling in dendritic cells promotes IL-10-mediated suppression of antitumor immunity. Cancer Res 2011; 71:5467-76; PMID:21730023; http://dx.doi.org/ 10.1158/0008-5472.CAN-10-3988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Adams S, O'Neill DW, Nonaka D, Hardin E, Chiriboga L, Siu K, Cruz CM, Angiulli A, Angiulli F, Ritter E et al.. Immunization of malignant melanoma patients with full-length NY-ESO-1 protein using TLR7 agonist imiquimod as vaccine adjuvant. J Immunol 2008; 181:776-84; PMID:18566444; http://dx.doi.org/ 10.4049/jimmunol.181.1.776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilensky A, Tzach-Nahman R, Potempa J, Shapira L, Nussbaum G. Porphyromonas gingivalis gingipains selectively reduce CD14 expression, leading to macrophage hyporesponsiveness to bacterial infection. J Innate Immun 2015; 7:127-35; PMID:25228314; http://dx.doi.org/ 10.1159/000365970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Palm E, Khalaf H, Bengtsson T. Suppression of inflammatory responses of human gingival fibroblasts by gingipains from Porphyromonas gingivalis. Mol Oral Microbiol 2015; 30:74-85; PMID:25055828; http://dx.doi.org/ 10.1111/omi.12073 [DOI] [PubMed] [Google Scholar]
- 10.Klarstrom Engstrom K, Khalaf H, Kalvegren H, Bengtsson T. The role of Porphyromonas gingivalis gingipains in platelet activation and innate immune modulation. Mol Oral Microbiol 2015; 30:62-73; PMID:25043711; http://dx.doi.org/ 10.1111/omi.12067 [DOI] [PubMed] [Google Scholar]
- 11.Beuth J. Proteolytic enzyme therapy in evidence-based complementary oncology: fact or fiction? Integr Cancer Ther 2008; 7:311-6; PMID:19116226; http://dx.doi.org/ 10.1177/1534735408327251 [DOI] [PubMed] [Google Scholar]
- 12.Guimaraes-Ferreira CA, Rodrigues EG, Mortara RA, Cabral H, Serrano FA, Ribeiro-dos-Santos R, Travassos LR. Antitumor effects in vitro and in vivo and mechanisms of protection against melanoma B16F10-Nex2 cells by fastuosain, a cysteine proteinase from Bromelia fastuosa. Neoplasia 2007; 9:723-33; PMID:17898868; http://dx.doi.org/ 10.1593/neo.07427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bersanetti PA, Park H-Y, Bae KS, Son K-H, Shin D-H, Hirata IY, Juliano MA, Carmona AK, Juliano L. Characterization of arazyme, an exocellular metalloprotease isolated from Serratia proteamaculans culture medium. Enzyme and Microbial Technology 2005; 37:574-81; http://dx.doi.org/ 10.1016/j.enzmictec.2005.01.041 [DOI] [Google Scholar]
- 14.Pereira FV, Ferreira-Guimaraes CA, Paschoalin T, Scutti JA, Melo FM, Silva LS, Tiago M, Matsuo AL, Juliano L, Juliano MA et al.. A natural bacterial-derived product, the metalloprotease arazyme, inhibits metastatic murine melanoma by inducing MMP-8 cross-reactive antibodies. PLoS One 2014; 9:e96141; PMID:24788523; http://dx.doi.org/ 10.1371/journal.pone.0096141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dobroff AS, Rodrigues EG, Moraes JZ, Travassos LR. Protective, anti-tumor monoclonal antibody recognizes a conformational epitope similar to melibiose at the surface of invasive murine melanoma cells. Hybrid Hybridomics 2002; 21:321-31; PMID:12470474; http://dx.doi.org/ 10.1089/153685902761022661 [DOI] [PubMed] [Google Scholar]
- 16.Sancho D, Joffre OP, Keller AM, Rogers NC, Martinez D, Hernanz-Falcon P, Rosewell I, Reis e Sousa C. Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature 2009; 458:899-903; PMID:19219027; http://dx.doi.org/ 10.1038/nature07750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dias BR, Rodrigues EG, Nimrichter L, Nakayasu ES, Almeida IC, Travassos LR. Identification of iGb3 and iGb4 in melanoma B16F10-Nex2 cells and the iNKT cell-mediated antitumor effect of dendritic cells primed with iGb3. Mol Cancer 2009; 8:116; PMID:19968878; http://dx.doi.org/ 10.1186/1476-4598-8-116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, Chaleff S, Kotb M, Gillies SD, King M, Mangada J et al.. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol 2005; 174:6477-89; PMID:15879151; http://dx.doi.org/ 10.4049/jimmunol.174.10.6477 [DOI] [PubMed] [Google Scholar]
- 19.Shultz LD, Ishikawa F, Greiner DL. Humanized mice in translational biomedical research. Nat Rev Immunol 2007; 7:118-30; PMID:17259968; http://dx.doi.org/ 10.1038/nri2017 [DOI] [PubMed] [Google Scholar]
- 20.Zaidi MR, Merlino G. The two faces of interferon-gamma in cancer. Clin Cancer Res 2011; 17:6118-24; PMID:21705455; http://dx.doi.org/ 10.1158/1078-0432.CCR-11-0482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liao YP, Schaue D, McBride WH. Modification of the tumor microenvironment to enhance immunity. Front Biosci 2007; 12:3576-600; PMID:17485323; http://dx.doi.org/ 10.2741/2336 [DOI] [PubMed] [Google Scholar]
- 22.Marchi LH, Paschoalin T, Travassos LR, Rodrigues EG. Gene therapy with interleukin-10 receptor and interleukin-12 induces a protective interferon-gamma-dependent response against B16F10-Nex2 melanoma. Cancer Gene Ther 2011; 18:110-22; PMID:20885448; http://dx.doi.org/ 10.1038/cgt.2010.58 [DOI] [PubMed] [Google Scholar]
- 23.Hiniker SM, Chen DS, Reddy S, Chang DT, Jones JC, Mollick JA, Swetter SM, Knox SJ. A systemic complete response of metastatic melanoma to local radiation and immunotherapy. Transl Oncol 2012; 5:404-7; PMID:23323154; http://dx.doi.org/ 10.1593/tlo.12280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pufnock JS, Cigal M, Rolczynski LS, Andersen-Nissen E, Wolfl M, McElrath MJ, Greenberg PD. Priming CD8+ T cells with dendritic cells matured using TLR4 and TLR7/8 ligands together enhances generation of CD8+ T cells retaining CD28. Blood 2011; 117:6542-51; PMID:21493800; http://dx.doi.org/ 10.1182/blood-2010-11-317966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Andersen BM, Ohlfest JR. Increasing the efficacy of tumor cell vaccines by enhancing cross priming. Cancer Lett 2012; 325:155-64; PMID:22809568; http://dx.doi.org/ 10.1016/j.canlet.2012.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Osorio F, Reis e Sousa C. Myeloid C-type lectin receptors in pathogen recognition and host defense. Immunity 2011; 34:651-64; PMID:21616435; http://dx.doi.org/ 10.1016/j.immuni.2011.05.001 [DOI] [PubMed] [Google Scholar]
- 27.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol 2004; 4:499-511; PMID:15229469; http://dx.doi.org/ 10.1038/nri1391 [DOI] [PubMed] [Google Scholar]
- 28.Mourao-Sa D, Robinson MJ, Zelenay S, Sancho D, Chakravarty P, Larsen R, Plantinga M, Van Rooijen N, Soares MP, Lambrecht B et al.. CLEC-2 signaling via Syk in myeloid cells can regulate inflammatory responses. Eur J Immunol 2011; 41:3040-53; PMID:21728173; http://dx.doi.org/ 10.1002/eji.201141641 [DOI] [PubMed] [Google Scholar]
- 29.Pagano RL, Sampaio SC, Juliano MA, Juliano L, Giorgi R. Involvement of proteinase-activated receptors 1 and 2 in spreading and phagocytosis by murine adherent peritoneal cells: modulation by the C-terminal of S100A9 protein. Eur J Pharmacol 2010; 628:240-6; PMID:19941849; http://dx.doi.org/ 10.1016/j.ejphar.2009.11.033 [DOI] [PubMed] [Google Scholar]
- 30.Dong C, Davis RJ, Flavell RA. MAP kinases in the immune response. Annu Rev Immunol 2002; 20:55-72; PMID:11861597; http://dx.doi.org/ 10.1146/annurev.immunol.20.091301.131133 [DOI] [PubMed] [Google Scholar]
- 31.Korhonen R, Huotari N, Hommo T, Leppanen T, Moilanen E. The expression of interleukin-12 is increased by MAP kinase phosphatase-1 through a mechanism related to interferon regulatory factor 1. Mol Immunol 2012; 51:219-26; PMID:22464096; http://dx.doi.org/ 10.1016/j.molimm.2012.03.019 [DOI] [PubMed] [Google Scholar]
- 32.Wiemann B, Starnes CO. Coley's toxins, tumor necrosis factor and cancer research: a historical perspective. Pharmacol Ther 1994; 64:529-64; PMID:7724661; http://dx.doi.org/ 10.1016/0163-7258(94)90023-X [DOI] [PubMed] [Google Scholar]
- 33.Herr HW, Morales A. History of bacillus Calmette-Guerin and bladder cancer: an immunotherapy success story. J Urol 2008; 179:53-6; PMID:17997439; http://dx.doi.org/ 10.1016/j.juro.2007.08.122 [DOI] [PubMed] [Google Scholar]
- 34.Bernardes N, Seruca R, Chakrabarty AM, Fialho AM. Microbial-based therapy of cancer: current progress and future prospects. Bioeng Bugs 2010; 1:178-90; PMID:21326924; http://dx.doi.org/ 10.4161/bbug.1.3.10903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer 2012; 12:265-77; PMID:22437871; http://dx.doi.org/ 10.1038/nrc3258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rodrigues EG, Garofalo AS, Travassos LR. Endogenous accumulation of IFN-gamma in IFN-gamma-R(−/−) mice increases resistance to B16F10-Nex2 murine melanoma: a model for direct IFN-gamma anti-tumor cytotoxicity in vitro and in vivo. Cytokines Cell Mol Ther 2002; 7:107-16; PMID:12850810; http://dx.doi.org/ 10.1080/13684730310000121 [DOI] [PubMed] [Google Scholar]
- 37.Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity 2004; 21:137-48; PMID:15308095; http://dx.doi.org/ 10.1016/j.immuni.2004.07.017 [DOI] [PubMed] [Google Scholar]
- 38.Wu R, Forget MA, Chacon J, Bernatchez C, Haymaker C, Chen JQ, Hwu P, Radvanyi LG. Adoptive T-cell therapy using autologous tumor-infiltrating lymphocytes for metastatic melanoma: current status and future outlook. Cancer J 2012; 18:160-75; PMID:22453018; http://dx.doi.org/ 10.1097/PPO.0b013e31824d4465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Haabeth OA, Tveita AA, Fauskanger M, Schjesvold F, Lorvik KB, Hofgaard PO, Omholt H, Munthe LA, Dembic Z, Corthay A et al.. How do CD4(+) T cells detect and eliminate tumor cells that either lack or express MHC class II molecules? Front Immunol 2014; 5:174; PMID:24782871; http://dx.doi.org/ 10.3389/fimmu.2014.00174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Steer HJ, Lake RA, Nowak AK, Robinson BW. Harnessing the immune response to treat cancer. Oncogene 2010; 29:6301-13; PMID:20856204; http://dx.doi.org/ 10.1038/onc.2010.437 [DOI] [PubMed] [Google Scholar]
- 41.Yong X, Xiao YF, Luo G, He B, Lu MH, Hu CJ, Guo H, Yang SM. Strategies for enhancing vaccine-induced CTL antitumor immune responses. J Biomed Biotechnol 2012; 2012:605045; PMID:23093850; http://dx.doi.org/ 10.1155/2012/605045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Piras V, Selvarajoo K. Beyond MyD88 and TRIF pathways in toll-like receptor signaling. Front Immunol 2014; 5:70; PMID:24605113; http://dx.doi.org/ 10.3389/fimmu.2014.00070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gandhapudi SK, Chilton PM, Mitchell TC. TRIF is required for TLR4 mediated adjuvant effects on T cell clonal expansion. PLoS One 2013; 8:e56855; PMID:23457630; http://dx.doi.org/ 10.1371/journal.pone.0056855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dadaglio G, Fayolle C, Zhang X, Ryffel B, Oberkampf M, Felix T, Hervas-Stubbs S, Osicka R, Sebo P, Ladant D et al.. Antigen targeting to CD11b+ dendritic cells in association with TLR4/TRIF signaling promotes strong CD8+ T cell responses. J Immunol 2014; 193:1787-98; PMID:25024388; http://dx.doi.org/ 10.4049/jimmunol.1302974 [DOI] [PubMed] [Google Scholar]
- 45.Wei F, Yang D, Tewary P, Li Y, Li S, Chen X, Howard OM, Bustin M, Oppenheim JJ. The alarmin HMGN1 contributes to antitumor immunity and is a potent immunoadjuvant. Cancer Res 2014; 74:5989-98; PMID:25205103; http://dx.doi.org/ 10.1158/0008-5472.CAN-13-2042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fang H, Ang B, Xu X, Huang X, Wu Y, Sun Y, Wang W, Li N, Cao X, Wan T. TLR4 is essential for dendritic cell activation and anti-tumor T-cell response enhancement by DAMPs released from chemically stressed cancer cells. Cell Mol Immunol 2014; 11:150-9; PMID:24362470; http://dx.doi.org/ 10.1038/cmi.2013.59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saenz R, Futalan D, Leutenez L, Eekhout F, Fecteau JF, Sundelius S, Sundqvist S, Larsson M, Hayashi T, Minev B et al.. TLR4-dependent activation of dendritic cells by an HMGB1-derived peptide adjuvant. J Transl Med 2014; 12:211; PMID:25123824; http://dx.doi.org/ 10.1186/1479-5876-12-211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chaganty BK, Murthy AK, Evani SJ, Li W, Guentzel MN, Chambers JP, Zhong G, Arulanandam BP. Heat denatured enzymatically inactive recombinant chlamydial protease-like activity factor induces robust protective immunity against genital chlamydial challenge. Vaccine 2010; 28:2323-9; PMID:20056182; http://dx.doi.org/ 10.1016/j.vaccine.2009.12.064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bonifait L, Grenier D. The SspA subtilisin-like protease of Streptococcus suis triggers a pro-inflammatory response in macrophages through a non-proteolytic mechanism. BMC Microbiol 2011; 11:47; PMID:21362190; http://dx.doi.org/ 10.1186/1471-2180-11-47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li X1, Syrovets T, Paskas S, Laumonnier Y, Simmet T. Mature dendritic cells express functional thrombin receptors triggering chemotaxis and CCL18/pulmonary and activation-regulated chemokine induction. J Immunol 2008; 181:1215-23; PMID:18606675; http://dx.doi.org/ 10.4049/jimmunol.181.2.1215 [DOI] [PubMed] [Google Scholar]
- 51.Ramelli G1, Fuertes S, Narayan S, Busso N, Acha-Orbea H, So A. Protease-activated receptor 2 signalling promotes dendritic cell antigen transport and T-cell activation in vivo. Immunology 2010; 129:20-7; PMID:19845798; http://dx.doi.org/ 10.1111/j.1365-2567.2009.03144.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fields RC1, Schoenecker JG, Hart JP, Hoffman MR, Pizzo SV, Lawson JH. Protease-activated receptor-2 signaling triggers dendritic cell development. Am J Pathol 2003; 162:1817-22; PMID:12759239; http://dx.doi.org/ 10.1016/S0002-9440(10)64316-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nishikawa H, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Curr Opin Immunol 2014; 27:1-7; PMID:24413387; http://dx.doi.org/ 10.1016/j.coi.2013.12.005 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.