

Abstract
A route to 3-benzylidene dihydrofurochromen-2-ones from 2H-chromenes is described. Lactonization of 2H-chromenes was achieved using a two-step cyclopropanation-rearrangement sequence. Subsequent conversion of these intermediates to the corresponding α-benzylidene lactones was achieved by lithium enolate Aldol reaction, followed by base-promoted elimination of the aldolate mesylates. The alkene geometry was found to be base-dependent. While KOtBu favored formation of the E-isomer, DBU showed a slight preference for the Z-isomer. In further studies, these 3-benzylidene dihydrofurochromen-2-ones were converted to polyaromatic structures possessing all the required functionality for biflavonoid synthesis.
Keywords: biflavonoids, donor-acceptor cyclopropanes, α-benzylidene lactones, chamaejasmine, isochamaejasmine
Graphical Abstract
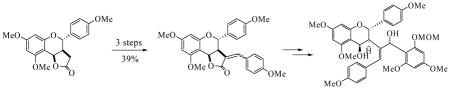
Introduction
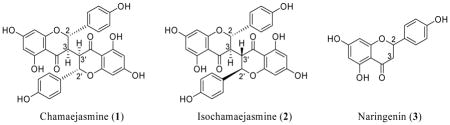
Biflavonoids represent a class of natural products that continue to attract attention both for their structural complexities and diverse biological activities. Two examples are chamaejasmine (1) and the related isochamaejasmine (2). Isolated from the root of Stellera chamaejasme L., a traditional Chinese herb, isochamaejasmine was found to alter several cell-signaling pathways, which could explain its use in the treatment of solid tumors, as well as tuberculosis.1 The same compound has been isolated from the yellow bark of B. zanguebarica, and was found to be active against several tumor cell lines including human myeloid leukemia cells.2 Structurally-related analogues of this compound have demonstrated antimalarial activity.3 Also isolated from more than one plant source, chamaejasmine has been shown to possess both anti-inflammatory activity and aldose reductase inhibiting activity,4 suggesting that it could be a potential treatment for the complications associated with diabetic neuropathy and related conditions. Structurally, chamaejasmine and isochamaejasmine are dimers of the flavanone narigenin (3) linked at C-3, giving them a C-3/C-3′ connectivity. Both natural products have a trans-trans configuration at the C-2/C-3 and C-2′/C-3′ positions, differing only in the absolute stereochemistry at C-2′ and C-3′.
In 2005, Li and Ma reported the first synthesis of dl-chamaejasmine using a biomimetic strategy.5 However, the key reductive dimerization step in this synthesis provided methyl-protected 1 in only a 10% yield. To date, a non-racemic synthesis of either compound has not been documented.
The goal of this research was to investigate a general strategy that could be used to target a range of biflavonoids, including compounds 1 and 2. Addressing this challenge, we viewed chalcones 5 as potential precursors to the basic biflavonoid templates 4 (Scheme 1). The key step would be an intramolecular oxa-Michael reaction of the deprotected alkoxide, which has been shown to occur with complete trans-selectivity in related systems.6 If these reactions prove to be stereospecific, cyclizations of the individual Z and E-chalcones could result in the formation of trans products, but with opposite stereochemistries at C-2′ and C-3′. Chalcone structures such as compounds 5, then, became our synthetic targets. Retrosynthetic analysis led us back to a new class of α-benzylidene lactones (6),7 which, we reasoned, should provide chalcones 5 upon cleavage with an o-alkoxy aryl lithium reagent.8 A number of strategies were considered for controlling the alkene stereochemistry in the α-benzylidene lactones, including the use of α-phosphonolactones such as 7. Yu and Wiemer have shown that the stereochemistry of Horner-Wadsworth-Emmons (HWE) olefinations of related α-phosphono-γ-lactones can be controlled to give either E- or Z-alkenes, depending on the reaction conditions used.9
Scheme 1.

Results and Discussion
Our starting materials were chromenes rac-8a and rac-8b (Scheme 2), which were prepared according to well-documented literature procedures.10 We have previously shown that 2-aryl 2H-chromenes can be stereoselectively lactonized in a two-step cyclopropanation-rearrangement sequence.11 Thus, treatment of chromene 8a with the diazo derivative 9a in the presence of catalytic Rh2(S-TBSP)4 gave the donor-acceptor cyclopropane 10, which rearranged to the α-carbomethoxy lactone 12 on treatment with Sn(OTf)2. Originally, we intended to use benzyl protective groups, but unfortunately chromene 8b was unreactive to cyclopropanation. An equal lack of reactivity was observed when MOM protective groups were used. A reaction we were particularly interested in was the cyclopropanation of 8a with phosphono-substituted diazo derivative 9b, as rearrangement of the product 11 would have given direct access to the target lactone 14. However, chromene 8a was completely unreactive to 9b under rhodium catalysis. Osipov and coworkers have reported some success in the CuI-mediated cyclopropanations of alkenes with α-trifluoromethyl-diazophosphonate in refluxing toluene,12 but this, too, failed in our hands. Instead, compound 14 was prepared by α-phosphorylation of lactone 13,13 obtained from compound 12 by decarboxylation with NaI in refluxing DMF.14
Scheme 2.

Unfortunately, all attempts at the HWE olefination of 14 with p-anisaldehyde using either the conditions reported by Yu and Wiemer (KHMDS, 18-crown-6)9 or other conditions (NaH, refluxing THF) failed to produce either of the desired α-benzylidene lactone products 16 (Scheme 3). Successful methylation of 14 to give 15, albeit in low yield, showed that enolate formation was occurring, which indicated that reaction with the aldehyde was problematic.
Scheme 3.

To try to circumvent this obstacle, we looked at an Aldol route to the same derivatives. We reasoned that if the aldol is stereoselective, a stereospecific elimination step would provide the α-benzylidene lactone diastereomerically enriched. However, this approach proved to be challenging. The boron enolate of lactone 13 provided a single aldol stereoisomer15 with p-anisaldehyde, but the yields were disappointing (Scheme 4).
Scheme 4.

By contrast, higher aldol yields were observed with the corresponding lithium enolate, although the reaction resulted in a mixture of stereoisomers 17 (Scheme 5). Moreover, base-induced elimination of the corresponding mesylates of this mixture was found to be base-dependent. With KOtBu, 16E was the predominant product, while DBU showed a slight preference for the formation of 16Z. Although the complete chromatographic separation of these stereoisomers was challenging, sufficient amounts of each were isolated to allow full spectral characterizations. Based on well-established literature precedent,9 16E was identified by the presence of an olefinic proton downfield (δ = 7.53 ppm) relative to the corresponding Z-isomer (δ = 5.89 ppm). Using this information, the E:Z isomeric ratio in product mixtures was determined from the integration of these olefinic protons in the NMR spectra.
Scheme 5.

The next goal was to transform these lactones into our chalcone targets using ring cleavage methodology (see Scheme 1). However, lactones 16 proved to be surprisingly resistant to modification. The mixture of lactones 16E and 16Z, for example, was completely unreactive to aryllithium reagents, thereby thwarting our original plans (see Scheme 1) and forcing us to consider alternative strategies. Hydrolysis with lithium hydroxide progressed to completion by tlc, but all attempts to isolate the hydroxy acid products led to ring closure back to the lactones. By comparison, reaction with LiAlH4 never went to completion, even after two days in the presence of a large excess of the reducing agent. Fortunately, however, reduction with Red-Al went to completion and gave a mixture of the diols 18E and 18Z in 83% yield (Scheme 6). These isomers were easily separable by column chromatography, and their stereochemical assignments were made by direct correlation with the previously determined structures of 16E and 16Z. All attempts to oxidize diol 18E to the corresponding keto-aldehyde resulted in conversion back to its lactone form. However, protection of both alcohol groups as their TES ethers, followed by DDQ oxidation gave aldehyde 19, by selective oxidation of the protected allylic alcohol.16 Addition of aryllithium reagent 2017 then provided compound 21 as an inseparable mixture of isomers, possessing all the necessary functionality required for biflavonoid synthesis. The 1H-NMR spectrum of this mixture was complex, but showed the expected loss of the aldehyde signal along with a gain of aromatic signals. The structure assignment was further supported by the HRMS data.
Scheme 6.

Conclusions
We have described the first synthesis of 3-benzylidene dihydrofurochromen-2-ones, and we have identified a potential strategy for the synthesis of complex biflavonoids such as chamaejasmine and isochamaejasmine. Because asymmetric routes to 2-aryl 2H-chromenes are well-documented,18 enantioselective syntheses of these natural products should eventually be possible using the methodologies described herein.
Experimental Details
All reactions were performed using oven-dried glassware under inert atmosphere (Ar or N2). Flash purification of compounds was conducted using Biotage-Isolera™ One. Anhydrous DMSO, DMF, and toluene were purchased from Sigma Aldrich in a SureSeal™ bottle and used without further purification. Other solvents and reagents (THF, DCM, Et3N, CH3CN) were dried over sodium benzophenone ketyl or calcium hydride and distilled before use. The 1H NMR and 13C NMR spectra were recorded on a Bruker-Avance 300 or 600 MHz instrument in CDCl3 and data are reported as chemical shift (δ) in ppm from tetramethylsilane as an internal standard. For 13C NMR spectra data are reported as δ in ppm from tetramethylsilane with the solvent as an internal indicator (CDCl3, 77.16 ppm). HRMS analyses were conducted on a Bruker Ultimate 3000, using ESI.
1-tert-butyl 1-methyl 5,7-dimethoxy-2-(4-methoxyphenyl)-1a,2-dihydrocyclopropa[c]chromene-1,1(7bH)-dicarboxylate (10)11a
To a solution of chromene 8a (175 mg, 0.55 mmol) and Rh2(S-TBSP)4 (8 mg, 0.055 mmol in DCM (5 mL) was added a solution of tert-butyl methyl malonate diazo (347 mg, 1.73 mmol) in DCM (3.3 mL) via syringe pump over a period of 3–4 hours. After the addition was complete the reaction mixture was stirred overnight. The solvent was removed under reduced pressure and the residue was purified by flash column chromatography using Et2O/hexane system as the eluent. Yield: 170 mg (66%); pale yellow oil; 1H NMR (300 MHz, CDCl3): δ 7.82 (d, J = 8.8 Hz, 2H), 6.82 (d, J = 8.7 Hz, 2H), 6.10 (d, J = 2.2 Hz, 1H), 5.84 (d, J = 2.2 Hz, 1H), 5.49 (s, 1H), 3.82 (s, 3H), 3.80 (s, 3H), 3.74 (s, 3H), 3.65 (s, 3H), 3.25 (d, J = 9.6 Hz, 1H), 2.52 (d, J = 9.6 Hz, 1H), 1.49 (s, 9H).
Methyl 7,9-dimethoxy-4-(4-methoxyphenyl)-2-oxo-3,3a,4,9b-tetrahydro-2H-furo[3,2-c]chromene-3-carboxylate (12)11a
To a solution of cyclopropane 10 (51 mg, 0.11 mmol) in dry DCM (2.2 mL) at 0 °C was added tin(II) triflate (23 mg, 0.054 mmol). The resulting solution was allowed to warm up to room temperature overnight. It was then quenched with water and the layers were separated. The aqueous layer was extracted with DCM (x3). The combined organic layers were washed with brine and dried over anhydrous Na2SO4. The solvent was removed in vacuo and the residue was purified by flash column chromatography. Yield: 45 mg (78%); yellow oil; 1H NMR (600 MHz, CDCl3): δ 7.36 (d, J = 8.6 Hz, 2H), 6.98 (d, J = 8.6 Hz, 2H), 6.14 (d, J = 2.1 Hz, 1H), 6.10 (d, J = 2.1 Hz, 1H), 5.83 (d, J = 4.8 Hz, 1H), 4.57 (d, J = 11.5 Hz, 1H), 3.86 (s, 3H), 3.84 (s, 3H), 3.76 (s, 6H), 3.29 (dd, J = 11.5, 4.9 Hz), 1H), 3.23 (s, 1H).
7,9-dimethoxy-4-(4-methoxyphenyl)-3,3a,4,9b-tetrahydro-2H-furo[3,2-c]chromen-2-one (13)
To a solution of lactone 12 (54 mg, 0.13 mmol) in DMF (2 mL) was added sodium iodide (58 mg, 0.39 mmol). Afte refluxing for 5 hours the solvent was removed under reduced pressure. The residue was then dissolved in water the extracted with EtOAc. The combined organic layers were washed with brine and dried over anhydrous sodium sulfate. The solvent was removed and the residue was purified by flash column chromatography. Yield: 42 mg (91%); yellow oil; 1H NMR (600 MHz, CDCl3): δ 7.34 (d, J = 8.6 Hz, 2H) 6.96-6.94 (d, J=8.6 Hz, 2H), 6.14 (d, J = 2.1Hz, 2H), 6.11 (d, J = 2.1 Hz, 2H), 5.60 (d, J = 4.8 Hz, 1H), 4.55 (d, J=11.5 Hz, 1H), 3.86 (s, 3H), 3.83 (s, 3H), 3.75 (s, 3H), 2.86-2.82 (m, 1H), 2.78 (dd, J = 17.8, 7.9 Hz, 1H), 2.25 (d, J = 17.8 Hz, 1H); 13C NMR (600MHz, CDCl3): δ 175.4, 162.5, 160.5, 160.2, 157.5, 129.2, 129.1, 114.3, 99.9, 93.1, 92.4, 72.2, 55.8, 55.4, 55.3, 39.1, 32.4; HRMS (ESI-TOF) m/z: [M+H]+ calcd for C20H21O6, 357.1333; found, 357.1328.
Diethyl (7,9-dimethoxy-4-(4-methoxyphenyl)-2-oxo-3,3a,4,9b-tetrahydro-2H-furo[3,2-c]chromen-3-yl)phosphonate (14)
To a solution of lactone 13 (22 mg, 0.062 mmol) in THF (0.5 mL) at −78 °C was added LHMDS (0.13 mL, 0.13 mmol, 1M in THF). After stirring the solution at that temperature for 2 hours TMEDA (20 μL, 0.13 mmol) and diethylchlorophosphite (20 μL, 0.14 mmol) was added and the reaction mixture was allowed to warm to room temperature over 3 hours. The reaction was then quenched by the slow addition of 1 M acetic acid in Et2O (2 mL). The resulted mixture was filtered through a celite pad and the pad was washed with Et2O. After concentration under reduced pressure the residue was stirred overnight with the flask was left open to air. The mixture was dissolved in Et2O, washed with saturated NaHCO3 and brine respectively and the organic layer was dried over anhydrous MgSO4. The solvent was removed under reduced pressure and the residue purified by flash column chromatography. Yield: 13 mg (43%) as a mixture of diastereomers; clear oil; 1H NMR (300 MHz, CDCl3): δ 7.37(d, J = 8.7 Hz, 2H), 6.96 (d, J = 8.6 Hz, 2H), 6.14 (d, J = 2.2 Hz, 1H), 6.09 (d, J = 2.2 Hz, 1H), 5.90 (d, J = 2.1 Hz, 1H), 4.53 (d, J = 5.0 Hz, 1H), 4.17-4.01 (m, 4H), 3.85 (s, 3H), 3.83 (s, 3H), 3.75 (s, 3H), 3.25-3.20 (m, 1H) 2.74 (d, J = 24.7, 1H), 1.31 (t, J = 7.1, 3 H), 1.23 (t, J = 7.0, 3H).
3-(hydroxy(4-methoxyphenyl)methyl)-7,9-dimethoxy-4-(4-methoxyphenyl)-3,3a,4,9b-tetrahydro-2H-furo[3,2-c]chromen-2-one (17)
To a solution of lactone 13 (295 mg, 0.83 mmol) in THF (2 mL) was added LHMDS (1.65 mL, 1.65 mmol, 1 M in THF) at −78 °C. After stirring for 2 hours at that temperature, p-anisaldehyde (200 μL, 1.65 mmol) was added. The solution was stirred for an additional 2 hours at −78 °C, after which the reaction was quenched by the addition of saturated aqueous NH4Cl and warmed to room temperature. The resultant mixture was then extracted with EtOAc (x3), and the combined organic layers were washed with brine and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the residue purified by flash column chromatography. Yield: 308 mg (76%) as a mixture of diastereomers; clear oil; 1H NMR (600 MHz, CDCl3): δ 6.91 (d, J = 8.3 Hz, 2H), 6.81 (d, J = 8.3 Hz, 2H), 6.65 (d, J = 8.3 Hz, 2H), 6.60 (d, J = 8.3 Hz, 2H), 6.11 (s, 1H), 6.03 (s, 1H), 5.76 (d, J = 5.5 Hz, 1H), 5.31 (s, 1H), 4.33 (d, J = 11.5 Hz, 1H), 3.84 (s, 3H), 3.80 (s, 3H), 3.78 (s, 3H), 3.72 (s, 3H), 2.83 (dd, J = 11.5, 5.5 Hz,1H), 2.46 (s, 1H), 2.25 (d, J = 3.5 Hz, 1H); 13C NMR (600MHz, CDCl3): 177.2, 162.2, 160.8, 159.6, 158.8, 157.3, 132.7, 128.8, 128.1, 125.8, 113.8, 113.7, 100.3, 99.9, 93.0, 92.4, 77.2, 72.9, 72.7, 55.7, 55.3, 55.1, 52.5, 39.1; other diastereomer: δ 6.97 (d, J = 7.7 Hz, 2H), 6.95 (d, J = 7.68 Hz, 2H), 6.77 (d, J = 7.7 Hz, 2H), 6.72 (d, J = 7.7 Hz, 2H), 6.10 (s, 1H), 6.04 (s, 1H), 5.50 (d, J = 5.3 Hz, 1H), 4.91 (d, J = 9.0 Hz, 1H), 4.42 (d, J = 10.9 Hz, 1H), 3.84 (s, 3H), 3.83 (s, 3H), 3.80 (s, 3H), 3.72 (s, 3H), 3.22 (s, 1H), 2.59 (d, J = 9.1, 1H), 2.56 (dd, J = 10.8, 5.4 Hz, 1H); 13C NMR (600MHz, CDCl3): 177.0, 162.5, 160.6, 160.0, 159.6, 157.2, 131.7, 129.0, 128.2, 127.6, 114.1, 114.0, 99.7, 93.1, 92.5, 76.7, 72.3, 71.5, 55.8, 55.4, 55.3, 55.2, 51.3, 41.7; HRMS (ESI-TOF) m/z: [M+H]+ calcd for C20H21O6, 493.1857; found, 493.1855.
7,9-dimethoxy-3-(4-methoxybenzylidene)-4-(4-methoxyphenyl)-3,3a,4,9b-tetrahydro-2H-furo[3,2-c]chromen-2-one (16)
Procedure A
To a solution of aldol product 17 (308 mg, 0.63 mmol) in DCM (4 mL) at 0 °C was added mesyl chloride (98 μL, 1.26 mmol) and Et3N (437 μL, 3.14 mmol). After stirring at room temperature for 1 hour, the reaction mixture was cooled to 0 °C and DBU (668 μL, 3.13 mmol) was added. The reaction was warmed to room temperature and stirred overnight. It was then diluted by the addition of distilled water and the layers were separated. The aqueous layer was extracted with DCM (x3), and the combined organic layers were washed with brine, dried over anhydrous Na2SO4 and filtered. The solvent was removed in vacuo and the residue purified by flash column chromatography. Yield: 242.9 mg (82%); yellow oil.
Procedure B
To a solution of of aldol product 17 (27 mg, 0.055 mmol) in DCM (0.5 mL) at 0 °C was added Et3N (38 μL, 0.27 mmol) and mesyl chloride (9 μL, 0.11 mmol). The mixture was warmed to room temperature and stirred for 1 hour. It was then quenched by the addition of distilled water. The layers were separated and the aqueous phase was extracted with DCM. The organic layer was then washed with brine, dried over anhydrous Na2SO4 and filtered. The solvent was the removed in vacuo and the residue dissolved in dry THF (7 mL). Potassium tert-butoxide (19 mg, 0.17 mmol) was then added and the mixture was refluxed until reaction was complete, as indicated by TLC. After completion the reaction mixture was diluted with distilled water and was extracted with EtOAc. The organic layer was washed with brine, dried over anhydrous MgSO4 and filtered. The solvent was removed in vacuo and the residue was purified by flash column chromatography. Yield: 16.2 mg (62%); pale yellow oil.
E- alkene (16E)
1H NMR (300 MHz, CDCl3): δ 7.53 (s, 1H), 7.18 (d, J = 8.6 Hz, 2H), 6.89 (d, J = 8.7 Hz, 2H), 6.59 (d, J = 8.8 Hz, 2H), 6.57 (d, J= 8.6 Hz, 2H), 6.17 (s, 2H), 5.5 (d, J = 4.7 Hz, 1H), 4.59 (d, J =10.8 Hz, 1H), 3.88 (s, 3H), 3.79 (s,3H), 3.77(s, 3H), 3.63 (s, 3H); 13C NMR (600 MHz, CDCl3): 172.0, 162.5, 160.7, 160.3, 159.7, 157.5, 139.8, 131.0, 129.4, 128.2, 126.2, 122.5, 113.6, 113.6, 99.9, 93.1, 92.5, 76.7, 70.1, 55.8, 55.4, 55.2, 55.0, 44.4; HRMS (ESI-TOF) m/z: [M+Na]+ calcd for C28H26NaO7, 497.1571; found, 497.1623. Z- alkene (16Z): 1H NMR (300 MHz, CDCl3): δ 7.66 (d, J = 8.7 Hz, 2H), 7.32 (d, J = 8.5 Hz, 2H), 6.94 (d, J = 8.6 Hz, 2H), 6.83 (d, J = 8.8 Hz, 2H), 6.16 (s, 2H), 5.90 (s, 1H), 5.60 (d, 5.1 Hz, 1H), 4.65 (d, J = 11.0 Hz, 1H), 3.88 (s, 3H), 3.85 (s,3H), 3.81 (s, 3H), 3.78 (s, 3H), 3.23 (dd, J = 11.0, 5.0 Hz, 1H); 13C NMR (600 MHz, CDCl3): 168.7, 162.4, 160.9, 160.8, 160.0, 157.6, 143.2, 133.0, 129.5, 129.1, 126.1, 121.3, 113.9, 113.5, 100.2, 93.1, 92.6, 77.6, 69.2, 55.8, 55.4, 55.4, 55.3, 48.5.
3-(3-hydroxy-1-(4-methoxyphenyl)prop-1-en-2-yl)-5,7-dimethoxy-2-(4-methoxyphenyl)chroman-4-ol (18)
Red-Al (0.66 mL, 2.05 mmol, 60% weight in toluene) was added dropwise to a solution of α-benzylidene lactone 16 (242.9 mg, 0.51 mmol) in THF (4.2 mL) at 0 °C. The reaction was warmed to room temperature and stirred for 3 hours. The reaction was then cooled to 0 °C and a saturated solution of Rochelle’s salt was added dropwise. The layers were separated and the organic layer was washed with a saturated solution of Rochelle’s salt. The combined aqueous layers were extracted with Et2O (x3), followed by washing of the combined organic layers with brine. The organic layer was dried over anhydrous MgSO4 and the solvent was removed under reduced pressure. The residue was purified by flash column chromatography. Yield: 204 mg (83%); clear oil (Z isomer: 73 mg; E isomer: 125 mg; mixture: 6 mg). E Isomer (18E): 1H NMR (600 MHz, CDCl3): δ 7.11 (d, J = 8.7 Hz, 2H), 7.03 (d, J = 8.3 Hz, 2H), 6.87 (d, J = 8.3 Hz, 2H), 6.82 (d, J = 8.7 Hz, 2H), 6.55 (s, 1H), 6.10 (d, J = 2.2 Hz, 1H), 6.02 (d, J = 2.2 Hz, 1H), 5.35 (d, J = 11.1 Hz, 1H), 5.12 (d, J = 3.3 Hz, 1H), 4.11 (d, J = 12.0 Hz, 1H), 3.89 (s, 3H), 3.81 (s, 3H), 3.80 (s, 3H), 3.77 (d, J = 12.4 Hz, 1H), 3.71 (s, 3H), 3.58 (dd, J = 11.0, 3.42 Hz, 1H); 13C NMR (600MHz, CDCl3): δ 161.4, 159.8, 158.8, 158.5, 155.9, 138.0, 134.8, 130.2, 129.8, 129.5, 129.4, 113.8, 113.7, 105.9, 93.1, 91.7, 75.8, 65.1, 64.1, 60.3, 55.6, 55.3, 55.2, 55.1, 44.3; HRMS (ESI-TOF) m/z: [M] + calcd for C28H30O7, 478.1992; found, 478.1963. Z Isomer (18Z): 1H NMR (600 MHz, CDCl3): δ 7.35 (d, J = 8.7 Hz, 2H), 7.27 (d, J = 8.8 Hz, 2H), 6.86 (d, J = 8.7 Hz, 2H), 6.82 (d, J = 8.8 Hz, 2H), 6.47 (s, 1H), 5.40 (d, J = 11.2 Hz, 1H), 5.11 (d, J = 3.4 Hz, 1H), 3.88 (s, 2H), 3.85 (s, 3H), 3.77 (s, 3H), 3.77 (s, 3H), 3.74 (s, 3H), 3.15 (dd, J = 11.2, 3.2 Hz, 1H); 13C NMR (600MHz, CDCl3): δ 161.5, 159.9, 158.9, 158.7, 155.9, 136.7, 135.3, 130.4, 130.4, 129.5, 129.1, 113.9, 113.5, 105.9, 93.2, 91.7, 75.6, 64.9, 58.2, 55.6, 55.4, 55.2, 52.7; HRMS (ESI-TOF) m/z: [M] + calcd for C28H30O7, 478.1992; found, 478.1963.
(E)-((5,7-dimethoxy-2-(4-methoxyphenyl)-3-(1-(4-methoxyphenyl)-3-((triethylsilyl)oxy)prop-1-en-2-yl)chroman-4-yl)oxy)triethylsilane
To a solution of diol 18E (125 mg, 0.26 mmol), imidazole (110 mg, 1.62 mmol), and 4-DMAP (12.7 mg, 0.1 mmol) in dry DMF (8 mL) at 0 °C was added TESCl (184 μL, 1.1 mmol). The reaction was then warmed to rt and stirred overnight. It was then poured into a saturated solution of NaHCO3 and extracted with Et2O (x3). The organic layer was then washed with brine and dried over anhydrous MgSO4. The solvent was removed and the residue was purified by flash column chromatography. Yield: 139 mg (75 %); clear oil. 1H NMR (600 MHz, CDCl3): 7.11 (d, J = 7.7 Hz, 2H), 6.97 (d, J = 7.8 Hz, 2H), 6.82-6.81 (m, 4H), 6.65 (s, 1H), 5.99 (s, 1H), 5.93 (s, 1H), 5.49 (d, J =11.6 Hz, 1H), 5.22 (s, 1H), 4.73 (d, J = 15.8 Hz, 1H), 3.91 (d, J = 15.8 Hz, 1H), 3.81 (s, 3H), 3.792 (s, 3H), 3.784 (s, 3H), 3.680 (s, 3H), 3.46 (d, J = 11.6 Hz, 1H), 0.90 (t, J = 7.7 Hz, 9H), 0.85 (t, J = 7.6 Hz, 9H), 0.63-0.53 (m, 6H), 0.463 (q, J = 7.8 Hz, 6H). 13C NMR (600 MHz, CDCl3): 161.1, 159.8, 157.8, 157.7, 155.9, 139.3, 130.9, 130.8, 129.8, 129.7, 125.1, 113.8, 113.7, 106.8, 92.8, 90.6, 75.3, 64.8, 55.3, 55.2, 55.1, 54.8, 43.7, 6.9, 6.7, 5.0, 4.3; HRMS (ESI-TOF) m/z: [M+Na]+ calcd for C40H50NaSi2, 729.3613; found, 729.3606.
((E)-2-(5,7-dimethoxy-2-(4-methoxyphenyl)-4-((triethylsilyl)oxy)chroman-3-yl)-3-(4-methoxyphenyl)acrylaldehyde (19)
To a solution of bis-protected triethylsiyl ether (36.4 mg, 0.05 mmol) in DCM at 0 °C was added DDQ (12.8 mg, 0.06 mmol) and phosphate pH 7 buffer (0.18 mL). The solution was warmed to room temperature and monitored for completion. Upon completion, saturated NaHCO3 was added and the layers were separated. The aqueous phase was extracted with Et2O (x3). The combined organic layers were then washed with brine and dried over MgSO4. The solvent was then removed under reduced pressure and the residue was purified by flash column chromatography. Yield: 20.7 mg (68%; 79% based on recovered starting material); clear oil. 1H NMR (600 MHz, CDCl3): δ 9.57 (s. 1H), 7.30 (s, 1H), 7.22 (d, J = 8.0 Hz, 2H), 7.18 (d, J = 7.9 Hz, 2H), 6.91 (d, J = 8.3 Hz, 2H), 6.77 (d, J = 7.9 Hz, 2H), 6.14 (d, J = 11.0 Hz, 1H), 6.05 (s, 2H), 5.20 (d, J = 2.1 Hz, 1H), 3.83 (s, 3H), 3.82 (s, 3H), 3.76 (s, 3H), 3.73 (s, 3H), 3.66 (d, J = 10.9 Hz, 1H), 0.85 (t, J = 8.0, 9 H), 0.62-0.49 (m, 6H); 13C NMR (600 MHz, CDCl3): 193.0, 161.3, 160.3, 159.7, 159.7, 158.4, 145.2, 131.2, 130.7, 129.6, 114.4, 113.8, 106.3, 92.9, 91.0, 74.8, 64.1, 59.7, 55.3, 55.2, 55.2, 54.7, 6.9, 4.8; HRMS (ESI-TOF) m/z: [M+H]+ calcd for C34H43O7Si, 591.2773; found, 591.2721.
(E)-2-(5,7-dimethoxy-2-(4-methoxyphenyl)-4-((triethylsilyl)oxy)chroman-3-yl)-1-(2,4-dimethoxy-6-(methoxymethoxy)phenyl)-3-(4-methoxyphenyl)prop-2-en-1-ol (21)
To a solution of aldehyde 19 (24.5 mg, 0.04 mmol) in toluene (0.3 mL) at −78 °C, was added a freshly prepared solution of aryllithium 20 (0.41 mL, 0.3 M in toluene, 0.12 mmol). The reaction mixture was stirred at −78 °C for 15 mins and then warmed to −50 °C over 30 mins, and then to rt over 2 hours. The reaction was then quenched by the addition of a saturated solution of NH4Cl and extracted with EtOAc (x3). The combined organic layers were then washed brine and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the residue purified by flash column chromatography. Yield: 23.1 mg as a mixture of inseparable diastereomers (71%); HRMS (ESI-TOF) m/z: [M+Na]+ calcd for C44H56NaO11Si, 811.3490; found, 811.3447.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health, through Grant R15 GM088840-01, for financial support of this research. The NMR data were made possible through NSF CRIF Award 0840220, which supported the acquisition of the 600 MHz NMR.
References
- 1.Tian Q, Li J, Xie X, Sun M, Sang H, Zhou C, An T, Hu L, Ye RD, Wang M-W. Stereospecific induction of nuclear factor-κ B activation by isochamaejasmin. Mol Pharmacol. 2005;68:1534–1542. doi: 10.1124/mol.105.014720. [DOI] [PubMed] [Google Scholar]
- 2.Moeller M, Suschke U, Nolkemper S, Schneele J, Distl M, Sporer F, Reichling J, Wink M. Antibacterial, antiviral, antiproliferative and apoptosis-inducing properties of Brackenridgea zanguebarica (Ochnaceae) J Pharm Pharmacol. 2006;58:1131–1138. doi: 10.1211/jpp.58.8.0015. [DOI] [PubMed] [Google Scholar]
- 3.Ichino C, Kiyohara H, Soonthornchareonnon N, Chuakul W, Ishiyama A, Sekiguchi H, Namatame M, Otoguro K, Omura S, Yamada H. Antimalarial activity of biflavonoids from Ochna integerrima. Planta Medica. 2006;72:611–614. doi: 10.1055/s-2006-931569. [DOI] [PubMed] [Google Scholar]
- 4.Feng B, Wang T, Zhang Y, Hua H, Jia J, Zhang H, Pei Y, Shi L, Wang Y. Aldose reductase inhibitors from Stellera chamaejasme. Pharmaceutical Biol. 2005;43:12–14. [Google Scholar]
- 5.Li W-DZ, Ma B-C. A Simple Biomimetic Synthesis of dl-Chamaejasmine, a Unique 3,3′-Biflavanone. Org Lett. 2005;7:271–274. doi: 10.1021/ol047718u. [DOI] [PubMed] [Google Scholar]
- 6.Donnelly DMX, Fitzpatrick BM, Ryan SM, Finet J-P. Aryllead Triacetates as Synthons for the Synthesis of BiflavonoidsPart 2. Synthesis of a Garcinia-Type Biflavonoid. J Chem Soc Perkin Trans 1. 1994:1797–1801. [Google Scholar]
- 7.For the synthesis of a related α-methylene lactone see: Ozaki Y, Mochida K, Kim SW. Total Synthesis of Sophorapterocarpan A, Maackian, and Anhydropisatin: Application of a 1,3-Michael-Claisen Annulation to Aromatic Synthesis. J Chem Soc, Perkin Trans 1. 1989;7:1219–1224.
- 8.For an example of a related reaction, see: Cromwell MEM, Gebhard R, Li X-Y, Batenburg ES, Hopman JCP, Lugtenburg J, Mathies RA. Synthesis and Vibrational Analysis of a Locked 14-s-cis Conformer of Retinal. J Am Chem Soc. 1992;114:10860–10869.
- 9.Yu JS, Wiemer DF. Temperature Effects on Stereocontrol in the Horner-Wadsworth-Emmons Condensation of α-Phosphono Lactones. J Org Chem. 2007;72:6263–6265. doi: 10.1021/jo070722+. [DOI] [PubMed] [Google Scholar]
- 10.8a: Aponte JC, Verastegui M, Malaga E, Zimic M, Quiliano M, Vaisberg AJ, Gilman RH, Hammond GB. Synthesis, Cytotoxicity, and Anti-Trypanosoma cruzi Activity of New Chalcones. J Med Chem. 2008;51:6230–6234. doi: 10.1021/jm800812k.Stokes S, Mustain R, Pickle L, Mead KT. Rhodium-catalyzed cyclopropanations of 2-aryl-2H-chromenes with dialkyl malonate esters. A comparison of a-diazo derivatives and phenyliodonium ylides. Tetrahedron Lett. 2012;53(30):3890–3893.8b: Hatakeyama K, Ohmori K, Suzuki K. Synthesis of (±)-Lotthanongine, a Novel Natural Product with a Flavan-Indole Hybrid Structure. Synlett. 2005:1311–1315.
- 11.(a) Stokes S, Mustain R, Pickle L, Mead KT. Rhodium-catalyzed cyclopropanations of 2-aryl-2H-chromenes with dialkyl malonate esters. A comparison of a-diazo derivatives and phenyliodonium ylides. Tetrahedron Lett. 2012;53(30):3890–3893. [Google Scholar]; (b) Stokes S, Spears B, Laseter C, Barker B, Mead KT. γ-Lactonizations of 2H-Chromenes via Cyclopropanation. Tetrahedron Lett. 2010;51(31):4003–4006. [Google Scholar]
- 12.Titanyuk ID, Beletskaya IP, Peregudov AS, Osipov SN. Trifluoromethylated cyclopropanes and epoxides from CuI-mediated transformations of α-trifluoromethyl-diazophosphate. J Fluorine Chem. 2007;128(7):723–728. [Google Scholar]
- 13.Lee K, Wiemer DF. Reaction of diethyl phosphorochloridite with enolates: a general method for synthesis of beta-keto phosphonates and alpha-phosphono esters through carbon-phosphorus bond formation. J Org Chem. 1991;56(19):5556–60. [Google Scholar]
- 14.Krapcho AP, Ciganek E. Krapcho Dealkoxycarbonylation Reaction of Esters with α-Electron-Withdrawing Substituents. Org Reactions. 2013;81:1–535. [Google Scholar]
- 15.The stereochemistry was assigned by comparison to related Aldol reactions. See: Ito H, Momose T, Konishi M, Yamada E, Watanabe K, Iguchi K. Enantioselective total synthesis of both diastereomers of preclavulone-A methyl ester. Tetrahedron. 2006;62:10425–10433.
- 16.Paterson I, Cowden CJ, Rahn VS, Woodrow MD. A Facile Oxidation/Deprotection of Electron Rich Silyl Ethers Using DDQ. Synlett. 1998:915–917. [Google Scholar]
- 17.Hodgetts KJ. Inter- and intramolecular Mitsunobu reaction based approaches to 2-substituted chromans and chroman-4-ones. Tetrahedron. 2005;61:6860–6870. [Google Scholar]
- 18.Wipf P, Weiner WS. Enantioselective Synthesis and Photoracemization Studies of (+)-2-Cyclopropyl-7,8-dimethoxy-2H-chromene-5-carboxylic Acid Methyl Ester, an Advanced Intermediate of a Dihydrofolate Reductase Inhibitor. J Org Chem. 1999;64:5321–5324. doi: 10.1021/jo990352s. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.