

Abstract
Objective
To assess the efficacy/safety of the B-lymphocyte stimulator inhibitor belimumab/standard-of-care (SOC) versus placebo/SOC in active systemic lupus erythematosus (SLE).
Methods
In a multicenter, randomized, controlled, phase 3 trial, 819 antinuclear antibody- or anti-dsDNA-positive SLE patients with Safety of Estrogens in Lupus Erythematosus National Assessment–SLE Disease Activity Index (SELENA-SLEDAI) ≥ 6 were randomized (1:1:1 ratio) to receive intravenous belimumab 1 or 10 mg/kg, or placebo on days 0, 14, and 28, and then every 28 days for 72 weeks. Primary efficacy analyses: SLE Responder Index (SRI) at week 52 (≥ 4-point reduction in SELENA-SLEDAI; no new British Isles Lupus Assessment Group A and < 2 new B organ domain scores; no worsening in Physician’s Global Assessment).
Results
Belimumab 10 mg/kg plus SOC met the primary efficacy endpoint: significantly greater SRI response at week 52 than placebo (43.2% versus 33.5%; P = 0.017); the rate with belimumab 1 mg/kg was 40.6% (P = 0.089). Week-76 response rates: 32.4%, 39.1%, and 38.5% with placebo, and belimumab 1 and 10 mg/kg, respectively. In post-hoc sensitivity analyses evaluating higher SELENA-SLEDAI thresholds, belimumab 10 mg/kg achieved better discrimination at weeks 52/76. Risk of severe SELENA-SLEDAI flares over 76 weeks was reduced with belimumab 1 mg/kg (34%; P = 0.023) and 10 mg/kg (23%; P = 0.13). Serious and severe adverse events including infections, laboratory abnormalities, malignancies, and deaths, were comparable across groups.
Conclusion
Belimumab plus SOC significantly improved SRI response rate, reduced SLE disease activity and severe flares, and was generally well-tolerated in SLE.
Systemic lupus erythematosus (SLE) is a chronic autoimmune disorder that affects a variety of organ systems and markedly impairs health-related quality of life (1–4). While available therapies, such as corticosteroids, hydroxychloroquine, and immunosuppressive drugs, have improved the outcomes of patients with SLE, there remains a significant unmet need for safe and more effective treatments. A novel approach to address immune abnormalities in SLE is to inhibit B-lymphocyte stimulator (BLyS; also known as B-cell activating factor of the tumor necrosis factor ligand family), a key survival cytokine for B cells (5–8). Overexpression of BLyS promotes survival of B cells (including autoreactive B cells), whereas inhibition of BLyS results in autoreactive B-cell apoptosis (9,10). Elevated circulating BLyS levels are common in SLE, and correlate with increased SLE disease activity and elevated anti–double-stranded DNA (anti-dsDNA) antibody concentrations (11–13).
Belimumab is a fully human immunoglobulin (Ig)–G1-λ monoclonal antibody that binds soluble human BLyS and inhibits its biologic activities. The clinical and pharmacodynamic effects of belimumab were evaluated in a phase 2 study in patients with active SLE who were receiving standard of care (SOC) therapies (14). Reductions in circulating CD20+ B lymphocytes, short-lived plasma cells, and anti-dsDNA antibody titers were observed. A post-hoc analysis identified a subset of seropositive (antinuclear antibody [ANA] ≥ 1:80 and/or anti-dsDNA ≥ 30 IU/mL) patients (71.5% of the original cohort) in whom belimumab reduced SLE disease activity compared with placebo. In a 5-year, open-label extension of this study, improvement in SLE disease activity was sustained in the seropositive subset remaining on treatment (15). Furthermore, flare frequencies and autoantibody levels declined, and rates of adverse events (AEs), including infectious and serious AEs, remained stable or decreased over the 5-year period. Post-hoc analysis of the phase 2 results led to the development of the SLE Responder Index (SRI), which reflects improvement in disease activity using a global scoring system, while simultaneously requiring that there be no worsening of the disease in any organ system or by physician judgment (16).
With the SRI at week 52 as the primary endpoint, belimumab was evaluated in two phase 3 trials comparing belimumab 1 and 10 mg/kg plus SOC with placebo plus SOC in patients with seropositive active SLE. In BLISS-52, a 52-week trial conducted primarily in Asia, South America, and Eastern Europe, belimumab was well tolerated, reduced SLE disease activity, prevented flares, improved serologic activity in serologically active patients, and reduced corticosteroid use (17). BLISS-76, the second phase 3 clinical trial of belimumab in SLE, was conducted primarily in North America and Europe. Treatment continued through week 72, with the final evaluation at week 76. The results on efficacy, safety, tolerability, and biologic markers are presented from the BLISS-76 trial.
PATIENTS AND METHODS
Study design
In this phase 3, multicenter, randomized, double-blind, placebo-controlled trial, patients with SLE on SOC therapy were assigned to receive placebo, or belimumab 1 or 10 mg/kg by intravenous (IV) infusion over 1 hour on days 0, 14, and 28, and every 28 days through week 72. While the initiation of an immunosuppressive (IS) drug was prohibited during the trial, the addition of a new antimalarial (AM) drug and dose increases of concomitant IS or AM drugs were permitted until week 16. After week 16, however, the maximum doses of IS or AM drugs could be no greater than the higher of the baseline or week-16 dose. For corticosteroids, any dose was permitted through week 24; thereafter through week 44, the dose had to be within 25% or 5 mg of baseline. Between weeks 44 and 52, no increase over the higher of the baseline or week-44 dose was permitted. From weeks 52 through 68, the dose had to be within 25% or 5 mg of baseline, and an increase over the higher of the baseline or week-68 dose was prohibited after week 68. Prednisone could be reduced at the discretion of the investigator. As in the companion phase 3 BLISS-52 trial, the addition of a new biologic agent at any time, an inhibitor of the renin-angiotensin system after 4 months, or a new 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitor after 6 months was prohibited; other antihypertensive or lipid-lowering agents were allowed during the study (17). The Safety of Estrogens in Lupus Erythematosus National Assessment–SLE Disease Activity Index (SELENA-SLEDAI) (18), Physician’s Global Assessment (PGA) (18), British Isles Lupus Assessment Group (BILAG) (19,20), and SLE Flare Index (SFI) (21) were evaluated every 4 weeks (except weeks 56 and 64), as were AEs, vital signs, concomitant medications, and laboratory and pregnancy tests.
Entry criteria
Enrolled patients were required, at a minimum, to meet the following criteria: 1) age ≥ 18 years; 2) a diagnosis of SLE according to the revised American College of Rheumatology criteria (22); 3) active disease (SELENA-SLEDAI score ≥ 6) at screening (18); and 4) seropositivity as defined by 2 positive ANA or anti-dsDNA test results (ANA titers ≥ 1:80 and/or anti-dsDNA antibodies ≥ 30 IU/mL), of which ≥ 1 test result had to be obtained during screening. The study entry criteria were identical to those in BLISS-52 (17). BLISS-76 enrolled patients from Europe and North/Central America (136 centers in 19 countries). A stable treatment regimen was required for ≥ 30 days before the first study dose; stable treatment could include prednisone (or equivalent) alone (7.5 to 40 mg/d) or combined (0 to 40 mg/d) with AM drugs, nonsteroidal anti-inflammatory drugs, and/or IS therapies. Exclusion criteria included serious intercurrent illness, severe active lupus nephritis, severe central nervous system manifestations, and pregnancy. Prior treatment with a B-cell–targeted therapy was exclusionary, as was any investigational biologic agent within 1 year of screening or investigational nonbiologic agent within 60 days. Additional medication exclusions included IV cyclophosphamide within 6 months, and a tumor necrosis factor inhibitor, anakinra, IV Ig, prednisone > 100 mg/d, or plasmapheresis within 3 months of screening, as well as immunization with a live vaccine within 1 month. Each site was required to obtain ethics committee/institutional review board approval of the final study protocol. Patients’ rights, safety, and well-being were protected based on the principles of the Declaration of Helsinki. Informed consent was obtained from each patient prior to study screening. The first patient was randomized in February 2007, and follow-up of the last patient was completed in March 2010.
Randomization
With the exception of unblinded site pharmacists or designees whose responsibilities were restricted solely to receiving, preparing, and dispensing the study agent, all study site personnel and patients, as well as sponsor and clinical research organization personnel, were blinded to trial agent assignments. Separate study monitors were responsible for the blinded (clinical) and unblinded (study agent preparation) components of the trial. After screening, eligible patients were randomly assigned via a centralized interactive voice response system to 1 of 3 treatment groups in a 1:1:1 ratio. At randomization, patients were stratified by screening SELENA-SLEDAI score (6 to 9 versus ≥ 10), proteinuria (< versus ≥ 2 g/24 h), and race (African or indigenous American descent versus other).
Efficacy measures
The primary efficacy endpoint was the SRI response rate at week 52. An SRI response was defined as a ≥ 4-point reduction in SELENA-SLEDAI score, no new BILAG A organ domain score and no more than 1 new BILAG B score (19,20), and no worsening (increase < 0.3) in PGA score versus baseline (18). Major secondary endpoints were SRI response rate at week 76, percentage of patients with a ≥ 4-point reduction from baseline in SELENA-SLEDAI score at week 52, change in PGA at week 24, change in SF-36® v2 Health Survey physical component summary (PCS) score at week 24, and percentage of patients with a mean prednisone dose that was decreased ≥ 25% from baseline and was ≤ 7.5 mg/d during weeks 40 to 52. Disease activity was also assessed with the SFI (21), modified to exclude the single criterion of increased SELENA-SLEDAI score to > 12 as defining severe flare (18).
Biologic markers
Assessed biologic markers included serum Ig, complement (C3 and C4), autoantibodies (eg, anti-dsDNA and ANA), and B- and T-cell subsets. Serologic assays were performed using enzyme-linked immunosorbent assay, except ANA, which was determined by indirect immunofluorescence on HEP-2 cells (Quest Diagnostics, Van Nuys, California). Peripheral blood lymphocytes were collected at baseline, and weeks 8, 24, 52, and 76, forwarded to a central fluorescence-activated cell-sorting facility (Nichols Laboratory, La Jolla, California), and stained with antibodies to quantitate B cells and B-cell subsets, and T cells and T-cell subsets (CD3, CD4, and CD8).
Safety
Adverse events were coded according to Medical Dictionary for Regulatory Activities version 12.0 preferred term or system organ class and were graded for severity using the Adverse Event Severity Grading Tables, modified from the Division of Microbiology and Infectious Diseases Adult Toxicity Tables (23).
Statistics
The primary efficacy endpoint compared SRI response rates at week 52 between each belimumab treatment group and the placebo group using a logistic-regression model adjusted for baseline randomization stratification factors. Patients who withdrew from the study or had changes in concomitant medications that were restricted by the protocol were considered treatment failures. Analysis was performed in a modified intention-to-treat population defined as all randomized patients who received ≥ 1 dose of study agent. The target sample size of 810 patients (270/group) was based on providing ≥ 90% power at the 5% significance level to detect ≥ 14% absolute improvement in SRI response rate at week 52 for belimumab 10 mg/kg relative to placebo. The primary efficacy analyses used a step-down procedure to control for the overall type 1 error (2-sided α = 0.05), comparing belimumab 10 mg/kg with placebo and then 1 mg/kg with placebo if 10 mg/kg was superior. Four sensitivity analyses of the primary endpoint at week 52 were prespecified in the protocol and analysis plan, including an unadjusted analysis, last-observation-carried-forward analysis, completer analysis, and per-protocol analysis (which excluded patients with major protocol violations). For secondary endpoints, analyses of categorical variables were performed using a logistic-regression model. Analysis of covariance was used for continuous variables, such as PGA changes from baseline to week 24. The analyses were adjusted for baseline stratification factors.
RESULTS
Patient disposition and demographics
Of 1353 patients screened, 826 were randomized, and 819 received ≥1 dose of study treatment (Figure 1). Baseline demographics, SLE disease characteristics, and medications were generally well balanced across treatment groups (Table 1). There were no differences among groups in discontinuation rates. Withdrawal rates in the placebo, and belimumab 1- and 10-mg/kg groups were 25.5%, 20.3%, and 23.4%, respectively, at week 52, and 32.4%, 26.6%, and 30.0%, respectively, at week 76; reasons for discontinuation were similar across groups.
Figure 1.
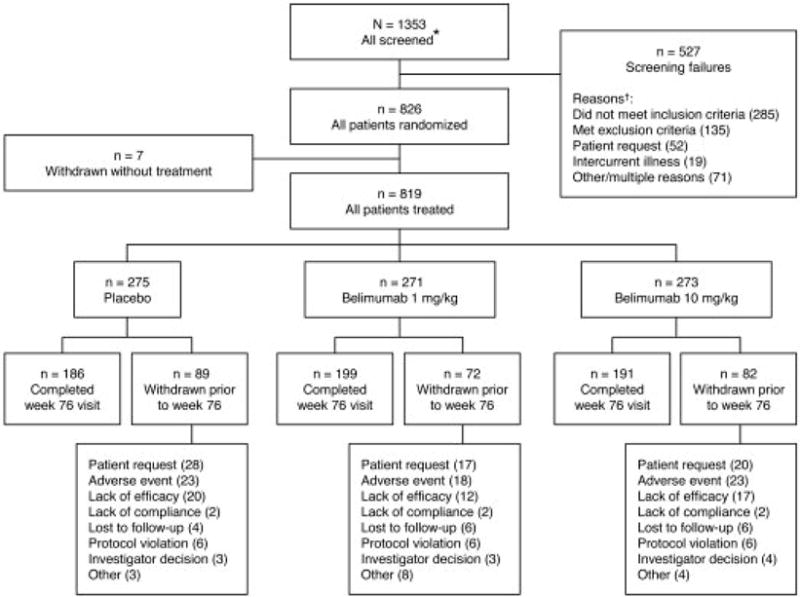
Flow diagram of patient disposition during study. * May count patients more than once if rescreened. † Multiple reasons for some patients.
Table 1.
Baseline demographics and clinical characteristics of treated patients
| Characteristics | Placebo (n = 275) |
Belimumab 1 mg/kg (n = 271) |
Belimumab 10 mg/kg (n = 273) |
|---|---|---|---|
| Female sex, n (%) | 252 (91.6) | 253 (93.4) | 259 (94.9) |
| Race, n (%)* | |||
| Indigenous American† | 36 (13.1) | 33 (12.2) | 34 (12.5) |
| White/Caucasian | 188 (68.4) | 192 (70.8) | 189 (69.2) |
| Black/African-American | 39 (14.2) | 40 (14.8) | 39 (14.3) |
| Asian | 11 (4.0) | 6 (2.2) | 11 (4.0) |
| Hispanic or Latino origin‡ | 55 (20.0) | 62 (22.9) | 56 (20.5) |
| Age, mean ± SD (y) | 40.0 ± 11.9 | 40.0 ± 11.4 | 40.5 ± 11.1 |
| SLE disease activity | |||
| Disease duration, mean ± SD (y) | 7.4 ± 6.7 | 7.9 ± 7.1 | 7.2 ± 7.5 |
| SELENA-SLEDAI, mean ± SD | 9.8 ± 4.0 | 9.7 ± 3.7 | 9.5 ± 3.6 |
| SELENA-SLEDAI ≥10, n (%) | 140 (50.9) | 144 (53.1) | 136 (49.8) |
| ≥1 BILAG A or 2 B scores, n (%) | 187 (68.0) | 173 (63.8) | 160 (58.6) |
| PGA score, mean ± SD | 1.5 ± 0.5 | 1.4 ± 0.5 | 1.4 ± 0.5 |
| Organ involvement | |||
| BILAG A/B organ domain scores at baseline, n (%) | |||
| Musculoskeletal | 195 (70.9) | 177 (65.3) | 179 (65.6) |
| Mucocutaneous | 178 (64.7) | 159 (58.7) | 141 (51.6) |
| Hematology | 35 (12.7) | 40 (14.8) | 35 (12.8) |
| General | 38 (13.8) | 30 (11.1) | 38 (13.9) |
| Vasculitis | 30 (10.9) | 23 (8.5) | 18 (6.6) |
| Renal | 21 (7.6) | 14 (5.2) | 24 (8.8) |
| Cardiovascular/respiratory | 9 (3.3) | 13 (4.8) | 15 (5.5) |
| Neurological | 6 (2.2) | 7 (2.6) | 7 (2.6) |
| SLICC damage index score, mean ± SD | 1.0 ± 1.5 | 1.0 ± 1.4 | 1.0 ± 1.4 |
| Proteinuria, mean ± SD (g/24 h) | 0.4 ± 0.8 | 0.3 ± 0.7 | 0.4 ± 0.7 |
| Proteinuria ≥2g/24 h, n (%) | 11 (4.0) | 7 (2.6) | 15 (5.5) |
| Medications | |||
| Daily prednisone use, n (%) | 212 (77.1) | 211 (77.9) | 200 (73.3) |
| >7·5 mg/d at baseline, n (%) | 126 (45.8) | 130 (48.0) | 120 (44.0) |
| Prednisone, mean ± SD (mg/d) | 9.4 ± 8.9 | 8.7 ± 7.6 | 8.4 ± 7.9 |
| Any immunosuppressive use, n (%)§ | 154 (56.0) | 153 (56.5) | 148 (54.2) |
| Mycophenolate | 42 (15.2) | 45 (16.6) | 50 (18.3) |
| Azathioprine | 57 (20.7) | 52 (19.2) | 58 (21.2) |
| Methotrexate | 60 (21.9) | 53 (19.6) | 39 (14.3) |
| Antimalarial (aminoquinolone) use, n (%) | 180 (65.5) | 171 (63.1) | 168 (61.5) |
| Biomarkers | |||
| BLyS ALOD, n (%)‖ | 269 (98.9) | 268 (98.9) | 263 (98.1) |
| ANA ≥ 1:80, n (%) | 253 (92.0) | 256 (94.5) | 245 (89.7) |
| Anti-dsDNA ≥30 IU/mL, n (%) | 174 (63.3) | 171 (63.1) | 179 (65.6) |
| Anti-dsDNA, mean ± SD (IU/mL)¶ | 556 ± 931 | 451 ± 748 | 551 ± 911 |
| C3, mean ± SD (mg/L) | 958 ± 303 | 995 ± 321 | 973 ± 325 |
| C3 below LLN (<900 mg/L), n (%) | 116 (42) | 100 (37) | 115 (42) |
| C4, mean ± SD (mg/dL) | 16 ± 9 | 17 ± 10 | 16 ± 10 |
| C4 below LLN (<16 mg/dL), n (%) | 143 (52) | 141 (52) | 147 (54) |
| IgG, mean ± SD (g/L) | 16 ± 6 | 16 ± 7 | 15 ± 6 |
| IgA, mean ± SD (g/L) | 3.0 ± 1.5 | 2.9 ± 1.5 | 3.0 ± 1.5 |
| IgM, mean ± SD (g/L) | 1.1 ± 0.7 | 1.1 ± 0.7 | 1.2 ± 0.9 |
| B-cell subsets | |||
| CD19+, mean ± SD (/mm3) | 137 ± 140 | 132 ± 188 | 134 ± 125 |
| CD20+, mean ± SD (/mm3) | 136 ± 140 | 128 ± 187 | 131 ± 125 |
| CD20-/CD27BRIGHT (short-lived plasma cells), mean ± SD (/mL)# | 568 ± 770 | 473 ± 694 | 674 ± 1009 |
| T-cell subsets | |||
| CD3+, mean ± SD (GI/L) | 0.80 ± 0.48 | 0.78 ± 0.44 | 0.82 ± 0.50 |
| CD3+/CD4+, mean ± SD (GI/L) | 0.48 ± 0.33 | 0.48 ± 0.31 | 0.49 ± 0.35 |
| CD3+/CD8+, mean ± SD (GI/L) | 0.31 ± 0.24 | 0.30 ± 0.19 | 0.33 ± 0.22 |
ALOD = above limit of detection (0.5 ng/mL). ANA = antinuclear antibodies. anti-dsDNA = anti–double-stranded DNA. BILAG = British Isles Lupus Assessment Group. BLyS = B-lymphocyte stimulator. C = complement. Ig = immunoglobulin. LLN = lower limit of normal. PGA = Physician’s Global Assessment. SD = standard deviation. SE = standard error, SELENA-SLEDAI = Safety of Estrogens in Lupus Erythematosus National Assessment–Systemic Lupus Erythematosus Disease Activity Index. SLE = systemic lupus erythematosus. SLICC = Systemic Lupus International Collaborative Clinics.
Patients could be categorized in more than 1 race subgroup.
Indigenous American refers to Alaska Native or American Indian from North/South/Central America).
These patients are also accounted for in the race subgroups.
Excluding aminoquinoline antimalarials (hydroxychloroquine, chloroquine, quinacrine).
Serum BLyS levels were only determined prior to belimumab dosing because interference from belimumab precluded an accurate measurement of BLyS levels.
In patients who were positive at baseline with anti-dsDNA (IgG) assay with a detectable range of 30–3600 IU/mL.
Rare subset count per mL = (rare cell event count)/(CD19+ event count) * CD19+ count per mm3 * 1000.
Primary efficacy endpoint
SRI response at week 52
There were more SRI responders at week 52 in the belimumab 10-mg/kg group than in the placebo group (43.2% versus 33.5%; P = 0.017; Table 2 and Figure 2 A). The percentage of SRI responders in the belimumab 1-mg/kg group (40.6%) was also numerically greater than in the placebo group (P = 0.089). Consistent with the primary analysis, the four prespecified sensitivity analyses were also statistically significant with belimumab 10 mg/kg, but not with 1 mg/kg. The duration of SRI response in week-52 responders was significantly greater with belimumab 10 mg/kg than with placebo for 1 to 6 months before or after the week-52 visit (Figure 2 B). Compared with patients treated with placebo, more patients treated with belimumab 10 mg/kg met the criterion for the SELENA-SLEDAI component of the SRI (P = 0.006). Belimumab 1 mg/kg was associated with a greater likelihood of patients meeting the no-worsening criteria for the BILAG (P = 0.013) and PGA (P = 0.012) component of the SRI.
Table 2.
Clinical and biomarker outcomes
| Efficacy parameter | Placebo (n = 275) |
Belimumab 1 mg/kg (n = 271) |
Belimumab 10 mg/kg (n = 273) |
|---|---|---|---|
| SRI response rate at week 52, n (%)* | 92 (33.5) | 110 (40.6) | 118 (43.2)† |
| ≥4-point reduction in SELENA-SLEDAI‖ | 97 (35.3) | 116 (42.8) | 127 (46.5) ‡ |
| No worsening by BILAG¶ | 180 (65.5) | 203 (74.9)† | 189 (69.2) |
| No worsening by PGA | 173 (62.9) | 197 (72.7)† | 190 (69.6) |
| SRI by SELENA-SLEDAI score improvement at week 52, n (%) | |||
| SRI 5, n (%)# | 56 (20.4) | 84 (31.0) ‡ | 89 (32.6) § |
| SRI 6, n (%)# | 52 (18.9) | 78 (28.8)‡ | 84 (30.8)‡ |
| SRI 7, n (%)** | 29/216 (13.4) | 42/217 (19.4) | 46/216 (21.3)† |
| SRI 8, n (%)** | 28/210 (13.3) | 39/211 (18.5) | 45/210 (21.4)† |
| SRI 9, n (%)** | 12/147 (8.2) | 21/150 (14.0) | 22/143 (15.4) |
| SRI 10, n (%)** | 12/140 (8.6) | 20/144 (13.9) | 21/136 (15.4) |
| SRI response rate at week 76, n (%)*‖ | 89 (32.4) | 106 (39.1) | 105 (38.5) |
| ≥4-point reduction in SELENA-SLEDAI‖ | 93 (33.8) | 114 (42.1)† | 113 (41.4) |
| No worsening by BILAG¶ | 162 (58.9) | 187 (69.0)† | 173 (63.4) |
| No worsening by PGA | 160 (58.2) | 178 (65.7) | 172 (63.0) |
| SRI by SELENA-SLEDAI score improvement at week 76, n (%) | |||
| SRI 5, n (%)# | 60 (21.8) | 77 (28.4) | 84 (30.8)† |
| SRI 6, n (%)# | 56 (20.4) | 73 (26.9) | 79 (28.9)† |
| SRI 7, n (%)** | 30/216 (13.9) | 47/217 (21.7)† | 47/216 (21.8)† |
| SRI 8, n (%)** | 27/210 (12.9) | 42/211 (19.9)† | 46/210 (21.9)‡ |
| SRI 9, n (%)** | 7/147 (4.8) | 22/150 (14.7)‡ | 22/143 (15.4)‡ |
| SRI 10, n (%)** | 7/140 (5.0) | 21/144 (14.6)‡ | 19/136 (14.0)† |
| Corticosteroid-sparing activity | |||
| Prednisone reduced by ≥ 25% to ≤ 7.5 mg/d during weeks 40–52, n (%)‖ | 16/126 (12.7) | 25/130 (19.2) | 21/120 (17.5) |
| Prednisone reduced by ≥ 25% to ≤ 7.5 mg/d during weeks 64–76, n (%) | 22/126 (17.5) | 35/130 (26.9) | 29/120 (24.2) |
| Severe SFI flares | |||
| Patients with flare over 76 weeks, n (%) | 73 (26.5) | 50 (18.5)† | 56 (20.5) |
| Hazard ratio (95% CI) | 0.66 (0.46, 0.94) | 0.77 (0.54, 1.09) | |
| Patients with flare from weeks 24 to 76, n/N (%) | 52/239 (21.8) | 31/245 (12.7)‡ | 37/236 (15.7) |
| Hazard ratio (95% CI) | 0.55 (0.35, 0.86) | 0.70 (0.46, 1.07) | |
| Biologic markers | |||
| Normalization of low C3, n (%)†† | |||
| Week 52 | 16/77 (20.8) | 24/74 (32.4) | 37/85 (43.5)‡ |
| Week 76 | 13/70 (18.6) | 19/70 (27.1) | 40/78 (51.3)§ |
| Normalization of low C4, n (%)†† | |||
| Week 52 | 17/99 (17.2) | 35/105 (33.3)‡ | 52/112 (46.4)§ |
| Week 76 | 17/93 (18.3) | 36/98 (36.7)‡ | 52/102 (51.0)§ |
| Anti-dsDNA positive to negative, n (%)‡‡ | |||
| Week 52 | 10 (8.3) | 23 (17.0)† | 19 (14.5) |
| Week 76 | 11 (9.8) | 31 (24.8)‡ | 23 (19.2)† |
P values are for pair-wise comparison of the placebo group with the belimumab 1- or 10-mg/kg treatment group. anti-dsDNA = anti–double-stranded DNA. BILAG = British Isles Lupus Assessment Group. C = complement. PCS = physical component summary. PGA = Physician’s Global Assessment. SE = standard error. SELENA-SLEDAI = Safety of Estrogens in Lupus Erythematosus National Assessment–Systemic Lupus Erythematosus Disease Activity Index. SFI = Systemic Lupus Erythematosus Flare Index. SLE = systemic lupus erythematosus. SRI = SLE Responder Index.
Percentage of patients with reduction in SELENA-SLEDAI ≥ 4 and no worsening by BILAG index (no new A/2B flares) and no worsening by PGA score (< 0.3-point increase).
P < 0.05.
P < 0.01.
P < 0.001.
Major secondary endpoint.
No new BILAG 1A/2B flares.
SRI modified based on 5–6-point reduction in SELENA-SLEDAI included all patients for analysis.
SRI modified based on 7–10-point reduction in SELENA-SLEDAI evaluated patients who had ≥ 7–10-point score at baseline.
Low C3: < 90 mg/dL, low C4: < 16 mg/dL.
In patients who were positive at baseline with anti-dsDNA (immunoglobulin G) assay with a detectable range of 30–3600 IU/mL.
Figure 2.
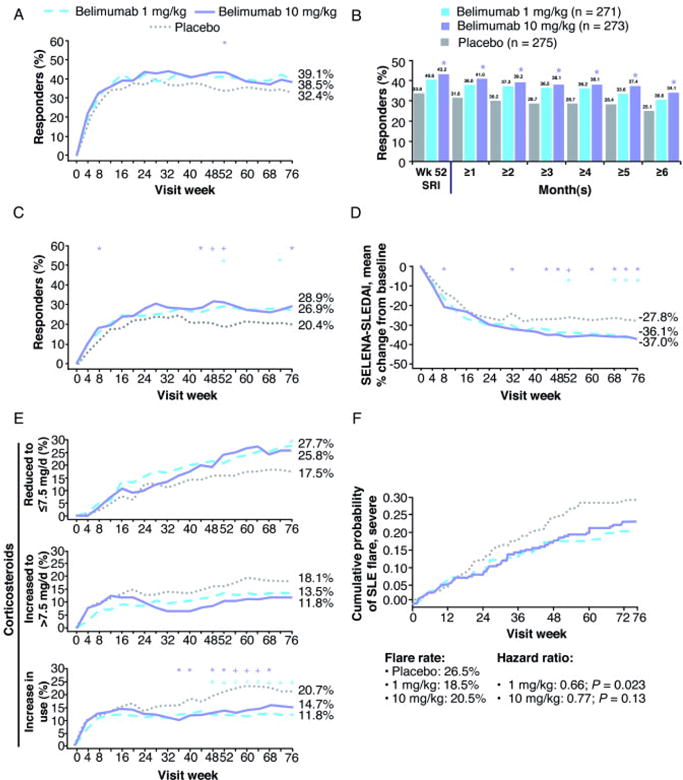
Select clinical outcomes. A. Systemic Lupus Erythematosus Responder Index (SRI) response rate over 76 weeks. B. Durability of week-52 SRI response for 1 to 6 months before or after the week-52 visit. C. Modified SRI response rate (≥ 6-point reduction) over 76 weeks. D. Mean percent change in Safety of Estrogens in Lupus Erythematosus National Assessment– Systemic Lupus Erythematosus Disease Activity Index (SELENA-SLEDAI) (last-observation-carried-forward analysis). E. Percents of patients with corticosteroid dose reduced to ≤ 7.5 mg/d from > 7.5 mg/d at baseline (top; n = 376), and increased to > 7.5 mg/d from ≤ 7.5 mg/d at baseline (middle; n = 443), and with increased corticosteroid use over 76 weeks (bottom). F. Cumulative probability of severe systemic lupus erythematosus (SLE) flare. * P < 0.05. + P < 0.01. # P < 0.001.
Major secondary efficacy endpoints
SRI response at week 76
The SRI response rates were numerically greater with belimumab 10 mg/kg (38.5%; P = 0.13) and 1 mg/kg (39.1%; P = 0.11) versus placebo (32.4%) at week 76 (Table 2 and Figure 2 A).
Post-hoc sensitivity analysis with higher SELENA-SLEDAI score thresholds
The effect of belimumab using a modified SRI that required higher thresholds for the SELENA-SLEDAI component (i.e., starting from a ≥ 5-point reduction to a ≥ 10-point reduction) was evaluated. These more stringent response criteria increased the differentiation of belimumab treatment from placebo at both weeks 52 and 76, with belimumab 10 mg/kg achieving a significant difference from placebo for every SELENA-SLEDAI threshold at week 76 (all P < 0.05; Table 2 and Figure 2 C).
SELENA-SLEDAI reduction ≥ 4 points at week 52
Significantly more patients receiving belimumab 10 mg/kg had a ≥ 4-point reduction in SELENA-SLEDAI score at week 52 versus placebo (46.5% versus 35.3%; P = 0.006; Table 2). Evaluating SELENA-SLEDAI changes (supporting analysis) using the last-observation-carried-forward imputation method revealed significant improvements in mean percent change in SELENA-SLEDAI score for belimumab 10 mg/kg from weeks 44 through 76 and for 1 mg/kg from weeks 52 through 76 (except at week 60; Figure 2 D).
Change in PGA score at week 24
There were no significant differences in mean change in PGA score at week 24 between the placebo (−0.49) and belimumab (−0.47 for 1 mg/kg and −0.44 for 10 mg/kg) groups.
Corticosteroid dose reduction between weeks 40 and 52
A subgroup of 376 patients (46%) was receiving prednisone (or equivalent) > 7.5 mg/d at baseline. Greater proportions of patients receiving belimumab 1 mg/kg (19%) and 10 mg/kg (18%) were able to reduce corticosteroids by ≥ 25% and to ≤ 7.5 mg/d between weeks 40 and 52 compared with placebo (13%), but these differences were not statistically significant (Table 2). Between weeks 64 and 76, a similar proportional prednisone reduction was observed. More patients treated with belimumab who were receiving prednisone > 7.5 mg/d at baseline were able to lower their dose to ≤ 7.5 mg/d over time through week 76 (Figure 2 E). In addition, fewer patients on belimumab required dose increases to > 7.5 mg/d over 76 weeks, although the differences in these subgroups did not achieve statistical significance (Figure 2 E).
Corticosteroid usage
There was a greater incidence of treatment failures at week 76 for violating prednisone dosing rules in the placebo group (14.9%) than in the belimumab 1-mg/kg (7.5% [P = 0.005]) and 10-mg/kg (8.1% [P = 0.011]) groups. There was also a trend toward a greater proportion of patients treated with placebo increasing their prednisone dose during most of the trial, including weeks 52–76 (Figure 2 E).
Change in SF-36 score at week 24
There were no significant differences in mean change in SF-36 PCS score at week 24 between the belimumab groups (+3.78 for 1 mg/kg and +3.21 for 10 mg/kg) and the placebo group (+3.35). SF-36 PCS score improvements at week 52 were +4.37 (P = 0.012) for belimumab 1 mg/kg and +3.44 for 10 mg/kg versus +2.85 for placebo, and at week 76 were +4.26 and +3.95 versus +3.37, respectively.
Supporting secondary efficacy analyses
Reduction in risk of severe flare over 76 weeks based on the modified SFI
The risks of severe flares over 76 weeks were reduced by 34% with belimumab 1 mg/kg (P = 0.023) and by 23% with 10 mg/kg (P = 0.13; Table 2 and Figure 2 F). The magnitude of reduction in the risk of severe flares was even greater with belimumab treatment from weeks 24 to 76 (Table 2).
Biologic markers
Belimumab treatment produced rapid and sustained reductions in anti-dsDNA antibody levels, as well as increases in C3 and C4 concentrations, compared with placebo (Figure 3 A and B). Normalization of low C3 and C4, and positive anti-dsDNA was more likely with belimumab 10 mg/kg than with placebo (Table 2). Of patients with baseline anti-dsDNA values of 30–99 IU/mL, treatment with placebo, and belimumab 1 and 10 mg/kg resulted in normalization at week 52 in 26%, 48%, and 40%, respectively; the corresponding rates for patients with baseline values >99 IU/mL were 1.1%, 2.2%, and 2.3%. Patients without anti-dsDNA antibodies at baseline converted to positive at week 52 at rates of 9.1%, 4.0%, and 1.4% in the placebo, and belimumab 1- and 10-mg/kg groups, respectively.
Figure 3.
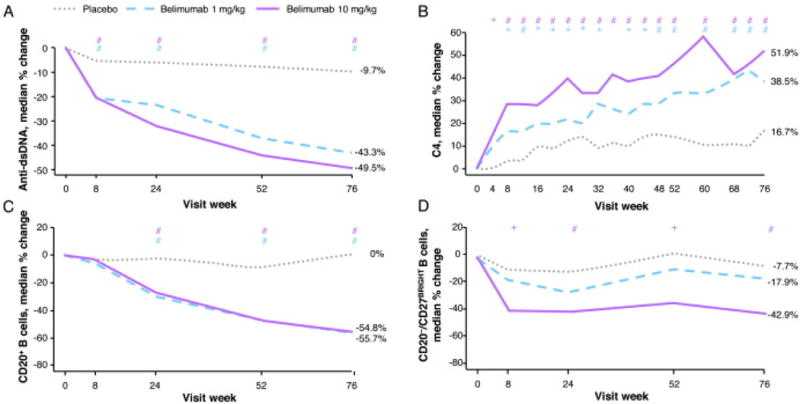
Select biomarker analyses. A. Median percent change in anti-double-stranded DNA (anti-dsDNA) in patients positive for anti-dsDNA at baseline. B. Median percent change in complement 4 (C4) in patients with low values (< 16 mg/dL) at baseline. (Note: the median percent change values at week 76 in complement 3 in patients with low values [< 90 mg/dL] at baseline were 4.8%, 18.9%, and 21.1% for placebo, and belimumab 1 and 10 mg/kg, respectively.) C. Median percent change in CD20+ B-cell subset. D. Median percent change in CD20-/CD27BRIGHT short-lived plasma B-cell subset. * P < 0.05. + P < 0.01. # P < 0.001.
Both doses of belimumab were associated with reductions in B cells (CD20+) at weeks 24, 52, and 76 (P < 0.001; Figure 3 C). These reductions were observed in several B-cell subsets, including activated (CD20+/CD69+) and naïve (CD20+CD27−; data not shown) cells, similar in pattern to CD20+ B cells (Figure 3 C). Plasma-cell subsets, including SLE-specific subsets (CD19+/CD27BRIGHT/CD38BRIGHT; data not shown) similar to the short-lived plasma cell subset (CD20−/CD27BRIGHT), were significantly lower after treatment with belimumab 10 mg/kg than with placebo (Figure 3 D). Similar to the results seen in the phase 2 trial of belimumab (14,24), no changes in T cells (CD3+/CD4+ and CD3+/CD8+) or memory B cells (CD20+/CD27+) occurred in any group at week 52 or 76 (data not shown).
Safety and tolerability
The incidence of adverse events, laboratory abnormalities, and infections, as well as serious and/or severe AEs, including infections, malignancies, and deaths, was similar across groups (Table 3).
Table 3.
Treatment-emergent adverse events and pregnancy outcomes during 76 weeks of study
| Treatment-emergent AEs* | Placebo (n = 275) |
Belimumab 1 mg/kg (n = 271) |
Belimumab 10 mg/kg (n = 273) |
|---|---|---|---|
| ≥1 AE | 253 (92.0) | 253 (93.4) | 253 (92.7) |
| ≥1 serious AE | 54 (19.6) | 63 (23.2) | 61 (22.3) |
| ≥1 severe AE | 52 (18.9) | 51 (18.8) | 54 (19.8) |
| Discontinuations due to AEs | 23 (8.4) | 18 (6.6) | 23 (8.4) |
| Deaths | 0 | 2 (0.7) | 1 (0.4) |
| Malignant neoplasm, all | 1 (0.4) | 4 (1.5) | 2 (0.7) |
| Solid organ† | 1 (0.4) | 3 (1.1) | 1 (0.4) |
| Nonmelanoma skin‡ | 0 | 1 (0.4) | 1 (0.4) |
| Infections | |||
| All | 190 (69.1) | 202 (74.5) | 202 (74.0) |
| ≥1 serious infection AE | 16 (5.8) | 19 (7.0) | 20 (7.3) |
| ≥1 severe infection AE§ | 11 (4.0) | 8 (3.0) | 7 (2.6) |
| Opportunistic infection | 0 | 0 | 1 (0.4) |
| Treatment-emergent AEs (≥ 10% of any treatment group) | |||
| Upper respiratory tract infection | 58 (21.1) | 53 (19.6) | 54 (19.8) |
| Headache | 38 (13.8) | 56 (20.7) | 44 (16.1) |
| Urinary tract infection | 43 (15.6) | 50 (18.5) | 44 (16.1) |
| Arthralgia | 43 (15.6) | 43 (15.9) | 41 (15.0) |
| Nausea | 27 (9.8) | 43 (15.9) | 46 (16.8) |
| Diarrhea | 28 (10.2) | 35 (12.9) | 33 (12.1) |
| Nasopharyngitis | 24 (8.7) | 29 (10.7) | 43 (15.8) |
| Sinusitis | 28 (10.2) | 21 (7.7) | 31 (11.4) |
| Back pain | 21 (7.6) | 26 (9.6) | 27 (9.9) |
| Fatigue | 25 (9.1) | 27 (10.0) | 21 (7.7) |
| Pyrexia | 21 (7.6) | 23 (8.5) | 29 (10.6) |
| Bronchitis | 21 (7.6) | 19 (7.0) | 32 (11.7) |
| Insomnia | 13 (4.7) | 27 (10.0) | 17 (6.2) |
| Infusion reactions‖ | |||
| All (including hypersensitivity) | 27 (9.8) | 42 (15.5) | 37 (13.6) |
| Requiring medical intervention¶ | 9 (3.3) | 16 (5.9) | 17 (6.2) |
| Severe | 1 (0.4) | 1 (0.4) | 3 (1.1) |
| Laboratory abnormalities of grade 3 or 4 in > 2% of patients | |||
| White blood cells (< 2 × 109 L) | 12 (4.4) | 11 (4.1) | 11 (4.0) |
| Neutrophils (< 1 × 109 L) | 20 (7.3) | 18 (6.7) | 16 (5.9) |
| Lymphocytes (< 5 × 108 L) | 80 (29.1) | 78 (29.2) | 76 (27.9) |
| Hemoglobin (≤ 80 g/L) | 15 (5.5) | 7 (2.6) | 5 (1.8) |
| Prothrombin time (17·25 s)# | 31 (11.6) | 36 (13.6) | 30 (11.2) |
| Proteinuria (> 2 g/24 h)** | 21 (7.7) | 19 (7.1) | 28 (10.4) |
| Hypogammaglobulinemia (< 4 g/L)†† | 1 (0.4) | 1 (0.4) | 1 (0.4) |
| Median % change in Ig from baseline at week 76‡‡ | |||
| IgG | −0.8 | −15.1 | −16.4 |
| IgA | −2.1 | −18.0 | −20.4 |
| IgM | −3.8 | −31.8 | −35.0 |
| Pregnancy, n | |||
| All | 0 | 3 | 2 |
| Live birth without congenital anomaly | 0 | 2§§ | 1‖‖ |
AEs = adverse events. Ig = immunoglobulin.
Except where indicated otherwise, values are number of patients and percentage (%) or proportion of patients who experienced AE or laboratory abnormality over 76 weeks of study.
Placebo: carcinoid stomach tumor; belimumab 1 mg/kg: stage 0 cervix carcinoma, breast cancer, and ovarian cancer; and belimumab 10 mg/kg: basal cell carcinoma.
Squamous cell carcinoma.
Grade 3 or 4.
Infusion reactions that occurred on the day of an infusion and resolved within 7 days, and all hypersensitivity reactions that occurred on the day of infusion.
Study agent interrupted or discontinued, or drug given.
81%, 81%, and 87% of patients receiving placebo, and belimumab 1 and 10 mg/kg, respectively, with grade 3/4 prothrombin time were receiving warfarin or other vitamin K antagonists.
Spot urine protein/creatinine ratio mg/mg.
Grade 3 = IgG < 4 g/L; grade 4 = IgG < 2.5 g/L.
Collected at weeks 8, 16, 24, 32, 40, 52, 64, and 76.
The third outcome was an elective termination.
The second pregnancy was lost to follow-up.
Three deaths occurred in patients receiving belimumab (2 in the 1-mg/kg group and 1 in the 10-mg/kg group) during the study. One death was due to unknown causes, 1 to ovarian cancer, and 1 to cardiac arrest after the onset of a multi-organ severe SLE flare (belimumab 10 mg/kg). None of the deaths were considered related to study treatment. Depression was reported more frequently with belimumab (6% to 7%) than with placebo (4%). There were no suicides or suicide attempts in any treatment group during this study.
Seven patients aged 52 to 65 years were diagnosed with malignancies, including 1 on placebo (carcinoid tumor of the stomach), 4 on belimumab 1 mg/kg (carcinoma of the cervix, breast cancer, ovarian cancer, and squamous cell carcinoma of the skin), and 2 on 10 mg/kg (basal cell carcinoma and squamous cell carcinoma of the skin).
Rates of serious or severe infection were similar in all treatment groups. No patients died from infection. One opportunistic infection—disseminated cytomegalovirus infection—occurred in a patient at 3 weeks after the third dose of belimumab 10 mg/kg and resolved with antiviral medication. This patient had history of high-dose steroid use and was also being treated with azathioprine.
Infusion reactions (including hypersensitivity) were more common with belimumab than with placebo (14% to 16% versus 10%), with no apparent dose relationship. The rates of serious and/or severe infusion reactions were 0.7%, 0.7%, and 1.5% with placebo, and belimumab 1 and 10 mg/kg, respectively, and no anaphylaxis was reported. There was a decrease in the incidence of infusion reactions after the second or third study dose. Hypersensitivity reactions on the day of an infusion occurred in 4 patients receiving belimumab 1 mg/kg: 2 were allergic reactions attributed to products or medications other than study medication, and 2 were cases of angioedema (one of which was considered related to belimumab). All infusion and hypersensitivity reactions resolved with antihistamine and/or prednisone treatment on the day of infusion.
In patients receiving placebo, and belimumab 1 and 10 mg/kg, respectively, the proportions with shifts in immunoglobulin levels from high or normal at baseline to low by week 52 were as follows: 3.0%, 6.0%, and 9.7% for IgG; 0.7%, 1.9%, and 4.5% for IgA; and 7.4%, 20.8%, and 20.9% for IgM. One patient in each treatment group had grade 3 IgG reduction, which was not associated with infection.
Five pregnancies occurred during the study in patients who received belimumab (3 on 1 mg/kg and 2 on 10 mg/kg); 3 pregnancies resulted in normal live births and 1 in elective termination, and 1 patient was lost to follow-up.
DISCUSSION
This randomized clinical trial of patients with a broad range of active SLE manifestations met its primary efficacy endpoint by demonstrating a reduction in SLE disease activity with belimumab 10 mg/kg added to the SOC without worsening of patients’ overall conditions or the development of significant disease activity in new organ systems. All four sensitivity analyses of the primary endpoint were statistically significant for belimumab 10 mg/kg at week 52; however, none were significant for 1 mg/kg, although the SRI response rate with that dose was numerically greater than that achieved with SOC alone. These results are consistent with those from the recently published BLISS-52 trial, although the effect size in BLISS-52 was greater (17). The two studies evaluated the same biologic agent with similar patient entry criteria, but they were performed in different regions of the world with differences in available and accepted SLE therapies. Taken together, they provide compelling evidence of the efficacy of belimumab in patients with active, autoantibody-positive SLE.
The SRI response rates were numerically higher with belimumab than with placebo at week 76, but the differences were not statistically significant. The trial was designed and powered to evaluate response rates at week 52. It is likely that the ability to discriminate between doses was compromised at week 76 because of an additional 7% dropout rate that occurred in each group between weeks 52 (23%) and 76 (30%). In addition, the more liberal use of prednisone in the placebo group over 76 weeks could have reduced disease activity more than in the belimumab group. Durability of the benefit of belimumab in reducing SLE disease activity, however, was supported by several findings in the study. For patients with a week-52 SRI response, a greater percentage receiving belimumab 10 mg/kg than SOC alone had a durable response lasting 1 to 6 months before or after week 52. Incorporating more stringent thresholds (5 to 10 points) of SELENA-SLEDAI reduction into the SRI revealed that these modified SRI response rates were even more differentiated in the belimumab treatment groups from those in the group receiving SOC alone at weeks 52 through 76. Improvements in the percent change from baseline in SELENA-SELDAI were observed with both belimumab doses at week 52 and persisted through week 76 compared with SOC alone. Trends for greater reduction in SFI severe flares were observed through week 76 with both belimumab doses compared with SOC. The reduction in severe flares and decreased steroid use observed with belimumab may reduce long-term damage, potentially resulting in reduced disease costs and improved health-related quality of life (25–28). In fact, improvement in the SF-36 PCS score was observed at week 52 in patients treated with belimumab.
Belimumab treatment resulted in sustained improvements in serologic activity. Significant reductions in anti-dsDNA antibody titers and increases in C3 and C4 concentrations were observed with both belimumab doses at week 8, and persisted throughout week 76. The reductions with belimumab in median anti-dsDNA IgG antibody levels (44%–49%) were greater than the reductions in total IgG (15%-16%), suggesting a selective effect on autoantibody-producing cells (29,30). The significant reductions with belimumab in B-cell subsets observed in this trial were similar to those noted in the phase 2 study (14,24). Reduction of BLyS-dependent activated B cells may decrease production of pro-inflammatory cytokines (31), while reduction of SLE-specific short-lived plasmablasts can reduce autoantibody production (24,29,30). In BLISS-76, reductions were observed in activated and naïve B cells (both belimumab doses), and short-lived plasma cells including SLE-specific plasma cells (10 mg/kg), but not in T cells (24) and memory B cells (14). By neutralizing BLyS, which is overexpressed in SLE and promotes B-cell survival, belimumab may restore the ability of autoimmune B cells to undergo apoptosis in a partially selective manner and ultimately lead to reduced SLE disease activity (7,12).
Belimumab treatment was generally well tolerated, and AEs reported with both doses plus SOC were similar to those with SOC alone. Malignancies, occurring in 6 patients aged > 50 years who received belimumab, varied in the organ of origin. The rate of malignancies (excluding non-melanoma skin cancers) per 100 patient-years in all patients with SLE treated with belimumab as of December 31, 2009 (0.45 [95% confidence interval 0.27, 0.72]) is consistent with that reported in the literature for patients with SLE (0.53 [0.48, 0.59]) (32,33). Infusion and hypersensitivity reactions, although more frequent with belimumab than with placebo, were uncommon, responded to standard medical management, and decreased after the first three infusions.
Until larger numbers of patients are treated with belimumab for longer durations, the incidence of rare, severe, or serious AEs will remain unknown. It should also be noted that the BLISS-76 study design was not powered to compare risks against the background of different SOC agents, nor could some patient subpopulations be evaluated—eg, pediatric patients, patients with severe active lupus nephritis, and those with severe active central nervous system involvement—since these groups were excluded from the trial.
In conclusion, this international phase 3 trial in patients with ANA- or anti-dsDNA-positive SLE treated with SOC plus belimumab versus placebo met the 1-year SRI primary efficacy endpoint and provides supportive evidence of the durability of effect until 76 weeks.
Acknowledgments
The study was supported by Human Genome Sciences and GlaxoSmithKline. Editorial support was provided by Matt Stenger and Geoff Marx of BioScience Communications, New York, NY, and was funded by Human Genome Sciences and GlaxoSmithKline.
Supported by Human Genome Sciences, Inc., Rockville, Maryland, and GlaxoSmithKline, Uxbridge, Middlesex, United Kingdom. Supported in part by the National Institutes of Health, Bethesda, Maryland (grant M01- RR00043 to the General Clinical Research Center at the University of Southern California Keck School of Medicine).
Dr Furie has received research or grant support, travel support, and payment for review activities, board membership, and consultancy from Human Genome Sciences and GlaxoSmithKline, and is a member of the BLISS-76 steering committee. Dr Petri has received research or grant support, travel support, and payment for review activities, board membership, and consultancy from Human Genome Sciences and GlaxoSmithKline, and is a member of the BLISS-76 steering committee. Dr Cervera has received payment for board membership and consultancy from Human Genome Sciences and GlaxoSmithKline, and is a member of the BLISS-76 steering committee. Dr Merrill has received consulting fees and grant support from Human Genome Sciences and GlaxoSmithKline. Dr Chatham has received research or grant support and travel support from Human Genome Sciences. Dr Stohl has received research or grant support and consultancy fees from Human Genome Sciences and/or GlaxoSmithKline. Drs Hough, Zhong, and Freimuth are employed by and own stock in Human Genome Sciences. Dr van Vollenhoven has received consultancy fees and honoraria from Human Genome Sciences and GlaxoSmithKline, and is a member of the BLISS-76 steering committee.
Footnotes
The other authors declare that they have no conflicts of interest.
ClinicalTrials.gov identifier NCT00410384.
References
- 1.ACR Ad Hoc Committee on Systemic Lupus Erythematosus Guidelines. Guidelines for the referral and management of systemic lupus erythematosus in adults. Arthritis Rheum. 1999;42:1785–96. doi: 10.1002/1529-0131(199909)42:9<1785::AID-ANR1>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 2.Bernatsky S, Boivin JF, Joseph L, Manzi S, Ginzler E, Gladman DD, et al. Mortality in systemic lupus erythematosus. Arthritis Rheum. 2006;54:2550–7. doi: 10.1002/art.21955. [DOI] [PubMed] [Google Scholar]
- 3.D’Cruz DP, Khamashta MA, Hughes GRV. Systemic lupus erythematosus. Lancet. 2007;369:587–96. doi: 10.1016/S0140-6736(07)60279-7. [DOI] [PubMed] [Google Scholar]
- 4.Lau CS, Mak A. The socioeconomic burden of SLE. Nat Rev Rheumatol. 2009;5:400–4. doi: 10.1038/nrrheum.2009.106. [DOI] [PubMed] [Google Scholar]
- 5.Browning JL. B cells move to centre stage: novel opportunities for autoimmune disease treatment. Nat Rev Drug Disc. 2006;5:564–76. doi: 10.1038/nrd2085. [DOI] [PubMed] [Google Scholar]
- 6.Cancro MP. Signalling crosstalk in B cells: Managing worth and need. Nat Rev Immunol. 2009;9:657–61. doi: 10.1038/nri2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cancro MP, D’Cruz DP, Khamashta MA. The role of B lymphocyte stimulator (BLyS) in systemic lupus erythematosus. J Clin Invest. 2009;119:1066–73. doi: 10.1172/JCI38010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moore PA, Belvedere O, Orr A, Pieri K, LaFleur DW, Feng P, et al. BLyS: member of the tumor necrosis factor family and B lymphocyte stimulator. Science. 1999;285:260–3. doi: 10.1126/science.285.5425.260. [DOI] [PubMed] [Google Scholar]
- 9.Baker KP, Edwards BM, Main SH, Choi GH, Wager RE, Halpern WG, et al. Generation and characterization of LymphoStat-B, a human monoclonal antibody that antagonizes the bioactivities of B lymphocyte stimulator. Arthritis Rheum. 2003;48:3253–65. doi: 10.1002/art.11299. [DOI] [PubMed] [Google Scholar]
- 10.Halpern WG, Lappin P, Zanardi T, Cai W, Corcoran M, Zhong J, Baker KP. Chronic administration of belimumab, a BLyS antagonist, decreases tissue and peripheral blood B-lymphocyte populations in cynomolgus monkeys: pharmacokinetic, pharmacodynamic, and toxicologic effects. Toxicol Sci. 2006;91:586–99. doi: 10.1093/toxsci/kfj148. [DOI] [PubMed] [Google Scholar]
- 11.Cheema GS, Roschke V, Hilbert DM, Stohl W. Elevated serum B lymphocyte stimulator levels in patients with systemic immune-based rheumatic diseases. Arthritis Rheum. 2001;44:1313–9. doi: 10.1002/1529-0131(200106)44:6<1313::AID-ART223>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 12.Petri M, Stohl W, Chatham W, McCune WJ, Chevrier M, Ryel J, et al. Association of plasma B-lymphocyte stimulator (BLyS) levels and disease activity in systemic lupus erythematosus. Arthritis Rheum. 2008;58:2453–9. doi: 10.1002/art.23678. [DOI] [PubMed] [Google Scholar]
- 13.Zhang J, Roschke V, Baker KP, Wang Z, Alarcon GS, Fessler BJ, et al. Cutting edge: a role for B lymphocyte stimulator in systemic lupus erythematosus. J Immunol. 2001;166:6–10. doi: 10.4049/jimmunol.166.1.6. [DOI] [PubMed] [Google Scholar]
- 14.Wallace DJ, Stohl W, Furie RA, Lisse JR, McKay JD, Merrill JT, et al. A phase II, randomized, double-blind, placebo-controlled, dose-ranging study of belimumab in patients with active systemic lupus erythematosus. Arthritis Rheum. 2009;61:1168–78. doi: 10.1002/art.24699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Merrill JT, Wallace DJ, Furie RA, Petri MA, Stohl W, Chatham WW, et al. Five-Year Experience with Belimumab, a BLyS-Specific Inhibitor, in Patients with Systemic Lupus Erythematosus (SLE) Arthritis Rheum. 2010;62(suppl):S608. Abstr 1457. [Google Scholar]
- 16.Furie RA, Petri MA, Wallace DJ, Ginzler EM, Merrill JT, Stohl W, et al. Novel evidence-based systemic lupus erythematosus responder index. Arthritis Rheum. 2009;61:1143–51. doi: 10.1002/art.24698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Navarra SV, Guzmán RM, Gallacher AE, Hall S, Levy RA, Jimenez RE, et al. Belimumab, a BLyS-specific inhibitor, reduced disease activity, flares and prednisone use in patients with active SLE: efficacy and safety results from the phase 3 BLISS-52 study. Lancet. 2011;377:721–31. doi: 10.1016/S0140-6736(10)61354-2. [DOI] [PubMed] [Google Scholar]
- 18.Petri M, Kim MY, Kalunian KC, Grossman J, Hahn BH, Sammaritano LR, et al. Combined oral contraceptives in women with systemic lupus erythematosus. N Engl J Med. 2005;353:2550–8. doi: 10.1056/NEJMoa051135. [DOI] [PubMed] [Google Scholar]
- 19.Hay EM, Bacon PA, Gordon C, Isenberg DA, Maddison P, Snaith ML, et al. The BILAG index: a reliable and valid instrument for measuring clinical disease activity in systemic lupus erythematosus. Q J Med. 1993;86:447–58. [PubMed] [Google Scholar]
- 20.Isenberg DA, Gordon C, on behalf of the BILAG group From BILAG to BLIPS–disease activity assessment in lupus past, present and future. Lupus. 2000;9:651–4. doi: 10.1191/096120300672904669. [DOI] [PubMed] [Google Scholar]
- 21.Petri M, Buyon J, Kim M. Classification and definition of major flares in SLE clinical trials. Lupus. 1999;8:685–91. doi: 10.1191/096120399680411281. [DOI] [PubMed] [Google Scholar]
- 22.Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus Erythematosus. Arthritis Rheum. 1997;40:1725. doi: 10.1002/art.1780400928. [DOI] [PubMed] [Google Scholar]
- 23.Adult Toxicity Tables. Division of Microbiology and Infectious Diseases (DMID) 2001 http://www.niaid.nih.gov/labsandresources/resources/dmidclinrsrch/pages/toxtables.aspx (accessed July 15, 2010)
- 24.Jacobi AM, Huang W, Wang T, Freimuth W, Sanz I, Furie R, et al. Effect of long-term belimumab treatment on B cells in systemic lupus erythematosus. Extension of a phase II, double-blind, placebo-controlled, dose-ranging study. Arthritis Rheum. 2010;62:201–10. doi: 10.1002/art.27189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chatham WW, Kimberly RP. Treatment of lupus with corticosteroids. Lupus. 2001;10:140–7. doi: 10.1191/096120301675075008. [DOI] [PubMed] [Google Scholar]
- 26.Gladman DD, Urowitz MD, Rahman P, Ibañez D, Tam L-S. Accrual of organ damage over time in patients with systemic lupus erythematosus. J Rheumatol. 2003;30:1955–9. [PubMed] [Google Scholar]
- 27.Zhu TY, Tam LS, Lee VW, Lee KK, Li EK. The impact of flare on disease costs of patients with systemic lupus erythematosus. Arthritis Rheum. 2009;61:1159–67. doi: 10.1002/art.24725. [DOI] [PubMed] [Google Scholar]
- 28.Zonana-Nacach A, Barr SG, Magder LS, Petri M. Damage in systemic lupus erythematosus and its association with corticosteroids. Arthritis Rheum. 2000;43:1801–8. doi: 10.1002/1529-0131(200008)43:8<1801::AID-ANR16>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 29.Avery DT, Kalled SL, Ellyard JI, Ambrose C, Bixler SA, Thien M, et al. BAFF selectively enhances the survival of plasmablasts generated from human memory B cells. J Clin Invest. 2003;112:286–97. doi: 10.1172/JCI18025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jacobi AM, Odendahl M, Reiter K, Bruns A, Burmester GR, Radbruch A, et al. Correlation between circulating CD27high plasma cells and disease activity in patients with systemic lupus erythematosus. Arthritis Rheum. 2003;48:1332–42. doi: 10.1002/art.10949. [DOI] [PubMed] [Google Scholar]
- 31.Dorner T. Crossroads of B cell activation in autoimmunity: rationale of targeting B cells. J Rheumatol Suppl. 2006;77:3–11. [PubMed] [Google Scholar]
- 32.Bernatsky S, Boivin JF, Joseph L, Rajan R, Zoma A, Manzi S, et al. An international cohort study of cancer in systemic lupus erythematosus. Arthritis Rheum. 2005;52:1481–90. doi: 10.1002/art.21029. [DOI] [PubMed] [Google Scholar]
- 33.Wallace D, Navarra S, Gallacher A, Gúzman R, Thomas M, Furie R, et al. for the BLISS-52 and -76, and LBSL02/99 Study Groups Safety profile of belimumab, a BLyS-specific inhibitor, in patients with active systemic lupus erythematosus (SLE): pooled data from phase 2 and 3 studies. Arthritis Rheum. 2010;62(suppl):S491. Abstr 1172. [Google Scholar]