

Mitochondria infiltrate leading edge lamellipodia, increasing local mitochondrial mass and relative ATP concentration. AMPK regulates infiltration of mitochondria into the leading edge of 2D lamellipodia and 3D invadopodia, coupling local metabolic sensing to subcellular targeting of mitochondria during cell movement.
Abstract
Cell migration is a complex behavior involving many energy-expensive biochemical events that iteratively alter cell shape and location. Mitochondria, the principal producers of cellular ATP, are dynamic organelles that fuse, divide, and relocate to respond to cellular metabolic demands. Using ovarian cancer cells as a model, we show that mitochondria actively infiltrate leading edge lamellipodia, thereby increasing local mitochondrial mass and relative ATP concentration and supporting a localized reversal of the Warburg shift toward aerobic glycolysis. This correlates with increased pseudopodial activity of the AMP-activated protein kinase (AMPK), a critically important cellular energy sensor and metabolic regulator. Furthermore, localized pharmacological activation of AMPK increases leading edge mitochondrial flux, ATP content, and cytoskeletal dynamics, whereas optogenetic inhibition of AMPK halts mitochondrial trafficking during both migration and the invasion of three-dimensional extracellular matrix. These observations indicate that AMPK couples local energy demands to subcellular targeting of mitochondria during cell migration and invasion.
INTRODUCTION
Cell movement is a complex, highly dynamic process that integrates myriad diverse biochemical events to iteratively reshape and relocate the entire cell (Ridley et al., 2003). Execution of these events involves considerable expenditure of cellular energy, and recent studies demonstrate a critical role for energy-sensing pathways in various modes of migration (Nakano and Takashima, 2012). Within the protrusive leading edge alone, the consumption of ATP through sequestration and/or hydrolysis by actin monomers, molecular motors, and regulatory kinases could, if not replenished, generate a local ATP deficit that could halt cytoskeletal dynamics and cell migration. This paradigm can be further extended to invadopodia, lobopodia, and similar structures formed during migration and invasion in three dimensions, wherein the increased complexity of altering and penetrating through a three-dimensional (3D) extracellular matrix (ECM) requires increased coordination of multiple intracellular signaling networks with dynamic metabolic demands (Green and Yamada, 2007; Han et al., 2013). It is reasonable, then, to expect that such localized energy demands may be most efficiently met by a localized energy source rather than reliance on diffusion from central sources.
Mitochondria are essential eukaryotic organelles responsible for, among other functions, generation of reactive oxygen species, regulation of cytosolic Ca2+ transients, and production of the largest pool of cellular ATP (Nunnari and Suomalainen, 2012; Sena and Chandel, 2012). Mitochondria form a reticular network that radiates from the microtubule-organizing center near the nucleus to supply central and distal regions of the cell with essential energy and metabolites. The morphology of this network is dynamically regulated through fission, fusion, and trafficking events that integrate mitochondrial structure and function with subcellular distribution (Boldogh and Pon, 2007; Soubannier and McBride, 2009; Hoppins, 2014). Indeed, mitochondrial distribution is often quite heterogeneous, showing enrichment at sites where the demand for energy is high (Boldogh and Pon, 2007; Soubannier and McBride, 2009; Hoppins, 2014). This suggests that mitochondria can be actively recruited to subcellular regions of high-energy demand. To this end, considerable work has been done to demonstrate the active trafficking of mitochondria along microtubules to support energy-expensive processes and regions within neuronal cells (Cai et al., 2011). More recently, a number of studies have also convincingly correlated the dynamics and subcellular distribution of mitochondria in the leading edge with the migration and invasion of various cancer cells, including those derived from breast, prostate, lung, and glia (Arismendi-Morillo et al., 2012; Desai et al., 2013; Zhou et al., 2014; Caino et al., 2015). However, the underlying signals that couple mitochondrial dynamics to cell migration are poorly understood.
In this work, we extended the analysis of mitochondrial trafficking during migration and invasion to investigate the signaling events governing this mitochondrial movement. We demonstrate, with fine spatiotemporal resolution, the active infiltration of mitochondria to leading edge structures during migration and, for the first time, to protrusive structures formed during 3D matrix invasion. We also show that mitochondria within cellular protrusions support localized ATP production and metabolism. Finally, we identify the AMP-activated protein kinase (AMPK), an essential energy sensor and metabolic regulator, as an important mediator of directional mitochondrial movement during migration and invasion.
RESULTS
Mitochondria rapidly and actively traffic into the leading edge of migrating cells
In ongoing studies of cell migration (McKenzie et al., 2011) and mitochondrial dynamics (Cunniff et al., 2013), we routinely observed mitochondria rapidly trafficking from the cell body into the leading edge of migrating SKOV-3 human ovarian adenocarcinoma cells (Figure 1, A–C, and Supplemental Movie S1). This behavior is not limited to a particular cell type, tissue origin, or pathological condition, as B16F10 mouse melanoma cells, human malignant mesothelioma (H-MESO) cells, HeLa cells, immortalized nontumorigenic human mesothelial (LP9) cells, and REF52 rat embryo fibroblasts also displayed rapid infiltration of mitochondria into lamellipodia (Supplemental Movie S2). For the ensuing studies, we focused our attention on SKOV-3 cells as a model system. Their migration and invasion dynamics are well characterized (McKenzie et al., 2011), and, despite being predisposed to using aerobic glycolysis (i.e., the Warburg effect) for ATP production (Zhang et al., 2006), they still contain and rely on functional mitochondria for metabolism, survival, and invasion (Yang et al., 2014).
FIGURE 1:

Mitochondria traffic to the leading edge of migrating cells. (A) SKOV-3 cells expressing mito-dsRed were imaged over time. Representative images from the indicated times (mitochondria are shown in yellow; cell outline is depicted as a dotted line). (B) Individual frames of mitochondria in insets from the leading edge (LE1) and trailing edge (TE) from A at the indicated time points. Far right, temporal color-coded overlays in which the mitochondria from each time point are colored as indicated, overlaid, and displayed as a maximum-intensity projection to better visualize movement over time. (C) Maximum intensity projections of temporal color-coded stacks showing the cumulative flux of mitochondria over 120 min in the two leading edges (green and blue boxes) and the trailing edge (red box) depicted in A. All three images are oriented such that the peripheral cell membrane is at the top, to better demonstrate mitochondrial movement toward the membrane in the leading edge and away from the membrane in the trailing edge. (D) The form factor (a function of both mitochondrial length and branching, equal to perimeter2/4π × area) was calculated for mitochondria within cell body (CB) and leading edge (LE) regions (∼50 measurements from ∼20 cells; box, 25th–75th quartiles; whiskers, minimum and maximum; p < 0.001). (E) Tracks of individual mitochondria from leading and trailing edges plotted with respect to the leading edge and trailing edge membranes, respectively. (F) Absolute values of trajectory angles of leading and trailing edge mitochondria relative to the cell membrane (as shown in D). Each bin = 10o; dotted line represents Gaussian-fit curve. (G) The maximum instantaneous velocity (the fastest observed velocity over the period of observation, regardless of the duration of movement), as well as the mean velocity of the leading edge membrane (Mem) and leading edge mitochondria (Mito). For the maximum instantaneous velocities, boxes are 25th−75th quartiles, whiskers represent minimum and maximum, and p < 0.0001. Mean velocities ± SD (p < 0.005). (H) Motile and nonmotile SKOV-3 cells expressing mito-dsRed and mTurquoise-LifeAct were imaged at the indicated times. (I) The average relative flux (± SD) of mitochondria was measured in leading edges (LE), trailing edges (TE), and cell bodies (CB) from motile cells and peripheral (P1, P2) and midbody (Mid) regions of nonmotile cells (p < 0.001). (J) The average relative mitochondrial flux (± SD) was measured in leading edges of untreated (Untd) cells and cells treated with (+) or after washout of (wo) nocodazole (Noc), Taxol (Tax), or cytochalasin D (cytoD).
Gross observation of mitochondrial morphology and formal quantification of mitochondrial reticulation (i.e., form factor, F, a measure of both mitochondrial length and degree of branching, equal to perimeter2/4π × area) showed that leading edge mitochondria were smaller and less networked than those located in the cell body (Figure 1, A–C, and Supplemental Movies S1 and S2), consistent with reports that mitochondrial fission facilitates motility (Liu et al., 2009). Further analysis of individual mitochondrial trajectories showed that mitochondrial trafficking during migration is directional and polarized. Specifically, mitochondria infiltrate the leading edge with trajectories directed toward the leading edge membrane, whereas mitochondria in the trailing edge move away from the trailing edge membrane and inward toward the cell center (Figure 1, A–F, and Supplemental Movies S1 and S2). Furthermore, both the maximal instantaneous velocity and the mean velocity at which mitochondria infiltrated the leading edge were significantly higher than the leading edge velocity (Figure 1G). In addition, individual mitochondria were often recruited to the leading edge from central and/or perinuclear clusters, bypassing mitochondria that were initially closer to the periphery (Supplemental Movies S1 and S2). This polarized trafficking in motile cells is in stark contrast to the dynamics seen in stationary cells, in which mitochondria are less active and show no net accumulation or positive flux in those regions (Figure 1, H and I, and Supplemental Movie S3). These data indicate that leading edge recruitment of mitochondria is active, regulated, and deterministic rather than stochastic or simply proportional to cell speed or bulk cytoplasmic flow. These observations significantly strengthen prior observations of dynamic but bulk rearrangement of mitochondrial mass toward the anterior of migrating cells (Desai et al., 2013) and with more static observations of individual mitochondria within leading edge structures (Zhao et al., 2013; Caino et al., 2015).
Because both microtubules and actin microfilaments play fundamental roles in leading edge dynamics (Etienne-Manneville, 2013; Blanchoin et al., 2014) and have been implicated in mitochondrial transport (Anesti and Scorrano, 2006; Pon, 2011), we examined the effect of perturbation of these cytoskeletal structures on mitochondrial trafficking to the leading edge. Disruption of microtubule dynamics with nocodazole or Taxol rapidly inhibited mitochondrial flux into the leading edge and promoted retrograde flux into the cell body (Figure 1J). Leading edge mitochondrial flux was rapidly and completely restored upon nocodazole or Taxol washout (Figure 1J). Of interest, mitochondrial flux was unaffected by acute inhibition of microfilament dynamics with low-dose cytochalasin D but was effectively eliminated by longer treatments (Figure 1J). Thus mitochondrial flux into the leading edge requires intact and dynamic microtubules but seems to be only indirectly influenced by actin microfilament dynamics.
Mitochondria drive pseudopodial metabolism and ATP production: subcellular reversal of the Warburg effect
Although mitochondria have many functions, they are principally regarded as generators of cellular ATP through oxidative phosphorylation (Nunnari and Suomalainen, 2012). ATP can, however, be synthesized through several other pathways, albeit with lower efficiency. Of particular relevance is the tendency of many, if not most, cancer cells to manifest the Warburg effect and greatly increase their ATP production through aerobic glycolysis (Ward and Thompson, 2012; Chen et al., 2015). Despite having been discovered nearly a century ago, the true function of Warburg effect remains unclear. Although Warburg-shifted cancer cells do exhibit greatly increased aerobic glycolysis, considerable evidence suggests that the effect is not a wholesale “switch” from oxidative phosphorylation but instead an up-regulation of secondary metabolic pathway that is well suited to provide anabolic and anaplerotic intermediates to other pathways (e.g., the pentose-phosphate shunt), a tenet supported by the fact that many Warburg-shifted cell types still maintain and require functional mitochondria (Ward and Thompson, 2012; Chen et al., 2015).
Mitochondrial trafficking into the lamellipodia of Warburg-shifted SKOV-3 cells prompted us to assess subcellular energy metabolism in the leading edge versus the remainder of the cell and the relative importance of mitochondrial function in these regions. Initial attempts using previously described fluorescence-based energy biosensors (e.g., the Perceval biosensor; Berg et al., 2009) did not provide sufficient dynamic range or spatiotemporal resolution to reliably discriminate leading edge versus cell body energy flux (unpublished data). Thus we parsed subcellular energetics by measuring metabolic flux in chemotactic pseudopodia and cell bodies using custom, Transwell-like cell culture inserts compatible with the Seahorse XF24 analyzer (see Materials and Methods and Figure 2, A and B). Specifically, we measured extracellular acidification rate (ECAR) and oxygen consumption rate (OCR) to assess glycolysis and mitochondrial function, respectively, and ATP levels in cell bodies and pseudopodia as a function of increasing concentration of 3-bromopyruvate (to inhibit hexokinase and glycolytic flux) and oligomycin (to inhibit mitochondrial ATP synthase). As expected, analysis of SKOV-3 cell bodies revealed a metabolic profile consistent with the Warburg effect. Addition of oligomycin to inhibit mitochondrial function (evidenced by decreased OCR) promoted increased glycolytic flux (evidenced by elevated ECAR) and sustained levels of ATP synthesis (Figure 2C). Conversely, addition of 3-bromopyruvate inhibited glycolysis and ATP synthesis while increasing mitochondrial respiration (Figure 2C). Analysis of pseudopodia, however, revealed a striking reversal of this trend: inhibition of glycolysis had no effect on either mitochondrial respiration or ATP synthesis, whereas inhibition of mitochondrial function decreased ATP synthesis without affecting glycolytic flux (Figure 2C). Although a reversal of the Warburg effect has been seen at tumor subpopulation- and whole-cell levels (Sotgia et al., 2012; Lu et al., 2015), the present data are, to our knowledge, the first observations of a subcellular Warburg reversal. These observations establish that even in the context of a Warburg-shifted cell, mitochondria are the driving force for ATP synthesis within protrusive structures formed during chemotaxis.
FIGURE 2:
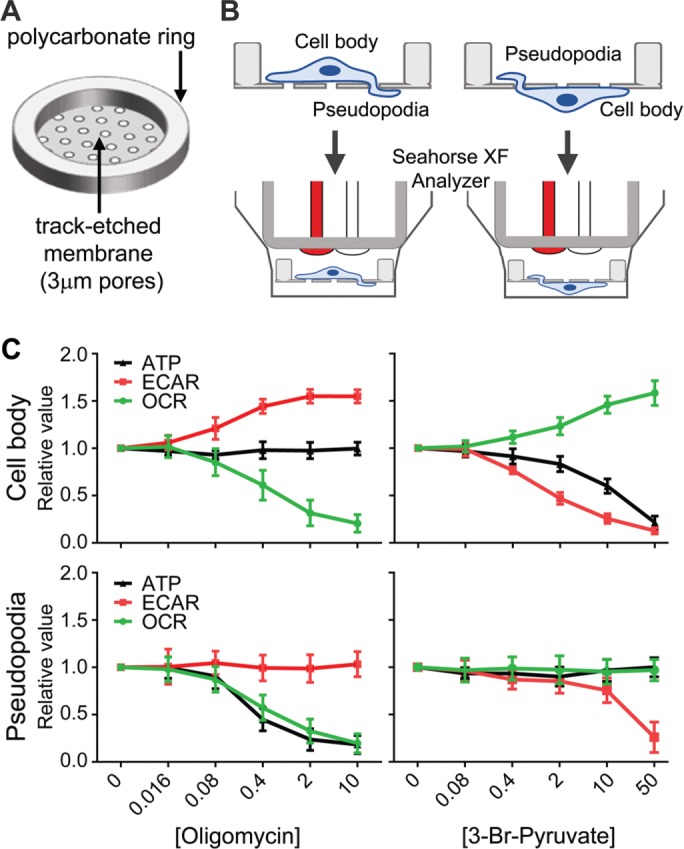
Mitochondria drive pseudopodial metabolism and ATP production—subcellular reversal of the Warburg effect. (A, B) Schematic of custom culture insert and its use for distinct metabolic analysis of cell bodies and pseudopodia. A thin membrane with track-etched 3-μm pores was cut to size and bonded to polycarbonate support rings using a laser cutter (see Materials and Methods for details), forming mini–Transwell-like culture inserts compatible with the Seahorse XF24 metabolic analyzer. Cells can be cultured on the obverse or converse of the inserts and induced to form pseudopodia through to the opposite side, allowing metabolic analysis of cell bodies or pseudopodia. (C) Metabolic analyses of glycolysis (measured by ECAR), mitochondrial oxidative phosphorylation (measured by OCR), and ATP in cell bodies and pseudopodia as a function of increasing concentration of oligomycin to inhibit mitochondrial function or 3-bromopyruvate to inhibit glycolysis (n = 6; average values [relative to untreated conditions] ± SD).
Energy demand and AMPK activity are elevated in the leading edge
The previous analyses profiled the levels of ATP in cell body and pseudopodia separately. When compared directly, we found a significant increase in ATP levels (normalized to total protein) within pseudopodia compared with cell bodies (Figure 3, A–C). This localized increase was ablated by treatment of pseudopodia with rotenone, an inhibitor of complex I in the mitochondrial electron transport chain (Figure 3B), confirming that the source of ATP in pseudopodia was mitochondrial. The localized increase in ATP was also eliminated by treatment of disruption of pseudopodial microtubules with nocodazole (Figure 3B), consistent with the evacuation of mitochondria from leading edge observed under similar conditions (Figure 1I). Taken together, these data corroborate the subcellular metabolic analyses discussed earlier and underscore the importance of mitochondria in generating ATP within leading edge structures.
FIGURE 3:
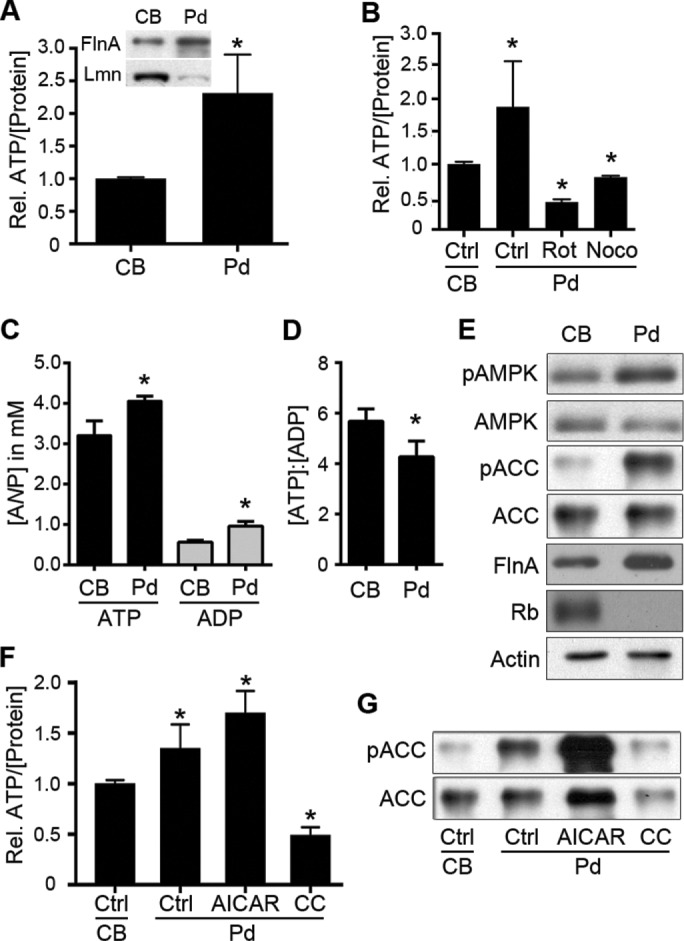
Pseudopodia harbor altered nucleotide levels, ATP/ADP ratio, and AMPK activity compared with cell bodies. (A) Relative levels of ATP (per microgram of protein) were assayed from equal amounts of extracts from purified cell bodies (CBs) and pseudopodia (Pd; n = 4, *p < 0.001). Inset, enrichment of CB and Pd fractions was confirmed by immunoblotting for the leading edge marker filamin A (FlnA) and the nuclear marker lamin A/C (Lmn) in purified Pd and CBs, respectively. (B) Relative ATP levels in Pd relative to respective cell bodies were determined in control conditions (Ctrl; p < 0.05) or after treatment of Pd with rotenone (Rot, 2.5 μM; p < 0.001) or nocodazole (Noco, 0.1 nM; p < 0.01) for 20 min (n = 3 for all samples). (C) Actual ATP and ADP concentrations in Pd relative to respective cell bodies. ATP and ADP concentrations in CB and Pd extracts were determined and used, along with averaged measurements of CB and Pd volumes, to determine true subcellular nucleotide concentrations (average ± SD from three experiments, nCB = 6 and nPd = 15; p = 0.017 and 0.0052 for ATP and ADP, respectively). (D) Average ATP-to-ADP ratio (± SD) in CB and Pd determined from the data in C (p = 0.0365). (E) CB and Pd extracts were immunoblotted to assess levels of active, phospho-T172 AMPK (pAMPK) and AMPK-phosphorylated ACC (pACC), as well as total AMPK and ACC, as indicated. Filamin A (FlnA), retinoblastoma protein (Rb), and actin were immunoblotted to show pseudopod enrichment, cell body enrichment, and equal protein loading, respectively. (F) Relative levels of ATP in Pd relative to cell bodies determined in control conditions (Ctrl; p < 0.005) or after treatment of Pd with AICAR (0.5 mM; p < 0.001) or compound C (CC; 10 μM; p < 0.001; n = 4 for all samples). (G) Extracts from control-treated CBs, control-treated Pd (Ctrl) or Pd treated with AICAR or compound C (CC) as in F were immunoblotted to assess levels of AMPK-phosphorylated or total ACC, as indicated.
The elevated ATP in pseudopodia (∼4.1 vs. ∼3.2 mM in the cell body) suggests a high energy balance or surplus in the leading edge. Of interest, however, the ratio of ADP to protein was also significantly higher in pseudopodia relative to cell bodies (0.96 vs. 0.56 mM; Figure 3C), yielding a significantly lower ATP:ADP ratio in pseudopodia (Figure 3D) and suggesting, instead, a higher rate of ATP hydrolysis and a localized energy deficit. The AMP-activated protein kinase (AMPK) is an important sensor of cellular energy balance; it is inhibited by high concentrations of ATP and activated by elevated ADP and AMP, and, once activated, it positively and negatively regulates energy-producing and energy-consuming processes, respectively (Maldonado and Lemasters, 2014; Hardie et al., 2016). Of interest, several studies also established AMPK as a key regulator of cytoskeletal dynamics, cell shape, and cell migration (Lee et al., 2007; Nakano et al., 2010; Kim et al., 2011; Caino et al., 2013). Furthermore, recent work demonstrated AMPK as a regulator of directed mitochondrial trafficking in axons (Tao et al., 2014). These factors prompted us to investigate a possible role for AMPK in regulating mitochondrial recruitment to the leading edge.
Given the distinct nucleotide balances in pseudopodia versus cell bodies, we first examined the relative AMPK activity in these fractions. AMPK activity, as assessed by phosphorylation of Thr-172 in the AMPK activation loop and by phosphorylation of Ser-79 in acetyl-CoA carboxylase (ACC; an AMPK substrate), was significantly enriched in pseudopodia compared with cell bodies (Figure 3D), consistent with the relative increase in ADP in these structures (Figure 3C). These data establish that the increased flux of mitochondria and altered energy metabolism in leading edge structures are paralleled by a localized increase in the activity of AMPK, a key energy sensor with roles in metabolism, cell migration, and mitochondrial trafficking. This prompted us to investigate the role of AMPK in mediating leading edge mitochondrial trafficking and cell migration dynamics.
Localized activation of AMPK increases ATP levels, mitochondrial flux, and membrane ruffling in cell protrusions
If mitochondria are recruited to the leading edge to generate ATP in response to energy-sensing signals, then manipulation of the AMPK energy-sensing pathway should have demonstrable effects on ATP levels and mitochondrial recruitment. In support of this hypothesis, treatment of migrating cells with 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside (AICAR), an adenosine analogue widely used to stimulate AMPK activity, increased the density and dynamics of mitochondria in the leading edge, as well as leading edge velocity (Supplemental Figure S1). Of greater importance, specific treatment of pseudopodia with AICAR hyperactivated AMPK (Figure 3F) and increased relative pseudopodial ATP levels (Figure 3E).
We then investigated whether this localized, AMPK-mediated increase in ATP correlated with an AMPK-mediated increase in mitochondrial recruitment to the leading edge. For this, we monitored mitochondrial dynamics in live cells while locally manipulating AMPK activity using a microfluidic delivery system. This approach uses delivery and withdrawal micropipettes held in close opposition to create moveable, picoliter- to nanoliter-scale fluidic streams, allowing the delivery of pharmacological compounds with subcellular spatial resolution (Figure 4A and Supplemental Movie S4). Localized activation of AMPK by transient treatment of one side of a migratory leading edge with 1 mM AICAR promoted the targeted infiltration of mitochondria into the treated area of the cell within 20 min, whereas no recruitment was observed into untreated, contralateral cell areas (Figure 4B and Supplemental Movie S5). Even in cells with considerable, preexisting penetration of mitochondria into the leading edge, localized application of AICAR significantly increased leading edge mitochondrial flux (Figure 4, C and D, and Supplemental Movie S6). This effect was corroborated biochemically by immunoblotting lysates from purified, control- and AICAR-treated pseudopodia, which showed an increase in the mitochondrion-specific protein peroxiredoxin-3 (Cunniff et al., 2013) in AICAR-treated pseudopodia relative to untreated pseudopodia (Figure 4E).
FIGURE 4:

Localized activation of AMPK induces mitochondrial recruitment. (A) Snapshot of localized drug delivery technique, showing a rhodamine-dextran solution flowing from a delivery micropipette (bottom) into a withdrawal micropipette (top) positioned ∼10 μm away. (B) Distribution of mitochondria in the leading edge of an SKOV-3 cell expressing mito-dsRed and mTurquoise-LifeAct before and after localized application of 1 mM AICAR (green fluorescence [left] and green dashed line [right]). To better visualize mitochondrial recruitment, images on the right show thresholded mitochondrial fluorescence from the raw data on the left (cell outline is depicted by the dashed line). (C) Time course of mitochondrial flux into the leading edge before (top) and after (bottom) localized application of AICAR (magenta fluorescence). Multicolor images on the right depict temporal color-coded stack projections of mitochondria from the indicated time points, with the localized AICAR stream depicted by the dotted magenta line. (D) Quantification of mitochondrial flux into the leading edge of cells treated with control medium (Ctrl) or medium + AICAR (box, 25th–75th quartiles; whiskers, minimum and maximum; p < 0.001). (E) Extracts from purified, untreated Pd or Pd treated with 1 mM AICAR for 10 min were blotted with antibodies against the mitochondria-specific protein peroxiredoxin-3 (PRX3) or the cytoplasmic protein ACC.
To determine whether the AMPK-mediated increase in local mitochondrial recruitment had any effects on leading edge dynamics, we monitored actin dynamics in live cells after nanofluidic delivery of AICAR. Local application of AICAR significantly increased the frequency, amplitude, and velocity of actin-rich membrane ruffles in the treated area, with no observable effects in untreated regions (Figure 5 and Supplemental Movie S7). Of importance, this effect was ablated by coapplication of rotenone (Figure 5 and Supplemental Movie S7), confirming the requirement of localized mitochondrial function for the effect. These data show that localized activation of AMPK in the leading edge causes a rapid and targeted recruitment of mitochondria, which support protrusion dynamics through localized synthesis of ATP.
FIGURE 5:

Localized activation of AMPK increases membrane ruffling in a manner dependent on mitochondrial ATP synthesis. (A) Actin cytoskeletal dynamics before and after local application of AICAR (blue fluorescence) and then AICAR + rotenone (Rot; dashed magenta outline). (B) Kymographic analysis of drug-treated leading edge and contralateral, untreated edge (demarcated by the dotted line in A) before and after application of AICAR (blue line) and AICAR + rotenone (magenta line). (C) Quantification of membrane protrusion frequency, amplitude, and velocity in leading edges treated with control medium (Ctrl), medium containing AICAR, and medium containing AICAR + rotenone. Frequencies are depicted as average values ± SD. Magnitude and velocities are depicted in box-and-whisker plots (boxes, 25th–75th quartiles; whiskers, minimum and maximum; 20 measurements from five cells; *p < 0.005).
Optogenetic inhibition of AMPK arrests mitochondrial movement and leading edge dynamics
The foregoing experiments correlate AMPK activation in the leading edge with local increases in ATP content, recruitment of mitochondria, and protrusion dynamics. Although the effects are compelling, experiments inhibiting AMPK are required to establish a causal relationship between AMPK, mitochondrial flux, and leading edge dynamics. However, acute inhibition of AMPK is far more difficult than activation, given the lack of specific inhibitors. Much work has been done using compound C, an often-used but nonselective inhibitor of AMPK (Bain et al., 2007). Indeed, we too found that treatment of pseudopodia with compound C inhibited local AMPK activity and decreased local ATP levels (Figure 3, E and F), whereas treatment of migrating cells with compound C resulted in fission and immobilization of leading edge mitochondria and decreased leading edge dynamics (Supplemental Figure S1). However, given strong reservations about the specificity of this reagent (Bain et al., 2007), we wanted to refine our approach using a more specific method of inhibition. The de rigueur approaches using dominant negatives, antisense, RNA interference, or clustered regularly interspaced short palindromic repeats/Cas9 were not suitable for our needs; they are slow, taking tens of hours or multiple days to manifest full loss of function, and long-term loss of AMPK function has significant consequences for cell morphology and cell migration (Supplemental Figure S2; Williams and Brenman, 2008; Kim et al., 2011; Hardie et al., 2012; Nakano and Takashima, 2012; Caino et al., 2015).
We therefore developed a new approach for rapid, specific inhibition of AMPK by fusing two novel and powerful approaches: genetically encoded, highly specific peptide inhibition of AMPK (Miyamoto et al., 2015) and optogenetic control of kinase function using the photoswitchable light-oxygen-voltage-2 (LOV2) domain (Yi et al., 2014). Specifically, we created constructs that fused an AMPK-inhibitory peptide (AIP) or a scrambled control peptide (AIPscr) to the Jα helix of LOV2 (Figure 6, A and B), thereby generating a photoactivatable (PA) inhibitor (and its negative control) that is sterically blocked under dark conditions but rapidly unfolds to inhibit AMPK upon exposure to blue light (Figure 6C). Two notes of technical importance: first, the PA-AIP and PA-AIPscr inhibitors are fused to the mVenus fluorescent protein, allowing microscopic identification of positive cells; second, the fluorescent tag of the mitochondrial marker used throughout these studies (mito-DsRed) is excited at a wavelength that does not efficiently unfold the LOV2 domain or photoactivate the inhibitor (Yi et al., 2014). Thus cotransfection of plasmids encoding PA-AIP and mito-DsRed enabled us to examine mitochondrial dynamics in live, migrating cells before and after rapid, specific inhibition of AMPK. Initial experiments (in which PA-AIP was “preactivated” by a 1-min exposure to 445-nm blue light, followed by “maintenance” exposures every 10 s and acquisition of mitochondria images every 90 s) showed rapid and nearly complete ablation of mitochondrial trafficking in migrating cells upon inhibition of AMPK (Supplemental Figure S3 and Supplemental Movie S8). Comparable but more graded patterns were seen using “softer” (i.e., less frequent and/or without prolonged preactivation) photoactivation protocols. Specifically, increasing the frequency of photoactivation pulses led to a “dose-dependent” decrease in mitochondrial velocity and net flux in cells expressing PA-AIP (Figure 6, D and E, and Supplemental Movie S9), whereas no effect was seen in control, PA-AIPscr–expressing cells (Figure 6E). These data demonstrate that AMPK activity is indeed required for mitochondrial trafficking in migrating cells.
FIGURE 6:
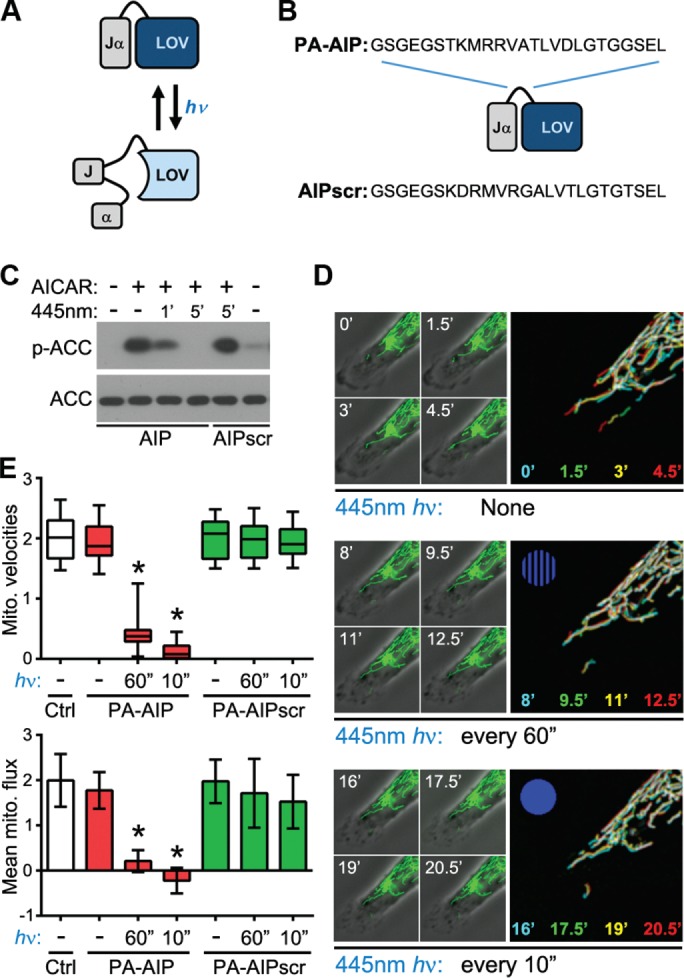
Optogenetic inhibition of AMPK arrests mitochondrial trafficking. (A, B) Schematic of basis and design of PA constructs comprising the AIP or AIPscr, which can be regulated by exposure to blue light (hν). See the text for details. (C) Immunoblot of total and AMPK-phosphorylated ACC in cells expressing PA-AIP or PA-AIPscr treated with 0.5 mM AICAR then exposed to blue light as indicated. (D) Time course of mitochondrial dynamics in cells expressing PA-AIP before (top) and after exposure to 445-nm blue light every 60 s (middle) followed by every 10 s (bottom). Large panels images are temporal color-coded stack projections of mitochondria at the indicated time points (with increasing frequency of blue light exposure symbolized by the blue dot). (E) Quantification of mitochondrial velocity (top) and flux (bottom) in the leading edges of control cells (Ctrl) or cells expressing PA-AIP or PA-AIPscr and exposed to 445-nm light as indicated (boxes, 25th–75th quartiles; whiskers, minimum and maximum; bars, mean ± SD; 12 cells from four experiments; *p < 0.0005).
Inhibition of AMPK blocks trafficking of mitochondria during 3D matrix invasion
As discussed earlier, studies have correlated the subcellular distribution of mitochondria not only with enhanced cell migration but also with ECM invasion and metastasis (Arismendi-Morillo et al., 2012; Desai et al., 2013; Zhou et al., 2014; Caino et al., 2015). However, there are no reports examining the localization and trafficking of mitochondria in live cells during matrix invasion. To this end, we analyzed mitochondrial dynamics in SKOV-3 cells embedded in 3D collagen gels and found that, as in the leading edges of cells migrating in two dimensions, mitochondria rapidly trafficked into the distal tips of protrusive invadopodia (Figure 7A and Supplemental Movie S10). This infiltration was even more striking in more highly metastatic and invasive SKOV-3ip cells (Figure 7, B and C, and Supplemental Movie S10) and B16F10 mouse melanoma cells (Figure 7D and Supplemental Movie S10). To determine whether mitochondrial flux into 3D invadopodia is, as in two-dimensional lamellipodia, dependent on AMPK activity, we cotransfected SKOV-3 cells with mito-DsRed and either PA-AIP or PA-AIPscr and cultured them in 3D collagen. Mitochondrial infiltration into invadopodia was unaffected by photoactivation of PA-AIPscr but was arrested and actually reversed by inhibition of AMPK by photoactivation of PA-AIP (Figure 7, E and F, and Supplemental Movie S11). To our knowledge, these are the first observations of polarized mitochondrial trafficking during cancer cell invasion and are certainly the first demonstration of the regulation of mitochondrial dynamics during invasion by AMPK.
FIGURE 7:

Mitochondria traffic into the distal tips of 3D invadopodia in an AMPK-dependent manner. (A) SKOV-3, (B, C), SKOV-3ip, and (D) B16F10 cells expressing mTurquoise-LifeAct and mito-DsRed (yellow) were embedded in 3D collagen I gels overnight and then imaged over the indicated times. Scale bars, 10 μm (A, B, D), 20 μm (C). (E) Time course of mitochondrial dynamics in 3D collagen-embedded SKOV-3 cells expressing PA-AIP or PA-AIPscr before (Ctrl) and after photoactivation (+PA) by exposure to 445-nm blue light, as described for Figure 6 (bar, 5 μm). Multicolored images are temporal color-coded stack projections of mitochondrial movement over the indicated time points. (F) Quantification of mitochondrial velocities in the invading leading edges of SKOV-3 cells expressing PA-AIP or PA-AIPscr, embedded in 3D collagen gels, before and after photoactivation by 445-nm light (boxes, 25th–75th quartiles; whiskers, minimum and maximum; bars, mean ± SD; 30 measurements from three experiments; *p < 0.001).
DISCUSSION
The data presented here demonstrate that cellular energy balance is not homogeneous and that the leading edge of migrating cells is an energy-hungry subcellular structure. Moreover, they demonstrate that mitochondria rapidly traffic into leading edge structures during cell migration and matrix invasion and that this trafficking is driven by AMPK, a master regulator of cellular energy. These observations couple the subcellular dynamics of mitochondria to the localized energy demands of cell migration and support a model in which high utilization of ATP within the leading edge of a migrating cell promotes local activation of AMPK, which in turn stimulates directional microtubule-based trafficking of mitochondria to replenish ATP, which fuels the leading edge migration machinery. There is evidence that different subcellular compartments are likely to synthesize and hydrolyze ATP at different rates, leading to localized regions of altered energy demand and balance (Metallo and Vander Heiden, 2013). However, it is not the bulk concentration of ATP but instead the ratio of ATP to ADP that is believed to be the principal parameter for determining energy balance (Metallo and Vander Heiden, 2013; Yuan et al., 2013). Thus high local abundance of ATP-hydrolyzing or -sequestering proteins may exert profound influence on local energy balance. In this context, it is interesting, albeit somewhat paradoxical, that pseudopodia appear to persist in a state of “constant” energy deficit, as evidenced by the lower ATP:ADP ratio than in cell bodies (Figure 3). This observation fits well with the observed activation of AMPK but is at odds with the idea that the mitochondria recruited into the leading edge fully meet this energy demand. One possible explanation is that the “constant” energy deficit implied by the lower ATP:ADP ratio in purified pseudopodia is not truly constant but instead represents a snapshot of a rapidly changing metabolic state. However, the pseudopod purification technique used here to determine this ratio is not amenable to high- time-resolution analyses. Another possibility is that the overall lower ATP:ADP ratio in bulk, purified pseudopodia masks smaller subregions of higher ratios (presumably, these would exist nearest the infiltrating mitochondria). Investigation of these possibilities requires the ability to monitor ATP and/or ATP:ADP ratios rapidly and with subcellular spatial resolution. This effort is best served by the Yellen laboratory’s Perceval ATP:ADP biosensor (and its recently improved version, Perceval-HR; Tantama et al., 2013), and efforts to implement this approach are underway. Nonetheless, the AMPK delivery of mitochondria to leading and invading edge structures strongly implicates the existence of localized structures or processes that are best served by close proximity to mitochondrial output.
The role of AMPK in cell migration is complex and poorly understood. AMPK has been shown to promote (Chiu et al., 2009; Cai et al., 2010; Nakano et al., 2010; Kim et al., 2011; Caino et al., 2013) or inhibit (Caino et al., 2013; Roy et al., 2015; Yan et al., 2015) migration. Of note, all work showing a negative correlation between AMPK and cell migration examined the effects of long-term, whole-cell modulation of AMPK activity. Prior, elegant work using novel actuators and biosensors established the importance of spatiotemporal regulation of AMPK (Miyamoto et al., 2015). This paradigm has strong precedent, as myriad enzymes require tight spatiotemporal regulation to fulfill specific needs (Kholodenko, 2006; Kinkhabwala and Bastiaens, 2010), especially during cellular events such as cell migration and invasion, which comprise numerous spatially disparate processes (Rafelski and Theriot, 2004). Our results strongly support this contention by demonstrating enrichment of AMPK activity within protrusive structures formed during chemotaxis, which in turn is supported by our observations on subcellular metabolic heterogeneity in migrating cells. The many inputs of AMPK signaling allow it to differentiate localized and/or acute energy fluxes from whole-cell energy crisis, and its many outputs can instigate specific proportional responses to distinct metabolic stimuli (Hardie et al., 2016). It appears, then, that the role of AMPK as a master energy sensor and regulator of metabolism should be refined with the perspective that cellular energy is not homogeneous, and the entire cell need not mount a global response to a localized need.
One intriguing question arising from this work is how polarized trafficking of mitochondria during direction migration might be regulated or affected by LKB1, the direct upstream activator of AMPK, as well as a tumor suppressor and master regulator of cell polarity (Nakano and Takashima, 2012). LKB1 regulates cell migration in many contexts and through several distinct pathways (Zhang et al., 2008; Kline et al., 2013; Chan et al., 2014). In addition, LKB1 has strong but complex regulatory effects on mitochondrial dynamics (Courchet et al., 2013; Shackelford et al., 2013; Tanner et al., 2013; Pooya et al., 2014; Swisa et al., 2015). Consider also that microtubules are important for establishing and maintaining cell polarity and regulating cell migration (Etienne-Manneville, 2013; Akhshi et al., 2014) and that AMPK activity regulates polarized, microtubule-dependent trafficking of mitochondria to the leading edge during migration (the present results). This confluence of facts underscores the interest in LKB1. Although it is clear that LKB1 and AMPK each can function without the other (Hawley et al., 2005; Chan et al., 2014), it is compelling to consider how localized metabolic needs, AMPK, and mitochondrial trafficking might be regulated in tumor cells in which LKB1, as a tumor suppressor, is functionally altered or lost. Obvious possibilities include compensatory involvement of alternate kinases upstream of AMPK (e.g., CaMKKβ or TAK1) or functional uncoupling and dysregulation of the pathway.
A second question concerns the targets for AMPK that regulate mitochondrial trafficking. As discussed earlier, global and persistent activation of AMPK can elicit a quantitatively and qualitatively different cellular response than does localized, dynamic AMPK activity. This suggests regulation of a quantitatively and/or qualitatively distinct set of substrates in each case. Several screens have identified myriad AMPK substrates with roles in nearly every conceivable cellular function, with an intriguingly large number involved in cell migration (e.g., Schaffer et al., 2015; reviewed in Hardie et al., 2016). What, specifically, are the substrates that evoke increased transport of mitochondria on microtubules? Ostensibly, the most intriguing candidates would be those that have been both implicated in some aspect of mitochondrial (or general organelle) trafficking or microtubule transport and identified as putative or bona fide AMPK substrates. Detailed consideration of these possibilities and their ramifications is beyond the scope of this discussion; however, among those we find most intriguing are various kinesins, kinesin light chains, and dynein heavy chains, the p150Glued dynactin subunit, CLASP1 and CLIP-170, and APC (Nakano et al., 2010; Moon et al., 2014; Ducommun et al., 2015; Hoffman et al., 2015; Schaffer et al., 2015). In addition, the recent report of AMPK presiding over mitochondrial fission by phosphorylation of mitochondrial fission factor (Ducommun et al., 2015; Toyama et al., 2016) and observations that mitochondrial fission facilitates their motility (Liu et al., 2009) offer an interesting mechanism in this regard. Clearly, a focused effort will be needed to cull possibilities from definitively relevant targets.
A third question is potentially the most important: are the observations made here applicable to all cells? In this work, we delineated mechanistic details using cancer cells; this raises the question of whether this is a behavior of all cells or just cancer cells. Our data reinforce the notion established by prior reports (Desai et al., 2013; Zhou et al., 2014) that leading edge recruitment of mitochondria is a fundamental aspect of cell migration rather than an adaption to therapeutic inhibition of certain signaling pathways (Caino et al., 2015). It is possible, then, that this fundamentality may extend beyond transformed cells and into normal cells. Although we did observe rapid infiltration of mitochondria into lamellipodia in normal mesothelial cells and fibroblasts, considerable additional efforts are required to fully test the regulation and involvement of AMPK in additional cellular backgrounds.
Another aspect of this question is whether this behavior is specific to one type of cancer. To say that cancer is a heterogeneous disease and thus cells from different tumor types are likely to have distinct behaviors is an understatement. However, the core constituents of the pathway delineated here—principally AMPK activity and functional mitochondria—are important not only for ovarian cancer but also for many other cancer types (Jeon and Hay, 2012; Wallace, 2012; Liang and Mills, 2013; Weinberg and Chandel, 2015), and we underscore the fact that AMPK-dependent mitochondrial flux during 3D ECM invasion was demonstrated in other cancer cell types in this study. Nonetheless, there is considerably more work to be done to fully address the question. In addition, it is not surprising that different tumor types may evolve different mechanisms to achieve the same ends. For example, whereas some studies suggested that invadopodia metabolism is driven by glycolysis (Nguyen et al., 2000; Attanasio et al., 2011), other studies strongly support an important role for mitochondrial localization in invadopodia and metastasis (Arismendi-Morillo et al., 2012; Sotgia et al., 2012; Desai et al., 2013; Zhao et al., 2013; Caino et al., 2015). Our data demonstrating robust infiltration of mitochondria into the distal tips of 3D invadopodia favor the latter. The spatial heterogeneity of cellular bioenergetics demonstrated here is consistent with the significant degree of metabolic plasticity exhibited by tumor cells both inherently and in response to cell-level threats such as pathway inhibition (Caino et al., 2015; Ghosh et al., 2015) or irradiation (Lu et al., 2015). Thus it is quite likely that cells of differing backgrounds and pathological states and in different microenvironmental conditions may invoke transient or permanent reprogramming of metabolic signaling. This, in turn, could change the mechanistic details of mitochondrial recruitment. Given this underlying complexity, there is clearly more work to be done to fully understand the role of mitochondrial dynamics and its regulation by AMPK during cell movement.
MATERIALS AND METHODS
Reagents and materials
Compound C was purchased from Calbiochem/EMD Millipore (Billerica, MA), and AICAR was from Toronto Research Chemicals (Toronto, Canada). All other chemicals (unless otherwise stated) were purchased from Sigma-Aldrich (St. Louis, MO).
Cell culture
SKOV-3 human ovarian adenocarcinoma and B16-F10 mouse melanoma cells were originally obtained from the American Type Culture Collection (Manassas, VA) and maintained in antibiotic-free DMEM (Invitrogen/Thermo Fisher, Carlsbad, CA) supplemented with 10% (vol/vol) fetal bovine serum (FBS; HyClone, Logan, UT) at 37°C in a humidified incubator containing 5% CO2. Human malignant mesothelioma cells and immortalized, nontumorigenic mesothelial cells (LP9) were cultured as previously described (Cunniff et al., 2013). For culture in 3D collagen gels, a “sandwich” approach was used. Briefly, collagen gels were formed by mixing (per milliliter, in the following order) 500 μl of collagen I (4 mg/ml; Advanced Biomatrix, Carlsbad, CA), 125 μl of 5× neutralizing solution (250 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], pH 7.4, in 5× phosphate-buffered saline [PBS]), 5 μl of 2 N NaOH, and 375 μl of CO2-independent medium (ThermoFisher, Waltham, MA) containing 5% FBS. The pH of the gel was checked with pH paper and adjusted to ∼7.4 with additional 2 N NaOH as needed. A thin (∼50 μm) layer of this mixture was formed by spreading an appropriate volume on the bottom of a glass-bottom dish (35- or 50-mm FluoroDish; World Precision Instruments, Sarasota, FL) and incubating at 37°C for 30 min. Cells suspended in DMEM plus 5% FBS were added to the dishes at a density of 5 × 103/cm2, allowed to attach for 1 h at 37°C, and then overlaid with 250–750 μl of the same collagen gel mixture as described. Cells were imaged 8–12 h after embedding.
Immunoblotting
Cell lysates were prepared by collecting cells in NP-40 lysis buffer (150 mM NaCl, 1% NP-40, 50 mM Tris, pH 8, 1 μg/ml leupeptin, 1 μg/ml aprotinin, 1 mM NaF, 1 mM NaVO3, 1 mM phenylmethylsulfonyl fluoride) and cleared by centrifugation at 14,000 rpm. Protein concentration was determined by bicinchoninic acid assay (Bio-Rad, Hercules, CA). Lysates (10–20 μg protein/well) were resolved by SDS–PAGE and transferred to polyvinylidene fluoride membranes for immunoblotting. Blots were blocked with 5% bovine serum albumin (BSA) in Tris-buffered saline containing 0.1% Tween 20 (BSA/TBST) and incubated at 4°C overnight with rabbit anti-pAMPK (1:1000; Cell Signaling, Danvers, MA), anti-AMPK (1:2000; Cell Signaling), anti–peroxiredoxin 3 (1:2000; Abfrontier/Axxora, Farmingdale, NY), anti-ACC (1:1000; Cell Signaling) anti-pACC (1:1000; Cell Signaling), anti–retinoblastoma protein (pRB, 1:1000; Cell Signaling), anti–filamin A (1:1000; EMD Millipore, Billerica, MA), or anti–lamin A/C (1:1000; Cell Signaling) in 5% BSA/TBST. Blots were incubated with horseradish peroxidase (HRP)–conjugated secondary antibodies (1:2500; EMD Millipore) for 30 min at room temperature. Enhanced chemiluminescent substrate from Millipore was used to detect HRP-conjugated secondary antibodies on x-ray film.
Plasmids and transfections
Cloning strategies and products were managed using SnapGene software. The following plasmids were used: Mito-DsRed (from Henry Higgs, Dartmouth College, Hanover, NH); mCherry-actin (from Roger Tsien, University of California at San Diego, La Jolla, CA); pLifeAct-mTurquoise2 (plasmid 36201; Addgene, Cambridge, MA); and mCherry-tubulin (generated by swapping mCherry for the enhanced green fluorescent protein [EGFP] in pEGFP-tubulin, a gift from Lee Ligon, Rensselaer Polytechnic Institute, Troy, NY). Plasmids encoding PA-AIP or PA-AIPscr were constructed using mVenus-PA-PKI (encoding a photoactivatable PKA inhibitor peptide fused to mVenus; a kind gift from K. Hahn, University of North Carolina at Chapel Hill, Chapel Hill, NC; Yi et al., 2014) as a backbone. Coding sequences for the LOV2 domain fused to AIP (Miyamoto et al., 2015) or AIPscr were purchased as phosphorylated, double-stranded synthetic fragments (GeneArt strings; ThermoFisher) with overhanging 5′ XhoI and 3′ EcoRI sites and used to replace the LOV2-PKI sequence in PA-PKI. All constructs were confirmed by sequencing, and a complete map of any newly constructed plasmid is available upon request. Cells were transfected with plasmid DNAs using Fugene6 (Promega, Madison, WI) according to the manufacturer’s protocol. Briefly 6 μl of Fugene6 was added to 100 μl of serum and antibiotic-free DMEM, mixed, and incubated at room temperature for 5 min. A total of 1.5 μg DNA was added to the mixture, mixed, and incubated at room temperature for 15 min. The mixture was then added dropwise to a 35-mm dish containing cells at ∼85% confluence, allowed to stand for 2–3 min, and then returned to the incubator. Cells were imaged 48–72 h posttransfection.
Live-cell imaging
Twenty-four hours after transfection, cells were either plated on fibronectin (10 μg/ml)-coated, 35-mm glass-bottom imaging dishes (MatTek, Ashland, MA) or FluoroDish cell culture dishes (35 or 50 mm; World Precision Instruments, Sarasota, FL) or were embedded in collagen gels as described and imaged the next day. Before imaging, growth medium was replaced with CO2-independent imaging medium containing 134 mM NaCl, 5.4 mM KCl, 1.0 mM MgSO4, 1.8 mM CaCl2, 20 mM HEPES, and 5 mM d-glucose (pH 7.4). A stage warmer (NevTek Airstream, Williamsville, VA) was used to maintain cells at 37°C, and cells were imaged on an Eclipse TE-2000E inverted microscope (Nikon) equipped with a 40×/1.3 numerical aperture (NA) Plan Fluor or 60×/1.4 NA Plan Apo oil-immersion objective, the appropriate fluorophore-specific filters (Chroma Technology, Bellows Falls, VT), a Spectra X LED light engine (Lumencor, Beaverton, OR), and a Clara charge-coupled device camera (Andor, Concord, MA). Images were acquired every 5–60 s with 500- to 800-ms exposure times.
Custom culture inserts and subcellular metabolic analyses
For subcellular metabolic analyses, custom Transwell-like culture inserts were made essentially as described elsewhere (Xu et al., 2014). Briefly, a sheet of clear polycarbonate, ∼1 mm thick (0.040 × 12 × 36–inch clear, cell-cast acrylic sheet; ePlastics.com), was placed onto the bed of a CO2 laser cutter (Mini 24; Epilog Laser, Golden, CO; resolution range, 75–1200 dpi), and arrays of 4-mm holes were made (90% laser power at 2700 Hz). Next the holes were covered with 25 × 75–mm sheets of track-etched membrane (3-μm pore size, 25 × 80 mm, polyvinylpyrrolidone free; PFB3-50; Neuro Probe, Gaithersburg, MD), and cuts of 6.25-mm outer diameter were made through the two layers, fusing the membrane to the acrylic and releasing the ring-shaped inserts. After manufacture, inserts were sterilized using ultraviolet light. Before use, membranes were coated on both sides with 10 μg/ml fibronectin overnight at 4°C and then washed with sterile PBS.
To culture cells to analyze metabolism in cell bodies, inserts were placed membrane-side down on a pool of DMEM (3 ml in a 6-cm tissue culture dish), 7 × 105 or 8 × 105 cells (for the B16F10 or SKOV-3 line, respectively) in 50 μl of DMEM were seeded in the top or “well” of the inserts, and the dishes were placed in a tissue culture incubator for 2 h. Epidermal growth factor (EGF) was added to the 3-ml pool of DMEM to a final concentration of 25 ng/ml, and the inserts were cultured for an additional 60 min. Culture for analysis of pseudopodial metabolism was similar, except that inserts were placed membrane-side up onto the pool of DMEM, and a 50-μl drop of cell suspension was seeded directly on top of the membrane. After a 2-h incubation, inserts were inverted, submerging the cells and exposing the well of the inserts, to which was added 50 μl of DMEM plus EGF. The efficiency of pseudopod formation in both orientations was confirmed in preliminary experiments by fluorescence microscopy of phalloidin- and 4′,6-diamidino-2-phenylindole–stained cells after fixation.
For metabolic analyses, cells were refed with buffer-free, assay-compatible culture medium (XF DMEM; 102353-100; Seahorse Bioscience, North Billerica, MA) and incubated at 37°C without CO2 for 1 h before analysis. Cell-seeded inserts were placed membrane-side down into Seahorse Islet Plates, and ECAR (in mpH/min per 106 cells) and OCR (in nmol O2/min per 106 cells) were measured using a Seahorse XF24 Analyzer. The analysis protocol consisted of mix, incubation, and read times of 2 min each for cell bodies or 2, 2, and 5 min for pseudopodia. Control experiments using 2-deoxyglucose or oxamate were performed to determine the average level of nonglycolytic acidification (∼15% of ECAR baseline), which was subtracted before calculating relative experimental values. Owing to limitations in assay sensitivity, analysis of ATP levels in cell bodies and pseudopodia was performed using standard Transwell six-well culture plate inserts, pseudopod/cell body isolation, and nucleotide analysis as described later. In these assays, the indicated concentrations of oligomycin or 3-bromopyruvate were added 5 min before harvest.
Cell body and pseudopod isolation
Pseudopods were isolated from cell bodies as previously described (Howe et al., 2005). Briefly, 3.0-μm-pore polycarbonate membrane cell culture inserts were coated with 10 μg/ml fibronectin (BD Biosciences) overnight at 4°C. The inserts were washed in PBS to remove unbound fibronectin and blocked in DMEM plus 1% BSA for 1 h at 37°C. Cells were starved for 16 h, trypsinized, neutralized with an equal volume of 1 mg/ml soybean trypsin inhibitor, resuspended in DMEM plus 1% BSA to final cell count of 1.5 × 106/ml, and rocked at 37°C for 1 h. After rocking, 1.5 × 106 cells were seeded onto fibronectin-coated inserts and allowed to adhere for 2 h. EGF (40 ng/ml; Sigma-Aldrich) was added to the bottom of the Transwell chamber to stimulate migration for 45 min. After pseudopods were formed, compounds were added to the bottom of the chamber to selectively treat the pseudopods. Pseudopods were harvested by scraping the top of the insert with a cotton swab and were collected in lysis buffer. To collect cell bodies, the undersides of the inserts were scraped to remove the pseudopods and the cell bodies collected in lysis buffer.
Measurement of ATP and ADP levels
ATP and ADP levels were measured using either the ATPlite assay kit from PerkinElmer (Waltham, MA) or the respective kits from Abcam (Cambridge, MA) according to the manufacturers’ instructions. Samples were read on a Synergy HT plate reader (BioTek, Winooski, VT). After luminescence values were obtained, protein concentrations for each sample were measured; the nucleotide levels are expressed as relative luminescence/[protein].
To determine actual nucleotide concentrations (mM, rather than nucleotide concentration relative to protein concentration), volumes for cell bodies (CBs) and pseudopodia (Pd) for SKOV-3 cells were calculated in two ways. The first was a literature-based method in which the values for the average amount of CB or Pd protein yielded per Transwell insert (472.4 and 49.2 μg, respectively) were divided by 100 mg/ml, an approximate value for the concentration of soluble protein in the cytoplasm of mammalian cells (e.g., www.bionumbers.org). The result was divided by the number of cells plated per insert (i.e., 1.5 × 106) to yield a nominal CB volume of ∼3149 μm3 (femtoliters) and a per-cell Pd volume of ∼328 μm3. Given that the average number of Pd per cell in our preparations is 3.6, the volume of an individual Pd is ∼91 μm3. The second calculation involved physical measurement of CB and Pd volumes from photomicrographs of cells cultured on Transwell inserts and then fixed and stained with fluorescent phalloidin or CellTracker Green BODIPY. A total of 90 cells (∼5 cells/insert from six different inserts from three different Pd preparations) were imaged by wide-field or confocal microscopy, and CB and Pd volumes were measured by hand using the MeasureStack plug-in for ImageJ (imagej.nih.gov/ij/). This approach yielded a per-cell Pd volume of ∼385 μm3 and a CB volume of ∼3485 μm3. The average nuclear volume (∼215 μm3) was subtracted from the measured CB volume to give a cytoplasmic CB volume of ∼3270 μm3 (this is an appropriate adjustment, as the nuclei are spun out during the harvesting of CBs from a typical preparation). Thus the volumes of 3149 and 3270 μm3 for the CB and 328 and 385 μm3 for total Pd per cell measured by two very distinct methods are in excellent agreement with each other and within the range of volumes reported for other mammalian cell types. Furthermore, dividing the average yield of CB or Pd protein from a preparation by these values gives CB and Pd protein concentrations of 96.3 and 85.3 mg/ml, respectively; these values are also in fair agreement with the aforementioned assumption of 100 mg/ml. The values derived from direct volumetric measurements were subsequently used as nominal volumes for determining actual concentrations of ATP and ADP in CBs and Pd. ATP and ADP concentrations in extracts (5 μg of protein) were determined from separate aliquots of the same extracts using the described ATPlite assay. ADP levels were determined by conversion of ADP to ATP by inclusion of pyruvate kinase and phosphoenolpyruvate in the extract (using reagents and protocols from the ADP Assay Kit [Abcam]) and subtraction of the parallel values for unconverted ATP. Assay concentrations (μM) were converted into empirical concentrations (mM) by the formula ((([assay]/1000) × 200)/5) × [protein]CB or Pd, where division by 1000 converts μM to mM, 200 is the assay volume (in μl), and 5 is the amount of extract protein in the reaction (in μg), which reduces to ([assay]/25) × [protein]CB or Pd.
Localized drug delivery
Delivery and withdrawal pipettes were pulled from glass capillary tubes of inner diameter 1.5 and 1.0 mm, respectively, using the default setting of a two-stage pipette puller (World Precision Instruments) and polished using a microforge to yield an aperture of 1–2 and 5–10 μm, respectively. Delivery pipettes were loaded with test solutions spiked with fluorescein (F6377; Sigma-Aldrich) or trypan blue (Sigma-Aldrich) using a microloader (WPI). The pipettes were connected to a FemtoJet (Eppendorf, Hauppauge, NY) to expel and a syringe pump for withdrawal and were mounted in an HD-21 dual-micropipette holder (Narishige, East Meadow, NY) mounted on a Leitz micromanipulator; this allows additional control of the fluidic stream through independent positioning of the micropipettes. The pipettes were lowered onto the glass-bottom dish, fluid flow was initiated, and the size, shape, and rate of the flow stream were controlled by adjusting the distance between the delivery and withdrawal pipettes and the settings of the FemtoJet and syringe pump (typically, 0.06 psi of backpressure and 500 μl/h withdrawal, respectively). Once a steady stream was generated, it was lowered and placed directly over the leading edge of a target, motile cell. Drug concentrations were routinely determined by plotting the fluorescence intensity of the fluorescein from the opening of the delivery pipette to a fluorescein-free area over the cells, and the slope of the line was used to calculate the specific concentration of the drug. The concentration of the stream at the opening of the pipette was assumed to be the same as the concentration of the solution loaded into the pipette. For sequential delivery of AICAR and AICAR plus rotenone, dual-barrel theta capillaries were pulled into delivery pipettes, and each barrel was filled with one or the other drug solution. Switching between the two chambers was done using a PolyFil multibarrel micropipette coupling kit (WPI), placing a switchable three-way manifold between the FemtoJet pressure source and the entrances of each barrel.
Donut migration/invasion assay
Clean, sterile silicone donuts were allowed to air dry before being placed into sterile 35-mm, fibronectin-coated cell culture dishes as previously described (McKenzie et al., 2011). Briefly, cells were serum starved for 24 h before trypsinization, resuspended in serum-free medium, and counted using a hemocytometer. Cells were plated at confluence (6000 cells/donut), in a 10-μl volume using a sterile gel-loading tip, into the center of the donut. After adhering overnight, the donuts were carefully removed with sterile forceps. The nuclei were stained using Hoescht 33342 (1:2000 in medium; Invitrogen) for 15 min at 37°C. The staining medium was removed and replaced with fresh medium containing 40 ng/ml EGF with or without indicated compounds. Images were acquired as described using a 2× Plan Apo objective, and the number of migrated cells was determined using the DonutQuant ImageJ plug-in (McKenzie et al., 2011).
Mitochondrial tracking, velocity, rendering, and flux analysis
Time-coded stacks of mitochondria over time were generated using ImageJ. Briefly, images were uniformly adjusted for brightness/contrast, and a “top-hat” filter was applied to isolate bright mitochondria from background (Cunniff et al., 2013). Filtered images were used create binary masks, which were then multiplied by the original fluorescence image. The resulting image stack was color-coded as a function of time using the Temporal-Color Code function and a modified Rainbow Smooth look-up table. Stack projections were rendered as either maximum intensity projections or progressive overlays using the Z Code Stack plug-in (Supplemental Movie S1).
Mitochondrial tracks were calculated using the MTrack2 ImageJ plug-in, and vectors were calculated by plotting the final position as a function of normalizing the starting position of each mitochondrion to (0, 0). We have cos θ = 1 when the angle of mitochondrial motility vectors (θ) is equal to the angle from the center of the cell to the cell periphery proximal to the path of migration (leading edge). Conversely, cos θ = −1 when the angle of mitochondrial motility vectors (θ) is opposite the angle from the center of the cell to the cell periphery distal to the path of migration (trailing edge). The instantaneous velocity for an individual mitochondrion was defined as the Euclidean distance traveled between any two successive frames of a time-lapse series divided by the acquisition interval. For each mitochondrion analyzed, the largest of these values was designated the maximum instantaneous velocity, and the average of these values represented the mean velocity. The same approach was used to determine the instantaneous and mean velocities of proximal membrane segments. Mitochondrial flux was determined using the standard mass flux equation, jm = (Δm/Δt)/A, where m is the thresholded mitochondrial fluorescence area (a surrogate for mitochondrial mass), t is time (specifically, Δt = 3× the image acquisition interval [typically 30–90 s]), and A is the area (specifically, a region of interest that is ≤20% of the total cellular area and encompasses the leading edge, trailing edge, cell body, or periphery, as indicated).
Supplementary Material
Acknowledgments
We thank Jason Stumpff (University of Vermont, Burlington, VT) for many helpful discussions; Mercedes Rincon (University of Vermont) for use of the Seahorse X24 flux analyzer; Octavio Araujo (University of Vermont) for custom insert fabrication; and Roger Tsien, Henry Higgs, Lee Ligon, and Klaus Hahn for supplying plasmids. This work was supported by National Institutes of Health Grants GM097495 and GM117490 to A.K.H. and funds from the University of Vermont College of Medicine to A.K.H. and N.H.H.
Abbreviations used:
- 3D
three-dimensional
- ACC
acetyl coenzyme-A carboxylase
- AICAR
5-aminoimidizole-4-carboxyamide-1-β-d-ribofuranoside
- AMPK
AMP-activated protein kinase
- CB
cell body
- ECAR
extracellular acidification rate
- ECM
extracellular matrix
- LKB1
liver kinase B1
- LOV2
light/oxygen/voltage domain-2
- OCR
oxygen consumption rate
- PA-AIP
photoactivatable AMPK inhibitory peptide
- PA-AIPscr
scrambled PA-AIP
- Pd
pseudopodia
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-05-0286) on July 6, 2016.
REFERENCES
- Akhshi TK, Wernike D, Piekny A. Microtubules and actin crosstalk in cell migration and division. Cytoskeleton (Hoboken) 2014;71:1–23. doi: 10.1002/cm.21150. [DOI] [PubMed] [Google Scholar]
- Anesti V, Scorrano L. The relationship between mitochondrial shape and function and the cytoskeleton. Biochim Biophys Acta. 2006;1757:692–699. doi: 10.1016/j.bbabio.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Arismendi-Morillo G, Hoa NT, Ge L, Jadus MR. Mitochondrial network in glioma’s invadopodia displays an activated state both in situ and in vitro: potential functional implications. Ultrastruct Pathol. 2012;36:409–414. doi: 10.3109/01913123.2012.694582. [DOI] [PubMed] [Google Scholar]
- Attanasio F, Caldieri G, Giacchetti G, van Horssen R, Wieringa B, Buccione R. Novel invadopodia components revealed by differential proteomic analysis. Eur J Cell Biol. 2011;90:115–127. doi: 10.1016/j.ejcb.2010.05.004. [DOI] [PubMed] [Google Scholar]
- Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR, Cohen P. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg J, Hung YP, Yellen G. A genetically encoded fluorescent reporter of ATP:ADP ratio. Nat Methods. 2009;6:161–166. doi: 10.1038/nmeth.1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchoin L, Boujemaa-Paterski R, Sykes C, Plastino J. Actin dynamics, architecture, and mechanics in cell motility. Physiol Rev. 2014;94:235–263. doi: 10.1152/physrev.00018.2013. [DOI] [PubMed] [Google Scholar]
- Boldogh IR, Pon LA. Mitochondria on the move. Trends Cell Biol. 2007;17:502–510. doi: 10.1016/j.tcb.2007.07.008. [DOI] [PubMed] [Google Scholar]
- Cai Q, Davis ML, Sheng ZH. Regulation of axonal mitochondrial transport and its impact on synaptic transmission. Neurosci Res. 2011;70:9–15. doi: 10.1016/j.neures.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai XJ, Chen L, Li L, Feng M, Li X, Zhang K, Rong YY, Hu XB, Zhang MX, Zhang Y, et al. Adiponectin inhibits lipopolysaccharide-induced adventitial fibroblast migration and transition to myofibroblasts via AdipoR1-AMPK-iNOS pathway. Mol Endocrinol. 2010;24:218–228. doi: 10.1210/me.2009-0128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caino MC, Chae YC, Vaira V, Ferrero S, Nosotti M, Martin NM, Weeraratna A, O’Connell M, Jernigan D, Fatatis A, et al. Metabolic stress regulates cytoskeletal dynamics and metastasis of cancer cells. J Clin Invest. 2013;123:2907–2920. doi: 10.1172/JCI67841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caino MC, Ghosh JC, Chae YC, Vaira V, Rivadeneira DB, Faversani A, Rampini P, Kossenkov AV, Aird KM, Zhang R, et al. PI3K therapy reprograms mitochondrial trafficking to fuel tumor cell invasion. Proc Natl Acad Sci USA. 2015;112:8638–8643. doi: 10.1073/pnas.1500722112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan KT, Asokan SB, King SJ, Bo T, Dubose ES, Liu W, Berginski ME, Simon JM, Davis IJ, Gomez SM, et al. LKB1 loss in melanoma disrupts directional migration toward extracellular matrix cues. J Cell Biol. 2014;207:299–315. doi: 10.1083/jcb.201404067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Qian Y, Wu S. The Warburg effect: evolving interpretations of an established concept. Free Radic Biol Med. 2015;79:253–263. doi: 10.1016/j.freeradbiomed.2014.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu YC, Shieh DC, Tong KM, Chen CP, Huang KC, Chen PC, Fong YC, Hsu HC, Tang CH. Involvement of AdipoR receptor in adiponectin-induced motility and alpha2beta1 integrin upregulation in human chondrosarcoma cells. Carcinogenesis. 2009;30:1651–1659. doi: 10.1093/carcin/bgp156. [DOI] [PubMed] [Google Scholar]
- Courchet J, Lewis TL, Jr, Lee S, Courchet V, Liou DY, Aizawa S, Polleux F. Terminal axon branching is regulated by the LKB1-NUAK1 kinase pathway via presynaptic mitochondrial capture. Cell. 2013;153:1510–1525. doi: 10.1016/j.cell.2013.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunniff B, Benson K, Stumpff J, Newick K, Held P, Taatjes D, Joseph J, Kalyanaraman B, Heintz NH. Mitochondrial-targeted nitroxides disrupt mitochondrial architecture and inhibit expression of peroxiredoxin 3 and FOXM1 in malignant mesothelioma cells. J Cell Physiol. 2013;228:835–845. doi: 10.1002/jcp.24232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai SP, Bhatia SN, Toner M, Irimia D. Mitochondrial localization and the persistent migration of epithelial cancer cells. Biophys J. 2013;104:2077–2088. doi: 10.1016/j.bpj.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducommun S, Deak M, Sumpton D, Ford RJ, Nunez Galindo A, Kussmann M, Viollet B, Steinberg GR, Foretz M, Dayon L, et al. Motif affinity and mass spectrometry proteomic approach for the discovery of cellular AMPK targets: identification of mitochondrial fission factor as a new AMPK substrate. Cell Signal. 2015;27:978–988. doi: 10.1016/j.cellsig.2015.02.008. [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville S. Microtubules in cell migration. Annu Rev Cell Dev Biol. 2013;29:471–499. doi: 10.1146/annurev-cellbio-101011-155711. [DOI] [PubMed] [Google Scholar]
- Ghosh JC, Siegelin MD, Vaira V, Faversani A, Tavecchio M, Chae YC, Lisanti S, Rampini P, Giroda M, Caino MC, et al. Adaptive mitochondrial reprogramming and resistance to PI3K therapy. J Natl Cancer Inst. 2015;107, dju502 doi: 10.1093/jnci/dju502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green JA, Yamada KM. Three-dimensional microenvironments modulate fibroblast signaling responses. Adv Drug Deliv Rev. 2007;59:1293–1298. doi: 10.1016/j.addr.2007.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han T, Kang D, Ji D, Wang X, Zhan W, Fu M, Xin HB, Wang JB. How does cancer cell metabolism affect tumor migration and invasion. Cell Adh Migr. 2013;7:395–403. doi: 10.4161/cam.26345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG, Schaffer BE, Brunet A. AMPK: an energy-sensing pathway with multiple inputs and outputs. Trends Cell Biol. 2016;26:190–201. doi: 10.1016/j.tcb.2015.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, Hardie DG. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2:9–19. doi: 10.1016/j.cmet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Hoffman NJ, Parker BL, Chaudhuri R, Fisher-Wellman KH, Kleinert M, Humphrey SJ, Yang P, Holliday M, Trefely S, Fazakerley DJ, et al. Global phosphoproteomic analysis of human skeletal muscle reveals a network of exercise-regulated kinases and AMPK substrates. Cell Metab. 2015;22:922–935. doi: 10.1016/j.cmet.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppins S. The regulation of mitochondrial dynamics. Curr Opin Cell Biol. 2014;29:46–52. doi: 10.1016/j.ceb.2014.03.005. [DOI] [PubMed] [Google Scholar]
- Howe AK, Baldor LC, Hogan BP. Spatial regulation of the cAMP-dependent protein kinase during chemotactic cell migration. Proc Natl Acad Sci USA. 2005;102:14320–14325. doi: 10.1073/pnas.0507072102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon SM, Hay N. The dark face of AMPK as an essential tumor promoter. Cell Logist. 2012;2:197–202. doi: 10.4161/cl.22651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kholodenko BN. Cell-signalling dynamics in time and space. Nat Rev Mol Cell Biol. 2006;7:165–176. doi: 10.1038/nrm1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EK, Park JM, Lim S, Choi JW, Kim HS, Seok H, Seo JK, Oh K, Lee DS, Kim KT, et al. Activation of AMP-activated protein kinase is essential for lysophosphatidic acid-induced cell migration in ovarian cancer cells. J Biol Chem. 2011;286:24036–24045. doi: 10.1074/jbc.M110.209908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinkhabwala A, Bastiaens PI. Spatial aspects of intracellular information processing. Curr Opin Genet Dev. 2010;20:31–40. doi: 10.1016/j.gde.2009.12.006. [DOI] [PubMed] [Google Scholar]
- Kline ER, Shupe J, Gilbert-Ross M, Zhou W, Marcus AI. LKB1 represses focal adhesion kinase (FAK) signaling via a FAK-LKB1 complex to regulate FAK site maturation and directional persistence. J Biol Chem. 2013;288:17663–17674. doi: 10.1074/jbc.M112.444620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Koh H, Kim M, Kim Y, Lee SY, Karess RE, Lee SH, Shong M, Kim JM, Kim J, et al. Energy-dependent regulation of cell structure by AMP-activated protein kinase. Nature. 2007;447:1017–1020. doi: 10.1038/nature05828. [DOI] [PubMed] [Google Scholar]
- Liang J, Mills GB. AMPK: a contextual oncogene or tumor suppressor. Cancer Res. 2013;73:2929–2935. doi: 10.1158/0008-5472.CAN-12-3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Weaver D, Shirihai O, Hajnoczky G. Mitochondrial “kiss-and-run”: interplay between mitochondrial motility and fusion-fission dynamics. EMBO J. 2009;28:3074–3089. doi: 10.1038/emboj.2009.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu CL, Qin L, Liu HC, Candas D, Fan M, Li JJ. Tumor cells switch to mitochondrial oxidative phosphorylation under radiation via mTOR-mediated hexokinase II inhibition—a Warburg-reversing effect. PLoS One. 2015;10:e0121046. doi: 10.1371/journal.pone.0121046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado EN, Lemasters JJ. ATP/ADP ratio, the missed connection between mitochondria and the Warburg effect. Mitochondrion. 2014;19(Pt A):78–84. doi: 10.1016/j.mito.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie AJ, Campbell SL, Howe AK. Protein kinase A activity and anchoring are required for ovarian cancer cell migration and invasion. PLoS One. 2011;6:e26552. doi: 10.1371/journal.pone.0026552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metallo CM, Vander Heiden MG. Understanding metabolic regulation and its influence on cell physiology. Mol Cell. 2013;49:388–398. doi: 10.1016/j.molcel.2013.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto T, Rho E, Sample V, Akano H, Magari M, Ueno T, Gorshkov K, Chen M, Tokumitsu H, Zhang J, et al. Compartmentalized AMPK signaling illuminated by genetically encoded molecular sensors and actuators. Cell Rep. 2015;11:657–670. doi: 10.1016/j.celrep.2015.03.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon S, Han D, Kim Y, Jin J, Ho WK, Kim Y. Interactome analysis of AMP-activated protein kinase (AMPK)-alpha1 and -beta1 in INS-1 pancreatic beta-cells by affinity purification-mass spectrometry. Sci Rep. 2014;4:4376. doi: 10.1038/srep04376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano A, Kato H, Watanabe T, Min KD, Yamazaki S, Asano Y, Seguchi O, Higo S, Shintani Y, Asanuma H, et al. AMPK controls the speed of microtubule polymerization and directional cell migration through CLIP-170 phosphorylation. Nat Cell Biol. 2010;12:583–590. doi: 10.1038/ncb2060. [DOI] [PubMed] [Google Scholar]
- Nakano A, Takashima S. LKB1 and AMP-activated protein kinase: regulators of cell polarity. Genes Cells. 2012;17:737–747. doi: 10.1111/j.1365-2443.2012.01629.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TN, Wang HJ, Zalzal S, Nanci A, Nabi IR. Purification and characterization of beta-actin-rich tumor cell pseudopodia: role of glycolysis. Exp Cell Res. 2000;258:171–183. doi: 10.1006/excr.2000.4929. [DOI] [PubMed] [Google Scholar]
- Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148:1145–1159. doi: 10.1016/j.cell.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pon LA. Organelle transport: mitochondria hitch a ride on dynamic microtubules. Curr Biol. 2011;21:R654–R656. doi: 10.1016/j.cub.2011.07.035. [DOI] [PubMed] [Google Scholar]
- Pooya S, Liu X, Kumar VB, Anderson J, Imai F, Zhang W, Ciraolo G, Ratner N, Setchell KD, Yoshida Y, et al. The tumour suppressor LKB1 regulates myelination through mitochondrial metabolism. Nat Commun. 2014;5:4993. doi: 10.1038/ncomms5993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafelski SM, Theriot JA. Crawling toward a unified model of cell mobility: spatial and temporal regulation of actin dynamics. Annu Rev Biochem. 2004;73:209–239. doi: 10.1146/annurev.biochem.73.011303.073844. [DOI] [PubMed] [Google Scholar]
- Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT, Horwitz AR. Cell migration: integrating signals from front to back. Science. 2003;302:1704–1709. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- Roy I, McAllister DM, Gorse E, Dixon K, Piper CT, Zimmerman NP, Getschman AE, Tsai S, Engle DD, Evans DB, et al. Pancreatic cancer cell migration and metastasis is regulated by chemokine-biased agonism and bioenergetic signaling. Cancer Res. 2015;75:3529–3542. doi: 10.1158/0008-5472.CAN-14-2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffer BE, Levin RS, Hertz NT, Maures TJ, Schoof ML, Hollstein PE, Benayoun BA, Banko MR, Shaw RJ, Shokat KM, Brunet A. Identification of AMPK phosphorylation sites reveals a network of proteins involved in cell invasion and facilitates large-scale substrate prediction. Cell Metab. 2015;22:907–921. doi: 10.1016/j.cmet.2015.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell. 2012;48:158–167. doi: 10.1016/j.molcel.2012.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shackelford DB, Abt E, Gerken L, Vasquez DS, Seki A, Leblanc M, Wei L, Fishbein MC, Czernin J, Mischel PS, et al. LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell. 2013;23:143–158. doi: 10.1016/j.ccr.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotgia F, Whitaker-Menezes D, Martinez-Outschoorn UE, Flomenberg N, Birbe RC, Witkiewicz AK, Howell A, Philp NJ, Pestell RG, Lisanti MP. Mitochondrial metabolism in cancer metastasis: visualizing tumor cell mitochondria and the “reverse Warburg effect” in positive lymph node tissue. Cell Cycle. 2012;11:1445–1454. doi: 10.4161/cc.19841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soubannier V, McBride HM. Positioning mitochondrial plasticity within cellular signaling cascades. Biochim Biophys Acta. 2009;1793:154–170. doi: 10.1016/j.bbamcr.2008.07.008. [DOI] [PubMed] [Google Scholar]
- Swisa A, Granot Z, Tamarina N, Sayers S, Bardeesy N, Philipson L, Hodson DJ, Wikstrom JD, Rutter GA, Leibowitz G, et al. Loss of liver kinase B1 (LKB1) in beta cells enhances glucose-stimulated insulin secretion despite profound mitochondrial defects. J Biol Chem. 2015;290:20934–20946. doi: 10.1074/jbc.M115.639237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner CB, Madsen SR, Hallowell DM, Goring DM, Moore TM, Hardman SE, Heninger MR, Atwood DR, Thomson DM. Mitochondrial and performance adaptations to exercise training in mice lacking skeletal muscle LKB1. Am J Physiol Endocrinol Metab. 2013;305:E1018–E1029. doi: 10.1152/ajpendo.00227.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tantama M, Martinez-Francois JR, Mongeon R, Yellen G. Imaging energy status in live cells with a fluorescent biosensor of the intracellular ATP-to-ADP ratio. Nat Commun. 2013;4:2550. doi: 10.1038/ncomms3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao K, Matsuki N, Koyama R. AMP-activated protein kinase mediates activity-dependent axon branching by recruiting mitochondria to axon. Dev Neurobiol. 2014;74:557–573. doi: 10.1002/dneu.22149. [DOI] [PubMed] [Google Scholar]
- Toyama EQ, Herzig S, Courchet J, Lewis TL, Jr, Loson OC, Hellberg K, Young NP, Chen H, Polleux F, Chan DC, Shaw RJ. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science. 2016;351:275–281. doi: 10.1126/science.aab4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC. Mitochondria and cancer. Nat Rev Cancer. 2012;12:685–698. doi: 10.1038/nrc3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell. 2012;21:297–308. doi: 10.1016/j.ccr.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg SE, Chandel NS. Targeting mitochondria metabolism for cancer therapy. Nat Chem Biol. 2015;11:9–15. doi: 10.1038/nchembio.1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams T, Brenman JE. LKB1 and AMPK in cell polarity and division. Trends Cell Biol. 2008;18:193–198. doi: 10.1016/j.tcb.2008.01.008. [DOI] [PubMed] [Google Scholar]
- Xu W, Janocha AJ, Leahy RA, Klatte R, Dudzinski D, Mavrakis LA, Comhair SA, Lauer ME, Cotton CU, Erzurum SC. A novel method for pulmonary research: assessment of bioenergetic function at the air-liquid interface. Redox Biol. 2014;2:513–519. doi: 10.1016/j.redox.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Moss T, Mangala LS, Marini J, Zhao H, Wahlig S, Armaiz-Pena G, Jiang D, Achreja A, Win J, et al. Metabolic shifts toward glutamine regulate tumor growth, invasion and bioenergetics in ovarian cancer. Mol Syst Biol. 2014;10:728. doi: 10.1002/msb.20134892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y, Tsukamoto O, Nakano A, Kato H, Kioka H, Ito N, Higo S, Yamazaki S, Shintani Y, Matsuoka K, et al. Augmented AMPK activity inhibits cell migration by phosphorylating the novel substrate Pdlim5. Nat Commun. 2015;6:6137. doi: 10.1038/ncomms7137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi JJ, Wang H, Vilela M, Danuser G, Hahn KM. Manipulation of endogenous kinase activity in living cells using photoswitchable inhibitory peptides. ACS Synth Biol. 2014;3:788–795. doi: 10.1021/sb5001356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan HX, Xiong Y, Guan KL. Nutrient sensing, metabolism, and cell growth control. Mol Cell. 2013;49:379–387. doi: 10.1016/j.molcel.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Schafer-Hales K, Khuri FR, Zhou W, Vertino PM, Marcus AI. The tumor suppressor LKB1 regulates lung cancer cell polarity by mediating cdc42 recruitment and activity. Cancer Res. 2008;68:740–748. doi: 10.1158/0008-5472.CAN-07-2989. [DOI] [PubMed] [Google Scholar]
- Zhang XD, Deslandes E, Villedieu M, Poulain L, Duval M, Gauduchon P, Schwartz L, Icard P. Effect of 2-deoxy-D-glucose on various malignant cell lines in vitro. Anticancer Res. 2006;26:3561–3566. [PubMed] [Google Scholar]
- Zhao J, Zhang J, Yu M, Xie Y, Huang Y, Wolff DW, Abel PW, Tu Y. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene. 2013;32:4814–4824. doi: 10.1038/onc.2012.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G, Wang J, Zhao M, Xie TX, Tanaka N, Sano D, Patel AA, Ward AM, Sandulache VC, Jasser SA, et al. Gain-of-function mutant p53 promotes cell growth and cancer cell metabolism via inhibition of AMPK activation. Mol Cell. 2014;54:960–974. doi: 10.1016/j.molcel.2014.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.