

A tripartite split-GFP system faithfully reports the order of the subunits in septin hetero-octamers (and thus can serve as a “molecular ruler”), conversely yields little or no false signal even with very highly expressed cytosolic proteins, and detects authentic interactions of other cellular proteins that are bona fide septin-binding proteins.
Abstract
Various methods can provide a readout of the physical interaction between two biomolecules. A recently described tripartite split-GFP system has the potential to report by direct visualization via a fluorescence signal the intimate association of minimally tagged proteins expressed at their endogenous level in their native cellular milieu and can capture transient or weak interactions. Here we document the utility of this tripartite split-GFP system to assess in living cells protein–protein interactions in a dynamic cytoskeletal structure—the septin collar at the yeast bud neck. We show, first, that for septin–septin interactions, this method yields a robust signal whose strength reflects the known spacing between the subunits in septin filaments and thus serves as a “molecular ruler.” Second, the method yields little or no spurious signal even with highly abundant cytosolic proteins readily accessible to the bud neck (including molecular chaperone Hsp82 and glycolytic enzyme Pgk1). Third, using two proteins (Bni5 and Hsl1) that have been shown by other means to bind directly to septins at the bud neck in vivo, we validate that the tripartite split-GFP method yields the same conclusions and further insights about specificity. Finally, we demonstrate the capacity of this approach to uncover additional new information by examining whether three other proteins reported to localize to the bud neck (Nis1, Bud4, and Hof1) are able to interact physically with any of the subunits in the septin collar and, if so, with which ones.
INTRODUCTION
In eukaryotic cells, structures built with septins—a conserved family of GTP-binding proteins—serve a number of functions, including associating with and deforming the plasma membrane (PM; Bridges and Gladfelter, 2015), erecting a barrier to restrict diffusion and establish subcellular compartments (Saarikangas and Barral, 2011), and providing a three-dimensional scaffold to localize the action of enzymes and other proteins spatially and temporally (McMurray and Thorner, 2009; Oh and Bi, 2011). The first septin-based structure was visualized by electron microscopy (EM) in budding yeast (Saccharomyces cerevisiae) as an hourglass-shaped collar of circumferential bands located at the bud neck between a mother and daughter cell undergoing mitosis (Byers and Goetsch, 1976). Genetic and cytological analysis demonstrated that products of four mitotically expressed genes (CDC3, CDC10, CDC11, and CDC12; Hartwell, 1971, 1974) are necessary to form this structure, are integral components of it (Haarer and Pringle, 1987; Cid et al., 1998), and are required for the execution of cytokinesis and cell septation (Wloka and Bi, 2012). A fifth mitotically expressed septin gene (SHS1) was identified by other means (Carroll et al., 1998; Mino et al., 1998) and appears to be a recently evolved nonessential paralogue of CDC11 (Iwase et al., 2007; Garcia et al., 2011).
The products of these yeast septin genes assemble into two classes of linear hetero-octamers (Cdc11-Cdc12-Cdc3-Cdc10-Cdc10-Cdc3-Cdc12-Cdc11 and Shs1-Cdc12-Cdc3-Cdc10-Cdc10-Cdc3-Cdc12-Shs1). These subunit arrangements were determined by EM examination of recombinant septin complexes containing tagged subunits (Bertin et al., 2008; Garcia et al., 2011) and amply confirmed in vivo by numerous genetic experiments (McMurray et al., 2011; Finnigan et al., 2015b). In humans, the basic building blocks of septin structures are also hetero-octamers that have the capacity to self-assemble into higher-order ensembles (Hall and Russell, 2012; Mostowy and Cossart, 2012; Fung et al., 2014).
In vitro, Cdc11-capped hetero-octamers polymerize end to end into long, straight filaments that pair in register via cross-filament interactions mediated by the C-terminal extensions (CTEs) of both Cdc3 and Cdc12 (Versele et al., 2004; Bertin et al., 2008). Shs1-capped hetero-octamers, by contrast, bundle laterally to form arcs, spirals, and rings (Garcia et al., 2011). Together these two types of hetero-octamers, in conjunction with the collective actions of scores of other bud neck-localized proteins (Gladfelter et al., 2001; McMurray and Thorner, 2009), are responsible for the state of supramolecular assembly and function of septin structures during progression through the cell cycle (Bi and Park, 2012). In early G1, septins form a small patch at the incipient bud site that resolves into a ring and then expands, as the bud emerges, into the hourglass-shaped collar, which, at the onset of cytokinesis, is split (or collapses, like two accordions) into two, tighter, gasket-like bands that trap between them proteins that execute cell division.
Yeast proteins that localize at various times and to varying degrees in the near vicinity of septin structures include factors that participate in diverse processes, from cell polarity and vesicular transport to cell cycle control, actomyosin contractile ring assembly, and cell wall deposition. Similarly, septin-based structures and their associated proteins appear responsible for a range of specialized functions in mammalian cells, including the annulus in spermatozoa (Toure et al., 2011) and assemblies found at the base of the primary cilium (Malicki and Avidor-Reiss, 2014) and in the neck of the dendritic spines on neurons (Ewers et al., 2014).
For the majority of the proteins that reportedly colocalize with the septin collar at the bud neck, it is unclear whether their localization is septin dependent and, if so, whether they are recruited because they bind directly to a septin(s). Moreover, for those factors known to be direct interaction partners, it is not known whether they recognize a structural feature unique to a given septin or one generated only upon higher-order septin assembly.
A variety of methods have been developed to assess the physical encounter of two molecules in the cell, from Förster resonance energy transfer (FRET; Padilla-Parra and Tramier, 2012) to a number of protein-fragment complementation assays (PCAs; Remy and Michnick, 2015), with readouts as diverse as drug resistance, enzyme activity, colorimetric changes, and a fluorescence signal. Among the PCA approaches, bimolecular fluorescence complementation (BiFC) has gained rather wide acceptance (Magliery et al., 2005; Kerppola, 2009; Ohashi and Mizuno, 2014; Miller et al., 2015) because it can detect even weak or transient interactions for the reason that once two associating proteins bring the two halves of the fluorescent reporter protein together, reconstitution of the reporter stabilizes the complex. In addition, BiFC does not require cell fixation, cell lysis, or any special treatment with dyes or other reagents. However, a limitation of BiFC is that the proteins tested for interaction need to be fused to the halves of the fluorescent reporter (typically, a yellow variant of enhanced green fluorescent protein [eGFP]), which are rather bulky tags that may perturb the interaction under study or be fused to each partner in inappropriate orientations that prevent reconstitution.
Therefore, to commence a systematic examination of whether any given bud neck–localized protein intimately associates with any subunit(s) of the septin collar, we chose to implement a fluorescence-based tripartite split-GFP methodology (Cabantous et al., 2013) for assessing protein–protein interaction because, in this system, the tags appended to the test proteins are of minimal size and can be attached by flexible tethers. Binding of a protein bearing at its N-terminus the β10 strand (20 residues) of an evolved eGFP variant to a protein bearing at its C-terminus the β11 strand (21 residues) of this variant permits capture of the otherwise nonfluorescent GFPβ1-9 barrel (stable beta barrel of evolved eGFP variant of tripartite split-GFP system; 200 residues), allowing for reconstitution of eGFP structure and fluorophore formation, yielding a stable fluorescence readout.
As described here, we were able to use this approach in vivo to 1) robustly detect subunit–subunit interactions within the septin hetero-octamer, 2) document that illegitimate interactions with nonphysiological partners are minimal to undetectable, 3) use two septin-associated proteins with known septin-binding capacity to validate that this method authentically detects direct physical interaction, and 4) assess the septin-binding selectivity of three bud neck–localized proteins whose septin association per se has not been positively characterized previously. We also demonstrate that variation of the fluorescence signal with tether length provides a novel molecular ruler for measuring distances at the nanometer scale.
RESULTS
Rationale for using the tripartite split-GFP protein–protein interaction system
Various experimental strategies have been applied to determine the molecular organization of the septin superstructure that demarcates the division site in a budding yeast cell. Recombinant protein expression, in vitro reconstitution, and ultrastructural analysis by EM revealed the linear, apolar, hetero-octameric arrangement of the septin subunits in mitotic cells (Versele and Thorner, 2004; Farkasovsky et al., 2005; Bertin et al., 2008; Garcia et al., 2011). Genetic analyses confirmed subunit order in vivo and helped to delineate the contributions of individual subunits and their domains to the physiological functions of septins (McMurray et al., 2011; Finnigan et al., 2015a, b). Genome-wide cytological studies, including indirect immunofluorescence of epitope-tagged proteins (Kumar et al., 2002) and direct visualization of GFP-tagged proteins (Huh et al., 2003), provided a partial catalogue of proteins that localize to the bud neck. Other tactics for detecting protein–protein interaction have also been exploited to discern what other proteins associate with septins, including the two-hybrid screen (Drees et al., 2001), FRET analysis (Booth et al., 2015), a variant of the split-ubiquitin reconstitution system (dubbed SPLIFF; Dunkler et al., 2015), and affinity purification followed by mass spectrometry (Renz et al., 2016). Every approach has, however, its own inherent disadvantages and limitations in terms of sensitivity of detection, throughput, and/or application to live cells.
To initiate a systematic study to assess whether any given cellular protein is able to bind directly to a septin at the bud neck, we felt that a newly devised tripartite split-GFP system (Cabantous et al., 2013) had many advantages for assessing intimate physical contact between two proteins. First, this method involves the attachment of unstructured tags of minimal size to the target proteins of interest (N-terminal β10 [20 residues], C-terminal β11 [21 residues]; Supplemental Figure S1; also see Supplemental Tables S1 and S2). Unlike attachment of the much bulkier reporters required for other methods, this feature greatly reduces the likelihood that presence of the tag itself will compromise the structure or function of the target protein, interfere sterically with the binding interface between the two target proteins, or occlude a localization signal responsible for delivery to the proper cellular compartment in situ. Second, the tags can be anchored to the target proteins via flexible tethers (linker sequences) of variable length (Supplemental Figure S1). This property obviates the need for the tags to be installed in any particular orientation and, as we demonstrate, provides a means by which this system can be used to assess the separation distance between two target proteins on a biologically relevant length scale. Third, the readout in this system involves capture by the tags of a GFPβ1-9 barrel expressed as a free protein, an encounter that can occur at a detectable level only if the β10 and β11 tags are held in close proximity via association of the two target proteins. This three-way association reconstitutes eGFP, allows for formation of its fluorophore, and thereby further stabilizes interaction between the two target proteins. Thus this system can detect even weak or transient protein–protein interactions and yields a convenient fluorescence output for the positives.
Implementation of the tripartite split-GFP system for detecting septin interactions in vivo
As our initial test of the efficacy of the tripartite split-GFP method to detect authentic protein–protein interactions in live yeast cells, and given that the order of the subunits in septin complexes is well defined and invariant, we examined whether this method would accurately report the known arrangement of the septin subunits. We used the following experimental design (Figure 1A). First, in MATα cells, a protein of interest was tagged at its N-terminus with β10 and expressed from its endogenous promoter at its normal chromosomal locus (Table 1). To mark the bud neck, these same cells also expressed either Cdc10–monomeric red fluorescent protein mCherry (mCh) or Cdc11-mCh from its endogenous promoter at its normal chromosomal locus. Second, in MATa cells, another protein of interest was tagged at its C-terminus with β11 and expressed from its endogenous promoter at its normal chromosomal locus (Table 1). These same cells carried a CEN plasmid that expresses the GFPβ1-9 barrel under control of the inducible GAL1/10 promoter (Table 2), allowing for control of both the level and timing of its expression. Therefore, to assess the interaction between any two proteins (e.g., two septin subunits), the appropriate MATa and MATα haploids were mated, the resulting diploid grown on galactose-containing medium (thereby providing all three components of the tripartite split-GFP system in the same cell), and then examined under the fluorescence microscope.
FIGURE 1:

Implementation of the tripartite split-GFP system in live yeast. (A) A mating-based strategy to generate cells expressing all of the components of the tripartite split-GFP method. A haploid that expresses an N-terminally β10-tagged protein of interest from its endogenous promoter at its native chromosomal locus (and also expresses an independent marker for the subcellular location of interest, where available) is mated to a haploid of opposite mating type that expresses a C-terminally β11-tagged protein of interest from its endogenous promoter at its native chromosomal locus and also harbors a CEN plasmid that expresses from a regulatable promoter the GFPβ1-9 barrel. (B) All three components of the tripartite split-GFP system are required to generate a protein–protein interaction signal in vivo. Diploids strains (1–8; Supplemental Table S3) that express all three or only two, one, or no components of the tripartite split-GFP system, as indicated, were constructed as in A by two rounds of selection on minimal (SD)-Leu-His medium with 2% glucose. GFPβ1-9 was carried on a LEU2-marked CEN plasmid (pGF-IVL794) and expressed under control of the GAL1/10 promoter. Cultures of the indicated diploids were grown overnight to saturation in SD-Leu-His medium with 2% raffinose–0.2% sucrose, back-diluted into the same medium with 2% galactose, grown at 30°C for 4.5 h, harvested, washed, and imaged by fluorescence microscopy. All images were captured after the identical exposure time and processed using ImageJ. Dotted white line, cell periphery. Scale bar, 2 μm. E.V., empty vector (pRS315). Diploid 1 contained one copy of β10-(32-residue linker)-Cdc10 and one copy of Cdc10-(33-residue linker)-β11 (Supplemental Figure S1). (C) Quantification of the average GFP fluorescence at the bud neck in budded cells (25–100 per culture) for the strains in B. Error bar, SEM. Dashed red line, average intrinsic background fluorescence (∼35–40 pixels) at the bud neck in cells lacking the components of the tripartite split-GFP system for images taken at the identical exposure time. (D) Left, confirmation that the five proteins indicated are abundant cytosolic proteins. Strains expressing each of the indicated proteins (see Supplemental Table S4) as an eGFP fusion (GFY-1977, GFY-1981, GFY-2034, GFY-2030, and GFY-2033) were grown to saturation in rich (YPD) medium, back-diluted into fresh YPD, grown to mid exponential phase, and imaged as in A, except that the exposure times varied: Gpp1-eGFP (150 ms), Hsp82-eGFP (150 ms), Cdc19-eGFP (100 ms), Pgk1-eGFP (50 ms), and Tpi1-eGFP (50 ms). Right, none of five extremely abundant cytosolic proteins exhibits more than a very weak interaction with any septin. Representative examples for the interaction of the indicated β11-tagged proteins with β10-Cdc3 using the tripartite split-GFP method. In diploids 164, 170, 272, 282, and 277 expressing the indicated proteins, expression of GFPβ1-9 was induced and the cells imaged as in B. (E) Quantification, as in C, of the fluorescence signal at the bud neck in dividing diploids (25–100 per culture) that expressed each abundant β11-tagged cytosolic protein with each β10-septin (see Supplemental Table S3 for a complete list).
TABLE 1:
Yeast strains used in this study.
| Strain | Genotype | Reference |
|---|---|---|
| BY4741 | MATa leu2∆ ura3∆ met15∆ his3∆ | Brachmann et al. (1998) |
| BY4742 | MATα leu2∆ ura3∆ met15∆ his3∆ | Brachmann et al. (1998) |
| GFY-42 | BY4741; cdc10∆::CDC10::mCherry::ADH1(t)::SpHIS5 | Finnigan et al. (2015b) |
| GFY-59a | BY4741; cdc11∆::CDC11::mCherry::ADH1(t)::SpHIS5 | This study |
| GFY-1794b | BY4742; cdc10∆::GFPβ10::Linker(32)::CDC10::ADH1(t)::HygR cdc11∆::CDC11::mCherry::SpHIS5 | This study |
| GFY-1570c | BY4741; cdc10∆::CDC10::Linker(33)::GFPβ11::ADH1(t)::NatR | This study |
| GFY-1979d | BY4741; PGK1::Linker(33)::GFPβ11::ADH1(t)::KanR | This study |
| GFY-1983e | BY4741; HSP82::Linker(33)::GFPβ11::ADH1(t)::KanR | This study |
| GFY-2035f | BY4741; CDC19::Linker(43)::GFPβ11::SHS1(t)::KanR | This study |
| GFY-2036f | BY4741; GPP1::Linker(43)::GFPβ11::SHS1(t)::KanR | This study |
| GFY-2043f | BY4741; TPI1::Linker(43)::GFPβ11::SHS1(t)::KanR | This study |
| GFY-1793g | BY4742; cdc3∆::GFPβ10::Linker(18)::CDC3::ADH1(t)::HygR cdc10∆::CDC10::mCherry::SpHIS5 | This study |
| GFY-1851h | BY4741; cdc10∆::CDC10::GFPβ11::ADH1(t)::HygR | This study |
| GFY-1801i | BY4742; cdc10∆::GFPβ10::Linker(5)::CDC10::ADH1(t)::HygR cdc11∆::CDC11::mCherry::SpHIS5 | This study |
| GFY-1798 | BY4742; cdc3∆::GFPβ10::Linker(5)::CDC3::ADH1(t)::HygR cdc10∆::CDC10::mCherry::SpHIS5 | This study |
| GFY-1803 | BY4742; cdc12∆::GFPβ10::Linker(5)::CDC12::ADH1(t)::HygR cdc10∆::CDC10::mCherry::SpHIS5 | This study |
| GFY-1804 | BY4742; cdc11∆::GFPβ10::Linker(5)::CDC11::ADH1(t)::HygR cdc10∆::CDC10::mCherry::SpHIS5 | This study |
| GFY-1807 | BY4742; shs1∆::GFPβ10::Linker(5)::SHS1::ADH1(t)::HygR cdc10∆::CDC10::mCherry::SpHIS5 | This study |
| GFY-1796 | BY4742; cdc10∆::GFPβ10::Linker(18)::CDC10::ADH1(t)::HygR cdc11∆::CDC11::mCherry::SpHIS5 | This study |
| GFY-1793 | BY4742; cdc3∆::GFPβ10::Linker(18)::CDC3::ADH1(t)::HygR cdc10∆::CDC10::mCherry::SpHIS5 | This study |
| GFY-1797 | BY4742; cdc12∆::GFPβ10::Linker(18)::CDC12::ADH1(t)::HygR cdc10∆::CDC10::mCherry::SpHIS5 | This study |
| GFY-1795j | BY4742; cdc11∆::GFPβ10::Linker(18)::CDC11::ADH1(t)::HygR cdc10∆::CDC10::mCherry::SpHIS5 | This study |
| GFY-1806 | BY4742; shs1∆::GFPβ10::Linker(18)::SHS1::ADH1(t)::HygR cdc10∆::CDC10::mCherry::SpHIS5 | This study |
| GFY-1852k | BY4741; cdc10∆::CDC10::Linker(10)::GFPβ11::ADH1(t)::HygR | This study |
| GFY-1853l | BY4741; cdc10∆::CDC10::Linker(20)::GFPβ11::ADH1(t)::HygR | This study |
| GFY-1845 | BY4741; cdc3∆::CDC3::GFPβ11::ADH1(t)::HygR | This study |
| GFY-1848 | BY4741; cdc12∆::CDC12::GFPβ11::ADH1(t)::HygR | This study |
| GFY-1842 | BY4741; cdc11∆::CDC11::GFPβ11::ADH1(t)::HygR | This study |
| GFY-1839 | BY4741; shs1∆::SHS1::GFPβ11::ADH1(t)::HygR | This study |
| GFY-1809 | BY4742; bni5∆::GFPβ10::Linker(18)::BNI5::ADH1(t)::HygR cdc10∆::CDC10::mCherry::SpHIS5 | This study |
| GFY-1572 | BY4741; cdc3∆::CDC3::Linker(33)::GFPβ11::ADH1(t)::NatR | This study |
| GFY-1571 | BY4741; cdc12∆::CDC12::Linker(33)::GFPβ11::ADH1(t)::NatR | This study |
| GFY-1573 | BY4741; cdc11∆::CDC11::Linker(33)::GFPβ11::ADH1(t)::NatR | This study |
| GFY-1567 | BY4741; shs1∆::SHS1::Linker(33)::GFPβ11::ADH1(t)::NatR | This study |
| GFY-1735 | BY4741; bni5∆::BNI5::Linker(33)::GFPβ11::ADH1(t)::NatR | This study |
| GFY-1899 | BY4742; nis1∆::GFPβ10::Linker(18)::NIS1::ADH1(t)::HygR cdc10∆::CDC10::mCherry::SpHIS5 | This study |
| GFY-1854 | BY4741; nis1∆::NIS1::Linker(33)::GFPβ11::ADH1(t)::HygR | This study |
| GFY-1992m | BY4742; hsl1∆::GFPβ10::Linker(32)::HSL1(1-1518)::ADH1(t)::KanR cdc10∆::CDC10::mCherry::SpHIS5 | This study |
| GFY-1995n | BY4742; hsl1∆::GFPβ10::Linker(32)::HSL1(611-950 R635A R636A K645A H648A K649A R653A K654A K775A E776A N777A R828A L831A)::ADH1(t)::KanR cdc10∆::CDC10::mCherry::SpHIS5 | This study |
| GFY-1997o | BY4742; hsl1∆::GFPβ10::Linker(32)::HSL1(611-950; 1245-1518 R635A R636A K645A H648A K649A R653A K654A K775A E776A N777A R828A L831A)::ADH1(t)::KanR cdc10∆::CDC10::mCherry::SpHIS5 | This study |
| GFY-1846 | BY4741; cdc3∆::CDC3::Linker(10)::GFPβ11::ADH1(t)::HygR | This study |
| GFY-1849 | BY4741; cdc12∆::CDC12::Linker(10)::GFPβ11::ADH1(t)::HygR | This study |
| GFY-1843 | BY4741; cdc11∆::CDC11::Linker(10)::GFPβ11::ADH1(t)::HygR | This study |
| GFY-1840 | BY4741; shs1∆::SHS1::Linker(10)::GFPβ11::ADH1(t)::HygR | This study |
| GFY-1847 | BY4741; cdc3∆::CDC3::Linker(20)::GFPβ11::ADH1(t)::HygR | This study |
| GFY-2044 | BY4741; cdc12∆::CDC12::Linker(20)::GFPβ11::ADH1(t)::HygR | This study |
| GFY-1844 | BY4741; cdc11∆::CDC11::Linker(20)::GFPβ11::ADH1(t)::HygR | This study |
| GFY-1841 | BY4741; shs1∆::SHS1::Linker(20)::GFPβ11::ADH1(t)::HygR | This study |
| GFY-1996 | BY4742; hsl1∆::GFPβ10::Linker(18)::HSL1(611-950; 1245-1518 R635A R636A K645A H648A K649A R653A K654A K775A E776A N777A R828A L831A)::ADH1(t)::KanR cdc10∆::CDC10::mCherry::SpHIS5 | This study |
| GFY-1998 | BY4742; hsl1∆::GFPβ10::Linker(5)::HSL1(611-950; 1245-1518 R635A R636A K645A H648A K649A R653A K654A K775A E776A N777A R828A L831A)::ADH1(t)::KanR cdc10∆::CDC10::mCherry::SpHIS5 | This study |
| GFY-1977p | BY4741; PGK1::eGFP::ADH1(t)::KanR | This study |
| GFY-1981p | BY4741; HSP82::eGFP::ADH1(t)::KanR | This study |
| GFY-2031q | BY4741; TPI1::eGFP::ADH1(t)::HygR | This study |
| GFY-2033q | BY4741; GPP1::eGFP::ADH1(t)::HygR | This study |
| GFY-2034q | BY4741; CDC19::eGFP::ADH1(t)::HygR | This study |
| GFY-1318 | BY4741; CDC10::mCherry::KanR bni5∆::GFP::BNI5::SpHIS5 cdc11∆::CDC11::HygR SHS1 + pJT1520 | Finnigan et al. (2015a) |
aStrain was constructed by integrating CDC11::mCherry::ADH1(t)::SpHIS5 amplified from pGF-IVL1 into cdc11∆::KanR yeast (GFY-150). The strain was selected twice on 5-FOA–containing medium to counterselect for the WT CDC11-expressing covering vector. Unless otherwise noted, all strains were selected on 5-FOA to remove these covering vector(s) before diploid formation.
bStrain was constructed by integrating the tagged CDC10 allele (from pGF-IVL824) into cdc10∆ yeast (GFY-1603). The GFPβ10 sequence is MDLPDDHYLSTQTILSKDLN (Cabantous et al., 2013). The 32-residue linker sequence is DVGGGGSEGGGSGGPGSGGEGSAGGGSAGGGS. CDC11 was tagged with mCherry by amplifying the entire locus from chromosomal DNA from GFY-59 and transforming into GFY-1643. N-terminally tagged proteins were constructed using this strategy unless otherwise noted. All of the flexible linker sequences (N- or C-terminal) were modeled as previously described (Cabantous et al., 2013).
cStrain was constructed by integrating the tagged CDC10 allele (from pGF-IVL810) into cdc10∆ yeast (GFY-140). The GFPβ11 sequence is EKRDHMVLLEYVTAAGITDAS (Cabantous et al., 2013). The 33-residue linker is DYKDDDDKGSGAGGSPGGGSGGSGSSASGGSTS. C-terminally tagged strains were constructed using this strategy unless otherwise noted.
dStrain was constructed by creating an integrating vector containing the entire PGK1 ORF fused to the C-terminal tag including the drug cassette and, finally, 491 base pairs of 3′ UTR (pGF-IVL1054). The entire cassette was amplified and transformed into BY4741 yeast.
eStrain was constructed by first creating an integrating vector containing 471 base pairs of the HSP82 ORF fused to the C-terminal tag including the drug cassette, and finally, 500 base pairs of 3′ UTR (pGF-IVL1056). The entire cassette was amplified and transformed into BY4741 yeast.
fThese strains were constructed by amplifying the C-terminal cassette (from pGF-V763) including a slightly larger linker sequence (including an upstream GRRIPGLINP) with short 30–base pair oligonucleotide tails to each locus of interest. We used 469 base pairs of SHS1 3′ UTR sequence as the terminator, and the promoter of CCW12 (992 base pairs) replaced the Ptef sequence of the MX cassette. Strains were confirmed via diagnostic PCR and DNA sequencing of the full junction of the gene with the C-terminal tag.
gThe 18-residue linker sequence is DVGGGGSEGGGSGGPGSG.
hThere is no linker between the C-terminus of CDC10 and the GFPβ11 sequence.
iThe 5-residue linker has the sequence DVGGG.
jStrains with an N-terminal GFPβ10 tag with a 32-residue linker appended to CDC11 were lethal.
kThe 10-residue linker has the sequence GSSASGGSTS.
lThe 20-residue linker has the sequence GSPGGGSGGSGSSASGGSTS.
mThe tagged HSL1 construct was integrated into hsl1∆::HygR yeast (GFY-1902) by amplifying two PCR fragments that contained overlapping sequence within the HSL1 ORF from plasmid pGF-IVL1034.
nThis Hsl1 construct has a putative NLS sequence mutated to alanine to prevent nuclear import, as well as the KEN box (residues 775–781) and D-box (residues 828–836) destruction motifs to prevent APC-dependent degradation.
oThis Hsl1 construct contains both the septin-binding domain (residues 611–950) and sequence containing the C-terminal KA1 domain (residues 1245–1518), both of which are required for optimal localization of Hsl1 to the septin collar in vivo (Finnigan et al., 2016).
pStrains were constructed similar to GFY-1979 and GFY-1983 using full-length eGFP and confirmed via DNA sequencing.
qStrains were constructed similar to GFY-2035 using eGFP::ADH1(t)::HygR as template DNA. Proper integration was confirmed via DNA sequencing.
TABLE 2:
Plasmids used in this study.
| Plasmid | Description | Reference |
|---|---|---|
| pRS315 | CEN; LEU2 AMP | Sikorski and Hieter (1989) |
| pRS316 | CEN; URA3 AMP | Sikorski and Hieter (1989) |
| pGF-IVL794a | pRS315; prGAL1/10::GFPβ1-9(A)::ADH1(t)::KanR | This study |
| pGF-IVL795b | pRS315; prGAL1/10::GFPβ1-9(B)::ADH1(t)::KanR | This study |
| pGF-IVL553c | pRS315; prNIS1::NIS1::ADH1(t)::KanR | This study |
| pGF-IVL521 | pRS315; prCDC11::HSL1(1-1518)::eGFP::ADH1(t)::KanR | Finnigan et al. (2016) |
| pGF-IVL762d | pRS315; prCDC11::hsl1(611-950 R635A R636A K645A H648A K649A R653A K654A)::eGFP::ADH(1)::KanR | Finnigan et al. (2016) |
| pGF-IVL536e | pRS315; prCDC11::hsl1(611-950; 1245-1518)::eGFP::ADH1(t)::KanR | Finnigan et al. (2016) |
| pGF-IVL1095f | pRS315; prBUD4::BUD4(623-774)::eGFP::ADH1(t)::KanR | This study |
| pGF-IVL1110g | pRS315; prHOF1:: eGFP::HOF1(293-355)::ADH1(t)::KanR | This study |
| pGF-IVL1082 | pRS315; prBUD4::BUD4(623-774)::Linker(33)::GFPβ11::ADH1(t)::KanR | This study |
| pGF-IVL1084 | pRS315; prHOF1::HOF1(293-355)::Linker(33)::GFPβ11::ADH1(t)::KanR | This study |
| pGF-IVL1105 | pRS316; prGAL1/10::GFPβ1-9(A)::ADH1(t)::KanR | This study |
aThe evolved GFPβ1-9 was synthesized as a custom gene using a yeast codon bias. The A version designates the position of the last residue: this version ends with the sequence GPVLLPDNGS (Cabantous et al., 2013).
bA second variant of the GFPβ1-9 was created identical to the A version but ending with the sequence GPVLLP. The prGAL1/10 promoter includes 814 base pairs of 5′ UTR sequence.
cThe endogenous NIS1 promoter includes 576 base pairs of 5′ UTR sequence.
dThe putative NLS signal within the N-terminus of Hsl1(611-950) was experimentally determined to require residues R635 R636 K645 H648 K649 R653 K654 for constructs beginning at residue 611 (Finnigan et al., 2016). Mutation of these residues to alanine eliminates nuclear localization of this Hsl1 fragment.
eThe combination of the central Hsl1(611-950) septin-binding domain and the C-terminal KA1 domain contained within Hsl1(1245-1518) has been shown to be sufficient to efficiently target Hsl1 to the bud neck in vivo (Finnigan et al., 2016).
fThe Bud4(623-774) fragment has been shown to be sufficient to target to the septin collar in vivo (Wu et al., 2015). The BUD4 sequence was amplified from pGF-V416 and assembled by in vivo ligation and homologous recombination. Both N- and C-terminal eGFP fusions to this Bud4 fragment displayed localization to the septin collar.
gThe Hof1(293-355) fragment has been shown to be sufficient to target to the septin collar in vivo (Meitinger et al., 2013). The HOF1 sequence was amplified from pGF-V454 and assembled via in vivo ligation. Only the N-terminal eGFP fusion to this Hof1 fragment displayed localization to the septin collar (unpublished data).
As our first proof of principle, we asked whether we could readily detect the interaction between the two Cdc10 monomers that are juxtaposed at the center of the hetero-octamer (Supplemental Figure S1A). Indeed, in diploids coexpressing β10-Cdc10, Cdc10-β11, and GFPβ1-9, a very bright green fluorescence signal was generated in the vast majority (>90%) of the cells that was confined to and congruent with the septin collar at the bud neck, which was marked with Cdc11-mCh (Figure 1B). Of importance, when examined for the same exposure time in diploids produced from mating otherwise isogenic strains lacking any one of the tripartite split-GFP components, there was virtually no detectable signal above the background of the intrinsic fluorescence of a yeast cell measured with the cut-off filters we used (Figure 1B). Quantification of the average pixel intensity at the bud neck for the cells expressing β10-Cdc10, Cdc10-β11, and GFPβ1-9 indicated that the fluorescence signal was >20-fold brighter than any of the negative controls (Figure 1C). Using this same readout, we found that either of two different versions of the GFPβ1-9 barrel (Cabantous et al., 2013) that differ by four residues at their C-terminal end (i.e., just downstream of β9) yielded equally robust fluorescence signals (Supplemental Figure S2).
Our next concern was to eliminate the possibility that the tripartite split-GFP system might erroneously report adventitious and physiologically irrelevant interactions. Therefore we tested whether any of five gene products that are reportedly among the most abundant cytosolic proteins in S. cerevisiae (some estimates as high as 300,000–500,000 molecules/cell; Supplemental Table S4) could produce a significant degree of fluorescence by this method. Each of these candidates (Cdc19, Gpp1, Hsp82, Pgk1, and Tpi1), when expressed as a C-terminally GFP-tagged derivative from its endogenous promoter at its native chromosomal locus, exhibited a very prominent cytosolic distribution (Figure 1D, left) and thus should be readily accessible to the septin collar at the bud neck. Although none of these five test proteins has any interaction of any kind with any septin subunit recorded in the literature, with the single exception of a purported two-hybrid interaction between Cdc12 and Hsp82 (Millson et al., 2005), we reasoned that, if the tripartite split-GFP system were prone to false positives generated by random encounters, this would be revealed with such abundantly expressed proteins. To maximally challenge the tripartite split-GFP system and give it every opportunity to produce a spurious readout, we appended the β11 tag to the C-terminal end of each of the five test proteins via a flexible 33-residue linker and crossed strains producing them against a strain expressing N-terminally β10-tagged Cdc3, which, of all the mitotic septins, is the subunit with the longest N-terminal extension (Supplemental Figure S1B). Very reassuringly, with β10-Cdc3 (Figure 1D) and every other β10-tagged septin (Figure 1E), we found that the majority of the cells expressing Hsp82-β11 and Gpp1-β11 exhibited virtually no detectable signal at the bud neck above the intrinsic fluorescence of yeast cells and that the cells expressing Pgk1-β11, Cdc19-β11, and Tpi1-β11 displayed very weak fluorescence signals that were never more than twofold to threefold above the intrinsic background fluorescence.
Tripartite split-GFP readout accurately reflects distant constraints in septin complexes
Having found a robust readout for the interaction between the two tagged Cdc10 monomers (β10-Cdc10 and Cdc10-β11) that constitute the central core of the septin hetero-octamer, we reasoned that a further test of this system to report authentic protein–protein interactions would be to examine whether the readout obtained would accurately represent the known distance relationships between Cdc10 and the other subunits in the septin hetero-octamer. For this reason, and using the same overall strategy (Figure 1A), we generated diploids in which we assessed the ability of Cdc10-β11 to interact with N-terminally β10-tagged versions of each of the other septins (Figure 2). Gratifyingly, when the β10 or β11 tags were appended to a septin using linkers of minimal length, we found that Cdc10-β11 strongly interacted with only its immediately neighboring subunits, either β10-Cdc10 or β10-Cdc3, respectively, in the vast majority of the cells (80–90%; Figure 2, A and B).
FIGURE 2:

Influence of linker length on the output of the tripartite split-GFP system. (A) Left, diploid yeast (strains 24–28) were generated that express Cdc10-β11 along with each of the β10-septins (with the tags appended using the linker lengths given) as indicated, as well as a CEN plasmid (pGF-IVL794) to express GFPβ1-9 (unpublished data). Right, cultures of each of the corresponding diploids were grown and the cells imaged as in Figure 1B. Representative images are shown, and the percentage of the cells displaying the pattern shown is indicated in the upper right-hand corner. The fiducial marker for the septin collar is Cdc10-mCh expressed in the same cells, except in the cells expressing Cdc10-β11 and β10-Cdc10 (top), for which the marker is Cdc11-mCh. (B) Heat map depicting the percentage of cells (number in the box) in the population exhibiting a readily detectable green fluorescence signal for 40 diploids (strains 10–14, 17–21, 24–28, 31–35, 38–42, 45–49, 52–56, and 59x63) with the arrangements shown in A, but where the linker connecting the β11 tag to the C-terminus of Cdc10 was systematically varied from 0 to 10, 20, or 33 residues as indicated (at top) and the linker between the N-terminal β10 tag and the N-terminus of each septin was 5 or 18 residues as indicated (at left). (C) Quantification, as in Figure 1C, of data for the diploids for which the linker lengths for Cdc10-β11 were 0, 10, 20, or 33 residues as indicated and the linker between the N-terminal β10 tag and the N-terminus of each septin was five residues (strains 10–14, 17–21, 24–28, and 21–35).
We next explored the possibility that extending the length of the linker (Supplemental Figure S1A) might allow for detection of association with a stably held, but somewhat more distantly located, interaction partner. For this purpose, we varied the length of the linker in Cdc10-β11 from 0 to 10, 20, and 33 residues and also increased the length of the linker in the N-terminally β10-tagged versions of each of the other septins from five to 18 residues and generated the respective diploids. For the expected interactions (Cdc10-Cdc10 and Cdc10-Cdc3), we found that increasing the tether length had a negligible effect on the percentage of the cell population exhibiting an obvious fluorescence signal at the bud neck (Figure 2B) but did progressively enhance the strength of that fluorescence signal (Figure 2C). Moreover, and strikingly, when the tether length was increased, interaction of Cdc10-β11 with β11-Cdc12 became detectable, with both the fraction of the cells displaying a detectable signal and the pixel intensity of the signal increasing with increasing linker length (Figure 2C and Supplemental Figure S3). Nonetheless, on both absolute and relative scales, the output from the nearest-neighbor Cdc10-Cdc10 and Cdc10-Cdc3 interactions was much more robust (at least an order of magnitude brighter) than that from the longer-range Cdc10-Cdc12 interaction regardless of linker length (Figure 2C). Moreover, regardless of linker length, no interaction between the centrally disposed Cdc10 subunit and either of the alternative terminal subunits (Cdc11 and Shs1) was detectable. Thus our findings indicate that 1) the tripartite split-GFP system accurately reflects the spatial relationships among the subunits of the septin hetero-octamer and 2) because we were able to detect, albeit weakly, interaction between Cdc10-β11 with β11-Cdc12 by extending the linker length between each tag and its cognate protein (which we estimate to be ∼10 nm apart), systematic variation of the length of these tethers provides, in principle, a “molecular ruler” to estimate the maximum distance or limit of detection in this in vivo protein–protein interaction system, although such spacing constraints may vary significantly, depending on the size and flexibility of the native N- and C-termini in the two proteins being tested, as is the case for the septins (Supplemental Figure S1B). Moreover, for septin–septin interactions, especially at long tether lengths, the possibility of cross-filament interactions needs to be considered.
To further explore the inherent effects of the variability among the septins in the lengths of their native N- and C-termini (Supplemental Figure S1B) on the readout generated by the tripartite split-GFP system, we generated constructs in which the N-terminal β10 tag was appended to each septin by a minimal tether (five residues) and the β11 tag was appended to the C-terminus of each septin without any spacer at all. Similar to Cdc10-β11 tested against N-terminally β10-tagged versions of the other septins (Figure 2), β10-Cdc10 tested against C-terminal β11-tagged versions of the other septins yielded a robust signal with Cdc10-β11, as expected. However, compared with the nearest-neighbor interaction assessed by testing Cdc10-β11 against β10-Cdc3, β10-Cdc10 tested against Cdc3-β11 yielded a detectable but much weaker signal (Figure 3, A and E, left). In addition, β10-Cdc10 tested against Cdc12-β11 yielded a weak but readily detectable signal, quite akin to what we observed between Cdc10-β11 and β10-Cdc12 when we increased their linker lengths. Similarly, β10-Cdc3 tested against C-terminal β11-tagged versions of the other septins yielded the strongest output with the immediately juxtaposed subunits in the hetero-octamer (Cdc10-β11 and Cdc12-β11); however, β10-Cdc3 was also able to generate significant fluorescence signals with itself (Cdc3-β11) and the most distal subunits (Cdc11-β11 and Shs1-β11; Figure 3, B and E, middle left). We presume that this increased “promiscuity” is due to the fact that, unlike the other four septins, Cdc3 has a prominent (107 residues) N-terminal extension (Supplemental Figure S1B). By contrast, β10-Cdc12 generated its highest fluorescence signals only with its immediately juxtaposed subunits (Cdc3-β11, Cdc11-β11, and Shs1-β11), whereas its interaction with the most distant subunit (Cdc10-β11) was barely above background (Figure 3, C and E, middle right). The fact that Shs1 has the longest CTE of any septin (Supplemental Figure S1B) may explain why the combination of β10-Cdc12 and Shs1-β11 yielded the topmost signal. For β10-Cdc11, readily detectable signals were observed with its immediately adjacent subunit (Cdc12-β11) and with the subunits (Cdc11-β11 and Shs1-β11) with which it forms, respectively, both homotypic and heterotypic junctions when hetero-octamers polymerize into filaments (Booth et al., 2015; Finnigan et al., 2015b); however, β10-Cdc11 also displayed equivalent interaction with Cdc3-β11, presumably due to the length of the CTE present in Cdc3 (Figure 3, C and E, middle right). Finally, β10-Shs1 also displayed a detectable signal with its immediately adjacent subunit (Cdc12-β11) but significantly more robust signals with itself and with the subunit (Cdc11-β11) with which it forms a heterotypic junction in polymerized filaments (Supplemental Figure S4). As observed for β10-Cdc11, β10-Shs1 also displayed interaction with Cdc3-β11, again presumably due to the length of the CTE in Cdc3. Thus, in summary, although not obviating the ability to detect both authentic and nearby interaction partners, the positions of the tags clearly do affect their accessibility and hence the efficiency of their encounter and ability to capture the GFPβ1-9 barrel and produce a readout.
FIGURE 3:

Further analysis of septin–septin interactions using the tripartite split-GFP system. Each of the essential septins (Cdc3, Cdc10, Cdc11, and Cdc12) was tagged with an N-terminal β10 tag and a five-residue linker and then combined by mating, as in Figure 1A, with each of the five mitotic septins tagged at their immediate C-terminal end with β11, as indicated schematically in the left-hand diagrams. Except in the cases in which both copies of the same septin were tagged (e.g., β10-Cdc10 and Cdc10-β11), a second (WT) copy of each tagged subunit is present but has been omitted for clarity. The resulting diploids were grown, induced with galactose, washed, and imaged as in Figure 1B. (A) Diploids 73–77, (B) diploids 66–70, (C) diploids 101–105, and (D) diploid strains 80–84. For β10-Shs1 examined in the same way, see Supplemental Figure S4. The fiducial marker for the septin collar was Cdc10-mCh expressed in the same cells, except in the cells expressing β10-Cdc10 and Cdc10-β11, for which the marker was Cdc11-mCh. (E) Quantification, as in Figure 1C, of the data shown in A–D.
In this same regard, one potential drawback to placing the β10 tag on the N-terminus of either Cdc11 or Shs1 is that, even with the five-residue spacer used, these two septins natively lack a significant extension upstream of the polybasic tract present in their α0 helix (Supplemental Figure S1B). These basic residues are believed to mediate interaction with phosphatidylinositol-4,5-bis-phosphate (PtdIns4,5P2) on the inner leaflet of the PM (Bertin et al., 2010) and are required for the full in vivo function of these septins (Finnigan et al., 2015b). Thus access to a tag placed at the N-terminus of either of these two septins may be limited, resulting in a weaker signal in this assay format.
Validating the ability of the tripartite split-GFP method to detect septin-binding proteins
To verify that the tripartite split-GFP method can also faithfully report direct interaction of other cellular proteins with septins at the bud neck, we first examined Bni5 (448 residues). Bni5 is recruited to the bud neck before assembly of the actomyosin ring and is ejected from the bud neck by the time the septin collar has split (Fang et al., 2010; Schneider et al., 2013), as we also observed (Figure 4A). There is ample evidence from two-hybrid (Lee et al., 2002) and mutational analysis (Finnigan et al., 2015a) in vivo and in FRET (Booth et al., 2015) and EM (Patasi et al., 2015) analysis in vitro that Bni5 physically associates with septins Cdc11 and Shs1. We examined, first, the interaction of C-terminally tagged Bni5-β11 with N-terminally β10-tagged versions of the five mitotic septins. Despite the fact that, for the reasons mentioned earlier, the N-termini of Cdc11 and Shs1 are liable to be less accessible than for the other three septins, Bni5-β11 exhibited a readily detectable interaction with β10-Cdc11 and β10-Shs1 and displayed no detectable interaction with β10-Cdc3, β10-Cdc10, or β10-Cdc12 (Figure 4, B and C). These results are in agreement with the published subunit selectivity of Bni5 and with the fact that deletion analysis showed that only the C-terminal one-third of Bni5 is required for its function (Finnigan et al., 2015a). However, structure prediction indicates that Bni5 is likely a highly elongated protein comprising a long bundle of nearly all α-helical segments with significant separation between its N- and C-termini (Finnigan et al., 2015a). Therefore we also examined the interaction of N-terminally tagged β10-Bni5 with C-terminally β11-tagged versions of the five mitotic septins. Although the strongest interactions observed were once again between β10-Bni5 and Cdc11-β11 and Shs1-β11, nearly equivalent outputs were observed with the next-nearest subunit (Cdc12-β11) and even the more distantly positioned subunits (Cdc3-β11 and Cdc10-β11; Figure 4, D and E). Taken together, these data support the conclusion that the C-terminal end of Bni5 is anchored to Cdc11 and Shs1, whereas the N-terminal end of this highly elongated protein is more flexible and can sweep out a much larger conformational space. By contrast, we detected no interaction between β10-Bni5 and Bni5-β11, suggesting that this protein either does not readily self-associate or forms oligomers that are obligatorily in the parallel orientation.
FIGURE 4:
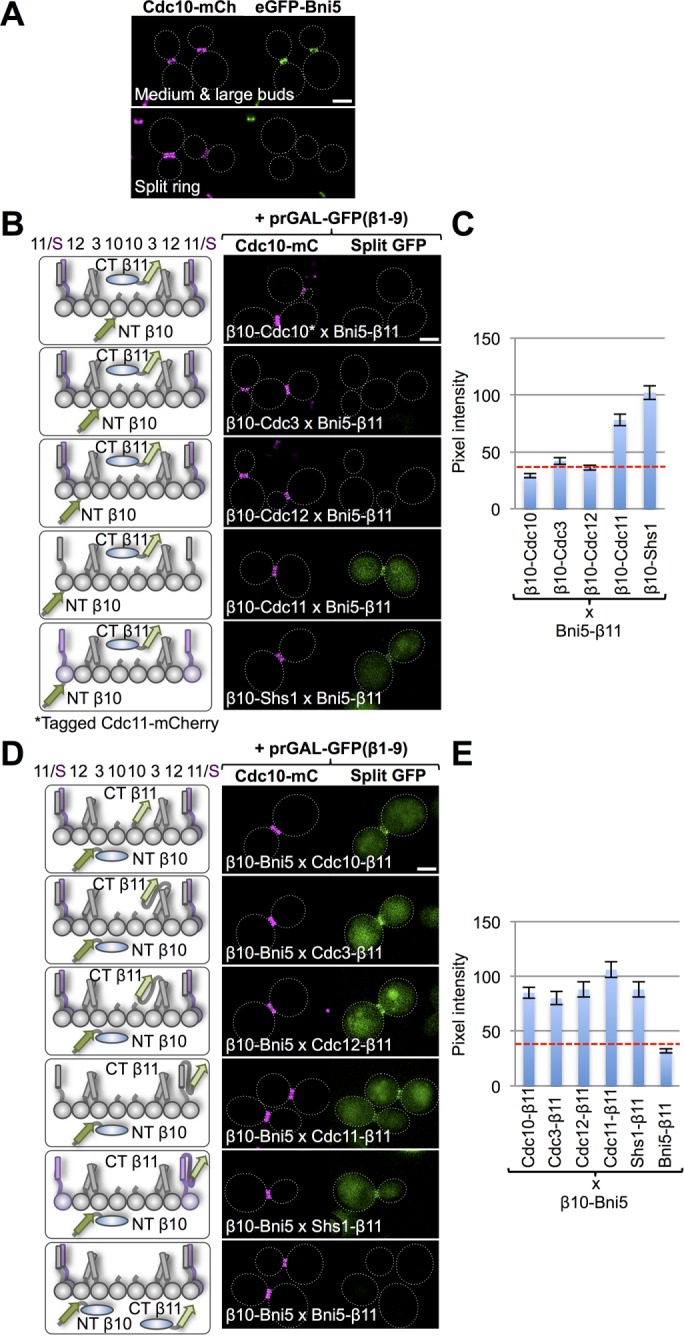
Interaction of Bni5 with the septin collar. (A) Cells (strain GFY-1318) expressing an eGFP-Bni5 fusion and coexpressing Cdc10-mCh were grown to mid exponential phase and visualized by fluorescence microscopy. Representative cells with a medium-to-large bud and an intact septin collar before cytokinesis (top) or with a split septin collar diagnostic of cells in cytokinesis (bottom). Dotted white line, cell periphery; scale bar, 2 μm. (B) Diploids (127, 120, 141, 134, and 148) expressing Bni5-(linker)33-β11 and N-terminally β10-(linker)18–tagged versions of the five mitotic septins (left) visualized by fluorescence microscopy as in Figure 1B (right). For the strain expressing β10-(linker)18-Cdc10 (top), the fiducial mark for the septin collar was Cdc11-mCh and, for all of the others, Cdc10-mCh. (C) Quantification, as in Figure 1C, of the data in B. (D) Diploids (151, 150, 153, 152, and 154) expressing β10-(linker)18-Bni5 and C-terminally (linker)33-β11–tagged versions of the five mitotic septins (left) visualized by fluorescence microscopy as in Figure 1B (right). Bottom, homotypic Bni5 interaction was assessed by examining a diploid (155) coexpressing Bni5-(linker)33-β11 and β10-(linker)18-Bni5. (E) Quantification, as in Figure 1C, of the data in D.
Another protein for which there is now incontrovertible evidence that it contains a domain by which it physically associates with septins is the large (1518 residues) protein kinase Hsl1 (Finnigan et al., 2016). Hsl1 is recruited to the bud neck at the onset of bud emergence (Shulewitz et al., 1999; Figure 5A, top) but then is eliminated before cytokinesis because it is targeted for proteasome-mediated destruction by the cell cycle–regulated protein-ubiquitin ligase (E3) known as the anaphase-promoting complex (APC; Burton and Solomon, 2000). Like the full-length enzyme (Figure 5A, top), a fragment of Hsl1 (611–950) containing its septin-binding domain is localized exclusively at the bud neck (especially when it is confined to the cytosol by mutational removal of a cryptic nuclear localization signal [NLS]; Figure 5A, middle), as documented recently (Finnigan et al., 2016). Moreover, as observed for full-length Hsl1 (Finnigan et al., 2016), presence of the C-terminal phosphatidylserine (PtdSer)-binding kinase associated–1 (KA1) domain of Hsl1 is able to override the effect of the NLS and greatly enhance the ability of the 611–950 fragment to engage the septins at the bud neck (Figure 5A, middle). Finally, because both the destruction box (D-box; motif for recognition of a substrate via mutual engagement by the Cdc10 and Cdc20 subunits of the APC) and KEN box (motif for substrate recognition by either the Cdc20 or Cdh1 subunit of the APC) by which the APC recognizes Hsl1 (Burton and Solomon, 2001) also reside within the 611–950 fragment, their mutational elimination increases the steady-state level of this fragment (Finnigan et al., 2016). Although the septin-binding region of Hsl1 has been delineated, the subunit specificity of Hsl1 binding to the septin collar, if any, has not yet been determined.
FIGURE 5:

Septin-binding domain of checkpoint kinase Hsl1 associates preferentially with Cdc3 and Cdc12. (A) Cells (strain GFY-42) expressing Cdc10-mCh and coexpressing from CEN plasmids, as indicated, 1) full-length Hsl1-eGFP (pGF-IVL521), 2) its septin-binding domain (residues 611–950) with a cryptic NLS mutated to alanine (pGF-IVL762; Finnigan et al., 2016), or 3) its septin-binding domain (611–950) fused to its C-terminal PtdSer-binding KA1 domain (residues 1245–1518; pGF-IVL536; Finnigan et al., 2016), each with eGFP fused to its C-terminus, were grown and visualized by fluorescence microscopy as in Figure 4A. Only cells with an intact septin collar were scored because, upon the onset of anaphase and formation of the split collar, Hsl1 is degraded (Burton and Solomon, 2000, 2001). (B) Diploids (176–190) expressing each of the three Hsl1 constructs in A that were N-terminally β10-(linker)32 tagged and each of the five mitotic septins C-terminally (linker)33-β11 tagged (left) visualized by fluorescence microscopy as in Figure 1B (right). For the 611–950 domain constructs (2 and 3) in these experiments, the cryptic NLS was mutated (R635A R636A K645A H648A K649A R653A K654A), as were both the KEN box (K775A E776A N777A) and D-box (R828A L831A) degradation motifs. (C) Quantification, as in Figure 1C, of the data shown in B, except that only budded cells with an intact septin collar (i.e., that had not entered cytokinesis) were scored. (D) Sixty diploids (186–190, 206–235, and 247–271) were generated in which the linker lengths in both the β10-tagged 611–950/1245–1518 fragment and in the septin-β11 constructs were systematically shortened, as indicated, visualized by fluorescence microscopy as in Figure 1B and quantified as in Figure 1C. Dashed green line, best fit trend line for the Cdc3 and Cdc12 data points; dashed blue line, best-fit trend line for Cdc10 data points.
For the preceding reasons, we tested, first, whether we could detect association of N-terminally β10-tagged full-length Hsl1 with C-terminally β11-tagged versions of the five mitotic septins. This arrangement yielded no detectable signal (Figure 5, B, left, and C, left), suggesting that, even when bound at the septin collar, the N-terminus of Hsl1 (which contains its catalytic domain) is very far from the C-terminus of every septin. We next tested an N-terminally β10-tagged version of the 611–950 fragment from which the NLS and the D-box and KEN box had been mutationally inactivated against the β11-tagged versions of the five mitotic septins. Satisfyingly, under these conditions, a readily detectable signal was observed only between β10-611-950 and Cdc12-β11 in the majority of cells (Figure 5B, middle), with more occasional and weaker interaction with Cdc3-β11 (Figure 5C, middle). Third, we examined the interaction of the same N-terminally β10-tagged NLS-less and D-box and KEN box–less 611–950 fragment to which the KA1 (residues 1245–1518 of Hsl1) was appended against the β11-tagged versions of the five mitotic septins. We again observed strongest interaction with Cdc12-β11, somewhat less robust interaction with Cdc3-β11 and Cdc10-β11, and much less with Cdc11-β11 and Shs1-β11 (Figure 5, B, right, and C, right).
To maximize the likelihood of capturing the 611–950 fragment or 611–950/1245–1518 fragment interaction with a septin(s), we used linkers of maximal length (the β10 tag was tethered by a 32-residue linker, and the β11 tag was appended to a 33-residue linker). We reasoned that we could make the test of the subunit-binding specificity of Hsl1 more stringent by systematically shortening the linker length in both the 611–950/1245–1518 fragment and the septin constructs and reassessing the resulting readouts. Reassuringly, using this approach, there was a clear trend in the 60 diploids constructed for this set of analyses (Figure 5D). As the linker lengths were shortened, the robustness of the output signal was progressively strengthened for the interaction of the β10 611–950/1245–1518 fragment with Cdc12-β11 and Cdc3-β11 and correspondingly greatly diminished for its interaction with the other three septin-β11 constructs. Hence it is clear that the septin-binding element in Hsl1 associates primarily with Cdc12 and Cdc3, a specificity not previously characterized.
Use of the tripartite split-GFP method to examine other bud neck–associated proteins
There are scores of cellular proteins that reportedly localize exclusively or to a detectable degree at the bud neck and do so with different spatiotemporal dynamics (Gladfelter et al., 2001; McMurray and Thorner, 2009; Finnigan et al., 2015a). For the majority of these proteins, there is little or no information about whether they localize to the bud neck because they directly bind to a septin(s) there, or not. Hence, as a final test of the usefulness of the tripartite split-GFP assay, we examined three bud neck–associated proteins whose capacity to physically associate with specific septins has, to our knowledge, not yet been definitively characterized.
The first such protein we examined was a poorly studied bud neck–localized factor, Nis1 (407 residues), whose purported ability to physically associate with septin Shs1 is based mainly on a two-hybrid interaction reported in a single study (Iwase and Toh-e, 2001). In our hands, Nis1-eGFP expressed under its endogenous promoter in a strain coexpressing Cdc10-mCh does not colocalize with the septin collar. In cells with small or large buds, Nis1 is found just adjacent to the division site in a small patch (Figure 5A, top); in cells undergoing cytokinesis, Nis1 is localized between the split septin rings (Figure 5A, bottom). An eGFP-Nis1 construct shows the identical localization pattern (G.C.F., unpublished results). Nonetheless, to determine whether the tripartite split-GFP system could detect any transient or weak interaction of Nis1 with any septin during cytokinesis (when Nis1 is localized between the split septin rings), we tested Nis1-β11 against β10-tagged versions of all five septins (Figure 6B) and also β10-Nis1 against β11-tagged versions of all five septins (Figure 6D; where, to maximize the likelihood of detection, the N-terminal β10 tag was tethered by an 18-residue linker and the β11 tag was appended via a 33-residue linker). We carefully examined all cells, but especially dividing cells that were actively undergoing cytokinesis (i.e., contained a split double ring marked by Cdc10-mCh or Cdc11-mCh). In neither arrangement (Nis1-β11 and β10-tagged septins or β10-Nis1 and β11-tagged septins) did we detect any signal above the intrinsic background fluorescence (Figure 6, C and E). We conclude, therefore, that Nis1 is sequestered between the split septins but does not make direct contact with any septin there. Thus recruitment of Nis1 to this location is not mediated by its binding to any septin. We cannot rule out, however, that the β10 or β11 tags (and/or the unstructured linkers) perturb the ability of Nis1 to interact with a septin, but such a concern seems unlikely, given that both eGFP-Nis1 and Nis1-eGFP, which are fused to much bulkier tags, display exclusive localization within the split septin rings at the bud neck.
FIGURE 6:
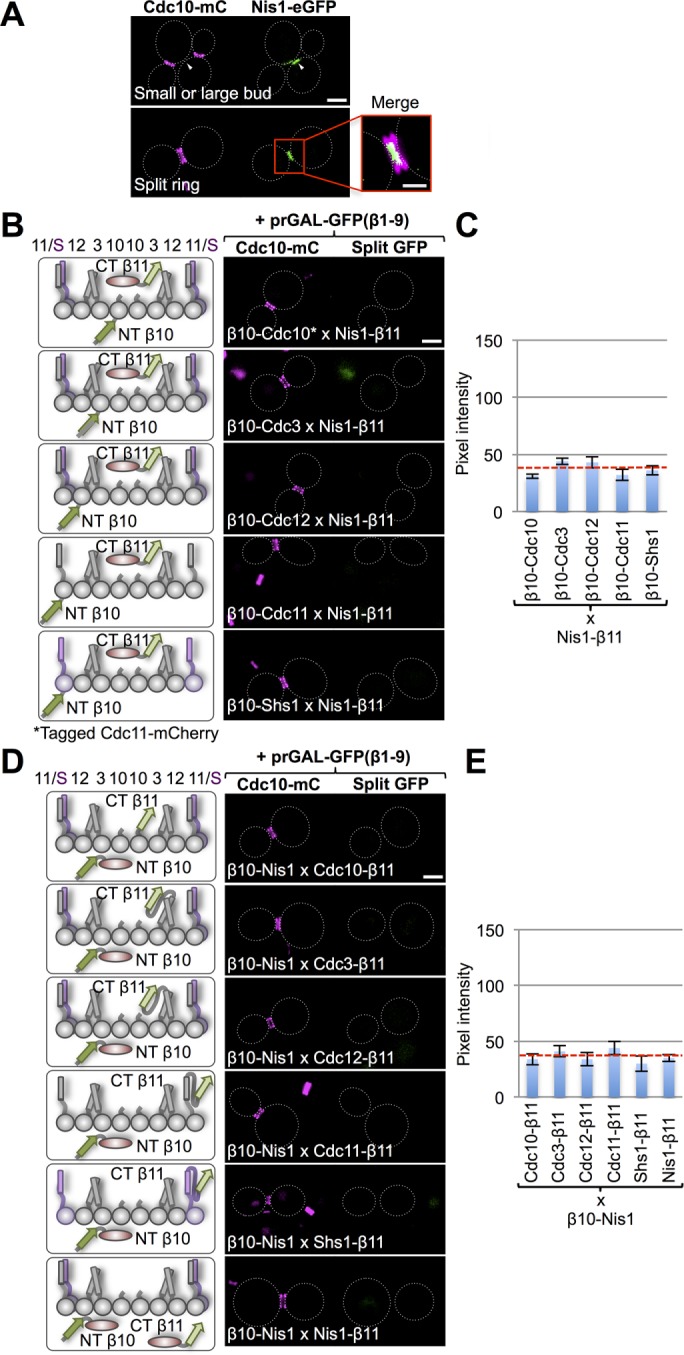
Bud neck–localized protein Nis1 does not interact directly with septins. (A) Cells (strain GFY-42) expressing Cdc10-mCh and coexpressing Nis1-eGFP under the control of its endogenous promoter from a CEN plasmid (pGF-IVL553) were grown and visualized as in Figure 4A. Top, GFP signal (white triangles) adjacent to the septin collar in budded cells before cytokinesis. Bottom, during cytokinesis, Nis1-eGFP localizes between the two rings generated by splitting of the septin collar. Inset, enlarged view of the merged image; scale bar, 1 μm. (B) Diploids (128, 121, 142, 135, and 149) expressing Nis1-(linker)33-β11 and the β10-(linker)18–tagged versions of each of the five mitotic septins (left), as well as either Cdc10-mCh or Cdc11-mCh (as indicated), visualized as in Figure 1B (right). (C) Quantification, as in Figure 1C, of the data in B. (D) Diploids (157–161 and 163) expressing β10-(linker)18-Nis1 and C-terminally (linker)33-β11–tagged versions of each of the five mitotic septins (left), as well as Cdc10-mCh, were visualized as in Figure 1B (right). (E) Quantification, as in Figure 1C, of the data in D.
The second bud neck–associated factor we examined was Bud4, a large (1447 residues) protein involved in bud site selection that is the putative yeast analogue of mammalian anillin (Eluere et al., 2012; Kang et al., 2013; Wu et al., 2015). Although prior work provided genetic and biochemical evidence that Bud4 makes direct contact with septins, the subunit specificity of its interaction was not defined. However, as for Hsl1 (Finnigan et al., 2016), domain scans across full-length Bud4 revealed a minimal domain (residues 623–774) that is largely sufficient for localization to the septin collar (Wu et al., 2015). Therefore, and as for Hsl1, we focused our analysis on this previously identified, putative septin-binding domain of Bud4. Indeed, in our hands, a Bud4(623-774)-eGFP construct strongly colocalized with the septin collar marked with Cdc10-mCh (although a detectable amount of this fragment was also found in the nucleus; Figure 7A). Tellingly, when Bud4(623-774)-β11 was tested against β10-tagged version of all five mitotic septins, prominent signals were observed with β10-Cdc3, β10-Cdc11, and β10-Shs1 (Figure 7, B and C), confirming that this region of Bud4 is indeed able to make intimate physical contact with these septins at the bud neck.
FIGURE 7:
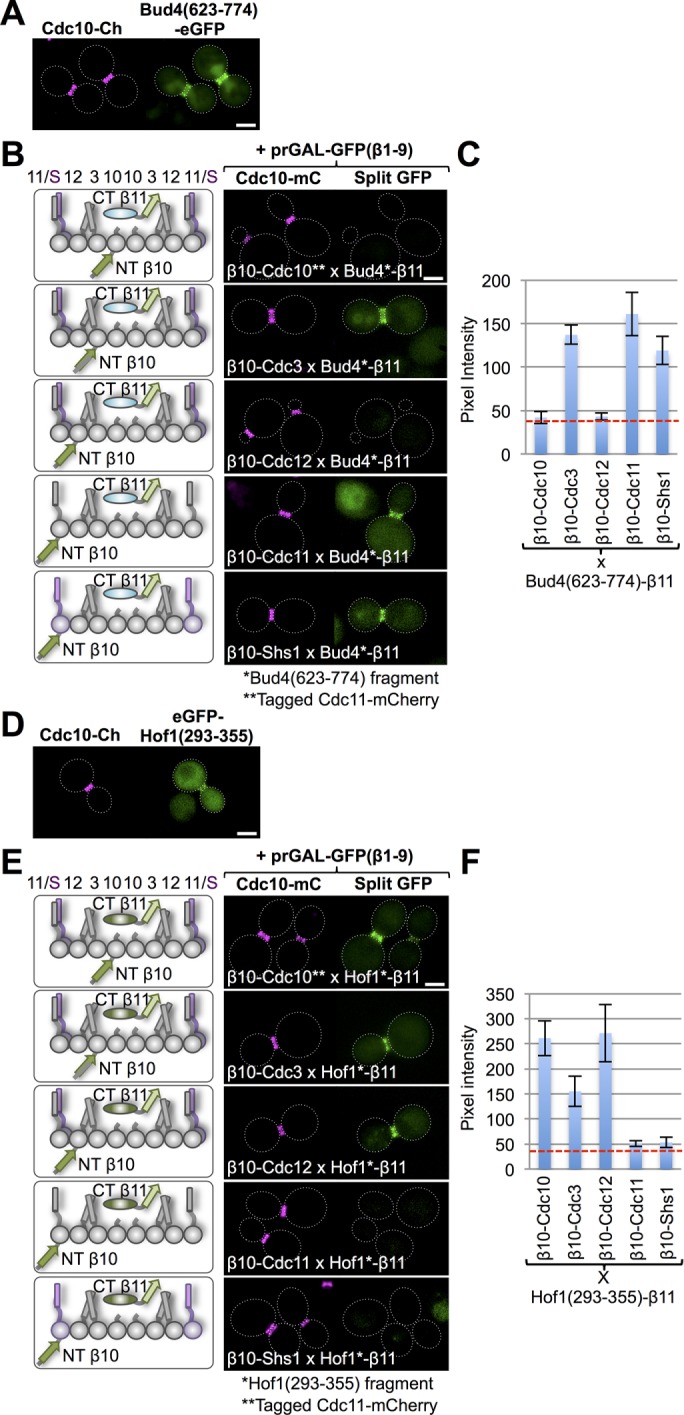
Septin-binding domains of Bud4 and Hof1 have distinct subunit preferences. (A) Cells (strain GFY-42) expressing Cdc10-mCh and coexpressing Bud4(623-774)-eGFP under the control of the BUD4 promoter from a CEN plasmid (pGF-IVL1095) were grown and visualized as in Figure 4A. (B) Diploids (287–291) expressing Bud4(623-774)-(linker)33-β11 from its native promoter on a LEU2-marked CEN plasmid (pGF-IVL1082), the β10-(linker)18–tagged versions of each of the five mitotic septins (left), a URA3-marked CEN plasmid (pGF-IVL1005) expressing GFPβ1-9 from the GAL1/GAL10 promoter (unpublished data), and either Cdc10-mCh or Cdc11-mCh (as indicated) were selected on SD-Ura-Leu medium, grown, induced with galactose, washed, and imaged as in Figure 1B. (C) Quantification, as in Figure 1C, of the data in B. (D) Cells (strain GFY-42) expressing Cdc10-mCh and coexpressing eGFP-Hof1(293-355) under the control of the HOF1 promoter from a CEN plasmid (pGF-IVL1110) were grown and visualized as in Figure 4A. (E) Diploids (292–296) expressing Hof1(293-355)-(linker)33-β11 from its native promoter on a LEU2-marked CEN plasmid (pGF-IVL1082), the β10-(linker)18–tagged versions of each of the five mitotic septins (left), a URA3-marked CEN plasmid (pGF-IVL1005) expressing GFPβ1-9 from the GAL1/GAL10 promoter (unpublished data), and either Cdc10-mCh or Cdc11-mCh (as indicated) were treated and examined as in B. (F) Quantification, as in Figure 1C, of the data in E.
The third bud neck–localized protein we interrogated was Hof1, an F-BAR domain–containing protein involved in membrane curvature and actomyosin ring coordination during cytokinesis (Nishihama et al., 2009; Meitinger et al., 2011, 2013; Oh et al., 2013). As for Bud4, prior genetic and biochemical findings indicated that Hof1 makes direct contact with septins at the bud neck, but its subunit selectivity was not clearly identified. Like Bud4, domain scans across full-length Hof1 (669 residues) delineated a minimal domain (residues 293–356) that is (partially) sufficient for localization to the septin collar (Meitinger et al., 2013). In our hands, an eGFP-Hof1(293-356) construct was found mainly in the cytosol but did localize detectably, although only rather weakly, at the bud neck, congruent with Cdc10-mCh (Figure 7D). However, consistent with the capacity of the tripartite split-GFP method to capture even weak or transient interactions, when Hof1(293-356)-β11 was tested against the β10-tagged version of all five mitotic septins, robust signals were observed with β10-Cdc12, β10-Cdc10, and β10-Cdc3 (Figure 7, E and F), confirming that this region of Hof1 is indeed able to make intimate physical contact with these septins at the bud neck.
Bud4(623-774)-β11 interacted with both terminal subunits of the hetero-octamer (β10-Cdc11 and β10-Shs1) but also with β10-Cdc3; by contrast, Hof1(293-355)-β11 interacted with β10-β10-Cdc12 and β10-Cdc10 but also with β10-Cdc3. Cdc3 is perhaps a bit more “promiscuous” in its ability to interact with other proteins docked at the septin collar, as judged by the tripartite split-GFP method, because its long N-terminal extension (Supplemental Figure S1B) may allow for greater flexibility and a longer “reach.” Of importance, however, when we swapped the tags and tested β10-Bud4(623-774) or β10-Hof1(293-356) against β11-tagged versions of all five mitotic septins, no fluorescence signal was generated by any combination (unpublished data), strongly suggesting that the septin-binding domains of Bud4 and Hof1 have a very strong preference for associating with the N-terminal “face” of their target septins in the hetero-octamer. We observed the converse with the septin-binding domain of Hsl1; we observed robust fluorescence signals only when β10-Hsl1(611-950) or β10-Hsl1(611-950/1245-1518) were combined with β11-tagged versions of all five mitotic septins (Figure 5), indicating that Hsl1 prefers association with the C-terminal ends of the septins in the hetero-octamer.
DISCUSSION
We demonstrated here that the tripartite split-GFP system (Cabantous et al., 2013) is a reliable, in vivo protein–protein interaction sensor in S. cerevisiae cells, for which this technology had not previously been applied. This method permitted successful interrogation of protein–protein associations in situ under near-native conditions at endogenous levels of expression. Any proteins examined are present in their normal modification state and in their natural intracellular location and milieu when their ability to physically interact is assessed. Given their small size, the tags (20 or 21 residues) have a minimal effect on the solubility and other behaviors of the tagged proteins, whose interaction was readily detectable even in diploid cells in which a wild-type (WT) copy of each tagged partner was also present (Supplemental Figure S1).
Regardless of when and where the β10-tagged and β11-tagged proteins interact, the readout is generated only upon expression of the complementary, otherwise nonfluorescent eGFPβ1-9 barrel. Hence background fluorescence is minimal. This arrangement allows for temporal control. For example, two β10- and β11-tagged proteins that interact transiently in only a particular phase of the cell cycle could be revealed, in principle, by arresting cells at different cell cycle stages with drugs or mutants and then inducing expression of the eGFPβ1-9 barrel. This arrangement also allows for spatial control. For example, to interrogate interaction of only PM-localized β10- and β11-tagged proteins, one could use as the detector a modified eGFPβ1-9 barrel targeted to the PM by an N-terminal myristoylation sequence or a C-terminal CaaX box (prenylation site consensus motif, where C is prenylated cysteine, a is any aliphatic residue, and residue at X dictates farnesylation or geranylgeranylation). Furthermore, by generating libraries of MATa cells expressing β10-tagged derivatives of every yeast gene and corresponding libraries of MATα cells expressing β11-tagged derivatives of every yeast gene (and vice versa), simple crosses among these collections and examination of the resulting diploids by fluorescence microscopy after induction of the eGFPβ1-9 barrel should, in theory, provide an independent means to generate a new global protein interactome map for the entire S. cerevisiae genome to expand upon and complement current data.
We addressed, first, interrelationships among the subunits in the linear, apolar septin hetero-octamer (Bertin et al., 2008). However, Cdc11-capped hetero-octameric rods polymerize end to end and also associate in register in a highly cooperative manner laterally (via cross-filament interaction mediated by coiled-coil formation between the C-termini of Cdc3 and Cdc12) to form long, paired filaments (Versele et al., 2004; Bertin et al., 2008; McMurray et al., 2011). Similarly, Shs1-capped hetero-octamers self-associate into arcs, spirals, and rings (Garcia et al., 2011). Together these septin complexes form higher-order arrays of various geometries in vivo (Rodal et al., 2005; Bertin et al., 2012; Ong et al., 2014). Hence one potential concern in interpreting the readout generated for septin–septin interactions using the tripartite split-GFP approach was that the observed fluorescence signal could arise from an amalgam of interactions occurring at four distinct levels of organization: 1) within hetero-octamers, 2) across the filaments within a pair, 3) between neighboring pairs of filaments, and/or 4) among higher-order assemblies in larger superstructures (Bertin et al., 2012).
We found, however, that the occurrence and intensity of the fluorescence signal observed between pairs of septins largely mirrors the known subunit order within the hetero-octamer. Thus our observations indicate that the tripartite split-GFP method mainly measures intimate short-range physical contacts. Even when the linkers tethering the β10 and β11 tags to their respective septins were extended, authentic nearest-neighbor subunit pairings generally yielded much more robust outputs than any longer-range, non–nearest-neighbor interactions. Nonetheless, by increasing linker length, we could begin to detect such readouts. For example, in the hetero-octamer, any given Cdc10 is adjacent to another Cdc10 and to Cdc3 and gives a very strong signal with either partner, and yet, at the longest linker lengths we used, interaction (albeit much weaker) of Cdc10 with Cdc12 (the next subunit over from Cdc3) was detectable. Hence systematic variation of the linkers that tether the β10 and β11 tags to their respective proteins provides a tunable “molecular ruler” for gauging in vivo the relative distances among protein components in a complex. The globular domain of a septin is ∼4 nm in diameter (Bertin et al., 2008); we estimate, therefore, that the longest linkers we used interrogated conformational space >10 nm from their point of attachment. Thus the shorter the linker length, the more stringent are the distance requirements for interaction; conversely, the longer the linker length, the more promiscuous are the associations that will be detected, up to a point. We saw the latter behavior for β10-Cdc3, presumably because it has a much longer N-terminal extension than any other mitotic septin. Conversely, we generally encountered less robust signals with β10-Cdc11 and β10-Shs1 because both have extremely short N-terminal extensions that include, just downstream, a tract of basic residues. By binding to PtdIns4,5P2 on the inner leaflet of the PM, these tracts in Cdc11, Shs1, and Cdc10 contribute to tight association between septin filaments and the PM and thus to septin function (Bertin et al., 2010; Bridges et al., 2014; Finnigan et al., 2015b). Membrane association may partially occlude access to the N-termini of these septins.
A primary purpose for implementing the tripartite split-GFP system was to address three questions about the ∼100 proteins that reportedly localize (in whole or in part) at the bud neck (either seemingly congruent with or otherwise in close proximity to the septin collar) at some point during cell cycle progression (Gladfelter et al., 2001; McMurray and Thorner, 2009; Finnigan et al., 2015a). 1) Which of these proteins actually binds directly to a septin and which are recruited to the bud neck by less direct mechanisms? 2) For proteins that associate intimately, do they contact a specific septin subunit, thereby providing insight into how these septin-binding proteins are organized at the bud neck? 3) For septin-binding proteins, does this approach allow for reliable parsing of the sequence elements necessary and sufficient for their septin interaction?
Examination of full-length (448 residues) Bni5, known to interact directly with Cdc11 and Shs1 (Lee et al., 2002; Booth et al., 2015; Finnigan et al., 2015a; Patasi et al., 2015), verified that the tripartite split-GFP method yields the same conclusion. Consistent with other evidence that the C-terminal end of Bni5 is required for its function (Finnigan et al., 2015a), Bni5-β11 interacted exclusively with N-terminally β10-tagged Cdc11 and Shs1 and no other septin. In agreement with structure predictions that Bni5 is an elongated, highly α-helical protein, β10-Bni5 had a longer “reach,” yielding a readily detectable fluorescence signal with every C-terminally β11-tagged septin. Thus this method provides a certain degree of insight into how septin-binding proteins are oriented when they dock on the septin collar (Figure 8).
FIGURE 8:

Diagrammatic summary of the interactions of bud neck–localized proteins with specific septin subunits detected using the tripartite split-GFP assay. The linear hetero-octamer is depicted as it resides in paired filaments (conjoined via formation of cross-filament coiled coils between the CTEs on Cdc3 and Cdc12). The N-terminal face of each septin is predicted to project 180° away from its CTE, based on the recently determined crystal structure of S. cerevisiae Cdc11 as a representative subunit (Brausemann et al., 2016). The C-terminus of Bni5 exhibited preferential interaction with the N terminal faces of Cdc11 and Shs1 (thick red arrows), whereas the N- terminus of Bni5 was able to interact weakly with the C-termini of all five septins (dashed red arrows). The N-terminus of the septin-binding domain of Hsl1 had preferential interaction with the C-termini of Cdc12 and Cdc3, whereas the C-terminus of the same fragment did not have a detectable interaction with any subunit. The C-terminus of the septin-binding domain of Bud4 exhibited preferential interaction with the N-termini of Cdc11, Shs1, and, weakly, Cdc3 (most likely due to the extremely long N-terminal extension on Cdc3), whereas the N-terminus of the same fragment did not have a detectable interaction with any subunit. The C-terminus of the septin-binding domain of Hof1 had preferential interaction with the N-termini of Cdc10, Cdc12, and, weakly, Cdc3, whereas the N-terminus of the same fragment did not have detectable interaction with any subunit.
To verify that the tripartite split-GFP method reports interaction of authentic septin-binding domains, we turned to the much larger (1518 residues) multidomain, septin collar–binding protein Hsl1. An internal segment of Hsl1 (residues 611–950) is both necessary and sufficient for targeting the enzyme to the septin collar in vivo and for direct binding to septin filaments in vitro (Finnigan et al., 2016). We found that the β10-611-950 fragment exhibited a marked preference for association with C-terminally β11-tagged Cdc12 and Cdc3, whereas 611-950-β11 yielded no detectable interaction with any N-terminally β10-tagged septin (Figure 8). Aside from confirming that this Hsl1 segment associates directly with the septin collar, the tripartite split-GFP method revealed that it docks via its N-terminal end to the C-terminal ends of Cdc12 and Cdc3. Thus this technique shed light on how a septin-binding domain within a larger septin-binding protein engages the septin collar. N-terminally β10-tagged full-length Hsl1 did not yield a detectable signal with any C-terminally β11-tagged septin, suggesting that, when bound to the septin collar, the N-terminal kinase domain likely faces away from the septin filaments and projects into the cytosol at the bud neck. C-terminally β11-tagged full-length Hsl1 also did not yield a detectable signal with any N-terminally β10-tagged septin. Given that the β11 tag is located downstream of the PtdSer-binding C-terminal KA1 domain, it was likely buried against the surface of the PM and, hence, inaccessible.
Although smaller than Bni5, neither Nis1-β11 combined with β10-tagged septins nor β10-Nis1 combined with β11-tagged septins displayed any signal above background, suggesting that Nis1 is located at the bud neck during cytokinesis by physical trapping between the two septin rings and not by direct binding to any septin. For Bud4 and Hof1, prior deletion analysis and fragment scanning identified segments that appear both necessary and sufficient for their septin association. However, whether Bud4 or Hof1 binding has any septin subunit specificity or any preference for which “face” of a septin filament they occupy had not been determined. When C-terminally β11-tagged, the Bud4 septin-binding domain interacted preferentially with β10-Cdc11 and β10-Shs1 (Figure 8), whereas when N-terminal β10-tagged, it did not interact detectably with any septin-β11. Similarly, when C-terminally β11-tagged, the Hof1 septin-binding element interacted preferentially with β10-Cdc12 and β10-Cdc10, but when N-terminal β10-tagged, it did not interact detectably with any septin-β11 (Figure 8). Thus the septin-binding domains in both Bud4 and Hof1 preferentially associate with distinct septins and do so via the N-terminal “face” of the septin filaments at the bud neck.
PM binding via its C-terminal KA1 domain greatly potentiates the ability of the Hsl1 septin-binding region to engage the septin collar in vivo (Finnigan et al., 2016). Similarly, Bud4 has a C-terminal, phosphoinositide-binding pleckstrin homology domain (residues 1305–1447; Gallego et al., 2010; Kang et al., 2013; Wu et al., 2015). Similarly, Hof1 contains an apparent N-terminal F-BAR domain (residues 1–280; Aspenström, 2009) required for its recruitment to the PM (Meitinger et al., 2013; Oh et al., 2013). Thus synergistic action of both PM-binding motifs and septin-binding elements in certain bud neck–localized proteins presumably coordinates their function with processes that influence the lipid composition of the PM.
Whether the subunit arrangement within septin hetero-octamers (and in the higher-order structures built from them) arose due to evolutionary selection for efficient assembly or as a means to dictate more effectively the positioning of associated septin-binding proteins (or both) has been an open question. There is some evidence for the latter. Localization of Bni5 to the terminal subunits (Cdc11 and Shs1) of the hetero-octamer is required for its function (Finnigan et al., 2015a). Artificially tethering Bni5 to that location (via gene fusions or nanobody-mediated recruitment) suffices, whereas Bni5 tethered by the same means to Cdc10 at the center of the hetero-octamer is unable to function (Finnigan et al., 2015a). Whether the subunit-specific position of any other septin-associated protein is crucial for its function has, to our knowledge, not been explored. Such information is important for understanding how these factors exert their physiological actions. Most studies reporting interaction between a septin and a cellular protein have not systematically examined specificity per se, for example, 1) testing a two-hybrid interaction between a protein of interest and one or two “representative” septin subunits rather than with all five mitotic septins (Nagaraj et al., 2008), 2) screening for synthetic lethality with mutations in one or a few septins rather than all five subunits (Costanzo et al., 2010), 3) coimmunoprecipitation or pull-down of a protein of interest with only a single subunit rather than all five septins (Meitinger et al., 2013; Renz et al., 2016), 4) scoring the phenotype of a fusion of a protein of interest to the C-terminus of one septin rather than constructing and testing fusions to all five (King et al., 2012; Kang et al., 2016), or 5) using a fluorescence PCA assay that pairs a tagged protein of interest with a single tagged septin rather than with each of the five septins (Renz et al., 2016). By contrast, the tripartite split-GFP method allows for facile assessment of the potential interaction between any two proteins of interest. In our study, we focused on the ability to detect initial protein–protein interactions because of the potentially complicating effects of long-term stabilization of dynamic protein–protein associations that arises from tethering the two proteins together “irreversibly” via formation of reconstituted eGFP.
MATERIALS AND METHODS
Yeast strains and plasmids
The yeast strains used in this study are listed in Table 1. Standard molecular biology techniques and protocols were followed (Sambrook and Russell, 2001). The tripartite split-GFP components we used are based on reagents described in detail elsewhere (Cabantous et al., 2013). Two gene constructs were synthesized (GenScript, Piscataway, NJ) with optimized yeast codon usage bias: 1) the evolved eGFPβ1-9 variant (Cabantous et al., 2013) and 2) the β10 and β11 strands of the evolved eGFP variant (Cabantous et al., 2013) in the form of a construct consisting of the sequence SpeI::β10::Linker(32)::SacI::Linker(33)::β11::ADH(t)::NotI. To generate the C-terminally tagged versions of the proteins examined, a common methodology was used that involves a PCR-based in vivo ligation and homologous recombination method in yeast (Finnigan and Thorner, 2015). In brief, plasmids were assembled that contain the native promoter region (usually 500 base pairs of the upstream 5′-untranslated region [UTR]), the open reading frame (ORF) of the gene of interest, a linker of 0, 10, 20, 33, or 43 residues, the C-terminal β11 sequence, the ADH1 terminator, and an MX-based drug cassette (Goldstein and McCusker, 1999). Each construct was confirmed via DNA sequencing. Next the entire cassette was PCR amplified using the plasmid DNA as the template, treated with DpnI, and introduced by DNA-mediated transformation into yeast to replace a chromosomal deletion of the gene of interest. To maintain viability for integration of the tagged version of any essential gene, the cells also harbored a URA3-based covering plasmid carrying a WT copy of the cognate gene, which was then removed by selection on medium containing 5-fluoro-orotic acid (5-FOA). Proper integration was confirmed by diagnostic PCRs on isolated chromosomal DNA and, when appropriate, Sanger sequencing of the manipulated loci (Barker Hall Sequencing Facility, University of California, Berkeley, CA).
To generate the N-terminally tagged versions of the proteins examined, a modified protocol was used. First, the promoter (present on a pRS315 plasmid) was tagged downstream with the β10::Linker(32)::SacI::Linker(33)::β11:::ADH1(t)::KanR sequence using in vivo ligation. Second, the promoter::β10::Linker(32) segment of the construct was subcloned to a separate pRS315 vector using a unique restriction site upstream of the promoter (typically Not1 or SpeI) and the unique SacI site. Third, a second round of in vivo assembly was performed to fuse the ORF of the gene of interest to the promoter::β10::Linker(32) element (with either the full-length linker or truncations of it resulting in linkers of 18 or five residues), followed by the ADH1 terminator and an MX-based drug resistance cassette. Fourth, the entire cassette was PCR amplified and integrated into the genome as described. For insertion into the genome of either CDC10::mCherry::SpHIS5 or CDC11::mCherry::SpHIS5 in the strains containing β10- or β11-tagged gene product, the mCherry-containing construct was PCR amplified using primers present in the flanking UTR (±300 for CDC11 and ±500 for CDC10), and the resulting product was introduced by DNA-meditated transformation and the desired derivatives selected on synthetic defined (SD)-His medium. For tagging of CDC19, GPP1, HSP82, PGK1, and TPI1, otherwise-WT yeast (BY4741) were transformed with either full-length eGFP or the β11 tag cassette flanked by appropriate PCR-generated segments of homology to the cognate chromosomal locus. DNA isolated from each resulting integrant was amplified with a high-fidelity polymerase and sequenced to verify correct construction. All DNA plasmids (Table 2) were constructed using in vivo ligation in yeast and confirmed by both diagnostic PCR and DNA sequencing of the entire coding region of each gene fusion and all junctions to UTR sequence.
Culture conditions
Yeast were grown in rich (YPD) medium (2% peptone, 1% yeast extract, 2% dextrose) or in synthetic medium. The drop-out medium contained the proper mixture of amino acids to maintain selection for all markers and/or plasmids and, as the carbon source, 2% dextrose, 2% raffinose–0.2% sucrose, or 2% galactose, as appropriate. During the construction of any strain that involved introduction of a tagged version of an essential septin gene, the cells harbored a URA3-marked covering plasmid expressing the corresponding WT septin gene. Only after confirmation of the integrated allele was the covering plasmid removed by two successive rounds of selection at 30°C on medium containing 5-FOA (Oakwood Products, West Columbia, SC; final concentration 0.5 mg/ml; heated for at least 30 min to 70°C and filter sterilized, not autoclaved). All of the haploid strains expressing integrated tagged copies of either septin subunits (or bud neck–localized proteins) were confirmed to be viable at 30°C on medium containing 5-FOA (for essential subunits) before they were used for mating and the selection of the resulting diploids. Conditions for diploid isolation involved two successive rounds of growth under conditions selective for both the mCherry::SpHIS5-marked septin introduced from the MATα partner (e.g., GFY-1794) and the LEU2-marked plasmid carrying the GAL1/10prom::GFPβ1-9 introduced from the MATa partner (e.g., GFY-1851). For the constructs involving Bud4 and Hof1, a modified diploid selection protocol was used. Each mating type received a plasmid expressing either 1) GAL1/10prom::GFPβ1-9 (URA3-marked) or 2) BUD4::β11 or HOF1::β11 (LEU2-marked). Diploids then were selected and propagated on SD-Ura-Leu medium.
Fluorescence microscopy and quantification
For imaging of strains not containing any plasmids (e.g., GFY-2034), yeast were grown overnight at 30°C in YPD to saturation, back-diluted in 10 ml of fresh YPD to an A600 nm of 0.25, and grown for an additional 4–5 h to an A600 nm of ∼1. Cells were harvested, washed with water, placed on a glass slide with a coverslip, and imaged within 5 min. Diploid strains were grown overnight to saturation at 30°C in synthetic medium lacking both Leu and His (to maintain selection) with 2% raffinose–0.2% sucrose. The cultures were back-diluted to an A600 nm of 0.3–0.4 in the same medium containing 2% galactose as the carbon source (to induce GFPβ1-9 expression) and grown for an additional 4.5–5 h at 30°C before being washed and imaged.
Yeast cells were examined using an Olympus BH-2 upright fluorescence microscope (Olympus, Tokyo, Japan) with 100× objective lens and equipped with an eGFP 49002 cut-off filter (Chroma Technology, Bellows Falls, VT) that allows for 94.65% transmission at 488 nm (the excitation maximum for eGFP) and 97.69% transmission at 509 nm (the emission maximum for eGFP) and an mCherry/Texas Red filter (Chroma) that allows for 97.82% transmission at 579 nm (mCherry has excitation maximum of 587 nm) and 97.84% transmission at 610 nm (the emission maximum for mCherry). A SOLO light source (Lumencore, Beaverton, OR), a CoolSNAP MYO charge-coupled device camera, and Micro-Manager software (Edelstein et al., 2010) were used to record the images. Postprocessing and analysis of images were done using ImageJ (National Institutes of Health, Bethesda, MD). All images were treated identically and rescaled together. For clarity, the periphery of yeast cells was labeled using white dotted lines from an overexposed image or a corresponding differential interference contrast image. Unless otherwise indicated, all images were taken for the same exposure time and using the same light source power level.
The results described are average values for each diploid strain derived from experiments conducted in triplicate. For quantification of the percentage of the cell population that displayed a detectable reconstituted eGFP signal at the bud neck, 25–100 cells in separate fields were scored and divided by the total number of cells in the same fields that exhibited a detectable mCherry signal (i.e., had an obvious septin collar). For quantification of the pixel intensity of the eGFP fluorescence at the bud neck, the box tool in ImageJ was used to carefully outline the bud neck in 25–100 cells that also clearly displayed a septin collar (as judged by the mCherry signal), and the average pixel intensity (with SEM) was calculated (after subtracting the average background fluorescence in any given image from each measurement taken in the same image). The average background fluorescence was determined by using the box tool to assess the pixel intensity of an equivalent area of five randomly chosen regions of each image that did not contain any cells. Note that this average background fluorescence is distinct from the pixel intensity present at the bud neck of otherwise-wild-type yeast due to the intrinsic fluorescence of cellular components (∼35–40, displayed as the dashed red line in the bar graphs).
For the experiments involving Hsl1 or Bni5, cells displaying a split septin ring (as judged by the mCherry signal) were excluded from the analysis because native Hsl1 is displaced from the bud neck at the onset of cytokinesis by its APC-mediated degradation (Burton and Solomon, 2000; Finnigan et al., 2016) and Bni5 is also ejected from this location before cytokinesis (Lee et al., 2002; Finnigan et al., 2015a). Conversely, for quantification of Nis1 at the bud neck, only budded cells that displayed a clear split septin ring were included, as native Nis1 is not present at the bud neck until cytokinesis (Figure 5A).
Supplementary Material
Acknowledgments
We thank the other members of the Thorner lab for useful suggestions and Geoffrey Waldo (Los Alamos National Laboratory, Santa Fe, NM) for providing invaluable research materials. This work was supported by a Postdoctoral Fellowship from the Adolph C. and Mary Sprague Miller Institute for Basic Research in Science (to G.C.F.) and National Institutes of Health Research Grants GM21841 (to J.T.) and GM101314 (to J.T. and colleague Eva Nogales).
Abbreviations used:
- APC
anaphase-promoting complex
- β10
10th beta strand of evolved eGFP variant of tripartite split-GFP system
- β11
11th beta strand of evolved eGFP variant of tripartite split-GFP system
- BiFC
bimolecular fluorescence complementation
- CTE
C-terminal extension
- D-box
destruction box (motif for recognition of a substrate via mutual engagement by the Cdc10 and Cdc20 subunits of the APC)
- eGFP
enhanced green fluorescent protein
- EM
electron microscopy
- 5-FOA
5-fluoro-orotic acid
- FRET
Förster resonance energy transfer
- GFPβ1-9
stable beta barrel of evolved eGFP variant of tripartite split-GFP system
- KA1
kinase associated–1 domain
- KEN box
motif for substrate recognition by either the Cdc20 or Cdh1 subunit of the APC
- mCh
monomeric red fluorescent protein mCherry
- NLS
nuclear localization signal
- PCA
protein-fragment complementation
- PM
plasma membrane
- PtdIns4,5P2
phosphatidylinositol-4,5-bis-phosphate
- PtdSer
phosphatidylserine.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-05-0337) on July 6, 2016.
REFERENCES
- Aspenström P. Roles of F-BAR/PCH proteins in the regulation of membrane dynamics and actin reorganization. Int Rev Cell Mol Biol. 2009;272:1–31. doi: 10.1016/S1937-6448(08)01601-8. [DOI] [PubMed] [Google Scholar]
- Bertin A, McMurray MA, Grob P, Park SS, Garcia G, 3rd, Patanwala I, Ng HL, Alber T, Thorner J, Nogales E. Saccharomyces cerevisiae septins: supramolecular organization of heterooligomers and the mechanism of filament assembly. Proc Natl Acad Sci USA. 2008;105:8274–8279. doi: 10.1073/pnas.0803330105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertin A, McMurray MA, Pierson J, Thai L, McDonald KL, Zehr EA, Garcia G, 3rd, Peters P, Thorner J, Nogales E. Three-dimensional ultrastructure of the septin filament network in Saccharomyces cerevisiae. Mol Biol Cell. 2012;23:423–432. doi: 10.1091/mbc.E11-10-0850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertin A, McMurray MA, Thai L, Garcia G, 3rd, Votin V, Grob P, Allyn T, Thorner J, Nogales E. Phosphatidylinositol-4,5-bisphosphate promotes budding yeast septin filament assembly and organization. J Mol Biol. 2010;404:711–731. doi: 10.1016/j.jmb.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi E, Park HO. Cell polarization and cytokinesis in budding yeast. Genetics. 2012;191:347–387. doi: 10.1534/genetics.111.132886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth EA, Vane EW, Dovala D, Thorner J. A Förster resonance energy transfer (FRET)-based system provides insight into the ordered assembly of yeast septin hetero-octamers. J Biol Chem. 2015;290:28388–28401. doi: 10.1074/jbc.M115.683128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachmann CB, Davies A, Cost GJ, Caputo E, Li J, Hieter P, Boeke JD. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 1998;14:115–132. doi: 10.1002/(SICI)1097-0061(19980130)14:2<115::AID-YEA204>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Brausemann A, Gerhardt S, Schott AK, Einsle O, Grosse-Berkenbusch A, Johnsson N, Gronemeyer T. Crystal structure of Cdc11, a septin subunit from Saccharomyces cerevisiae. J Struct Biol. 2016;193:157–161. doi: 10.1016/j.jsb.2016.01.004. [DOI] [PubMed] [Google Scholar]
- Bridges AA, Gladfelter AS. Septin form and function at the cell cortex. J Biol Chem. 2015;290:17173–17180. doi: 10.1074/jbc.R114.634444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges AA, Zhang H, Mehta SB, Occhipinti P, Tani T, Gladfelter AS. Septin assemblies form by diffusion-driven annealing on membranes. Proc Natl Acad Sci USA. 2014;111:2146–2151. doi: 10.1073/pnas.1314138111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton JL, Solomon MJ. Hsl1p, a Swe1p inhibitor, is degraded via the anaphase-promoting complex. Mol Cell Biol. 2000;20:4614–4625. doi: 10.1128/mcb.20.13.4614-4625.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton JL, Solomon MJ. D box and KEN box motifs in budding yeast Hsl1p are required for APC-mediated degradation and direct binding to Cdc20p and Cdh1p. Genes Dev. 2001;15:2381–2395. doi: 10.1101/gad.917901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byers B, Goetsch L. A highly ordered ring of membrane-associated filaments in budding yeast. J Cell Biol. 1976;69:717–721. doi: 10.1083/jcb.69.3.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabantous S, Nguyen HB, Pedelacq JD, Koraichi F, Chaudhary A, Ganguly K, Lockard MA, Favre G, Terwilliger TC, Waldo GS. A new protein-protein interaction sensor based on tripartite split-GFP association. Sci Rep. 2013;3:2854. doi: 10.1038/srep02854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll CW, Altman R, Schieltz D, Yates JR, Kellogg D. The septins are required for the mitosis-specific activation of the Gin4 kinase. J Cell Biol. 1998;143:709–717. doi: 10.1083/jcb.143.3.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cid VJ, Adamíková L, Cenamor R, Molina M, Sánchez M, Nombela C. Cell integrity and morphogenesis in a budding yeast septin mutant. Microbiology. 1998;144:3463–3474. doi: 10.1099/00221287-144-12-3463. [DOI] [PubMed] [Google Scholar]
- Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS, Ding H, Koh JL, Toufighi K, Mostafavi S, et al. The genetic landscape of a cell. Science. 2010;327:425–431. doi: 10.1126/science.1180823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drees BL, Sundin B, Brazeau E, Caviston JP, Chen GC, Guo W, Kozminski KG, Lau MW, Moskow JJ, Tong A, et al. A protein interaction map for cell polarity development. J Cell Biol. 2001;154:549–571. doi: 10.1083/jcb.200104057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunkler A, Rosler R, Kestler HA, Moreno-Andres D, Johnsson N. SPLIFF: a single-cell method to map protein-protein interactions in time and space. Methods Mol Biol. 2015;1346:151–168. doi: 10.1007/978-1-4939-2987-0_11. [DOI] [PubMed] [Google Scholar]
- Edelstein A, Amodaj N, Hoover K, Vale R, Stuurman N. Computer control of microscopes using microManager. Curr Protoc Mol Biol. 2010. Chapter 14, Unit 14.20. [DOI] [PMC free article] [PubMed]
- Eluere R, Varlet I, Bernadac A, Simon MN. Cdk and the anillin homolog Bud4 define a new pathway regulating septin organization in yeast. Cell Cycle. 2012;11:151–158. doi: 10.4161/cc.11.1.18542. [DOI] [PubMed] [Google Scholar]
- Ewers H, Tada T, Petersen JD, Racz B, Sheng M, Choquet D. A septin-dependent diffusion barrier at dendritic spine necks. PLoS One. 2014;9:e113916. doi: 10.1371/journal.pone.0113916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X, Luo J, Nishihama R, Wloka C, Dravis C, Travaglia M, Iwase M, Vallen EA, Bi E. Biphasic targeting and cleavage furrow ingression directed by the tail of a myosin II. J Cell Biol. 2010;191:1333–1350. doi: 10.1083/jcb.201005134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkasovsky M, Herter P, Voss B, Wittinghofer A. Nucleotide binding and filament assembly of recombinant yeast septin complexes. Biol Chem. 2005;386:643–656. doi: 10.1515/BC.2005.075. [DOI] [PubMed] [Google Scholar]
- Finnigan GC, Booth EA, Duvalyan A, Liao EN, Thorner J. The carboxy-terminal tails of septins Cdc11 and Shs1 recruit myosin-II binding factor Bni5 to the bud neck in Saccharomyces cerevisiae. Genetics. 2015a;200:821–840. doi: 10.1534/genetics.115.176503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnigan GC, Sterling SM, Duvalyan A, Liao EN, Sargsyan A, Garcia Gr, Nogales E, Thorner J. Coordinate action of distinct sequence elements localizes checkpoint kinase Hsl1 to the septin collar at the bud neck in Saccharomyces cerevisiae. Mol Biol Cell. 2016;27:2213–2233. doi: 10.1091/mbc.E16-03-0177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnigan GC, Takagi J, Cho C, Thorner J. Comprehensive genetic analysis of paralogous terminal septin subunits Shs1 and Cdc11 in Saccharomyces cerevisiae. Genetics. 2015b;200:841–861. doi: 10.1534/genetics.115.176495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnigan GC, Thorner J. Complex in vivo ligation using homologous recombina-tion and high-efficiency plasmid rescue from Saccharomyces cerevisiae. Bio Protoc. 2015;5:e1521. doi: 10.21769/bioprotoc.1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung KY, Dai L, Trimble WS. Cell and molecular biology of septins. Int Rev Cell Mol Biol. 2014;310:289–339. doi: 10.1016/B978-0-12-800180-6.00007-4. [DOI] [PubMed] [Google Scholar]
- Gallego O, Betts MJ, Gvozdenovic-Jeremic J, Maeda K, Matetzki C, Aguilar-Gurrieri C, Beltran-Alvarez P, Bonn S, Fernández-Tornero C, Jensen LJ, et al. A systematic screen for protein-lipid interactions in Saccharomyces cerevisiae. Mol Syst Biol. 2010;6:430.431–430.415. doi: 10.1038/msb.2010.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia G, 3rd, Bertin A, Li Z, Song Y, McMurray MA, Thorner J, Nogales E. Subunit-dependent modulation of septin assembly: budding yeast septin Shs1 promotes ring and gauze formation. J Cell Biol. 2011;195:993–1004. doi: 10.1083/jcb.201107123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladfelter AS, Pringle JR, Lew DJ. The septin cortex at the yeast mother-bud neck. Curr Opin Microbiol. 2001;4:681–689. doi: 10.1016/s1369-5274(01)00269-7. [DOI] [PubMed] [Google Scholar]
- Goldstein AL, McCusker JH. Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast. 1999;15:1541–1553. doi: 10.1002/(SICI)1097-0061(199910)15:14<1541::AID-YEA476>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Haarer BK, Pringle JR. Immunofluorescence localization of the Saccharomyces cerevisiae CDC12 gene product to the vicinity of the 10-nm filaments in the mother-bud neck. Mol Cell Biol. 1987;7:3678–3687. doi: 10.1128/mcb.7.10.3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall PA, Russell SE. Mammalian septins: dynamic heteromers with roles in cellular morphogenesis and compartmentalization. J Pathol. 2012;226:287–299. doi: 10.1002/path.3024. [DOI] [PubMed] [Google Scholar]
- Hartwell LH. Genetic control of the cell division cycle in yeast. IV. Genes controlling bud emergence and cytokinesis. Exp Cell Res. 1971;69:265–276. doi: 10.1016/0014-4827(71)90223-0. [DOI] [PubMed] [Google Scholar]
- Hartwell LH, Culotti J, Pringle JR, Reid BJ. Genetic control of the cell division cycle in yeast. Science. 1974;183:46–51. doi: 10.1126/science.183.4120.46. [DOI] [PubMed] [Google Scholar]
- Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O’Shea EK. Global analysis of protein localization in budding yeast. Nature. 2003;425:686–691. doi: 10.1038/nature02026. [DOI] [PubMed] [Google Scholar]
- Iwase M, Luo J, Bi E, Toh-e A. Shs1 plays separable roles in septin organization and cytokinesis in Saccharomyces cerevisiae. Genetics. 2007;177:215–229. doi: 10.1534/genetics.107.073007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwase M, Toh-e A. Nis1 encoded by YNL078W: a new neck protein of Saccharomyces cerevisiae. Genes Genet Syst. 2001;76:335–343. doi: 10.1266/ggs.76.335. [DOI] [PubMed] [Google Scholar]
- Kang PJ, Hood-DeGrenier JK, Park HO. Coupling of septins to the axial landmark by Bud4 in budding yeast. J Cell Sci. 2013;126:1218–1226. doi: 10.1242/jcs.118521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H, Tsygankov D, Lew DJ. Sensing a bud in the yeast morphogenesis checkpoint: a role for Elm1. Mol Biol Cell. 2016;27:1764–1775. doi: 10.1091/mbc.E16-01-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerppola TK. Visualization of molecular interactions using bimolecular fluorescence complementation analysis: characteristics of protein fragment complementation. Chem Soc Rev. 2009;38:2876–2886. doi: 10.1039/b909638h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King K, Jin M, Lew D. Roles of Hsl1p and Hsl7p in Swe1p degradation: beyond septin tethering. Eukaryot Cell. 2012;11:1496–1502. doi: 10.1128/EC.00196-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Agarwal S, Heyman JA, Matson S, Heidtman M, Piccirillo S, Umansky L, Drawid A, Jansen R, Liu Y, et al. Subcellular localization of the yeast proteome. Genes Dev. 2002;16:707–719. doi: 10.1101/gad.970902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PR, Song S, Ro HS, Park CJ, Lippincott J, Li R, Pringle JR, De Virgilio C, Longtine MS, Lee KS. Bni5p, a septin-interacting protein, is required for normal septin function and cytokinesis in Saccharomyces cerevisiae. Mol Cell Biol. 2002;22:6906–6920. doi: 10.1128/MCB.22.19.6906-6920.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magliery TJ, Wilson CG, Pan W, Mishler D, Ghosh I, Hamilton AD, Regan L. Detecting protein-protein interactions with a green fluorescent protein fragment reassembly trap: scope and mechanism. J Am Chem Soc. 2005;127:146–157. doi: 10.1021/ja046699g. [DOI] [PubMed] [Google Scholar]
- Malicki J, Avidor-Reiss T. From the cytoplasm into the cilium: bon voyage. Organogenesis. 2014;10:138–157. doi: 10.4161/org.29055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurray MA, Bertin A, Garcia G, 3rd, Lam L, Nogales E, Thorner J. Septin filament formation is essential in budding yeast. Dev Cell. 2011;20:540–549. doi: 10.1016/j.devcel.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurray MA, Thorner J. Septins: molecular partitioning and the generation of cellular asymmetry. Cell Div. 2009;4:18. doi: 10.1186/1747-1028-4-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meitinger F, Boehm ME, Hofmann A, Hub B, Zentgraf H, Lehmann WD, Pereira G. Phosphorylation-dependent regulation of the F-BAR protein Hof1 during cytokinesis. Genes Dev. 2011;25:875–888. doi: 10.1101/gad.622411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meitinger F, Palani S, Hub B, Pereira G. Dual function of the NDR-kinase Dbf2 in the regulation of the F-BAR protein Hof1 during cytokinesis. Mol Biol Cell. 2013;24:1290–1304. doi: 10.1091/mbc.E12-08-0608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KE, Kim Y, Huh WK, Park HO. Bimolecular fluorescence complementation (BiFC) analysis: advances and recent applications for genome-wide interaction studies. J Mol Biol. 2015;427:2039–2055. doi: 10.1016/j.jmb.2015.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millson S, Truman AW, King V, Prodromou C, Pearl LH, Piper PW. A two-hybrid screen of the yeast proteome for Hsp90 interactors uncovers a novel Hsp90 chaperone requirement in the activity of a stress-activated mitogen-activated protein kinase, Slt2p (Mpk1p) Eukaryot Cell. 2005;4:849–860. doi: 10.1128/EC.4.5.849-860.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mino A, Tanaka K, Kamei T, Umikawa M, Fujiwara T, Takai Y. Shs1p: a novel member of septin that interacts with Spa2p, involved in polarized growth in Saccharomyces cerevisiae. Biochem Biophys Res Commun. 1998;251:732–736. doi: 10.1006/bbrc.1998.9541. [DOI] [PubMed] [Google Scholar]
- Mostowy S, Cossart P. Septins: the fourth component of the cytoskeleton. Nat Rev Mol Cell Biol. 2012;13:183–194. doi: 10.1038/nrm3284. [DOI] [PubMed] [Google Scholar]
- Nagaraj S, Rajendran A, Jackson CE, Longtine MS. Role of nucleotide binding in septin-septin interactions and septin localization in Saccharomyces cerevisiae. Mol Cell Biol. 2008;28:5120–5137. doi: 10.1128/MCB.00786-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishihama R, Schreiter JH, Onishi M, Vallen EA, Hanna J, Moravcevic K, Lippincott MF, Han H, Lemmon MA, Pringle JR, et al. Role of Inn1 and its interactions with Hof1 and Cyk3 in promoting cleavage furrow and septum formation in S. cerevisiae. J Cell Biol. 2009;185:995–1012. doi: 10.1083/jcb.200903125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh Y, Bi E. Septin structure and function in yeast and beyond. Trends Cell Biol. 2011;21:141–148. doi: 10.1016/j.tcb.2010.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh Y, Schreiter J, Nishihama R, Wloka C, Bi E. Targeting and functional mechanisms of the cytokinesis-related F-BAR protein Hof1 during the cell cycle. Mol Biol Cell. 2013;24:1305–1320. doi: 10.1091/mbc.E12-11-0804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi K, Mizuno K. A novel pair of split Venus fragments to detect protein-protein interactions by in vitro and in vivo bimolecular fluorescence complementation assays. Methods Mol Biol. 2014;1174:247–262. doi: 10.1007/978-1-4939-0944-5_17. [DOI] [PubMed] [Google Scholar]
- Ong K, Wloka C, Okada S, Svitkina T, Bi E. Architecture and dynamic remodelling of the septin cytoskeleton during the cell cycle. Nat Commun. 2014;5:5698. doi: 10.1038/ncomms6698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padilla-Parra S, Tramier M. FRET microscopy in the living cell: different approaches, strengths and weaknesses. Bioessays. 2012;34:369–376. doi: 10.1002/bies.201100086. [DOI] [PubMed] [Google Scholar]
- Patasi C, Godocikova J, Michlikova S, Nie Y, Kacerikova R, Kvalova K, Raunser S, Farkasovsky M. The role of Bni5 in the regulation of septin higher-order structure formation. Biol Chem. 2015;396:1325–1337. doi: 10.1515/hsz-2015-0165. [DOI] [PubMed] [Google Scholar]
- Remy I, Michnick SW. Mapping biochemical networks with protein fragment complementation assays. Methods Mol Biol. 2015;1278:467–481. doi: 10.1007/978-1-4939-2425-7_31. [DOI] [PubMed] [Google Scholar]
- Renz C, Oeljeklaus S, Grinhagens S, Warscheid B, Johnsson N, Gronemeyer T. Identification of cell cycle-dependent interaction partners of the septins by quantitative mass spectrometry. PLoS One. 2016;11:e0148340. doi: 10.1371/journal.pone.0148340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodal AA, Kozubowski L, Goode BL, Drubin DG, Hartwig JH. Actin and septin ultrastructures at the budding yeast cell cortex. Mol Biol Cell. 2005;16:372–384. doi: 10.1091/mbc.E04-08-0734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saarikangas J, Barral Y. The emerging functions of septins in metazoans. EMBO Rep. 2011;12:1118–1126. doi: 10.1038/embor.2011.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual, 3rd ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- Schneider C, Grois J, Renz C, Gronemeyer T, Johnsson N. Septin rings act as a template for myosin higher-order structures and inhibit redundant polarity establish-ment. J Cell Sci. 2013;126:3390–3400. doi: 10.1242/jcs.125302. [DOI] [PubMed] [Google Scholar]
- Shulewitz MJ, Inouye CJ, Thorner J. Hsl7 localizes to a septin ring and serves as an adapter in a regulatory pathway that relieves tyrosine phosphorylation of Cdc28 protein kinase in Saccharomyces cerevisiae. Mol Cell Biol. 1999;19:7123–7137. doi: 10.1128/mcb.19.10.7123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toure A, Rode B, Hunnicutt GR, Escalier D, Gacon G. Septins at the annulus of mammalian sperm. Biol Chem. 2011;392:799–803. doi: 10.1515/BC.2011.074. [DOI] [PubMed] [Google Scholar]
- Versele M, Gullbrand B, Shulewitz MJ, Cid VJ, Bahmanyar S, Chen RE, Barth P, Alber T, Thorner J. Protein-protein interactions governing septin heteropentamer assembly and septin filament organization in Saccharomyces cerevisiae. Mol Biol Cell. 2004;15:4568–4583. doi: 10.1091/mbc.E04-04-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Versele M, Thorner J. Septin collar formation in budding yeast requires GTP binding and direct phosphorylation by the PAK, Cla4. J Cell Biol. 2004;164:701–715. doi: 10.1083/jcb.200312070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wloka C, Bi E. Mechanisms of cytokinesis in budding yeast. Cytoskeleton. 2012;69:710–726. doi: 10.1002/cm.21046. [DOI] [PubMed] [Google Scholar]
- Wu H, Guo J, Zhou YT, Gao XD. The anillin-related region of Bud4 is the major functional determinant for Bud4’s function in septin organization during bud growth and axial bud site selection in budding yeast. Eukaryot Cell. 2015;14:241–251. doi: 10.1128/EC.00268-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.