

SUMMARY
Enzymatic reactions involving bilayer lipids occur in an environment with strict physical and topological constraints. The integral membrane enzyme PagP transfers a palmitoyl group from a phospholipid to lipid A in order to assist Escherichia coli in evading host immune defenses during infection. PagP measures the palmitoyl group with an internal hydrocarbon ruler that is formed in the interior of the eight-stranded antiparallel β barrel. The access and egress of the palmitoyl group is thought to take a lateral route from the bilayer phase to the barrel interior. Molecular dynamics, mutagenesis, and a 1.4 Å crystal structure of PagP in an SDS/2-methyl-2, 4-pentanediol (MPD) cosolvent system reveal that phospholipid access occurs at the crenel present between strands F and G of PagP. In this way, the phospholipid head group can remain exposed to the cell exterior while the lipid acyl chain remains in a predominantly hydrophobic environment as it translocates to the protein interior.
INTRODUCTION
PagP is an integral membrane enzyme that transfers a palmitoyl group from the sn-1 position of a glycerophospholipid to the lipid A (endotoxin) moiety of lipopolysaccharide (LPS) resulting in the production of a palmitoylated lipid A and a lysophospholipid by-product. The donor phospholipid, LPS acceptor, and enzyme active site are all located in the external-facing leaflet of the Gram-negative outer membrane (Figure 1) (Bishop et al., 2000; Jia et al., 2004; Khan and Bishop, 2009; Smith et al., 2008). The enzyme provides bacterial resistance to host immune defenses involving cationic antimicrobial peptides (Guo et al., 1998), attenuates the host inflammatory response to infection triggered by the TLR4/MD2 pathway (Kawasaki et al., 2004; Muroi et al., 2002; Tanamoto and Azumi, 2000), and is a virulence determinant to infection by certain pathogens (Pilione et al., 2004; Preston et al., 2003; Robey et al., 2001). The protein belongs to the PhoP/PhoQ-regulon, which controls the expression of bacterial genes needed for covalent lipid A modifications induced during infection by antimicrobial peptides (Bader et al., 2005). PagP is a potential target for the development of anti-infective agents and a tool for the synthesis of lipid A-based vaccine adjuvants and endotoxin antagonists (Bishop, 2005; Raetz et al., 2007).
Figure 1. Reaction Catalyzed by PagP.

PagP catalyzes the transfer of a palmitate chain (shown in red) from the sn-1 position of phosphati-dylethanolamine (PtdEtn) to lipopolysaccharide (LPS), shown here as Kdo2-lipid A (Re endotoxin). This lipid consists of lipid A linked at the 6′ position with two units of 3-deoxy-D-manno-2-octulosonic acid (Kdo). Lipid A is a β-1′,6-linked disaccharide of glucosamine that is acylated with R-3-hydroxy-myristate chains at the 2, 3, 2′, and 3′ positions, and phosphorylated at the 1 and 4′ positions. Acyloxyacyl linkages with laurate and myristate chains at the 2′ and 3′ positions, respectively, provide the constitutive hexa-acylated lipid A, which is a potent endotoxin. A regulated proportion of lipid A in E. coli contains a palmitate chain (16:0) in acyloxyacyl linkage at position 2 (in red in the figure), which yields a hepta-acylated molecule with attenuated endotoxic properties.
As an enzyme of lipid metabolism, PagP is remarkable in its ability to select a 16-carbon saturated palmitate chain to the exclusion of all other acyl chain types present within the membrane pool of glycerophospholipids (Bishop et al., 2000). The enzyme is an eight-stranded β barrel (Hwang et al., 2002) whose catalytic mechanism likely involves cycling between two dynamically distinct states (Hwang et al., 2004), although the details of the reaction remain unknown. The crystal structure of PagP determined in the presence of the nondenaturing detergent lauryldimethylamine oxide (LDAO) (Ahn et al., 2004) revealed a detergent molecule bound in the interior of the barrel with the head group pointing toward the extracellular side of PagP. This detergent molecule marks the position of the hydrocarbon ruler, which selects acyl chains in the glycerophospholipid substrate with methylene unit resolution (Ahn et al., 2004; Khan et al., 2007, 2010). A recent simulation study of PagP has examined the solvation, but not the binding, of the enzyme by detergent and lipid molecules (Cox and Sansom, 2009).
We have recently reported that certain water miscible amphipathic alcohols, notably 2-methyl-2,4-pentanediol (MPD) can modulate the protein-binding properties of SDS so that the detergent behaves essentially as a “gentle” nondenaturing detergent (Michaux et al., 2008a; Privé, 2007). Under typical conditions, SDS is a strongly denaturing anionic detergent that is widely used in biochemical applications to solubilize proteins in a denatured state (Helenius and Simons, 1975). It is effective at millimolar concentrations and induces a nonnative structure with a high proportion of α helix, although its mechanism of action is not well understood (Guo et al., 1990; Manning and Colon, 2004; Mattice et al., 1976; Nielsen et al., 2007; Otzen, 2002; Otzen and Oliveberg, 2002; Reynolds and Tanford, 1970; Samso et al., 1995; Smith, 1994). MPD is best known as a protein precipitant in crystallization experiments (Anand et al., 2002). Concentrations of 1–2 M MPD protected both soluble and integral membrane proteins from SDS denaturation, and several proteins, including PagP, could be induced to fold from the SDS denatured state upon addition of MPD (Michaux et al., 2008a). PagP in SDS/MPD could be reversibly denatured and refolded upon heating and cooling cycles, confirming that the folded protein is at thermodynamic equilibrium in this detergent/cosolvent system.
Here, we report the 1.4 Å crystal structure of PagP crystallized directly from SDS/MPD. The barrel structure of PagP in SDS/MPD is similar to the previously determined crystal structure in LDAO (Ahn et al., 2004) and with the solution NMR structures in dodecyl-phosphocoline (DPC) and β-D-octyl-glucoside (OG) (Hwang et al., 2002). However, we observed differences in the conformations of the four extracellular loops, indicating regions of flexibility in the protein that may be important for substrate binding and catalysis. Like a parapet that surrounds the turret of a medieval castle, the PagP β barrel contains indentations known as the crenel and embrasure, which provide gateways for lateral lipid access between the membrane external leaflet and the hydrocarbon ruler. Cysteine crosslinking between these regions of diminished β strand hydrogen bonding has previously identified the crenel between strands F and G as the palmitoyl chain access route, whereas the embrasure at the β-bulge between strands A and B serves as the egress route for the chain following the coupling to lipid A acceptor (Khan and Bishop, 2009). We now use molecular dynamics simulations and mutagenesis to confirm a phospholipid access route via the F/G crenel coupled to a movement of extracellular loop L4. We identify Tyr147 in L4 as a potential gating residue controlling lateral access of phospholipids to the interior hydrocarbon ruler pocket of PagP.
RESULTS
Crystallization of PagP from SDS/MPD
E. coli PagP was directed to inclusion bodies by overexpressing the protein without a signal sequence (Ahn et al., 2004; Hwang et al., 2002). Washed inclusion bodies were dissolved in a buffer containing 1% SDS and 1 M MPD. The solution was heated to 100°C and the protein was refolded by slow cooling. Refolding was quantitative as judged by an SDS-PAGE assay (Michaux et al., 2008a). Circular dichroism confirmed that the refolded protein was predominantly β sheet and exhibited a characteristic exciton couplet at 232 nm indicating the correct formation of the hydrocarbon ruler (see Figure S1 available online) (Khan et al., 2007, 2010; Michaux et al., 2008a). No chromatographic steps were performed, and the only purification of the protein occurred during the initial preparation of the insoluble inclusion body material. The protein solution was concentrated to 5.8 mg/ml and contained 3.85% w/v SDS (135 mM) as measured by a methylene blue extraction assay (Arand et al., 1992). The critical micelle concentration (cmc) of SDS in 1 M MPD and 10 mM Tris-HCl is approximately 0.6 mM as measured by isothermal titration calorimetry (J. Holyoake and G.G.P., unpublished data), and so the SDS concentration in the final crystallization stock solution was over 250-fold above its cmc.
The protein was crystallized by the traditional hanging drop method by mixing equal volume solutions of the protein stock and a salt-rich reservoir containing 1 M MPD, and allowing vapor-phase equilibration between the protein droplet and the much larger volume of the reservoir solution. Two phases formed in the protein droplet as equilibration proceeded. Crystals nucleated at the interface of the two phases and grew exclusively into one of the phases. The fluorescent lipid NBD-DPPC (Avanti Polar Lipids) was added to a hanging drop containing crystals and the lipid dye partitioned preferentially into the phase in which the PagP crystals grew, suggesting that the SDS concentration in the phase that supported crystal growth was further enriched in detergent relative to the initial protein stock solution (Figure S2). Both the protein stock solution and the reservoir solution initially contained 1 M MPD, but because MPD is volatile, the MPD concentration in the phase containing the protein crystals is unknown. However, we located 10 MPD molecules in the final refined structure (see below), indicating a relatively high concentration of the cosolvent in the mother liquor surrounding the crystal.
Structure Overview
Full anisotropic refinement was performed to a resolution of 1.4 Å (Table 1). The structure is an eight-stranded β barrel that spans the membrane region with a leading amphipathic N-terminal α helix that lies at the interface between the periplasm and the outer membrane inner leaflet. The eight strands of the barrel are named A to H and the four extracellular loops are referred to as L1 to L4 (Figure 2). The hydrocarbon ruler pocket in the barrel interior is occupied by a SDS molecule in a position similar to the LDAO molecule found in the crystal structure determined in that detergent (Ahn et al., 2004).
Table 1.
Data Collection and Refinement Statistics
| Data Collection | |
|---|---|
| Space group | P6222 |
| Unit cell (Å) | 113.23, 113.23, 55.06 |
| Rsyma | 0.068 (0.531) |
| I/σ(I) | 28.1 (1.3) |
| Completeness (%) | 97.7 (80.4) |
| Redundancy | 8.2 (4.1) |
| Refinement | |
| Resolution (Å) | 20.0–1.40 |
| No. reflections | 40,392 |
| Rwork/Rfree | 0.171 (0.303)/0.208 (0.290) |
| Number of atoms | |
| Protein | 1321 |
| SDS | 97 (6 molecules) |
| MPD | 80 (10 molecules) |
| Ions | 32 |
| Water | 56 |
| B factors (Å2) | |
| Protein | 27.1 |
| SDS | 47.6 |
| MPD | 48.3 |
| Ions | 60.5 |
| Water | 38.1 |
| Rmsd bond length (Å) | 0.021 |
| Rmsd bond angles (°) | 2.12 |
Values in parentheses are for the high-resolution shell from 1.42–1.40 Å.
Figure 2. Differences between the SDS/MPD and the LDAO Crystal Structures of PagP.

(A) Superposition of the SDS/MPD (blue, red, green) and LDAO (gray) crystal structures of PagP. The thin red line in L1 connects residue 37 to 46 and indicates the disordered region of this loop. Only the β sheet residues were used in the least-squares fitting.
(B) Distances between the α carbons of the SDS/MPD and the LDAO crystal structures of PagP. Residues in β strands are in blue, and only those were used for the superposition. The N-terminal helix is shown in dark blue, the periplasm-facing loops are in green, and the extracellular loops L1–L4 are in red. See also Figure S1.
The high resolution of this structure allowed us to trace all of the amino acids in the N-terminal α helix (Huysmans et al., 2007), which extends from Asn-1 to Trp-17. Loop L1 links strand A to strand B and is partly disordered, and amino acids 38–45 from this loop are not included in the final model. This loop was also not fully modeled in the LDAO crystal structure (Ahn et al., 2004) and is highly dynamic in the NMR structures (Hwang et al., 2002, 2004), indicating that this region is intrinsically disordered, at least in the absence of a lipid bilayer.
The overall barrel structure of PagP in SDS/MPD resembles that of PagP in LDAO by X-ray crystallography (Ahn et al., 2004) and in DPC or OG micelles by NMR (Hwang et al., 2002). Least-squares superposition of the Cα atoms from the β sheet residues in strands A–H of PagP results in an RMSD of 0.70 Å between the SDS/MPD and the LDAO structures (Figure 2A). The structures of the short periplasm-facing loops are also well conserved between the structures; however, deviations greater than 1.2 Å are seen in the four extracellular loop regions (Figure 2B). These extracellular loops are also the most dynamic regions of the protein as measured by NMR (Hwang et al., 2002, 2004).
Lattice forces may influence some of the loop conformations, as surfaces on both the periplasmic and extracellular sides of the protein are involved in crystal packing contacts. This applies to both the SDS/MPD and LDAO structures, which adopt entirely different crystal lattices. The differences in the two structures are mostly in the extracellular loops, suggesting that these parts of the protein may be especially malleable. In fact, it is most likely the different lattice environments, rather than the influence of the SDS versus LDAO detergents per se, that have selected the different loop conformations. We suggest that the different crystal forms have selected two possible structural states for PagP. Such effects are common, and give insight into the flexibility of structures based on the static snapshots of single crystal structures. For example, a comparison of nine different structures of the HIV protease has been used to analyze the plasticity of the flap regions of the protein (Heaslet et al., 2007).
PagP/SDS Interactions
The final model contains six SDS molecules with an average B factor of 47 Å2. In all cases the B factors are higher for the head group than for the aliphatic chain. Five of the SDS molecules are in contact with the exterior of the protein in the membrane-spanning region of PagP and are bound into crevices in the surface of the molecule (Figure 3A; Movie S1). The sulfate head groups do not make any consistent types of interactions with the protein and are consequently less ordered in the crystal. Thus, bound SDS molecules behave like other detergents in which binding to the protein is determined primarily by the aliphatic chain (Hunte and Richers, 2008; Qin et al., 2006). The structure of PagP crystallized from LDAO contains five ordered detergent molecules, four of which are common with the SDS binding sites (Figure 3A). The detergent molecules seen in the SDS/MPD structure are thus bound in general detergent binding sites that likely correspond to lipid acyl chain binding sites in the natural outer membrane environment of PagP. No significant SDS-SDS contacts are observed in the crystal and any micellar structures that may be present in the crystal are not ordered.
Figure 3. SDS and MPD Environment in the Crystal.

(A) Externally associated SDS and MPD molecules are shown on the PagP solvent-accessible surface. PagP is colored with white carbons, red oxygens, blue nitrogens, and yellow sulfurs. The approximate position of the bilayer is indicated. The MPD molecules are shown with purple carbons and red oxygens. The SDS molecules are show with green aliphatic tails. The LDAO detergent coordinates from PDB ID 1THQ are included after superposition of the proteins and are shown with cyan carbons.
(B) The surface of PagP from the SDS/MPD structure is shown in pink, and a cut through the structure shows the pocket of the hydrocarbon ruler. The SDS molecule in the pocket is shown in stick representation with green carbons. The mesh shows the electron density for SDS in the 2Fo-Fc map contoured at 1.2σ. The LDAO molecule in the pocket of the LDAO crystal PDB ID 1THQ is shown with cyan carbons for comparison.
The remaining SDS molecule is found in the interior of PagP. The acyl chain binding pocket in the center of the β barrel is occupied by a detergent molecule in a similar fashion to that observed in LDAO (Ahn et al., 2004). This pocket is part of the hydrocarbon ruler of PagP that selects for palmitate chains in the sn-1 position of substrate phospholipids (Khan et al., 2007, 2010). The nine terminal carbons of the SDS molecule are found to be tightly held and have the lowest atomic B factors for this molecule. The B factors for carbons nearer to the head group increase significantly and the electron density becomes tubular in contrast to the zigzagging pattern of the lower part of the tail. The SDS in the pocket overlaps very well with the LDAO molecule in the terminal eight carbons in the acyl chain, but the paths diverge nearer to the head group region. As a result, the anionic SDS head group is located in a different position than the zwitterionic head group of LDAO (Figure 3B). The head group of SDS is closer to loop L1 and farther from L2, relative to the position of the LDAO head group.
PagP/MPD Interactions
The final model includes ten MPD molecules (Figure 3A; Movie S1). Seven of these are in conformations in which the two hydroxyl groups form intramolecular hydrogen bonds, such that the MPD molecules display a hydrophilic face and a hydrophobic face (Anand et al., 2002). Within the ordered molecules, there are no significant interactions between the MPD and SDS. A previous study also found no interactions between SDS and MPD in lysozyme crystals (Michaux et al., 2008b), and the present study expands this result to crystals of membrane proteins in which SDS is the stabilizing detergent.
Seven of the ten MPD molecules are bound to the hydrophobic membrane-exposed surface of PagP or to residues marking the transition between the hydrophobic and hydrophilic surfaces. These seven molecules are also in contact with hydrophilic residues of symmetry-related proteins and thus play important roles in building the crystal lattice. Overall, the MPD molecules are found in two types of environments in the crystal: they either fill in small deep pockets in the protein’s surface or are sandwiched between relatively flat surfaces of two symmetry equivalent molecules. Four of the MPD molecules stack directly with π electronic clouds of the Trp17, Tyr23, Trp89, and Trp117 aromatic side chains of PagP. MPD has been used in the crystallization of at least 20 other integral membrane proteins based on data contained in the membrane protein data bank (http://www.mpdb.tcd.ie) (Raman et al., 2006). Ordered MPD molecules have been reported in crystal structures of PagP in LDAO (Ahn et al., 2004), outer membrane phospholipase A (Snijder et al., 2001), OmpT (Vandeputte-Rutten et al., 2001), and BtuB crystallized from lipidic mesophase (Cherezov et al., 2006). In general, the MPD molecules in these structures occupy positions that are consistent with what we observe for PagP in SDS/MPD.
Catalytic Center
The conserved residues that are necessary for the catalytic activity of PagP include His33 in the extracellular loop L1 and Asp76 and Ser77 in L2 (Hwang et al., 2002). In the SDS/MPD structure, His33 is 13.0 Å away from Asp76 and 14.8 Å away from Ser77 (distances between α carbons; the distance between Oγ of Ser77 and Nε2 of His33 is 8.5 Å ). These residues are closer than in the LDAO structure where the Cα distances are 16.4 and 17.0 Å respectively. This difference is mostly due to a rigid-body tilting of loop L2 toward L1, but the residues in the SDS/MPD structure are still too far apart to form a catalytic center. Thus, the formation of a competent active site likely involves additional rearrangements of the surface loops, possibly triggered by the binding of donor phospholipid and/or lipid A acceptor (Bishop, 2005; Raetz et al., 2007).
Accessibility of the Hydrocarbon Ruler Pocket
The SDS crystal structure shows that extracellular loop L4 moves away from the barrel interior, resulting in a large gap (a crenel) (Khan and Bishop, 2009) between strands F and G (Figure 4A). This gap opens a path to the top part of the hydrocarbon ruler that was blocked by the side chain of Tyr147 in the LDAO structure (Ahn et al., 2004). A smaller crenel is present between strands B and C (Figure 4B). As a result, we sought to further analyze possible access routes into and out of the barrel interior.
Figure 4. Crenels as Possible Routes in and out of the Hydrocarbon Ruler Pocket.

PagP is shown in a surface representation with the β barrel and the periplasmic loops in white. Loop L1 is dark blue, L2 is yellow, L3 is cyan, and L4 is green. The SDS molecule buried within the hydrocarbon ruler pocket is shown as a space-filling model with black carbons, orange sulfur, and red oxygens. The SDS molecule is clearly visible through a crenel between strands F and G in (A) and through a crenel between strands B and C in (B). The F/G crenel is larger and deeper in the membrane than the B/C crenel.
We analyzed the possible acyl chain entry and exit routes by performing nonequilibrium molecular dynamics simulations. We chose this approach in order to gain insight into the route(s) of acyl chain entry and exit that are most easily accommodated by the protein. This is an early step in the overall reaction, and the subsequent steps along the catalytic pathway likely involve larger scale motions of the protein backbone that are beyond the scope of these simulations. The strategy was to test a large number of short nonequilibrium simulations in order to generate an ensemble of trajectories, rather than to generate a smaller number of longer simulations that would provide limited statistical sampling. The 100 ps length of the simulations was sufficient to observe productive insertions and extractions, yet short enough to allow a large number of repeats for statistical significance. Overall, we generated 1205 insertion attempts and 302 extraction attempts.
PagP in the conformation from the SDS/MPD crystal was placed in an explicit phospholipid bilayer. No significant reorganization of the PagP backbone was observed during the simulations. Acyl-chain entry was assessed by applying a biasing force to the sn-1 chain of a 1-palmitoyl-2-oleoyl-sn-glycero-3-phos-phocholine (POPC) molecule from the upper leaflet of the bilayer, directing it toward the empty PagP binding pocket. Of 1205 such simulations, 55 resulted in the sn-1 chain entering the binding pocket between strands F and G (the F/G route), five resulted in the sn-1 chain jumping out of the bilayer over the top of the protein near strands B to E, and in 1145 simulations the protein blocked the steered phospholipid from approaching the binding pocket more closely than other phospholipids in the first solvation shell (Figure 5A). Considering that the initial positions of the sn-1 acyl chain were distributed approximately uniformly around the barrel, roughly one-eighth of the 1205 binding simulations were directed at the F/G route. Thus, 55 of approximately 150 F/G attempts were successful, while none of the non-F/G route attempts were successful.
Figure 5. Plausible Routes of Ligand Entry and Exit to and from the PagP Binding Site Based on Steered MD Simulations.

The strands of the β barrel are labeled A–H and the extracellular loops L1, L2, L3, and L4 are shown in dark blue, yellow, light blue, and green, respectively. Steered acyl chains are overlaid in pink. Acyl chains that jumped out of the bilayer, and all unbiased lipids, are omitted for clarity. (A) The distal 13 carbons of the sn-1 chain of biased POPC molecules after 1205 insertion attempts. Only the F/G route is available for insertion. (B) Detergent heavy atoms within 6 Å of PagP during 302 extraction attempts. Both the F/G and the B/C routes can be used for extraction of acyl chains from the hydrocarbon ruler pocket. See also Figure S3.
Next, acyl-chain exit was assessed by applying a biasing force to a dodecyl phosphate (DP) molecule placed in the binding pocket of PagP, compelling it to exit in the bilayer plane. Of 302 such simulations, 95 resulted in detergent extraction by the F/G route, 26 by the B/C route, and 181 extraction attempts ended with the detergent still inside the binding pocket (Figure 5B). The fact that the B/C route was identified as a viable route of acyl-chain extraction, but not insertion, is likely due in part to the tilt of PagP in the bilayer plane, which lowers the F and G strands into the hydrophobic region of the bilayer more than it lowers the B and C strands. This tilt is predicted by the hydrophobic surface potential and arrangement of aromatic belt residues (Ahn et al., 2004) and confirmed experimentally by measuring oxygen contacts by NMR (Evanics et al., 2006). Analogous simulations starting from the LDAO crystal structure produced results similar to the simulations from the SDS structure, with additional rare phospholipid chain entry between the A and B strands (data not shown).
Gating of the F/G Crenel
In order to investigate the putative F/G route in the LDAO structure, we conducted MD simulations of the LDAO crystal structure in an explicit POPC bilayer using a nonequilibrium variant of the weighted ensemble method of Huber and Kim (1996). This evaluates the possibility of acyl-chain entry through the F/G route of the LDAO structure based on thermally driven conformational fluctuations and is conceptually similar to essential dynamics sampling (Amadei et al., 1996). In brief, the system was iteratively simulated in 0.2 ps segments, accepting simulation segments in which the donor chain spontaneously moved closer to the binding pocket, and discarding them otherwise. During this simulation, 350 of 400,000 simulation segments successfully progressed toward a bound state, yielding a biased trajectory of 70 ps. These simulations showed a disruption of the hydrogen bond between the phenolic oxygen of Tyr147 in L4 (immediately following strand G) and the amide hydrogen of Leu125 in strand F. This was followed by a rotation of the Tyr147 side chain away from strand F, opening a hole in the F/G route through which the sn-1 chain of the donor POPC molecule entered the PagP binding pocket (Figure 6; Movie S2). The progress of the acyl chain toward the pocket was impeded until Tyr147 moved away from Leu125. During the simulations outlined in this work, we did not observe any significant changes in the backbone H-bonding pattern of the β strands, with the exception of the Y147-L125 pair. Nevertheless, we do not rule out the possibility that such rearrangements are relevant to equilibrium binding modes.
Figure 6. Side View of the Putative Ligand Entry Route at the F/G Crenel.
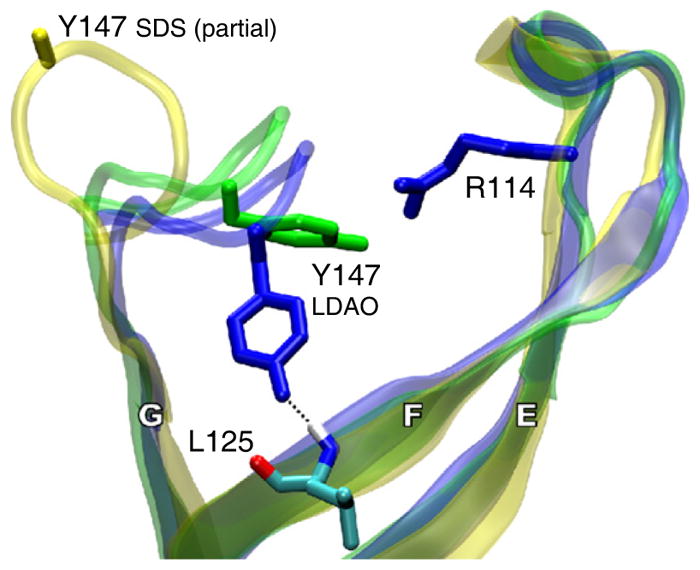
Only PagP strands E, F, and G, including loops L3 and L4, are shown. The phenolic oxygen of Tyr147 on loop L4 makes a hydrogen bond to the backbone amide hydrogen of Leu125 and obstructs the F/G route in a MD-equilibrated structure based on the LDAO (dark blue), but not the SDS (yellow), crystal form. During nonequilibrium MD sampling based on the weighted-sampling method (shown in green), Tyr147 rotates away from strand F, allowing the sn-1 chain of the donor POPC molecule to enter the binding pocket (not shown). The atoms of Leu125, shown only for the MD-equilibrated structure based on the LDAO crystal form, are colored red, dark blue, light blue, and white, for oxygen, nitrogen, carbon, and hydrogen, respectively. The hydrogen bond between the Tyr147 side chains and the main chain at Leu125 is shown as a broken black line. Residue Arg114 marks the position of loop L3. See also Movie S2.
Finally, we used functional assays to test the role of Tyr147 as a putative gating residue. Mutants Y147A and Y147F had similar CD spectra and denaturation profiles to wild-type PagP (Figures 7A and 7B), indicating that these mutations did not cause significant structural perturbations of the enzyme. PagP phospholipid: lipid A palmitoyltransferase activity was monitored in vitro using a defined detergent micellar enzymatic assay with TLC separation of radioactive lipid products (Khan et al., 2007, 2010). PagP selects the palmitate acyl chain from the phospholipid donor by a lateral lipid diffusion mechanism requiring access to the hydrocarbon ruler via the crenel located between β strands F and G (Khan and Bishop, 2009). In vitro reaction rates were measured for acylation of Kdo2-lipid A and for the phospholipase activity. The latter assay was carried out in the absence of lipid A acceptor, in which case the rate is four to five times slower than the lipid A acylation reaction rate (Khan and Bishop, 2009). For both the acyltransferase and the phospholipase activities, the reaction rate is 2- to 3-fold faster for the Y147A and Y147F mutants relative to the wild-type (Figures 7C and 7D), suggesting a role for the phenolic hydroxyl group of tyrosine 147 in controlling phospholipid gating through the F/G crenel.
Figure 7. Analysis of Wild-type PagP and the Y147A and Y147F Mutants.

Neither mutation has a significant effect on PagP structure or stability, as indicated by far UV CD spectra (A) and thermal denaturation by CD at 218 nm (B). Specific activities in vitro for lipid A palmitoyltransferase activity (C) and the phospholipase activity (D).
DISCUSSION
Crystallization from SDS/MPD
In this article, we report the use of SDS/MPD as a detergent system for membrane protein crystallization. We have previously used the SDS/MPD system to refold lysozyme (a β strand soluble protein), carbonic anhydrase (an α-helical soluble protein) and PagP (a β strand integral membrane protein) from the SDS-denatured state, indicating that the SDS/MPD system displays properties of a relatively mild detergent of wide applicability (Michaux et al., 2008a). In that study, we showed that MPD had little effect on the properties of LDAO and DM in the PagP refolding assay, and the unexpected observations were not simply due to the action of MPD on the protein. In fact, MPD is generally a mildly destabilizing osmolyte (Anand et al., 2002; Pittz and Timasheff, 1978). SDS alone has strongly denaturing properties, but the addition of MPD alters the physicochemical properties of the detergent such that the SDS micelles no longer associate strongly with polypeptides (Michaux et al., 2008a).
The mechanism of the cosolvent effect on the properties of SDS remains an area of speculation, although it is likely due to a partitioning of the cosolvent into the charged head group region of the SDS micelles thereby modifying the affinity of SDS micelles for the denatured states of the protein (Michaux et al., 2008a). The present crystal structure does not provide any insight into the SDS/MPD effect, however, since the effect depends on micellar states of the detergent. All of the ordered detergent and cosolvent molecules observed in this crystal structure associate as individual molecules with the protein surfaces, and we do not see any significant SDS-SDS or SDS-MPD interactions.
Access Route to the PagP Interior
In the structure of PagP in SDS/MPD, loops L2 and L3 have moved closer to the barrel axis relative to the LDAO structure. The largest difference, however, is the movement of loop L4 away from the acyl chain binding pocket. This creates a significant gap between strands F and G, opening a route to the internal hydrocarbon binding site. Our MD simulations are consistent in showing that this is the most favored route for acyl chain entry from the membrane phase. The increased phospholipase activity of the Tyr147 mutants provides independent experimental support that the phospholipid chain enters the barrel via the F/G route. The reaction rates for the phospholipase activity and the acyl transferase activity are affected at the same ratios by the Y147A and Y147F mutations, further suggesting that acyl chain entry is the rate limiting step in both reactions.
Additional mutants have been experimentally tested and recently reported. Hydrogen bonding between β strands F and G is disrupted by the presence of wild-type proline residues 127 and 144 which flank the PagP crenel. Mutation of each to Cys enabled crosslinking with a bifunctional reagent, which prevented the palmitoyltransferase reaction from occurring without compromising PagP structure and stability as measured by CD (Khan and Bishop, 2009). PagP has also been utilized recently as a model system to study outer membrane protein folding and assembly (Huysmans et al., 2010), and a role for weakened interstrand hydrogen bonding was suggested in identifying β barrel oligomerization interfaces (Naveed et al., 2009). Our results clearly indicate that in PagP these motifs fulfill an enzymatic role in lipid acyl chain selection.
Conclusion
A recent report on the FadL fatty acid transporter (Hearn et al., 2009) shows that hydrophobic compounds can exit this β barrel structure via a portal-type of opening in the side of the barrel wall. The structures of the FadL family of proteins are distinct from the PagP proteins, and the type of opening in the barrels is different. Nevertheless, in both cases, irregularities in the strand structures provide lateral access routes to and from the membrane hydrocarbon phase. This suggests that there are multiple ways in which β barrel membrane proteins can facilitate the passage of hydrophobic compounds, notably acyl chains, between the bulk lipid phase and the protein interiors. The portal-type of mechanism appears to be best suited for hydrophobic ligands with small head group or no polar moieties, while the crenel-type of mechanism described here allows for lateral access of amphipathic ligands with large head groups, including bilayer-forming lipids.
EXPERIMENTAL PROCEDURES
Overexpression and Purification of PagP Inclusion Bodies
E. coli PagPΔHΔS was cloned by the Quickchange procedure (Stratagene) from the plasmids described elsewhere (Hwang et al., 2002) and expressed into inclusion bodies. BL21(DE3) cells were transformed and cell cultures were grown in LB media at 20°C until the OD600 reached 0.6. Expression was induced in by adding IPTG to a concentration of 0.2 mM and growth was allowed to proceed for 5 hr at 37°C. Expression of PagP is driven into inclusion bodies under these conditions. The total production of protein as inclusion bodies was approximately 1 g of PagP from 6 L of LB media in shaker flasks. The cells were lysed in an Emulsiflex cell disrupter (Avestin) and inclusion bodies were collected by centrifugation of the lysed cells. The inclusion bodies were washed three times with a buffer consisting of 2% Triton X-100 and 10 mM Tris-HCl (pH 8) followed by centrifugation at 40,000 g. The final white pellet was dissolved in 6M guanidine hydrochloride and 10 mM Tris-HCl (pH 8) for storage. A 1 ml aliquot containing 13 mg of protein was precipitated by dilution into 25 ml of 10 mM Tris-HCl (pH 8). The precipitate was washed three times by centrifugation and resuspension in 25 ml of the same buffer. No further purification steps were carried on the sample that was used for crystallization.
Refolding
The white pellet from the washed inclusion bodies (13 mg) was dissolved in 20 ml of 1% SDS, 1M MPD, and 10 mM Tris-HCl (pH 8). The solution was heated for 2 min in a boiling water bath and allowed to cool down by letting the water bath slowly return to room temperature (approximately 2 hr). Refolding proceeded virtually to completion as judged by a gel shift assay (Michaux et al., 2008a) and the presence of a positive exciton band in the CD spectra at 232 nm (Khan et al., 2007). The PagP solution was concentrated in a 10 KDa MWCO spin filter (Amicon) resulting in 1.5 ml of a solution with 5.8 mg/ml of PagP. This solution was used directly for crystallization. The protein remained folded for months under these conditions as judged by the SDS-PAGE shift assay. The SDS concentration in this stock solution was 3.8% w/v by a SDS assay (Arand et al., 1992).
Crystallization
Crystals were grown by the hanging drop method. The reservoir solution consisted of 500 μl of 1M MPD, 0.1 M Na3Citrate buffer (pH 5.6), 1.7 M Li2SO4, and 0.3M (NH4)2SO4. This mixture separates spontaneously into two phases: a hanging drop was prepared by mixing 1 μl of the lower phase (rich in salt) with 1.5 μl of the PagP stock. The hanging drop was a homogeneous mixture at first but upon equilibration with the reservoir it also separated into two phases. Crystals were obtained only in one of the two phases in the hanging drop. Crystals were allowed to grow at 20°C for 3 months and then transferred to a temperature of 4°C for 4 days. A crystal of dimensions 0.4 × 0.2 × 0.2 mm was frozen directly in its mother liquor by plunging into liquid nitrogen.
Crystallographic Data Collection
Crystallographic data were collected at the Advanced Photon Source (APS) Argonne, Illinois at beamline 19ID (λ = 0.9800 Å) on an ADSC Q315 315 × 315 mm mosaic CCD detector at a temperature of 100 K. The data were integrated and scaled with the HKL3000 package (Minor et al., 2006). The diffraction was somewhat anisotropic, and extended to a resolution of approximately 1.55 Å along the a,b directions, but strong diffraction was obtained to well beyond 1.4 Å along the c axis. Details of the solution and refinement processes are given in the Supplemental Experimental Procedures. Figures were made with Pymol (DeLano, 2002).
Structure Solution and Refinement
The structure was solved by molecular replacement using EPMR (Kissinger et al., 1999) with the structure of PagP in LDAO, PDB accession code 1THQ (Ahn et al., 2004). The structure was refined and extended using alternate cycles of manual building in Coot (Emsley and Cowtan, 2004) and refinement in Refmac5 (Murshudov et al., 1997) using the CCP4i graphical interface (Potterton et al., 2003) with implicit riding hydrogens. The stereochemical quality of the final models was assessed with MolProbity (Davis et al., 2007) and Procheck (Laskowski et al., 1993). Restraints for the MPD molecules were modified from the standard Refmac 5 dictionary by removing the torsion and chiral restraints. Restraints for the SDS molecules were adapted from Coiro and Mazza (1991). Least-squares superpositions between the different models were made with the program LSQKab (Kabsch, 1976). Only the α carbons of the residues that are in β strands in the SDS/MPD structure as indicated by Procheck were used for the superpositions (88 atoms in total). The final model had the following Ramachandran statistics for the nonglycine and nonproline residues (Laskowski et al., 1993): 88.1% in the favored regions and 9.5% in the allowed regions.
Cloning, Expression, and Refolding PagP Mutants
Wild-type PagP, and the Y147A and Y147F mutants were constructed in the pETCrcAHΔS vector, with a C-terminal His6 tag and no signal peptide (Hwang et al., 2002) using the QuikChange protocol (Stratagene) and purified as described previously (Khan et al., 2007). The proteins were verified by electrospray ionization mass spectrometry. Precipitated protein samples were dissolved in a solution of 50:50 1% formic acid/acetonitrile just prior to ESI-MS. The sample concentration was maintained at 1 ng/μl and was injected directly onto a Waters/Micromass Q-Tof Ultima Global (a quadrupole time-of-flight) mass spectrometer. The spectra were reconstructed using MassLynx 4.0, MaxEnt 1 module. Wild-type PagP had an observed mass of 20175.3 ± 0.6 Da (theoretical mass 20175.5). Y147A PagP had an observed mass of 20082.1 ± 0.9 Da (theoretical mass 20083.4). Y147F PagP had an observed mass of 20159.5 ± 0.7 Da (theoretical mass 20159.5). The protein samples dissolved in 5 ml of 6 M Gdn-HCl, 10 mM Tris-HCl (pH 8.0) were diluted 10-fold into 10 mM Tris-HCl (pH 8.0), 0.1% LDAO. These samples were allowed to stir overnight at 4°C and applied to a NiNTA column. The samples were prepared from the columns as previously described in Khan et al. (2007). The concentration of the protein was determined using an extinction coefficient, ∈ 280 of 80,463 M−1 cm−1 for wild-type PagP and 78,211 M−1 cm−1 for PagPY147A and PagPY147F. The extinction coefficients were determined experimentally using the Edelhoch method (Edelhoch, 1967).
In Vitro Enzymatic Assays
Kdo2-lipid A and synthetic dipalmitoylphosphatidylcholine (DPPC) were obtained from Avanti Polar Lipids. A detailed description of the preparation of 32P-labeled Kdo2-lipid A, and thin layer chromatography (TLC)-based assays for lipid A palmitoyltransferase activity were previously described (Khan et al., 2007).
Phospholipase assays were carried out in a volume of 25 μl, with sufficient dipalmitoyl-1-14C-DPPC to achieve a final concentration of 20 μM (4000 cpm/μl). The lipid was dried under a stream of N2(g) and was dissolved in 22.5 μl reaction buffer containing 0.1M Tris-HCl (pH 8), 10 mM EDTA, and 0.25% dodecylmaltoside. The reactions were started by adding 2.5 μl of PagP and were conducted at 30°C. Reactions were stopped by adding 12.5 μl of the reaction mixture to 22.5 μl of a 1:1 CHCl3/MeOH. This generated a 2-phase solution, from which 5 μl of the lower phase was spotted on a silica gel 60 plate. The experiment was carried out over 5 hr, with the reaction being spotted once every hour. TLC plates were developed in a CHCl3:MeOH:H2O (65:25:4; v/v) solvent system, which was equilibrated in sealed glass tank for 4 hr. The plates were exposed overnight to a PhosphorImager screen and developed the following day with a Molecular Dynamics Typhoon 9200 PhosphorImager.
Circular Dichroism Spectroscopy
Samples to be analyzed by CD were maintained at a concentration of 0.3 mg/ml in 10 mM Tris-HCl (pH 8.0), 0.1% LDAO and were analyzed using a cuvette of 1 mm path length. The samples were analyzed using a Aviv 215 spectropolarimeter which was linked to a Peltier device Merlin Series M25 for temperature control. For each sample, three accumulations were averaged at a data pitch of 1 nm and a scanning speed of 10 nm/min. The temperature was maintained at 25°C and data were obtained from 200 to 260 nm. Thermal denaturation profiles were obtained by heating the samples from 20°C–100°C at 218 nm with a temperature slope of 1°C/min and a response time of 3 s.
Molecular Dynamics Simulations
The simulation system consisted of a solvated POPC bilayer in which 2 PagP molecules were embedded. The binding pocket of PagP was empty for steered insertion simulations and contained a DP detergent substrate analog for steered extraction simulations. Construction and 20–50 ns equilibration of the simulation systems are described in the Supplemental Experimental Procedures.
For each molecule of PagP in each starting conformation, a separate steered insertion simulation of 100 ps was carried out in which the sn-1 chain of a single phospholipid in the upper bilayer leaflet was forced toward the PagP binding pocket with a force of constant magnitude. Phospholipids were targeted for insertion if at least one of the distal 13 carbons in the sn-1 chain was within 3 nm of the protein. Similarly, steered extraction simulations forced the DP molecule away from its initial position in the PagP binding pocket. Additional details are provided in the Supplemental Experimental Procedures.
The Berger parameters (Berger et al., 1997) for POPC (Tieleman et al., 1998) and the OPLSAA parameters (Kaminski et al., 2001) for PagP were properly combined for the GROMACS simulation package (Lindahl et al., 2001) using the half-ε double-pairlist method (Chakrabarti et al., 2010) (Figure S3). In brief, the ε values of the 1–4 Lennard-Jones parameters of the lipids were multiplied by an additional factor of 0.5 in the pair-types section and the list of 1–4 interactions in the pairs section was duplicated. The regular OPLSAA combination rules were then applied. Additional details are provided in Supplemental Experimental Procedures and Figure S3.
Supplementary Material
Acknowledgments
G.G.P. was supported by CIHR operating grant MOP-13335 and IG1-93473. J.A.C.-S was supported by the CIHR Training Program in the Structural Biology of Membrane Proteins Linked to Disease. Computational studies were made possible by the facilities of the Centre for Computational Biology High Performance Facility (CCBHPF) at the Hospital for Sick Children and the Shared Hierarchical Academic Research Computing Network (SHARCNET). C.N. acknowledges support from the Research Training Centre at the Hospital for Sick Children and from the University of Toronto. R.P. is a CRCP chairholder and is supported by CIHR operating grant MOP-43998. Work in the laboratory of R.E.B. is supported by CIHR operating grant MOP-84329. This research was funded in part by the Ontario Ministry of Health and Long Term Care. The views expressed do not necessarily reflect those of the OMOHLTC.
Footnotes
ACCESSION NUMBERS
Atomic coordinates and structure factors have been deposited in the PDB (http://www.rcsb.org) under the accession code 3GP6.
Supplemental Information includes Supplemental Experimental Procedures, three figures, and two movies and can be found with this article online at doi:10.1016/j.str.2010.06.014.
References
- Ahn VE, Lo EI, Engel CK, Chen L, Hwang PM, Kay LE, Bishop RE, Privé GG. A hydrocarbon ruler measures palmitate in the enzymatic acylation of endotoxin. EMBO J. 2004;23:2931–2941. doi: 10.1038/sj.emboj.7600320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amadei A, Linssen AB, de Groot BL, van Aalten DM, Berendsen HJ. An efficient method for sampling the essential subspace of proteins. J Biomol Struct Dyn. 1996;13:615–625. doi: 10.1080/07391102.1996.10508874. [DOI] [PubMed] [Google Scholar]
- Anand K, Pal D, Hilgenfeld R. An overview on 2-methyl-2,4-pen-tanediol in crystallization and in crystals of biological macromolecules. Acta Crystallogr D Biol Crystallogr. 2002;58:1722–1728. doi: 10.1107/S0907444902014610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arand M, Friedberg T, Oesch F. Colorimetric quantitation of trace amounts of sodium lauryl sulfate in the presence of nucleic acids and proteins. Anal Biochem. 1992;207:73–75. doi: 10.1016/0003-2697(92)90502-x. [DOI] [PubMed] [Google Scholar]
- Bader MW, Sanowar S, Daley ME, Schneider AR, Cho U, Xu W, Klevit RE, Le Moual H, Miller SI. Recognition of antimicrobial peptides by a bacterial sensor kinase. Cell. 2005;122:461–472. doi: 10.1016/j.cell.2005.05.030. [DOI] [PubMed] [Google Scholar]
- Berger O, Edholm O, Jahnig F. Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys J. 1997;72:2002–2013. doi: 10.1016/S0006-3495(97)78845-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop RE. The lipid A palmitoyltransferase PagP: molecular mechanisms and role in bacterial pathogenesis. Mol Microbiol. 2005;57:900–912. doi: 10.1111/j.1365-2958.2005.04711.x. [DOI] [PubMed] [Google Scholar]
- Bishop RE, Gibbons HS, Guina T, Trent MS, Miller SI, Raetz CR. Transfer of palmitate from phospholipids to lipid A in outer membranes of gram-negative bacteria. EMBO J. 2000;19:5071–5080. doi: 10.1093/emboj/cdd507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti N, Neale C, Payandeh J, Pai EF, Pomés R. An iris-like mechanism of pore dilation in the CorA magnesium transport system. Biophys J. 2010;98:784–792. doi: 10.1016/j.bpj.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherezov V, Yamashita E, Liu W, Zhalnina M, Cramer WA, Caffrey M. In meso structure of the cobalamin transporter, BtuB, at 1.95 A resolution. J Mol Biol. 2006;364:716–734. doi: 10.1016/j.jmb.2006.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coiro V, Mazza F. Crystal phases of dodecyl sulfates obtained from aqueous solutions: structure of the hexaaquamagnesium salt. Crystal Structure Communications. 1991;47:1169–1173. [Google Scholar]
- Cox K, Sansom MS. One membrane protein, two structures and six environments: a comparative molecular dynamics simulation study of the bacterial outer membrane protein PagP. Mol Membr Biol. 2009;26:205–214. doi: 10.1080/09687680902788967. [DOI] [PubMed] [Google Scholar]
- Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, 3rd, Snoeyink J, Richardson JS, Richardson DC. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano WL. The PyMOL Molecular Graphics System 2002 [Google Scholar]
- Edelhoch H. Spectroscopic determination of tryptophan and tyrosine in proteins. Biochemistry. 1967;6:1948–1954. doi: 10.1021/bi00859a010. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Evanics F, Hwang PM, Cheng Y, Kay LE, Prosser RS. Topology of an outer-membrane enzyme: Measuring oxygen and water contacts in solution NMR studies of PagP. J Am Chem Soc. 2006;128:8256–8264. doi: 10.1021/ja0610075. [DOI] [PubMed] [Google Scholar]
- Guo L, Lim KB, Poduje CM, Daniel M, Gunn JS, Hackett M, Miller SI. Lipid A acylation and bacterial resistance against vertebrate antimicrobial peptides. Cell. 1998;95:189–198. doi: 10.1016/s0092-8674(00)81750-x. [DOI] [PubMed] [Google Scholar]
- Guo XH, Zhao NM, Chen SH, Teixeira J. Small-angle neutron scattering study of the structure of protein/detergent complexes. Biopolymers. 1990;29:335–346. doi: 10.1002/bip.360290206. [DOI] [PubMed] [Google Scholar]
- Hearn EM, Patel DR, Lepore BW, Indic M, van den Berg B. Transmembrane passage of hydrophobic compounds through a protein channel wall. Nature. 2009;458:367–370. doi: 10.1038/nature07678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaslet H, Rosenfeld R, Giffin M, Lin YC, Tam K, Torbett BE, Elder JH, McRee DE, Stout CD. Conformational flexibility inthe flap domains of ligand-free HIV protease. Acta Crystallogr D Biol Crystallogr. 2007;63:866–875. doi: 10.1107/S0907444907029125. [DOI] [PubMed] [Google Scholar]
- Helenius A, Simons K. Solubilization of membranes by detergents. Biochim Biophys Acta. 1975;415:29–79. doi: 10.1016/0304-4157(75)90016-7. [DOI] [PubMed] [Google Scholar]
- Huber GA, Kim S. Weighted-ensemble Brownian dynamics simulations for protein association reactions. Biophys J. 1996;70:97–110. doi: 10.1016/S0006-3495(96)79552-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunte C, Richers S. Lipids and membrane protein structures. Curr Opin Struct Biol. 2008;18:406–411. doi: 10.1016/j.sbi.2008.03.008. [DOI] [PubMed] [Google Scholar]
- Huysmans GH, Radford SE, Brockwell DJ, Baldwin SA. The N-terminal helix is a post-assembly clamp in the bacterial outer membrane protein PagP. J Mol Biol. 2007;373:529–540. doi: 10.1016/j.jmb.2007.07.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huysmans GH, Baldwin SA, Brockwell DJ, Radford SE. The transition state for folding of an outer membrane protein. Proc Natl Acad Sci USA. 2010;107:4099–4104. doi: 10.1073/pnas.0911904107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang PM, Choy WY, Lo EI, Chen L, Forman-Kay JD, Raetz CR, Privé GG, Bishop RE, Kay LE. Solution structure and dynamics of the outer membrane enzyme PagP by NMR. Proc Natl Acad Sci USA. 2002;99:13560–13565. doi: 10.1073/pnas.212344499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang PM, Bishop RE, Kay LE. The integral membrane enzyme PagP alternates between two dynamically distinct states. Proc Natl Acad Sci USA. 2004;101:9618–9623. doi: 10.1073/pnas.0402324101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia W, El Zoeiby A, Petruzziello TN, Jayabalasingham B, Seyedirashti S, Bishop RE. Lipid trafficking controls endotoxin acylation in outer membranes of Escherichia coli. J Biol Chem. 2004;279:44966–44975. doi: 10.1074/jbc.M404963200. [DOI] [PubMed] [Google Scholar]
- Kabsch W. A solution for the best rotation to relate two sets of vectors. Acta Cryst. A32. 1976:922–923. [Google Scholar]
- Kaminski G, Friesner R, Tirado-Rives J, Jorgensen W. Evaluation and Reparametrization of the OPLS-AA Force Field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptides. J Phys Chem B. 2001;105:6474–6487. [Google Scholar]
- Kawasaki K, Ernst RK, Miller SI. 3-O-deacylation of lipid A by PagL, a PhoP/PhoQ-regulated deacylase of Salmonella typhimurium, modulates signaling through Toll-like receptor 4. J Biol Chem. 2004;279:20044–20048. doi: 10.1074/jbc.M401275200. [DOI] [PubMed] [Google Scholar]
- Khan MA, Bishop RE. Molecular mechanism for lateral lipid diffusion between the outer membrane external leaflet and a beta-barrel hydrocarbon ruler. Biochemistry. 2009;48:9745–9756. doi: 10.1021/bi9013566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MA, Neale C, Michaux C, Pomés R, Privé GG, Woody RW, Bishop RE. Gauging a hydrocarbon ruler by an intrinsic exciton probe. Biochemistry. 2007;46:4565–4579. doi: 10.1021/bi602526k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MA, Moktar J, Mott PJ, Bishop RE. A thiolate anion buried within the hydrocarbon ruler perturbs PagP lipid acyl chain selection. Biochemistry. 2010;49:2368–2379. doi: 10.1021/bi901669q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kissinger CR, Gehlhaar DK, Fogel DB. Rapid automated molecular replacement by evolutionary search. Acta Crystallogr D Biol Crystallogr. 1999;55:484–491. doi: 10.1107/s0907444998012517. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- Lindahl E, Hess B, van der Spoel D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J Mol Model. 2001;7:306–317. [Google Scholar]
- Manning M, Colon W. Structural basis of protein kinetic stability: resistance to sodium dodecyl sulfate suggests a central role for rigidity and a bias toward beta-sheet structure. Biochemistry. 2004;43:11248–11254. doi: 10.1021/bi0491898. [DOI] [PubMed] [Google Scholar]
- Mattice WL, Riser JM, Clark DS. Conformational properties of the complexes formed by proteins and sodium dodecyl sulfate. Biochemistry. 1976;15:4264–4272. doi: 10.1021/bi00664a020. [DOI] [PubMed] [Google Scholar]
- Michaux C, Pomroy NC, Privé GG. Refolding SDS-denatured proteins by the addition of amphipathic cosolvents. J Mol Biol. 2008a;375:1477–1488. doi: 10.1016/j.jmb.2007.11.026. [DOI] [PubMed] [Google Scholar]
- Michaux C, Pouyez J, Wouters J, Privé GG. Protecting role of cosolvents in protein denaturation by SDS: a structural study. BMC Struct Biol. 2008b;8:29. doi: 10.1186/1472-6807-8-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL-3000: the integration of data reduction and structure solution–from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr. 2006;62:859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- Muroi M, Ohnishi T, Tanamoto K. MD-2, a novel accessory molecule, is involved in species-specific actions of Salmonella lipid A. Infect Immun. 2002;70:3546–3550. doi: 10.1128/IAI.70.7.3546-3550.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macro-molecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Naveed H, Jackups R, Jr, Liang J. Predicting weakly stable regions, oligomerization state, and protein-protein interfaces in transmembrane domains of outer membrane proteins. Proc Natl Acad Sci USA. 2009;106:12735–12740. doi: 10.1073/pnas.0902169106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen MM, Andersen KK, Westh P, Otzen DE. Unfolding of beta-sheet proteins in SDS. Biophys J. 2007;92:3674–3685. doi: 10.1529/biophysj.106.101238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otzen DE. Protein unfolding in detergents: effect of micelle structure, ionic strength, pH, and temperature. Biophys J. 2002;83:2219–2230. doi: 10.1016/S0006-3495(02)73982-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otzen DE, Oliveberg M. Burst-phase expansion of native protein prior to global unfolding in SDS. J Mol Biol. 2002;315:1231–1240. doi: 10.1006/jmbi.2001.5300. [DOI] [PubMed] [Google Scholar]
- Pilione MR, Pishko EJ, Preston A, Maskell DJ, Harvill ET. pagP is required for resistance to antibody-mediated complement lysis during Bordetella bronchiseptica respiratory infection. Infect Immun. 2004;72:2837–2842. doi: 10.1128/IAI.72.5.2837-2842.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittz EP, Timasheff SN. Interaction of ribonuclease A with aqueous 2-methyl-2,4-pentanediol at pH 5. Biochemistry. 1978;17:615–623. doi: 10.1021/bi00597a009. [DOI] [PubMed] [Google Scholar]
- Potterton E, Briggs P, Turkenburg M, Dodson E. A graphical user interface to the CCP4 program suite. Acta Crystallogr D Biol Crystallogr. 2003;59:1131–1137. doi: 10.1107/s0907444903008126. [DOI] [PubMed] [Google Scholar]
- Preston A, Maxim E, Toland E, Pishko EJ, Harvill ET, Caroff M, Maskell DJ. Bordetella bronchiseptica PagP is a Bvg-regulated lipid A palmitoyl transferase that is required for persistent colonization of the mouse respiratory tract. Mol Microbiol. 2003;48:725–736. doi: 10.1046/j.1365-2958.2003.03484.x. [DOI] [PubMed] [Google Scholar]
- Privé GG. Detergents for the stabilization and crystallization of membrane proteins. Methods. 2007;41:388–397. doi: 10.1016/j.ymeth.2007.01.007. [DOI] [PubMed] [Google Scholar]
- Qin L, Hiser C, Mulichak A, Garavito RM, Ferguson-Miller S. Identification of conserved lipid/detergent-binding sites in a high-resolution structure of the membrane protein cytochrome c oxidase. Proc Natl Acad Sci USA. 2006;103:16117–16122. doi: 10.1073/pnas.0606149103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raetz CR, Reynolds CM, Trent MS, Bishop RE. Lipid A modification systems in gram-negative bacteria. Annu Rev Biochem. 2007;76:295–329. doi: 10.1146/annurev.biochem.76.010307.145803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman P, Cherezov V, Caffrey M. The Membrane Protein Data Bank. Cell Mol Life Sci. 2006;63:36–51. doi: 10.1007/s00018-005-5350-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds JA, Tanford C. The gross conformation of protein-sodium dodecyl sulfate complexes. J Biol Chem. 1970;245:5161–5165. [PubMed] [Google Scholar]
- Robey M, O’Connell W, Cianciotto NP. Identification of Legionella pneumophila rcp, a pagP-like gene that confers resistance to cationic antimicrobial peptides and promotes intracellular infection. Infect Immun. 2001;69:4276–4286. doi: 10.1128/IAI.69.7.4276-4286.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samso M, Daban JR, Hansen S, Jones GR. Evidence for sodium dodecyl sulfate/protein complexes adopting a necklace structure. Eur J Biochem. 1995;232:818–824. [PubMed] [Google Scholar]
- Smith BJ. SDS polyacrylamide gel electrophoresis of proteins. Methods Mol Biol. 1994;32:23–34. doi: 10.1385/0-89603-268-X:23. [DOI] [PubMed] [Google Scholar]
- Smith AE, Kim SH, Liu F, Jia W, Vinogradov E, Gyles CL, Bishop RE. PagP activation in the outer membrane triggers R3 core oligosaccharide truncation in the cytoplasm of Escherichia coli O157:H7. J Biol Chem. 2008;283:4332–4343. doi: 10.1074/jbc.M708163200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder HJ, Van Eerde JH, Kingma RL, Kalk KH, Dekker N, Egmond MR, Dijkstra BW. Structural investigations of the active-site mutant Asn156Ala of outer membrane phospholipase A: function of the Asn-His interaction in the catalytic triad. Protein Sci. 2001;10:1962–1969. doi: 10.1110/ps.17701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanamoto K, Azumi S. Salmonella-type heptaacylated lipid A is inactive and acts as an antagonist of lipopolysaccharide action on human line cells. J Immunol. 2000;164:3149–3156. doi: 10.4049/jimmunol.164.6.3149. [DOI] [PubMed] [Google Scholar]
- Tieleman DP, Forrest LR, Sansom MS, Berendsen HJ. Lipid properties and the orientation of aromatic residues in OmpF, influenza M2, and alamethicin systems: molecular dynamics simulations. Biochemistry. 1998;37:17554–17561. doi: 10.1021/bi981802y. [DOI] [PubMed] [Google Scholar]
- Vandeputte-Rutten L, Kramer RA, Kroon J, Dekker N, Egmond MR, Gros P. Crystal structure of the outer membrane protease OmpT from Escherichia coli suggests a novel catalytic site. EMBO J. 2001;20:5033–5039. doi: 10.1093/emboj/20.18.5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.