

Abstract
The Escherichia coli outer membrane phospholipid:lipid A palmitoyltransferase PagP is normally a latent enzyme, but it can be directly activated in outer membranes by lipid redistribution associated with a breach in the permeability barrier. We now demonstrate that a lipid A myristate deficiency in an E. coli O157:H7 msbB mutant constitutively activates PagP in outer membranes. The lipid A myristate deficiency is associated with hydrophobic antibiotic sensitivity and, unexpectedly, with serum sensitivity, which resulted from O-antigen polysaccharide absence due to a cytoplasmically determined truncation at the first outer core glucose unit of the R3 core oligosaccharide. Mutational inactivation of pagP in the myristate-deficient lipid A background aggravated the hydrophobic antibiotic sensitivity as a result of losing a partially compensatory increase in lipid A palmitoylation while simultaneously restoring serum resistance and O-antigen attachment to intact lipopolysaccharide. Complementation with either wild-type pagP or catalytically inactive pagPSer77Ala alleles restored the R3 core truncation. However, the intact lipopolysaccharide was preserved after complementation with an internal deletion pagPΔ5–14 allele, which mostly eliminates a periplasmic amphipathic α-helical domain but fully supports cell surface lipid A palmitoylation. Our findings indicate that activation of PagP not only triggers lipid A palmitoylation in the outer membrane but also separately truncates the R3 core oligosaccharide in the cytoplasm. We discuss the implication that PagP might function as an apical sensory transducer, which can be activated by a breach in the outer membrane permeability barrier.
Like most enteric Gram-negative bacteria, Escherichia coli surrounds its cytoplasmic membrane with a reticulated peptidoglycan exoskeleton (murein) and an outer membrane (OM),5 which demarcates the so-called periplasmic space. The enterobacterial OM is an asymmetric lipid bilayer in which lipopolysaccharide (LPS) exclusively lines the external leaflet, whereas phospholipids line the inner leaflet (1, 2). The asymmetric lipid organization provides a permeability barrier to hydrophobic antibiotics and detergents encountered in the natural and host environments. Although hydrophobic antibiotics can freely permeate through phospholipid bilayers, negative charges in LPS are bridged by Mg2+ ions to create tight lateral packing interactions, which largely prevent permeation (3, 4). According to current models, perturbations of OM lipid asymmetry can result from the migration of phospholipids into the external leaflet to create localized rafts of phospholipid bilayers, which render bacteria susceptible to hydrophobic antibiotics (5, 6).
The LPS is a tripartite molecule consisting of the hydrophobic anchor lipid A (endotoxin), the core oligosaccharide, which is divided into the inner and outer core regions, and the O-antigen polysaccharide (7). The so-called rough LPS includes only the lipid A-core and is usually distinguished from the smooth LPS that also includes O-antigen. The O-antigen can provide bacterial resistance to serum by preventing deposition of the complement cascade’s membrane attack complex (8).
The entire LPS structure is assembled within three distinct subcellular compartments, namely the cytoplasmic membrane, the periplasmic space, and the OM (9). The Raetz pathway for lipid A biosynthesis includes nine Lpx enzymes, which convert UDP-GlcNAc into a β-1′,6-linked disaccharide of GlcN (7). The lipid A molecule is phosphorylated at positions 1 and 4′ and is acylated with R-3-hydroxymyristate in ester linkage at positions 3 and 3′ and in amide linkage at positions 2 and 2′ (Fig. 1). Attachment at position 6′ of two 3-deoxy-D-manno-oct-2-ulosonic acid (Kdo) sugars, which belong to the innermost region of the inner core, is followed by secondary acylation reactions to create the so-called acyloxyacyl linkages. Attachment of laurate at position 2′ is usually followed by attachment of myristate at position 3′, which is catalyzed by the myristoyltransferase MsbB (LpxM) (10). Each of these enzymatic reactions takes advantage of cytoplasmic energy-rich biosynthetic precursors (7).
FIGURE 1. Structure of lipid A and the inner R3 core oligosaccharide of E. coli O157:H7.

Lipid A is an acylated and phosphorylated disaccharide of GlcN linked by a β-1′,6-glycosidic bond. Three of the four primary R-3-hydroxymyristate chains can be modified with secondary acyloxyacyl groups. MsbB can incorporate a myristate chain, and a palmitate chain can be partially incorporated by PagP (red). Partial modification of the lipid A phosphate groups with diphosphate, PEtN, and L-Ara4N can also be observed. The inner core consists of two Kdo and three Hep sugars, which can be substituted with phosphate and PEtN groups. GlcNAc is a unique feature of the R3 inner core of E. coli O157:H7, but the remaining inner core structures and lipid A are virtually identical in E. coli K-12, which has a distinctly different outer core structure and no O-antigen. Structures shown in blue were found in this study to be absent in the msbB-deficient mutant of E. coli O157:H7. The dashed bonds specify partial substitutions.
The waa (rfa) operons encode cytoplasmic enzymes needed for the stepwise assembly of any one of the five known core oligosaccharides (K-12 and R1–R4) that can exist in E. coli. Although ~170 distinct O-antigens have been identified in E. coli alone, they are all assembled on the lipid carrier analog of dolichol phosphate known as bactoprenol phosphate. Translocation of the lipid A-core and bactoprenol diphosphate-O-antigen to the periplasmic surface of the inner membrane can be followed by polymerization of the O-antigen polysaccharide with its subsequent en bloc ligation to the outer core. At this periplasmic stage, several regulated partial modifications can occur on both lipid A and the inner core and include the addition of phosphoethanolamine (PEtN), derived from phosphatidylethanolamine, and 4-amino-4-deoxy-L-arabi-nose (L-Ara4N), derived from bactoprenol phosphate-L-Ara4N (11). These modifications provide resistance to poly-myxin B and can be induced by mildly acidic growth conditions, antimicrobial peptides, or Mg2+ limitation, which together activate the PhoP/PhoQ and PmrA/PmrB two-component signal transduction pathways (12, 13).
E. coli K-12 strains generally synthesize rough LPS lacking O-antigen polysaccharide unless certain genetic factors are exogenously provided (14). In contrast, smooth LPS with attached O157 polysaccharide is synthesized in enterohemorrhagic E. coli O157:H7. This most common serotype of Shiga toxin-producing E. coli is associated with hemorrhagic colitis and hemolytic-uremic syndrome in humans (15). The core oligosaccharide of E. coli O157:H7 is of the R3 type, which is distinctly different from K-12 in the outer core regions (16). The lipid A and inner core structures of the R3 and K-12 LPS are largely identical, with the exception of a few important differences that can be attributed to enzymes encoded in the plasmid pO157 shf locus.
The conserved inner core includes, in addition to the two Kdo units, three units of L-glycero-D-manno-heptose (Hep), which can be modified with phosphate and/or PEtN moieties at key positions (Fig. 1). A defining feature of the R3 inner core is the partial modification of HepIII by an α-1,7-linked GlcNAc unit, which is controlled by the shf locus-encoded glycosyl-transferase WabB (16). Additionally, under normal laboratory growth conditions, where the corresponding enzymes of E. coli K-12 are found to be latent (17), E. coli O157:H7 introduces significant amounts of PEtN into the lipid A phosphate groups (18). Finally, E. coli O157:H7 possesses two homologues of the msbB gene: msbB1 encoded on the chromosome, which is equivalent to the single msbB gene of E. coli K-12, and msbB2 encoded on the shf locus. Both msbB orthologues must be inactivated to create a myristate deficiency in the lipid A of E. coli O157:H7, and this is associated with reduced virulence (19), as similarly occurs in msbB-deficient Shigella flexneri (20). The only ascribed function for the single msbB2 gene is in the persistence of E. coli O157:H7 in its agricultural and bovine reservoirs (21).
The entire LPS structure is transported across the periplasmic space and delivered to the OM external leaflet, where lipid A can be further modified (11). PagP is the only known OM enzyme of LPS biosynthesis in E. coli, and it is controlled by PhoP/PhoQ (22). PagP acylates the lipid A position 2 R-3-hydroxymyristate chain with a phospholipid-derived palmitoyl group (23, 24), which provides resistance to cationic antimicrobial peptides (25–27) and attenuates the ability of LPS to trigger host defenses through the TLR4 pathway (28, 29). PagP structure and dynamics demonstrate that the palmitate recognition pocket, known as the hydrocarbon ruler, is only accessible from the OM external leaflet and, thus, requires aberrant translocation of phospholipids into the external leaflet (30–32). Indeed, PagP remains dormant in the OM until perturbations to lipid asymmetry, which compromise the OM permeability barrier, directly trigger PagP activity (33). PagP has been proposed to function as a sentinel that can be activated by a breach in the OM permeability barrier (34). We now demonstrate that a lipid A myristoylation mutant of enterohemorrhagic E. coli O157: H7, but not a similar mutant from E. coli K-12, necessarily triggers PagP activity in the OM to exert control on cytoplasmic enzymes that determine its characteristic R3 core oligosaccharide structure. PagP sensory transduction is not controlled by its cell surface catalytic machinery but depends instead on its periplasmic amphipathic α-helix.
EXPERIMENTAL PROCEDURES
Materials
32Pi was purchased from PerkinElmer Life Sciences. Antibiotics and Gal were obtained from Sigma. Pyridine, methanol, and 88% formic acid were obtained from Mallinckrodt. Chloroform was purchased from EM Science. Glass-backed Silica Gel 60 TLC plates were from Merck. The QIA-prep spin miniprep, Qiaquick PCR purification, and QIAEX II gel extraction kits were obtained from Qiagen. High fidelity PCR was performed with a proofreading DNA polymerase (Advantage-HF2 PCR kit; BD Biosciences Clontech). Restriction endonucleases, T4 DNA ligase, and dNTP were obtained from Fermentas. Bacto MacConkey agar was obtained from Difco. All other materials were obtained from commercial sources.
Bacterial Strains, Plasmids, and Growth Conditions
The bacterial strains and plasmids used in this study are described in Table 1. Cells were generally grown at 37 °C in Luria-Bertani (LB) broth. Antibiotics were added when necessary at final concentrations of 12 μg/ml for tetracycline, 20 μg/ml for chloramphenicol and gentamycin (Gm), 100 μg/ml for ampicillin (Ap), and streptomycin (Str), and 40 μg/ml for kanamycin. Antibiotic concentrations were reduced by a factor of 10 during selection of the hypersensitive E. coli O157:H7 strain 4303-TM. Single colonies were inoculated from plates into 5 ml of liquid medium and grown at 37 °C overnight to stationary phase. A 1% inoculum was then subcultured into the same medium and allowed to resume growth at 37 °C. Cultures were adjusted with EDTA using a stock solution of 250 mM EDTA, pH 8.0, which had been sterilized by using a 0.2-μm filter.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strains/plasmids | Descriptiona | Source |
|---|---|---|
| E. coli O157:H7 | ||
| 4304 | Wild type (phage-type 14), (Strr) | Ref. 19 |
| 4304-DM | 4304 ΔmsbB1, ΔmsbB2 (Strr) [msbB1/2] | Ref. 19 |
| 4304-PM | 4304 pagP::aacC1 (Strr, Gmr) [pagP] | This study |
| 4304-TM | 4304 ΔmsbB1, ΔmsbB2, pagP::aacC1 (Strr, Gmr) [msbB1/2/pagP] | This study |
|
| ||
| E. coli K-12 | ||
| MC1061 | F-, λ-, araD139, Δ (ara-leu)7697, Δ (lac)X74, galU, galK, hsdR2 (rK-mK+), mcrB1, rpsL | Ref. 33 |
| WJ0124 | MC1061 pagP::amp [pagP] | Ref. 33 |
| SK1061 | MC1061 msbB::Tn5, pagP::amp [msbB/pagP] | This study |
| MC-msbB | MC1061 msbB::Tn5 [msbB] | Ref. 18 |
| BMS67C12 | msbB::Tn5 mutant of JM83 (Kmr) | Ref. 36 |
| SM10 | thr, leu, tonA, lacY, supE, recA::RP4-2-Tc::Mu, Kmr, λpir | Ref. 61 |
|
| ||
| Plasmids | ||
| pGEM-T | PCR product cloning vector | Promega |
| pUCGM | Gmr cassette (aacC1)-containing plasmid | Ref. 62 |
| pR7Crc-Gm | pRE107 carrying pagP::aacC1 | This study |
| pRE107 | A suicide vector (oriR6K, RP4 mob, sacB, Apr) | Ref. 61 |
| pCrcAT | pagPO157 in pGEM-T vector | This study |
| pWG24 | waaGO157 cloned into pBAD24 (AmpR) | This study |
| pGU184 | galUO157 cloned into pACYC184 (CmR) [pGalU] | This study |
| pAA101 | E. coli galETK cloned in pBR313 | Ref. 41 |
| pACPagP | E. coli pagP cloned in pACYC184 | Ref. 33 |
| pACPagPSer77Ala | Derivative of pACPagP | This study |
| pACPagPΔ5–14 | Previously pACPagPΔ30–39 (precursor numbering) | Ref. 33 |
| pWQ3 | K. pneumoniae rfbkp01 cloned in pRK404 | Ref. 48 |
| pBAD-B2 | E. coli msbB2 cloned in pBAD24 [pMsbB2] | Ref. 19 |
| pEP24 | pagPO157 in pBAD24 | This study |
Abbreviated descriptions used in some figures are included in square brackets.
DNA Manipulations
Restriction enzyme digestions, ligations, transformations, and DNA electrophoresis were performed as described (35). The oligonucleotide primers used for DNA sequencing and PCR gene amplification were manufactured by Invitrogen. Purification of plasmids, PCR products, and restriction fragments was performed with the QIAprep, QIAquick, and QIAEX II kits, respectively, according to the manufacturer’s instructions (Qiagen). Genomic DNA was purified using the Easy-DNA kit (Invitrogen). DNA sequencing was performed at the ACGT Corp. sequencing facility (Toronto, Canada).
Mutant Constructions
An allelic exchange method was employed for creation of a pagP::aacC1 mutation in the double msbB mutant (4304-DM) of E. coli O157:H7 strain 4304 (Table 1). In brief, the pagP gene was amplified from wild type strain 4304 by PCR with primers CrcA (forward, ATGAGCTCAGGTTGACGATA) and CrcR (reverse, TTGAATTCTTGCTGACGTATC) to yield a 1.3-kb product. The amplicon was cloned into the pGEM-T vector, and the recombinant plasmid (pCrcAT) was digested with KpnI, which cuts a single site near the middle of the pagP gene. For insertion of the nonpolar Gm-cassette, the KpnI fragment containing the aacC1 gene was purified from pUCGM carrying the aacC1 gene in the multiple cloning site. The plasmid resulting from ligation of the aacC1 gene into pCrcAT was used as template DNA for a high fidelity PCR with proofreading DNA polymerase and the primer pair CrcA and CrcR. The 2.1-kb amplicon was purified for blunt end ligation with the pRE107 vector digested with SmaI. The resulting suicide vector construct was named pR7Crc-Gm. The SM10 donor E. coli was transformed with the pR7Crc-Gm plasmid harboring the mutated (pagP::aacC1) allele. Strain SM10 (pR7Crc-Gm) was mated with 4304-DM and incubated at 37 °C overnight on blood agar plates. This mating procedure was subsequently repeated using the wild-type strain 4304 to generate a single pagP: aacC1 mutant. The exconjugants were selected on LB agar plates containing appropriate antibiotics (StrR + GmR). The resulting exconjugants were spread on LB agar plates containing 7% sucrose and Gm and incubated at 30 °C in order to select isolates that had undergone a double crossover. A few selected colonies were purified, and the potential pagP::aacC1 mutants (Gmr and Aps) were tested by PCR for confirmation of the mutated allele. The primer pair of CrcA and CrcR was used for amplification of the pagP::aacC1 allele from the mutants. The expected size (2.1 kb) of the amplicon in the mutants was compared with that of the wild type pagP gene (1.3 kb) by 1% agarose-gel electrophoresis. The resulting pagP::aacC1 mutant of E. coli 4304-DM was named 4304-TM, whereas that of E. coli 4304 was named 4304-PM (Table 1). The msbB::Tn5 allele in the E. coli K-12 donor strain BMS67C12 was transferred by P1 transduction to the E. coli K-12 strain WJ0124 to create strain SK1061 (Table 1). A temperature-sensitive P1 cmr-100 lysate of BMS67C12 (36) was prepared as described elsewhere (18) and then mixed with E. coli WJ0124. The resulting msbB::Tn5 mutant of WJ0124 was verified by PCR.
Analysis of Lipid A by TLC
Analysis of lipid A compositional profiles was done by TLC separation of 32P-labeled lipid A species released from a mild acid hydrolysis procedure, applied to bacteria cultured with or without EDTA treatment (33, 37).
LPS Preparation and SDS-PAGE Analysis
LPS was prepared on a small scale from SDS-proteinase K-treated whole cell lysates (38). Large scale LPS preparations were made by the phenol/chloroform/petroleum ether extraction procedure as described elsewhere (39). The LPS was then separated on a 16% Tricine SDS-polyacrylamide gel (Novex, San Diego, CA) and was visualized by silver staining (40). PAGE conditions were adjusted as recommended by the manufacturer.
NMR Spectroscopy
NMR spectra were recorded at 25 °C in D2O on a Varian UNITY INOVA 500 instrument, using acetone as a reference for proton (2.225 ppm) and carbon (31.5 ppm) spectra. Varian standard programs for COSY, NOESY (mixing time of 400 ms), TOCSY (spin lock time, 120 ms), HSQC, and gHMBC (long range transfer delay, 100 ms) were used.
Isolation of the Core Oligosaccharide
LPS (30 mg) was hydrolyzed with 2% acetic acid (3 h, 100 °C). Lipid was removed by centrifugation, and soluble products were separated by gel chromatography on Sephadex G-50 to yield core oligosaccharide (15 mg) and a low molecular mass fraction. The core was additionally purified by anion exchange chromatography on a Hitrap Q column (Amersham Biosciences) using a gradient of NaCl from 0 to 1 M over 1 h, and the major acidic core fraction was desalted by gel chromatography.
Hydrazine O-Deacylation of the LPS
LPS (20 mg) was dissolved in anhydrous hydrazine (1 ml) and kept at 60 °C for 1 h, cooled, and poured into acetone (50 ml). Precipitate was collected, washed with acetone, dissolved in water, and freeze-dried to give O-deacylated LPS (12 mg).
Monosaccharide Analysis
Hydrolysis was performed with 4 M trifluoroacetic acid (110 °C, 3h), and monosaccharides were conventionally converted into alditol acetates and analyzed by gas chromatography on an Agilent 6850 chromatograph equipped with a DB-17 (30 × 0.25 mm) fused silica column using a temperature gradient from 180 °C (2 min) to 240 °C at 2 °C/min.
Mass Spectrometry
Electrospray ionization/mass spectrometry spectra were obtained using a Micromass Quattro spectrometer in 50% acetonitrile with 0.2% formic acid at a flow rate of 15 μl/min with direct injection. A 5-kV electrospray ionization voltage was used.
Immunoblotting
The LPS samples of 4304, DM (msbB1/msbB2), and DM (pBAD-B2) were resolved by 16% Tricine SDS-PAGE and transferred to a nitrocellulose membrane. The membrane was blocked with 5% skim milk in standard Tris-buffered saline-Tween 20 buffer. Anti-O157 rabbit serum (Difco) and anti-rabbit IgG coupled with horseradish peroxidase (Sigma) were used as primary and secondary antibodies, respectively. For chemiluminescent detection, the ECL detection kit was used according to the manufacturer’s instructions (Amersham Biosciences).
Construction of Plasmids for Phenotypic Complementation
The waaG gene of E. coli O157:H7 was amplified by PCR using 4304-WT genomic DNA and the two primers called WG24-Kpn (forward, GACAGGTACGTCGTTATGGTACCTGCTTTTTG) (where underlined italics identify restriction sites) and WG24-H3 (reverse, CTTTACCGCGCCAAAGCTTGGCAAACGGCTC) and then cloned into an arabinose-inducible pBAD24 expression vector digested with KpnI and HindIII. The insertion was verified by agarose gel electrophoresis, and the construct was named pWG24. Also, the galU gene of E. coli O157:H7 was amplified by PCR using 4304-WT genomic DNA and the two primers called GalU-H3 (forward, TGCATTACAAGCTTATGTCGGCTGG) and GalU-Sal (reverse, GTCGATTGGTCGACGCCGTTTCGTG). The 1.1-kb amplicon, including the endogenous promoter region, was digested with HindIII and SalI and inserted into pACYC184 digested with the same restriction enzymes, and the resulting plasmid was named pGU184. The pagP gene of E. coli O157:H7 was amplified by a high fidelity PCR using genomic DNA and the two primers called Pag24-Kpn (forward, TGGTCACQAAATGGTACCGAGTAAATATGTCG) and Pag24-H3 (reverse, GAAGTTACTAAAGCTTCATTTGTCTCAA). The ~600-bp amplicon was digested with KpnI and HindIII and cloned into an arabinose-inducible pBAD24 expression vector digested with KpnI and HindIII. The insertion was verified by agarose gel electrophoresis, and the construct was named pEP24. LPS samples were prepared from the bacterial transformants carrying pEP24 cultured on LB plates containing 0.2% arabinose for the induction of expression of the cloned pagPO157 gene. Plasmid pACPagPSer77Ala was prepared by site-directed mutagenesis, as described previously (31), except using pACPagP as a template. Plasmid pAA101 containing the galETK genes was obtained and used for complementation of 4304-DM (41). Exogenous Gal (final concentration 0.5%) was added into LB agar plates and used for culturing derivatives of 4304-DM.
Serum Resistance Assay
A serum resistance assay was conducted by a method described elsewhere (42) with minor modifications. In brief, aliquots of 90 μl of nonimmune calf serum were prepared and kept on ice before use. A 10-μl suspension of bacteria in phosphate-buffered saline (pH 7.4) containing ~1 × 105 colony-forming units/ml was added to the 90 μl of normal serum and to heat-inactivated serum (56 °C for 30 min). A 10-μl volume of 0 h sample was immediately taken for 10-fold serial dilution in phosphate-buffered saline, and each dilution was plated onto trypticase-soy agar. The remaining mixture was incubated at 37 °C for 1 h. After a 1-h incubation, a series of 10-fold dilutions in phosphate-buffered saline was made from both sets of mixtures (normal and heat-inactivated serum). Each dilution was plated onto trypticase-soy agar and incubated overnight at 37 °C. Viable counts were calculated as mean values of at least two independent experiments with the same nonimmune calf serum. Viability (percentage) was normalized in proportion to the value (100%) obtained from the 0 h counts of each strain.
Antibiotic Sensitivity Assays
For the determination of minimal inhibitory concentrations for vancomycin and novobiocin, the microdilution method was exploited as recommended by the National Committee for Clinical Laboratory Standards (43). In brief, 2-fold serial dilutions of the antibiotic were made in Mueller-Hinton broth (pH 7.2) in 96-well microtiter plates. Then bacteria at a final concentration of 5 × 105 colony-forming units/ml in a volume equal to that of the antibiotic-containing Mueller-Hinton broth were added to each well. The minimal inhibitory concentrations were recorded as the lowest concentration of the antibiotic that did not allow visible bacterial growth after 20 h of incubation at 37 °C.
Membrane Extraction of PagP
Extraction of PagP from membranes was performed using lauroyldimethylamine-N-oxide (LDAO) as described previously (23), and specific activity was determined by a standard TLC assay (24).
RESULTS
MsbB Deficiency Triggers PagP Activity in the OM of E. coli O157:H7
We have employed a TLC-based mild acid hydrolysis procedure (33, 37) to analyze 32P-labeled lipid A from both E. coli K-12 and O157:H7 wild-type strains together with their myristate and/or palmitate-deficient lipid A mutants (Fig. 2). The procedure disrupts the labile ketosidic bond and liberates the first Kdo sugar from lipid A without affecting the distribution of acyl chains. One artifact of the procedure is the partial dephosphorylation of the anomeric lipid A carbon at position 1. Roughly two-thirds of lipid A in E. coli K-12 contains a mono-phosphate group, whereas the remaining third contains a diphosphate group at this position. The small amount of 1-de-phosphorylated lipid A migrates the farthest among the three species on the TLC plate (Fig. 2). E. coli O157:H7 shows a similar profile, except that an additional species containing PEtN at position 1 is also apparent (18). Each of these species represents a hexa-acylated lipid A that exhibits a typical 4 + 2 acyl chain distribution, with two acyloxylacyl groups on the distal GlcN unit and two unmodified primary R-3-hydroxymyristate chains on the proximal GlcN unit.
FIGURE 2. TLC analysis of 32P lipid A profiles after mild acid hydrolysis.

Lipid A was labeled with 32Pi and isolated from cells by mild acid hydrolysis. Wild-type E. coli O157:H7 (4304) and K-12 (MC1061) were analyzed in comparison with their msbB and/or pagP mutant derivatives. Cultures were grown for 150 min and adjusted with or without 25 mM EDTA for an additional 5 min. The isolated lipid A species were separated by TLC and visualized with a PhosphorImager. The main species of lipid A that were identified previously by mass spectrometry are indicated to the left and include the 4′-monophosphate (1-OH), the 1,4′-bis-phosphate (1-OP), the 1-diphosphate (1-O-PP), the 1-diphosphoryl-EtN (1-O-PPEtN), and the 4′-L-Ara4N lipid A species. The penta-, hexa-, and hepta-acylated derivatives of each lipid A species are also indicated to the left (A). Schematic representations (B) reveal the relative migration of certain lipid A molecular subtypes in the TLC plate. The fastest migrating species (hepta-acyl 1-OH) has the greatest number of acyl chains and the least number of polar groups. The acylation patterns for the 1-OH lipid A species are shown as an example, which reveals that the penta-acylated derivative migrates most slowly. Of the two faster migrating hexa-acylated species with 4 + 2 and 3 + 3 acyl chain distributions, the 4 + 2 species migrates perceptibly faster, and this difference becomes accentuated as more polar groups are added. The effect of adding various polar substituents is shown schematically only for the penta-acylated lipid A species, but relative migrations for all combinations of acyl chain distributions and polar substituents can be deduced by extrapolation from these selected examples. Relative migrations are not drawn exactly to scale, and palmitate is shown in red.
Despite the presence of PagP in the OM, the enzyme remains dormant when OM lipid asymmetry is maintained (33). EDTA can promote the migration of phospholipids into the OM external leaflet, which sensitizes cells to hydrophobic antibiotics, by chelating Mg2+ ions that normally neutralize negative charges and promote tight LPS-LPS packing interactions (4–6). PagP activity triggered by EDTA occurs nearly instantaneously, is dependent on lipid trafficking, and is independent of de novo protein synthesis (33). Less than 5% of the lipid A 1-phosphate contains palmitate under normal growth conditions, but ~20% palmitoylation is achieved after a brief EDTA treatment. The additional hepta-acylated lipid A species that are apparent after EDTA treatment are clearly absent in the pagP mutant derivatives of the wild-type strains (Fig. 2).
MsbB deficiency generates a penta-acylated lipid A characterized by the absence of myristate in E. coli K-12 (37). EDTA treatment of the K-12 msbB mutant promotes palmitoylation to a similar extent as in the wild-type strain, but it generates a new hexa-acylated lipid A species characterized by an atypical 3 + 3 acyl chain distribution. In this case, a single acyloxyacyl group and a single unmodified primary R-3-hydroxymyristate chain are found on each GlcN unit. The two types of hexa-acylated lipid A migrate at similar positions, but the 4 + 2 acyl chain distribution migrates perceptibly faster on the TLC plate (Fig. 2). The positions of the palmitoylated lipid A species in the msbB-deficient strains are made apparent by their absence in the mutant derivatives that also lack pagP. In addition to the PEtN-modified species, two new slowly migrating penta-acylated species are apparent in the msbB- and pagP-deficient E. coli O157:H7 mutant, which are not present in the corresponding E. coli K-12 mutant. The species migrating just above the 1-diphosphate coincides with the 4′ L-Ara4N derivative, and the most polar substituent probably coincides with two species doubly modified by L-Ara4N at position 4′ and either diphosphate or PEtN at position 1 (37, 44). Lipid A myristoylation is a requirement for L-Ara4N addition in E. coli K-12 (45), but this does not appear to be true in E. coli O157:H7. Interestingly, lipid A palmitoylation occurred constitutively in the msbB-deficient E. coli O157:H7 mutant (Fig. 2). This observation suggests that msbB-deficiency in E. coli O157:H7 serves to increase lipid A palmitoylation in the absence of EDTA by introducing its own perturbation of lipid asymmetry, which directly triggers the activity of preexisting PagP in the OM.
MsbB Deficiency Is Associated with an OM Permeability Defect in E. coli O157:H7
Structural considerations of PagP in relation to OM bilayer organization predict that phospholipids must migrate into the external leaflet during PagP catalysis (22). We find that the msbB deficiency in E. coli O157:H7, which is associated with PagP activation, is also associated with increased sensitivity to vancomycin and the hydrophobic antibiotic novobiocin. Mutation of pagP in this msbB-deficient background aggravates the antibiotic sensitivity (Table 2), which suggests that lipid A palmitoylation can partially compensate for the absence of myristate in lipid A. Previous studies of msbB mutants have revealed increased sensitivity to antibiotics in Salmonella but not in E. coli K-12 (46, 47), and our observations confirm these latter findings (Table 2). We also observed slow growth and poor growth of msbB- and msbB/pagP-deficient E. coli O157:H7, respectively, on lactose MacConkey agar plates, which contain bile salts that select against organisms with a compromised OM permeability barrier (not shown). The association of PagP activation with hydrophobic antibiotic and bile salt sensitivity is consistent with earlier predictions that OM permeability to hydrophobic compounds can result from the migration of phospholipids into the OM external leaflet (5, 6).
TABLE 2.
Minimal inhibitory concentrations of antibiotics in E. coli lipid A acylation mutants
| Strains | Minimal inhibitory concentrationa
|
|
|---|---|---|
| Vancomycin | Novobiocin | |
|
| ||
| μg/ml | ||
| Wild-type O157:H7 4304 | >100 | >100 |
| pagP::aacC1 | >100 | 100 |
| ΔmsbB1, ΔmsbB2 | 50 | 50 |
| ΔmsbB1, ΔmsbB2, pagP::aacC1 | 25 | 3.125 |
| Wild-type K-12 MC1061 | >100 | >100 |
| pagP::amp | >100 | >100 |
| msbB::Tn5 | >100 | >100 |
| msbB::Tn5, pagP::amp | >100 | 100 |
Minimal inhibitory concentration was determined by a microdilution procedure using Mueller-Hinton broth as described under “Experimental Procedures.” The results are mean values of at least two independent experiments.
MsbB Deficiency Truncates the R3 Core Oligosaccharide of E. coli O157:H7
In order to evaluate the status of LPS glycosylation in the wild-type and msbB-deficient E. coli O157:H7 bacteria, we extracted LPS from cells and visualized it by silver staining after Tricine SDS-PAGE (Fig. 3). The results identify a complete lipid A-core that includes the attached O-antigen repeats characteristic of smooth E. coli O157:H7 LPS. Unexpectedly, the msbB-deficient mutant completely lacks O-antigen repeats and appears to possess a truncated lipid A-core, which can be restored by complementation with an msbB2 expression plasmid. A minor species migrating above the truncated lipid A-core might suggest the presence of a single O-antigen unit, but this was ruled out after immunoblotting with O157 antiserum (Fig. 3C). We verified that the LPS core in our E. coli K-12 msbB mutant was intact by observing that it could be modified with a Klebsiella pneumoniae O-antigen polysaccharide (48), which can be attached to the outer core when the required genes are expressed in E. coli K-12 (data not shown).
FIGURE 3. Tricine SDS-PAGE analysis of E. coli O157:H7 LPS.
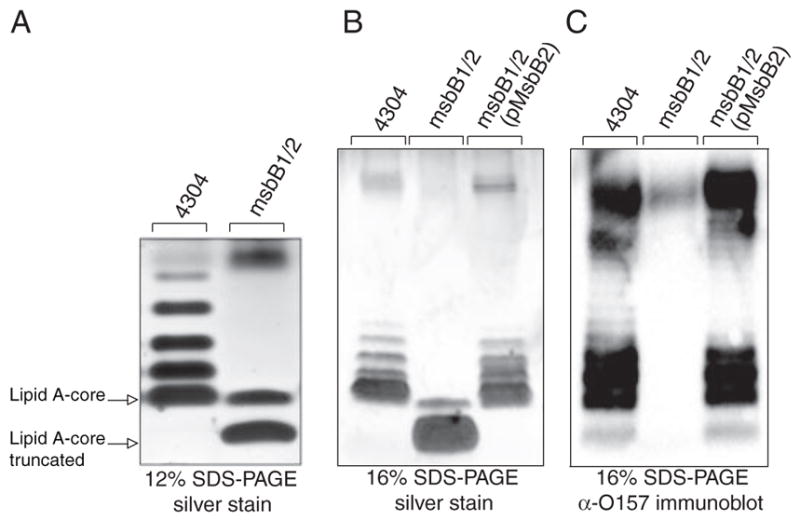
LPS was isolated from wild-type E. coli O157:H7, its msbB-deficient mutant, and the same mutant complemented with an MsbB2 expression plasmid. The LPS was resolved by Tricine SDS-PAGE and visualized by silver staining. Different resolution of the O-antigen ladder pattern was observed in 12% gels (A) and 16% gels (B). The position of O-antigen units was identified by immunoblotting with O157-specific antisera (C).
To further characterize the nature of the R3 core truncation, we purified it for biochemical analysis. Monosaccharide analysis of the whole LPS, by gas chromatography of alditol acetates, showed the presence of Hep and GlcN in an approximate 1:1 ratio. The core oligosaccharide appeared to contain only Hep and was further analyzed by NMR spectroscopy. A set of two-dimensional NMR spectra (COSY, TOCSY, NOESY, HSQC, and HMBC) was recorded, and all major proton and carbon signals were assigned. Spectra contained signals of two Hep residues and a Kdo as well as of PPEtN. Kdo was present in several forms with dominating α-pyranoside; Hep residue E also showed several sets of signals due to the attachment to Kdo variants. The sequence of the monosaccharides was determined using nuclear Overhauser effect and HMBC data, which contained correlations (H-H and H-C) F1-E3 and E1-C5. The 31P spectrum of the core contained two signals at −9 and −9.5 ppm, correlating with E4 and EtN-1 protons. This agrees with the attachment of PPEtN to O-4 of Hep E, which also led to the low field shift of H-4 and C-4 of Hep E to 4.58 and 72.6 ppm, respectively (compared with nonphosphorylated H-4 and C-4 of Hep F at 3.88 and 67.1 ppm) (Fig. 4). The electrospray ionization mass spectrum of the core contained peaks at m/z 824.3 (main species, full molecule with PEtN; calculated 824.5 Da), 806.3 (its anhydro-Kdo derivative; calculated 806.5 Da), 701.3 (same molecule without PEtN; calculated 701.5 Da), and 683.3 (its anhydro-Kdo derivative; calculated 683.4 Da). Thus, the core has the following structure.
FIGURE 4. NMR chemical shift assignments of truncated E. coli O157:H7 LPS.

Assignment of the HSQC correlation spectrum for the core oligosaccharide. Cross-peaks marked in green refer to CH groups, whereas those marked in red refer to CH2 groups (A). 1H and 13C NMR chemical shift values (δ, ppm) are shown for the core oligosaccharide (B).
In order to confirm the structure of the LPS, electrospray ionization mass spectrometry of O-deacylated LPS was recorded. It contained doubly and triply charged peaks of the expected full structure, (Hep)2(Kdo)2(GlcN)2(P)4(EtN)1(C14OH)2 (1980.5 Da), the structure without PEtN (1857.5 Da), and the structure without PPEtN (1777.5 Da). All peaks were accompanied by sodium adducts (+22). Thus, the chemical data show that the LPS had the following carbohydrate backbone.
These observations are consistent with a core truncated at the level of the first outer core Glc and with only a partial PPEtN modification at HepI (Fig. 1).
Both the R3 and K-12 core structures include three Hep units attached to KdoI (14). Partial modification of KdoII with PEtN is also observed when E. coli is cultured under calcium-enriched conditions (49, 50). The R3 and K-12 core structures share in common the first outer core Glc, which is attached by an α-1,3-linkage with HepII (Fig. 1). This first Glc unit is required for complete phosphorylation of HepI (51), which can be followed by partial modification with PEtN. HepI phosphorylation is a prerequisite for attachment of HepIII, which is itself a prerequisite for phosphorylation of HepII (52). Although HepII phosphorylation is nearly stoichiometric in E. coli K-12, its presence is only partial and mutually exclusive with the attachment of GlcNAc by WabB at HepIII in E. coli O157:H7 (Fig. 1) (16). Consequently, biosynthetic deficiencies in the first Glc will not only manifest the absence of the outer core and O-polysaccharide but will also necessarily exclude the inner core Glc-NAc, HepIII, and Hep phosphate groups. These findings demonstrate that our observed core truncation most likely represents a deficiency in the cytoplasmic incorporation of the first outer core Glc rather than the action of a series of previously unreported extracellular hydrolytic enzymes.
Mutation of pagP Restores R3 Core Structure in msbB-deficient E. coli O157:H7
We found that pagP mutations suppress the truncated core phenotype and restore smooth LPS to the msbB-deficient E. coli O157:H7 mutant (Fig. 5). A low copy recombinant pACPagP plasmid (33) introduced into the myristate- and palmitate-deficient lipid A mutant restores the truncated LPS observed in the absence of the pagP mutation. If the pagP mutation can restore smooth LPS to the msbB-deficient E. coli O157: H7, we reasoned that O-antigen-mediated resistance to serum should also be restored to wild-type levels as a consequence. We were able to demonstrate that only the msbB-deficient mutant, and not the wild-type E. coli O157:H7 or its mutant deficient in both msbB and pagP, displayed sensitivity to killing by serum that is characteristic of O-antigen-deficient E. coli K-12 (Table 3). These findings clearly dissociate hydrophobic antibiotic sensitivity (a consequence of lipid A underacylation) from serum sensitivity (a consequence of R3 core truncation).
FIGURE 5. Complementation of the truncated R3 core oligosaccharide of msbB-deficient E. coli O157:H7.

Silver-stained 16% Tricine SDS-PAGE analysis of LPS isolated from wild-type E. coli O157:H7 strain 4304 and its lipid A acylation mutants deficient in either msbB or both msbB and pagP. Bacteria transformed with pACPagP, pACPagPSer77Ala, and pACPagPΔ5–14 are indicated (A). The presence (+) or absence (−) of the PagP periplasmic amphipathic α-helix and of lipid A acylated by palmitate or myristate, as deduced from Figs. 2 and 6, is indicated (B). Shown is a structural model of PagP derived from the crystal structure (Protein Data Bank code 1THQ). The first seven N-terminal residues are disordered in the crystal structure, but the approximate position of the Δ5–14 deletion in the periplasmic amphipathic α-helix (red) and the cell surface catalytic residues are indicated. The bound LDAO detergent molecule (yellow) identifies the hydrocarbon ruler. The aromatic belts define the boundaries of the OM, where LPS occupies the external leaflet and phospholipids (PL) occupy the periplasmic leaflet (C).
TABLE 3.
Serum resistance associated with O157 polysaccharide
| Strains | Percentage of viable E. colia
|
||
|---|---|---|---|
| Time 0 | Time 1 h | Time 1 h (heat-inactivated) | |
|
| |||
| % | % | % | |
| Wild-type O157:H7 4304 | 100 | 96.6 | 97 |
| ΔmsbB1, ΔmsbB2 | 100 | 30 | 88.3 |
| ΔmsbB1, ΔmsbB2, pagP::aacC1 | 100 | 90.6 | 98.3 |
| Wild-type K-12 MC1061 | 100 | 27.4 | 76 |
The numbers of E. coli were calculated as mean values of at least two independent experiments with the same nonimmune calf serum as described under “Experimental Procedures,” and percentages were determined in relation to the time 0 viable counts.
Signal Transduction Is Mediated by the PagP Amphipathic α-Helix
Since overproduction of PagP could not produce an R3 core truncation in the wild-type strain (data not shown), we realized that R3 core truncation in E. coli O157:H7 is a consequence of an epistatic interaction between msbB and pagP. We were concerned that the production of an atypical lipid A structure with the 3 + 3 acyl chain distribution, which contains palmitate and lacks myristate, might somehow interfere with OM biogenesis and inadvertently trigger the periplasmic stress response. In E. coli K-12 subjected to a temperature shift from 30 to 42 °C, PagP-catalyzed lipid A palmitoylation has been implicated in the activation of the extracytoplasmic function or ECF sigma factor σE (53). However, we could show that a catalytically inactive pagPSer77Ala allele restores the R3 core truncation just as effectively as the wild-type pagP (Fig. 5), but only the latter is capable of palmitoylating lipid A in vivo (Fig. 6). Therefore, the observed R3 core truncation is not simply an artifact of producing an atypical lipid A molecular subtype.
FIGURE 6. TLC analysis of 32P lipid A profiles after mild acid hydrolysis.

Lipid A was labeled with 32Pi and isolated from cells by mild acid hydrolysis as described in the legend to Fig. 2. In this figure, hexa refers to the 3 + 3 acyl chain distribution.
PagP is an eight-stranded antiparallel β-barrel that is preceded by an amino-terminal amphipathic α-helix (30, 31). The active site is located at the cell surface, but the amphipathic α-helix lies along the periplasmic surface of the OM (Fig. 5C). We have previously created internal deletions that remove sections of the amphipathic α-helix but still allow the signal peptide to direct the export and assembly of PagP in the OM. We reported that OM lipid A palmitoylation, catalyzed by the amphipathic α-helix deletion constructs, could be detected at levels that were indistinguishable from a similar wild-type PagP construct (33). We now demonstrate that a pagPΔ5–14 allele, which removes most of the amphipathic α-helix, is fully active in the palmitoylation of lipid A under normal conditions (Fig. 6), but it is no longer capable of restoring the R3 core truncation (Fig. 5). Interestingly, the R3 core truncation correlates only with the presence of the PagP amphipathic α-helix and not with differences in the pattern of lipid A acylation (Fig. 5B). Since we have already established that OM permeability depends on the lipid A acylation pattern (Table 2), the R3 core truncation is not likely to be a secondary consequence of the influence of PagP on the barrier function of the OM. These findings suggest that PagP activation in the OM, initiated by the E. coli O157:H7 msbB deficiency, not only triggers lipid A palmitoylation at the cell surface but also separately triggers R3 core oligosaccharide truncation in the cytoplasm through the action of the PagP amphipathic α-helix.
The PagP Amphipathic α-Helix Is a Postassembly Membrane Clamp in Vivo
Huysmans et al. (54) have recently reported studies of PagP folding in liposomes, which verify our earlier conclusions that the PagP amphipathic α-helix is not essential for membrane assembly (33). Interestingly, the liposome folding studies also indicate that the PagP amphipathic α-helix functions as a postassembly clamp to stabilize PagP in membranes after folding is complete (54). To determine whether the pagPΔ5–14 allele encodes a protein that is more easily extracted from membranes in vivo, we took advantage of our previous findings that wild-type PagP can be solubilized from membranes by serial extractions with increasing amounts of the detergent LDAO (23). As predicted by the liposome studies, PagPΔ5–14 is much more sensitive to detergent extraction than is wild-type PagP (Fig. 7). The critical micellar concentration for LDAO is ~0.025%, and at the lowest concentration of LDAO tested (twice the critical micellar concentration), we observed nearly complete extraction of PagPΔ5–14 from membranes, whereas the wild-type PagP was only completely extracted at 20 times the critical micellar concentration (Fig. 7). Although the bulk of the wild-type PagP-specific activity was found in the soluble fraction, as previously reported (23), PagPΔ5–14 was not appreciably active in the soluble fraction, which suggests that the PagP amphipathic α-helix can have an important role in stabilizing PagP against detergent inactivation.
FIGURE 7. LDAO extraction of PagP specific activity from membranes.
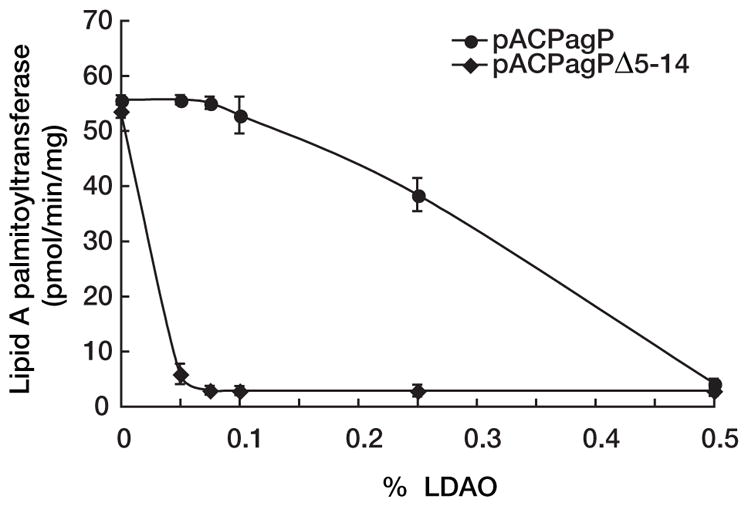
E. coli WJ0124 transformed with pACPagP or pACPagPΔ5–14 was serially extracted from membranes using increasing amounts of LDAO, and the specific activity remaining in the membranes was determined.
Furthermore, we have previously observed that increasing PagP expression by transformation with the pACPagP plasmid can increase the level of lipid A 1-phosphate palmitoylation to roughly 20%, but subsequent EDTA treatment results in hyperpalmitoylation up to 90% (33). Indeed, hyperpalmitoylation was borne out here during EDTA treatment of the pACPagP transformant shown in Fig. 6, but this was not the case for the pACPagPΔ5–14 transformant, which appears to be insensitive to EDTA activation in the msbB-deficient background. Importantly, under conditions where the R3 core truncation is induced (without detergent extraction of membranes and without EDTA treatment of cells), PagPΔ5–14 palmitoylates lipid A to a degree that cannot be distinguished from wild-type PagP (Figs. 6 and 7). Since lipid A palmitoylation dictates that the PagPΔ5–14 enzyme is properly assembled in the OM, cytoplasmic R3 core truncation is probably controlled by the PagP periplasmic amphipathic α-helix through a cell envelope sensory transduction mechanism.
UDP-Glc Depletion Is Linked to R3 Core Truncation in msbB-deficient E. coli O157:H7
We reasoned that the core truncation observed in msbB-deficient E. coli O157:H7 could be caused by a deficiency in the expression of the WaaG glucosyl-transferase responsible for the addition of the first outer core Glc residue (51). However, expression of WaaG from a recombinant plasmid did not correct the core truncation (data not shown). We then investigated the possibility that the WaaG donor substrate UDP-Glc might be limiting. Indeed, we found that expression from a recombinant GalU plasmid of UDP-Glc pyrophosphorylase, which replenishes the cytoplasmic pool of UDP-Glc (55, 56), could largely restore smooth LPS to the msbB-deficient E. coli O157:H7 mutant (Fig. 8). Similarly, exogenous Gal, which can be converted to UDP-Glc by the Leloir pathway (57), could also restore smooth LPS (Fig. 8). The Leloir pathway enzymes cloned on a recombinant GalETK plasmid did not appear to be limiting (data not shown), but this probably reflects the reversible nature of these reactions, which simultaneously produce and consume UDP-Glc. These observations support the hypothesis that the msbB-deficient E. coli O157:H7 mutant is limiting in UDP-Glc, which probably blocks WaaG and all reactions that depend on WaaG, ultimately leading to the observed R3 core truncation.
FIGURE 8. Complementation of the truncated R3 core oligosaccharide of msbB-deficient E. coli O157:H7.
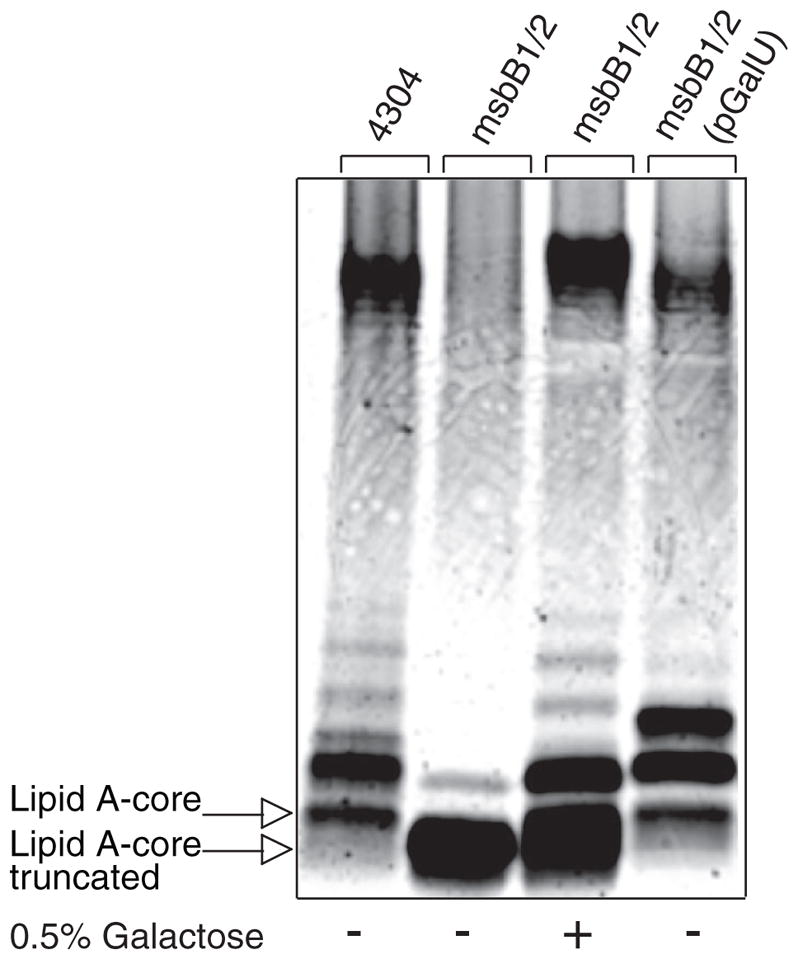
Silver-stained 16% Tricine SDS-PAGE analysis of LPS isolated from wild-type E. coli O157:H7 and its lipid A acylation mutant deficient in msbB. Bacteria were cultured in the presence and absence of exogenous Gal and with or without complementation by a plasmid encoding GalU.
DISCUSSION
Taken together, our observations demonstrate that PagP activation depends on msbB deficiency and that R3 core truncation depends on activated PagP. Signal transduction might connect PagP in the OM with cytoplasmic R3 core metabolism. In principle, regulation of the cytoplasmic pool of UDP-Glc could be exerted at the level of gene expression, through covalent enzyme modification, or through allosteric control of enzyme activity (56, 58). The notion that an OM protein can control gene transcription is not entirely unprecedented. The OM ferric citrate receptor FecA is known to control transcription of key components of the iron uptake machinery in response to ligand binding events that occur in the OM (59). At this stage, we can only speculate on how PagP influences the cytoplasmic pool of UDP-Glc or on the nature of the intrinsic differences between E. coli K-12 and O157:H7 cell envelopes that led us to our conclusions by necessarily studying the enterohemorrhagic bacterium.
Natural conditions that trigger PagP in wild-type cells, including E. coli K-12 and other species that encode PagP homologues, might include Mg2+ limitation, exposure to cationic amphipathic peptides, or any other condition known to perturb OM lipid asymmetry. The narrow distribution of PagP among mostly pathogenic organisms could indicate that the biological trigger is somehow associated with host-pathogen interactions (22). Conceivably, an R3 core truncation might be of a selective advantage if it facilitates exchange of host and pathogen factors and is appropriately triggered by prior contact made between host epithelial and E. coli O157:H7 cell surfaces. In bacillary dysentery, shortening the length of Shigella cell surface LPS by modifying the O-antigen structure is believed to facilitate the association of a key type-III secretion system with human epithelial cells (60).
Given that the R3 core truncation has not been observed previously in E. coli O157:H7 pathogenesis, we must concede that its biological significance remains unresolved. The possibility remains that constitutive activation of PagP in the msbB-deficient background excessively diverts the pool of UDP-Glc toward other cell surface components, such as L-Ara4N and colanic acid (58) or trehalose and membrane-derived oligosaccharides (56), and thereby truncates the R3 core as an unintended consequence. Regardless of its biological significance, we emphasize here that the R3 core truncation provides a useful phenotypic probe, which has revealed the apical component of what appears to be a previously unrecognized signal transduction pathway in pagP-encoding Gram-negative bacteria.
In the present study, we suspect that lipid A lacking myristate is initially glycosylated normally and transported to the OM, where it perturbs lipid asymmetry specifically in E. coli O157: H7. By activating PagP in the OM, the msbB deficiency initiates signal transduction to exert negative control on key cytoplasmic enzymes of R3 core biosynthesis, which only then leads to R3 core truncation (Fig. 9). The constitutive state of PagP activation serves to deplete the cell of all LPS bearing the attached O-antigen. Only this model has the power of predicting that a pagP mutation would restore smooth LPS and serum resistance to the msbB-deficient E. coli O157:H7 while simultaneously aggravating a defect in OM lipid asymmetry associated with lipid A underacylation and hydrophobic antibiotic sensitivity. We conclude that OM activation of PagP not only triggers lipid A palmitoylation, to partially compensate for lipid A underacylation in the E. coli O157:H7 msbB mutant, but also separately triggers signal transduction across three distinct cellular compartments through the action of the periplasmic amphipathic α-helix. Discovering the downstream signaling components that respond to the activated state of PagP will provide fertile ground for future research.
FIGURE 9. Model for PagP-mediated control of R3 core oligosaccharide structure.

PagP is the only known OM enzyme of LPS biosynthesis in E. coli, and it serves to transfer a palmitate chain from a phospholipid to lipid A with the production of a lysophospholipid by-product. Lipid A palmitoylation attenuates endotoxin signaling through the host TLR4 pathway and affords resistance to cationic antimicrobial peptides. PagP measures the phospholipid palmitate chain in its hydrocarbon ruler, which is only accessible from the OM external leaflet. A defect in cytoplasmic lipid A myristoylation triggers PagP activity in the OM of E. coli O157:H7. Activated PagP not only palmitoylates lipid A at the cell surface, to partially compensate for lipid A underacylation, but also separately initiates signal transduction through its periplasmic amphipathic α-helix. Signal transduction exerts negative control on the cytoplasmic UDP-Glc pool and leads to a truncation of the R3 core oligosaccharide.
SCHEME 1.

SCHEME 2.

Acknowledgments
We thank Janet Liao (University of Guelph) for excellent technical support. We gratefully acknowledge Miguel Valvano and Cristina Marolda for advice on LPS analysis and Chris Whitfield, Janet Wood, Jun Yu, and Richard Darveau for providing bacterial strains, plasmids, and phage.
Footnotes
The abbreviations used are: OM, outer membrane; L-Ara4N, 4-amino-4-deoxy-L-arabinose; Ap, ampicillin; Gm, gentamycin; Hep, L-glycero-D-manno-heptose; Kdo, 3-deoxy-D-manno-oct-2-ulosonic acid; LDAO, lauroyldimethylamine-N-oxide; LPS, lipopolysaccharide; PEtN, phosphoethanolamine; Str, streptomycin; Tricine, N-[2-hydroxy-1,1-bis(hydroxy-methyl)ethyl]glycine.
References
- 1.Kamio Y, Nikaido H. Biochemistry. 1976;15:2561–2570. doi: 10.1021/bi00657a012. [DOI] [PubMed] [Google Scholar]
- 2.Nikaido H. Microbiol Mol Biol Rev. 2003;67:593–656. doi: 10.1128/MMBR.67.4.593-656.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schindler M, Osborn MJ. Biochemistry. 1979;18:4425–4430. doi: 10.1021/bi00587a024. [DOI] [PubMed] [Google Scholar]
- 4.Leive L. Ann N Y Acad Sci. 1974;235:109–129. doi: 10.1111/j.1749-6632.1974.tb43261.x. [DOI] [PubMed] [Google Scholar]
- 5.Nikaido H, Nakae T. Adv Microb Physiol. 1979;20:163–250. doi: 10.1016/s0065-2911(08)60208-8. [DOI] [PubMed] [Google Scholar]
- 6.Nikaido H, Vaara M. Microbiol Rev. 1985;49:1–32. doi: 10.1128/mr.49.1.1-32.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raetz CR, Whitfield C. Annu Rev Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Joiner KA. Curr Top Microbiol Immunol. 1985;121:99–133. doi: 10.1007/978-3-642-45604-6_6. [DOI] [PubMed] [Google Scholar]
- 9.Bishop RE. Contrib Microbiol. 2005;12:1–27. doi: 10.1159/000081687. [DOI] [PubMed] [Google Scholar]
- 10.Clementz T, Zhou Z, Raetz CR. J Biol Chem. 1997;272:10353–10360. doi: 10.1074/jbc.272.16.10353. [DOI] [PubMed] [Google Scholar]
- 11.Raetz CR, Reynolds CM, Trent MS, Bishop RE. Annu Rev Biochem. 2007;76:295–329. doi: 10.1146/annurev.biochem.76.010307.145803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Groisman EA. J Bacteriol. 2001;183:1835–1842. doi: 10.1128/JB.183.6.1835-1842.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bader MW, Sanowar S, Daley ME, Schneider AR, Cho U, Xu W, Klevit RE, Le Moual H, Miller SI. Cell. 2005;122:461–472. doi: 10.1016/j.cell.2005.05.030. [DOI] [PubMed] [Google Scholar]
- 14.Heinrichs DE, Yethon JA, Whitfield C. Mol Microbiol. 1998;30:221–232. doi: 10.1046/j.1365-2958.1998.01063.x. [DOI] [PubMed] [Google Scholar]
- 15.Tarr PI, Gordon CA, Chandler WL. Lancet. 2005;365:1073–1086. doi: 10.1016/S0140-6736(05)71144-2. [DOI] [PubMed] [Google Scholar]
- 16.Kaniuk NA, Vinogradov E, Li J, Monteiro MA, Whitfield C. J Biol Chem. 2004;279:31237–31250. doi: 10.1074/jbc.M401879200. [DOI] [PubMed] [Google Scholar]
- 17.Gibbons HS, Kalb SR, Cotter RJ, Raetz CR. Mol Microbiol. 2005;55:425–440. doi: 10.1111/j.1365-2958.2004.04409.x. [DOI] [PubMed] [Google Scholar]
- 18.Kim SH, Jia W, Parreira VR, Bishop RE, Gyles CL. Microbiology. 2006;152:657–666. doi: 10.1099/mic.0.28692-0. [DOI] [PubMed] [Google Scholar]
- 19.Kim SH, Jia W, Bishop RE, Gyles C. Infect Immun. 2004;72:1174–1180. doi: 10.1128/IAI.72.2.1174-1180.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.D’Hauteville H, Khan S, Maskell DJ, Kussak A, Weintraub A, Mathison J, Ulevitch RJ, Wuscher N, Parsot C, Sansonetti PJ. J Immunol. 2002;168:5240–5251. doi: 10.4049/jimmunol.168.10.5240. [DOI] [PubMed] [Google Scholar]
- 21.Yoon JW, Lim JY, Park YH, Hovde CJ. Infect Immun. 2005;73:2367–2378. doi: 10.1128/IAI.73.4.2367-2378.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bishop RE. Mol Microbiol. 2005;57:900–912. doi: 10.1111/j.1365-2958.2005.04711.x. [DOI] [PubMed] [Google Scholar]
- 23.Bishop RE, Gibbons HS, Guina T, Trent MS, Miller SI, Raetz CR. EMBO J. 2000;19:5071–5080. doi: 10.1093/emboj/cdd507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khan MA, Neale C, Michaux C, Pomes R, Prive GG, Woody RW, Bishop RE. Biochemistry. 2007;46:4565–4579. doi: 10.1021/bi602526k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guo L, Lim KB, Poduje CM, Daniel M, Gunn JS, Hackett M, Miller SI. Cell. 1998;95:189–198. doi: 10.1016/s0092-8674(00)81750-x. [DOI] [PubMed] [Google Scholar]
- 26.Robey M, O’Connell W, Cianciotto NP. Infect Immun. 2001;69:4276–4286. doi: 10.1128/IAI.69.7.4276-4286.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pilione MR, Pishko EJ, Preston A, Maskell DJ, Harvill ET. Infect Immun. 2004;72:2837–2842. doi: 10.1128/IAI.72.5.2837-2842.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kawasaki K, Ernst RK, Miller SI. J Biol Chem. 2004;279:20044–20048. doi: 10.1074/jbc.M401275200. [DOI] [PubMed] [Google Scholar]
- 29.Miller SI, Ernst RK, Bader MW. Nat Rev Microbiol. 2005;3:36–46. doi: 10.1038/nrmicro1068. [DOI] [PubMed] [Google Scholar]
- 30.Ahn VE, Lo EI, Engel CK, Chen L, Hwang PM, Kay LE, Bishop RE, Prive GG. EMBO J. 2004;23:2931–2941. doi: 10.1038/sj.emboj.7600320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hwang PM, Choy WY, Lo EI, Chen L, Forman-Kay JD, Raetz CR, Prive GG, Bishop RE, Kay LE. Proc Natl Acad Sci U S A. 2002;99:13560–13565. doi: 10.1073/pnas.212344499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hwang PM, Bishop RE, Kay LE. Proc Natl Acad Sci U S A. 2004;101:9618–9623. doi: 10.1073/pnas.0402324101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jia W, Zoeiby AE, Petruzziello TN, Jayabalasingham B, Seyedirashti S, Bishop RE. J Biol Chem. 2004;279:44966–44975. doi: 10.1074/jbc.M404963200. [DOI] [PubMed] [Google Scholar]
- 34.Bishop RE. Biochim Biophys Acta. 2007 doi: 10.1016/j.bbamem.2007.07.021. [DOI] [Google Scholar]
- 35.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1989. [Google Scholar]
- 36.Somerville JE, Jr, Cassiano L, Darveau RP. Infect Immun. 1999;67:6583–6590. doi: 10.1128/iai.67.12.6583-6590.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou Z, Lin S, Cotter RJ, Raetz CR. J Biol Chem. 1999;274:18503–18514. doi: 10.1074/jbc.274.26.18503. [DOI] [PubMed] [Google Scholar]
- 38.Hitchcock PJ, Brown TM. J Bacteriol. 1983;154:269–277. doi: 10.1128/jb.154.1.269-277.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Galanos C, Luderitz O, Westphal O. Eur J Biochem. 1969;9:245–249. doi: 10.1111/j.1432-1033.1969.tb00601.x. [DOI] [PubMed] [Google Scholar]
- 40.Tsai CM, Frasch CE. Anal Biochem. 1982;119:115–119. doi: 10.1016/0003-2697(82)90673-x. [DOI] [PubMed] [Google Scholar]
- 41.Edwards-Jones B, Langford PR, Kroll JS, Yu J. Microbiology. 2004;150:1079–1084. doi: 10.1099/mic.0.26840-0. [DOI] [PubMed] [Google Scholar]
- 42.Helmuth R, Stephan R, Bunge C, Hoog B, Steinbeck A, Bulling E. Infect Immun. 1985;48:175–182. doi: 10.1128/iai.48.1.175-182.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.National Committee for Clinical Laboratory Standards. Performance Standards for Antimicrobial Disk and Dilution Susceptibility Tests for Bacteria Isolated from Animals Approved Standard. 2. Vol. 22. Wayne, PA: 2002. pp. M31–A32. [Google Scholar]
- 44.Zhou Z, Ribeiro AA, Raetz CR. J Biol Chem. 2000;275:13542–13551. doi: 10.1074/jbc.275.18.13542. [DOI] [PubMed] [Google Scholar]
- 45.Tran AX, Lester ME, Stead CM, Raetz CR, Maskell DJ, McGrath SC, Cotter RJ, Trent MS. J Biol Chem. 2005;280:28186–28194. doi: 10.1074/jbc.M505020200. [DOI] [PubMed] [Google Scholar]
- 46.Vaara M, Nurminen M. Antimicrob Agents Chemother. 1999;43:1459–1462. doi: 10.1128/aac.43.6.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Murray SR, Bermudes D, de Felipe KS, Low KB. J Bacteriol. 2001;183:5554–5561. doi: 10.1128/JB.183.19.5554-5561.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Clarke BR, Whitfield C. J Bacteriol. 1992;174:4614–4621. doi: 10.1128/jb.174.14.4614-4621.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kanipes MI, Lin S, Cotter RJ, Raetz CR. J Biol Chem. 2001;276:1156–1163. doi: 10.1074/jbc.M009019200. [DOI] [PubMed] [Google Scholar]
- 50.Reynolds CM, Kalb SR, Cotter RJ, Raetz CR. J Biol Chem. 2005;280:21202–21211. doi: 10.1074/jbc.M500964200. [DOI] [PubMed] [Google Scholar]
- 51.Yethon JA, Vinogradov E, Perry MB, Whitfield C. J Bacteriol. 2000;182:5620–5623. doi: 10.1128/jb.182.19.5620-5623.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yethon JA, Heinrichs DE, Monteiro MA, Perry MB, Whitfield C. J Biol Chem. 1998;273:26310–26316. doi: 10.1074/jbc.273.41.26310. [DOI] [PubMed] [Google Scholar]
- 53.Tam C, Missiakas D. Mol Microbiol. 2005;55:1403–1412. doi: 10.1111/j.1365-2958.2005.04497.x. [DOI] [PubMed] [Google Scholar]
- 54.Huysmans GH, Radford SE, Brockwell DJ, Baldwin SA. J Mol Biol. 2007;373:529–540. doi: 10.1016/j.jmb.2007.07.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marolda CL, Valvano MA. Mol Microbiol. 1996;22:827–840. doi: 10.1046/j.1365-2958.1996.01531.x. [DOI] [PubMed] [Google Scholar]
- 56.Bohringer J, Fischer D, Mosler G, Hengge-Aronis R. J Bacteriol. 1995;177:413–422. doi: 10.1128/jb.177.2.413-422.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Holden HM, Rayment I, Thoden JB. J Biol Chem. 2003;278:43885–43888. doi: 10.1074/jbc.R300025200. [DOI] [PubMed] [Google Scholar]
- 58.Grangeasse C, Obadia B, Mijakovic I, Deutscher J, Cozzone AJ, Doublet P. J Biol Chem. 2003;278:39323–39329. doi: 10.1074/jbc.M305134200. [DOI] [PubMed] [Google Scholar]
- 59.Harle C, Kim I, Angerer A, Braun V. EMBO J. 1995;14:1430–1438. doi: 10.1002/j.1460-2075.1995.tb07129.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.West NP, Sansonetti P, Mounier J, Exley RM, Parsot C, Guadagnini S, Prevost MC, Prochnicka-Chalufour A, Delepierre M, Tanguy M, Tang CM. Science. 2005;307:1313–1317. doi: 10.1126/science.1108472. [DOI] [PubMed] [Google Scholar]
- 61.Edwards RA, Keller LH, Schifferli DM. Gene (Amst) 1998;207:149–157. doi: 10.1016/s0378-1119(97)00619-7. [DOI] [PubMed] [Google Scholar]
- 62.Schweizer HD. BioTechniques. 1993;15:831–834. [PubMed] [Google Scholar]